

Abstract
Uridine-rich small nuclear ribonucleoproteins (U snRNPs) are components of the splicing machinery that removes introns from precursor mRNA. Like other splicing factors, U snRNPs are diffusely distributed throughout the nucleus and, in addition, are concentrated in distinct nuclear substructures referred to as speckles. We have examined the intranuclear distribution and mobility of the splicing factor U1 snRNP on a single-molecule level. Isolated U1 snRNPs were fluorescently labeled and incubated with digitonin-permeabilized 3T3 cells in the presence of Xenopus egg extract. By confocal microscopy, U1 snRNPs were found to be imported into nuclei, yielding a speckled intranuclear distribution. Employing a laser video-microscope optimized for high sensitivity and high speed, single U1 snRNPs were visualized and tracked at a spatial precision of 35 nm and a time resolution of 30 ms. The single-particle data revealed that U1 snRNPs occurred in small clusters that colocalized with speckles. In the clusters, U1 snRNPs resided for a mean decay time of 84 ms before leaving the optical slice in the direction of the optical axis, which corresponded to a mean effective diffusion coefficient of 1 μm2/s. An analysis of the trajectories of single U1 snRNPs revealed that at least three kinetic classes of low, medium, and high mobility were present. Moreover, the mean square displacements of these fractions were virtually independent of time, suggesting arrays of binding sites. The results substantiate the view that nuclear speckles are not rigid structures but highly dynamic domains characterized by a rapid turnover of U1 snRNPs and other splicing factors.
The splicing of precursor mRNA in the nucleus is catalyzed by supramolecular assemblies designated as spliceosomes, which comprise more than 70 different proteins and five uridine-rich small nuclear RNAs (snRNA; ref. 1). Most of these proteins and the snRNAs are organized in the Uridine-rich small nuclear ribonucleoproteins (U snRNPs), which are classified as U1, U2, U5, and U4/U6, according to the snRNAs they contain. The snRNAs U1, U2, U4, and U5 are synthesized in the nucleus with a 5′-terminal monomethyl-guanosine (m7G)-cap structure, transiently exported into the cytoplasm, where a common set of seven core proteins (Sm proteins) bind to the snRNAs Sm site and form a ribonucleoprotein complex called “Sm core” (2). Stable association of all Sm proteins is necessary for hypermethylation of the m7G cap to the 2,2,7-trimethyl-guanosine (m3G)-cap structure (3, 4). Also, several proteins associate specifically with the individual U snRNPs; in the case of U1, those proteins are 70K, U1-A, and U1-C (5). After cap modification and 3′end processing of the snRNAs (6), the mature snRNP particles are reimported into the nucleus by import receptors. The nuclear localization signal of U1 snRNPs is complex, with the m3G-cap structure representing one important signaling component (7, 8). A second component is located at the Sm core but has not been defined precisely yet (9). Recently, an adaptor called Snurportin-1, which interacts specifically with the m3G cap of U snRNPs and the receptor karyopherin β, has been identified (10).
The intranuclear distribution of splicing factors is a prominent example of the high degree of spatiotemporal organization of the nuclear contents (11–13). Immunostaining of cells with antibodies against splicing factors such as ASF/SF2 or U snRNPs produces images in which nuclei contain an abundance of tiny bright spots (speckles) dispersed on a more homogeneous and less bright background (11, 12). It is known that these speckles arise by the enrichment of splicing factors in diverse subnuclear structures such as interchromatin granule clusters and perichromatin fibrils; they are collectively designated as splicing factor compartments, whose function and generation are still unclear. It has been proposed (11, 14–18) that speckles are sites at which splicing factors are either reprocessed, stored to regulate the level of free factors, or assembled together with other components of the transcription and RNA-processing machinery into large complexes. It has been speculated that the generation of splicing factor compartments occurs by interaction with a putative karyoskeleton (16) or by self-assembly (19). The diffuse, nucleoplasmic staining probably is caused by splicing factors that are actively involved in the processing of nascent transcripts. Another important question is, therefore, how splicing factors can move fast enough through a condensed and crowded environment such as the nuclear interior to encounter effectively their sites of action. The problem is aggravated by the fact that splicing factors operate as parts of large supramolecular machines, the spliceosomes that have to be assembled at the right time and place (20). Therefore, the intranuclear dynamics of splicing factors and other important nuclear proteins recently have been studied intensively by a variety of techniques (15, 17, 21–23).
In early studies from this laboratory (24, 25), fluorescence microphotolysis (fluorescence recovery after photobleaching) was used to determine the apparent intranuclear diffusion coefficient of a homologous series of dextrans. The intranuclear diffusion coefficient was found to be reduced by a factor of 5–10, as compared with that in dilute aqueous solutions. Furthermore, the intranuclear diffusion coefficient depended only slightly on temperature within the range of 10–37°C, suggesting that the restriction of diffusion was caused mainly by an increase of viscosity. Similar conclusions were reached more recently by photobleaching studies and by fluorescence correlation spectroscopy (26, 27). On one hand, a diffusion coefficient nearly as large as that in aqueous solutions was observed for small oligodeoxynucleotides (21). On the other hand, green fluorescent protein (GFP) fusions of the nucleosome-binding protein HMG-17, fibrillarin, and ASF/SF2 showed diffusion coefficients that were ≈100 times smaller than those obtained for the respective molecules in aqueous solution (17). The GFP conjugate of ASF/SF2 showed this slow diffusion independently of whether it was associated with speckles or dispersed in the nucleoplasm (16). The significant restriction of mobility was interpreted to indicate frequent but transient interactions of the examined proteins with immobile intranuclear structures, whereas the speckled distribution of splicing factors was assumed to reflect spatial variations in the concentration of relatively immobile, yet unidentified binding sites. Altogether, it has been found that only a small fraction of the studied splicing factors is immobile, whereas the larger fraction is comparatively unrestricted and can “roam” the nucleus in search of sites of action (19).
Single-molecule detection combined with single-particle tracking represents a promising approach to the analysis of molecular transport in biological systems (28–30). Initially, in two-dimensional systems such as membranes and cell surfaces (reviewed in ref. 31), and more recently in three-dimensional systems (32) including cells (33, 34), single, fluorescently labeled protein molecules were identified and localized at a spatial precision below 50 nm and a time resolution in the millisecond range by employing fluorescence microscopes equipped with highly sensitive and high-speed charge-coupled device camera systems. Thus, it became possible to observe directly the pathways of single molecules in native cellular environments at the nanometer scale. In preceding studies, we have used single molecule detection and tracking to analyze the movement of GFP in solution (32) and of a recombinant β-galactosidase protein (34) inside the cell nucleus. Here, we have extended such studies to the intranuclear distribution and movement of the splicing factor U1 snRNP in semi-intact cells. We show that U1 snRNPs move with an effective diffusion constant of 1 μm2/s, a value that is ≈40 times lower than in aqueous solution. This result suggests a transient binding to immobile sites. For mobility outside splicing-factor compartments, a lower limit for the effective diffusion constant of D = 2 μm2/s was obtained. Our results show that single U1 snRNPs are associated predominantly with splicing factor compartments.
Materials and Methods
Reagents and Buffers.
Texas red-labeled 70-kDa dextran (TRD70) and fluorescein-labeled 150-kDa dextran (FD 150) were obtained from Molecular Probes. The extract of Xenopus laevis eggs was prepared according to ref. 35. Alexa 488- or Cy5-labeled U1 snRNPs were prepared as described (10). Purity, functionality, and integrity of labeled U1 snRNPs were confirmed as described (36).
Experimental Setup.
Single-molecule experiments were performed as recently described (32, 34). An inverted wide-field epi-fluorescence microscope was equipped with a 5W Ar+ laser and a 40 mW HeNe laser. After passing through λ/4 plates, the laser beams were combined by a dichromatic beam splitter and illuminated a rectangular diaphragm located in a conjugated image plane. The diaphragm was imaged into the object plane, yielding an illumination field of 8 × 6.5 μm2. The excitation intensity was adjusted to 2–5 kW/cm2 for each color. Dual-color fluorescence was separated from the excitation light by a double-dichromatic beam splitter (488/633 nm; PhotoMed GmbH, Seefeld, Germany). Green- and red-fluorescence images were separated by a dichromatic beam splitter (Dichroic BS XF 2016; PhotoMed GmbH) and respective emission filters (green, α-emitter XF 3084; red, custom α long-pass filter, edge 645 nm) and acquired by cooled slow-scan charge-coupled device cameras (Quantix, KAF 1400 Grade 1 CCD and Sensys, KAF 1401e Grade 2 CCD; Photometrics, Tucson, AZ). Trigger signals generated by the cameras during image acquisition were used to switch the lasers on and off by acousto-optical devices. Images were acquired with IPLAB 3.2.4 (Scanalytics, Billerica, MA) either in the slow-scan mode at 6 Hz or a high-speed-framing mode at up to 35 Hz (32, 37) with single-frame integration times of 20–50 ms.
U1 snRNP Nuclear Import.
3T3 cell samples were prepared as described (34). The transport solution contained Alexa 488- or Cy5-labeled U1 snRNPs (0.5 nM) and egg lysate. In dual-color fluorescence experiments, cells were preincubated with a high concentration (60 nM) of Alexa 488-labeled U1 snRNPs in the presence of egg lysate for 30 min. After washing, the transport solution containing Cy5-labeled U1 snRNPs (0.5 nM) and egg lysate was added. Nucleocytoplasmic transport of Alexa 488- or Cy5-labeled U1 snRNP and cell integrity were routinely checked by confocal laser-scanning microscopy (34).
Results and Discussion
Fluorescently Labeled U1 snRNPs Are Imported into the Nuclei of Permeabilized 3T3 Cells, Yielding a Speckled Intranuclear Distribution.
Isolated U1 snRNPs were labeled with the green-fluorescent dye Alexa488 or the red-fluorescent dye Cy5. Digitonin-permeabilized 3T3 cells were incubated with labeled U1 snRNP at a concentration of 60 nM (U1 snRNP-Alexa488) or 110 nM (U1 snRNP-Cy5) in the presence of an X. laevis oocyte extract. The import of labeled U1 snRNP into cell nuclei and the intranuclear distribution was followed by confocal microscopy. Fig. 1A illustrates that after 30 min of U1 snRNP-Alexa488 import at room temperature, the nuclei had acquired an appearance familiar from immunofluorescence studies in which a large number of speckles are found on a more homogeneous background. In Fig. 1B, contours are outlined that correspond to the half-maximum of intranuclear fluorescence intensity. A quantitative analysis (Fig. 1C) showed that the concentration of U1 snRNPs within speckles was 3- to 5-fold higher than in surrounding nucleoplasmic regions.
Figure 1.

Intranuclear accumulation of U1 snRNPs. Nuclear import of Alexa488- and Cy5-labeled U1 snRNPs was observed by confocal laser-scanning microscopy. A representative experiment using U1 snRNP-Alexa488 is shown. (A) After ≈30 min, U1 snRNPs accumulated in speckles. (B) Speckle borders are shown as a contour corresponding here to the half-maximum of intranuclear fluorescence intensity. (C) A quantitative analysis of the marked region in A shows that U1 snRNPs were concentrated up to 5-fold higher within speckles than in surrounding nucleoplasmic regions.
Single U1 snRNPs Can Be Visualized and Tracked in the Nucleus at High Spatial and Temporal Resolution.
The visualization of single intranuclear U1 snRNPs and their spatial relations to speckles are illustrated in Fig. 2. In this experiment, both Cy5-labeled and Alexa488-labeled U1 snRNPs were added to the extranuclear solution, however, at widely differing concentrations and at different times. A necessary condition for the wide-field microscopic detection of single molecules within an extended volume is that the molecules are present in exceedingly small concentrations (32). Therefore, for single-particle detection, we added the red-fluorescing U1 snRNP-Cy5 at a final concentration of 0.5 nM to the extracellular medium. At this concentration, a focal-volume element of 0.3 × 0.3 × 1 μm3 contains only 0.05 particles on the average. For the visualization of speckles, cells were preincubated as described for Fig. 1 with 60 nM U1 snRNP-Alexa488 for 30 min. After preincubation, the nuclei were brought into focus employing bright-field imaging. Cy5-labeled U1 snRNPs were added to the extranuclear solution, and single-particle imaging started 3 min later. Images were acquired at an integration time of 50 ms, with a lag time of 95 ms between the images.
Figure 2.

Time-lapse fluorescence images from single U1 snRNPs in the cell nucleus. (A) An image sequence showing single Cy5-labeled U1 snRNPs in a cell nucleus (single-frame integration time = 50 ms; lag time = 95 ms; bar = 2 μm). Several diffraction-limited spots corresponding to single U1 snRNPs are clearly discernible. The dashed line indicates the position of the nuclear envelope, which was detected in a bright-field image. Cells had been preincubated with a high concentration of Alexa488-labeled U1 snRNPs for visualization of the speckles. (B) Overlay of the green-speckled image and the positions of the U1 snRNPs determined from A. U1 snRNPs located close to the image borders and at the nuclear envelope were not evaluated.
Fig. 2A gives an example of the raw data obtained in the detection of single U1 snRNPs. In each image of the series, several discrete, diffraction-limited fluorescent spots can be recognized. Some of the spots are present already in the first image and can be followed over several images; other spots appear and disappear during the series. The identification of the spots as single U1 snRNPs was based on their number in the field of view and their intensity, as discussed (33, 34). From the raw data, the positions of the particles were determined by fitting their intensity distribution by a two-dimensional Gaussian function (34, 38). The average signal-to-noise ratio of U1 snRNP signals was 2–10, which corresponds (32) to a standard deviation of the position of σp = 10–60 nm (mean σp = 35 nm). In the following text, σp will be referred to as the “precision” of the position measurements.
In Fig. 2B, the positions of the particles in Fig. 2A are overlaid with an image of the speckles, which had been acquired before the time-lapse sequence. The appearance, sporadic movement, and disappearance of several U1 snRNPs can be followed clearly over time. Obviously, single U1 snRNPs and speckles frequently colocalize.
In a large number of further experiments, the intranuclear localization and movement of single U1 snRNPs were studied. Depending on the label on the U1 snRNPs, the red or the green channel of the video system was used. The frame-integration time was varied between 20 and 50 ms, and the lag time was varied between 7 and 95 ms. In the image series, each single U1 snRNP was localized, ignoring, however, such U1 snRNPs that apparently were attached to the nuclear envelope or situated too close to the image border.
Single U1 snRNPs Form Intranuclear Clusters that Colocalize with Speckles.
As is apparent in Fig. 2B, the distribution of U1 snRNPs in the nucleus was not a random one. This observation is demonstrated more clearly in Fig. 3A, in which are plotted the positions of all U1 snRNPs observed in one particular nucleus during a time of 5 min. A truly random distribution—obtained by a Monte Carlo simulation based on the same particle number and distribution space as Fig. 3A—is shown in Fig. 3B. By employing dual-color experiments as illustrated in Fig. 2B, it was verified that the discrete regions occupied by single U1 snRNPs were identical with speckles. Thus, long time exposures of the red channel in which single U1 snRNP-Cy5 were imaged with the corresponding images of speckles obtained in the green channel yielded overlapping intensity patterns (data not shown).
Figure 3.

U1 snRNPs were frequently observed in specific nuclear regions. (A) All positions that were occupied by single U1 snRNPs observed in a single nucleus over a time of 5 min are shown. U1 snRNPs appeared frequently at positions of increased concentration in the nucleus, which corresponded to speckles. (B) Randomly distributed spots in an ellipsoidal region with an identical object density as in A exhibit a considerably more uniform distribution.
Intranuclear U1 snRNP Clusters Are Dynamic Structures with a High Turnover Rate.
For the following analysis, it is essential to bring to mind that in our single-particle studies, only those molecules were monitored that were present in a thin slice centered at the focal plane. This condition occurred because the fluorescence signal of point objects rapidly decreases with increasing distance to the focal plane (39). In our experiments, the axial extension dz of the optical slice amounted to ≈1 μm; i.e., the imaged slice had a lateral extension 8 × 6.5 μm2 but a thickness of only 1 μm. Therefore, virtually all of the molecules that appeared in or disappeared from the images entered or left the optical slice in the direction of the z axis. We quantified the particle transport in the z direction by counting the number of U1 snRNPs present in the first image of a sequence and by determining how the number of these particles decreased with time. Such data were collected for a large number of image sequences and are plotted in normalized form in Fig. 4 (black circles). The data show that the particles rapidly disappeared from the observation slice with a decay time of 84 ± 5 ms. After 200 ms, only 10% remained of the initially present U1 snRNPs. For the interpretation of the effect, it was essential to elucidate the role of photobleaching effects. Therefore, we measured the intensity of those (few) particles that remained within the observed slice throughout the complete measuring time and found that their fluorescence signal decreased only by ≈11% (data not shown). We conclude that photobleaching was, indeed, negligible, and that the disappearance of particles was caused by their transport in the z direction.
Figure 4.

Movement of U1 snRNPs out of the observation slice in the nucleus. The total number of U1 snRNPs in the observation slice was plotted as a function of time (black circles, normalized data). The fit to a monoexponential decay function yielded a time constant of 84 ± 5 ms. Also shown is the theoretical reduction in particle number caused by diffusion out of the observation section by assuming three different effective diffusion constants, D1 = 0.2 μm2/s, D2 = 1 μm2/s, and D3 = 10 μm2/s, (dotted, solid, and dashed lines, respectively).
To test whether the rapid disappearance of U1 snRNPs from the observation slice can be explained simply by diffusion, we made model calculations in which the average movement of particles out of a slice of thickness dz caused by diffusion was computed according to ref. 40. The resulting theoretical decay curves for diffusion constants of D1 = 0.2 μm2/s, D2 = 1 μm2/s, and D3 = 10 μm2/s are plotted in Fig. 4 (dotted, solid, and dashed lines, respectively). It can be seen that the experimentally observed decrease of U1 snRNPs conformed closely to the curve computed on the basis of a diffusion constant of 1 μm2/s. U1 snRNPs have a ring-shaped main body with two protrusions at the top of the ring (41). This structure may be approximated roughly by a prolate ellipsoid of revolution with a semimajor axis a ≈7.5 nm and a semiminor axis b ≈4.5 nm. Such a structure has a diffusion coefficient of ≈40 μm2/s in aqueous solution (42). Furthermore, taking into account that the viscosity of the intranuclear aqueous phase may be 5–10 times larger than that of water (24, 25, 27), a diffusion coefficient of 4–8 μm2/s would be expected. The diffusional motion of the U1 snRNPs out of the clusters is apparently restricted, although the restricting forces cannot be very strong or long lasting.
The Quantitative Analysis of U1 snRNP Trajectories Suggests that U1 snRNPs Move Rapidly Within and Between Clusters of Binding Sites.
For a more sophisticated analysis, the trajectories of single U1 snRNP particles in the observation slice were taken into account. Thus, the trajectory for each observed U1 snRNP was computed as a set of coordinates {xi, yi}, where 1 ≤ i ≤ N, with N denoting the number of observations of an individual molecule. From each trajectory, a total of (N − 1)⋅N/2 square displacements, r2 (tlag), were obtained. A lag time was associated with each r2: tlag = n(till + tdelay). Here, till denotes the integration time, tdelay represents the delay time between two successive frame acquisitions, and n is the difference of the frame numbers. By averaging square displacements with identical lag times, the mean square displacements (MSD),
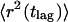 |
were obtained.
In the case of simple, two-dimensional Brownian motion, the diffusion coefficient D is related to the MSD by
 |
1 |
Thus, a linear relationship between MSD and lag time indicates Brownian motion and can be used to derive diffusion coefficients from single-molecule trajectories. However, if molecular transport is not based on free diffusion but, for instance, is based on confined diffusion or directed flow, the relation between MSD(tlag) and lag time is no longer linear (28). An analysis of molecular motion according to Eq. 1 also is not appropriate when the population contains different mobility fractions. Such heterogeneous populations can be analyzed much better and the mobility modes dissected to a certain degree by an analysis of jump-distance distributions (34, 43, 44). In this type of analysis, the probability p(r, t)dr that a particle starting at the origin will be encountered within a shell of radius r and a width dr at time t is considered. For a single species diffusing in two dimensions (Crank, 1975):
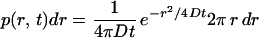 |
2 |
Experimentally, this probability distribution can be approximated by a frequency distribution obtained by counting the jump distances within respective intervals [r, r + dr] covered by single particles after a given lag time. An example of such a jump-distance frequency distribution comprising nearly 2,300 individual jumps is given in Fig. 5A (gray bars). The lag time was 27 ms.
Figure 5.

Analysis of U1 snRNP trajectories. (A) The frequency distribution of jumps for tlag = 27 ms comprises nearly 2,300 individual jumps. One transiently immobile fraction f1 (22%) and two mobile fractions f2 (50%) and f3 (28%) of U1 snRNPs were needed for an acceptable fit of the histogram (dotted, dashed, and dot-dash-dot line, respectively). The sum of the three fractions represent a good fit to the data (solid black line). (B) Normalized number of particles in fraction f1 plotted as a function of lag time. (C) Time dependence of the MSDi(tlag) for f2 (black squares) and f3 (black circles). After an initial increase from zero, both MSD2(tlag) and MSD3(tlag) remained constant. The slopes of the lines connecting the origin with the first measured points were used to derive lower limits for the diffusion constants.
The essential point in the jump-distance distribution analysis is that subpopulations can be detected and determined by curve fitting. Thus, the data shown in Fig. 5A could not be fitted satisfactorily by (2) with a single-diffusion coefficient. Rather, an expression containing three such terms had to be invoked for an acceptable fit (solid black line in Fig. 5A):
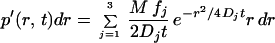 |
3 |
Here, M denotes the number of jumps considered in the analysis, and f1, f2, and f3 designate the fractions of subpopulations with diffusion constants D1, D2, and D3, respectively. The curves corresponding to f1, f2, and f3 are displayed in Fig. 5A as dotted, dashed, and dot-dash-dot lines, respectively.
Experimental jump-distance histograms for 14 different lag times were fitted by (3) to yield the magnitude of the fractions 1–3 and the corresponding mean square displacements by considering (1) for each fraction and lag time.
U1 snRNPs in fraction f1 (22%) did not jump beyond the average localization precision of σp = 35 nm (D1t = σ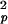 /4). The magnitude of f1 rapidly decayed with a time constant of 65 ms (Fig. 5B). Because this fraction was immobile on the time scale of the lag time, its decay indicates dissociation of U1 snRNPs from immobile, unidentified binding sites. Consequently, the decay time of f1 may be interpreted as a dissociation time.
/4). The magnitude of f1 rapidly decayed with a time constant of 65 ms (Fig. 5B). Because this fraction was immobile on the time scale of the lag time, its decay indicates dissociation of U1 snRNPs from immobile, unidentified binding sites. Consequently, the decay time of f1 may be interpreted as a dissociation time.
For particles in f2 (50%) and f3 (28%), the time dependence of the mean square displacement is shown in Fig. 5C. After an initial sharp increase, both MSD2(tlag) and MSD3(tlag) remained approximately constant. The initial slopes of the MSD curves from zero to the first time point were taken as lower limits for the corresponding diffusion constants. They indicated diffusion constants of D2 ≥ 1 μm2/s and D3 ≥ 0.1 μm2/s. After the initial increase, an MSD of 0.1 μm2 was not exceeded. This result suggested that the observed U1 snRNPs diffused in a restricted region (28). The square root of this value—0.3 μm—indicated the average maximum distance that could be jumped by the U1 snRNPs. Fig. 5A shows that the maximum jumps observed were over 0.7 μm.
We wish to stress here that the dissection of the jump-distance distribution into three fractions is arbitrary. Three fractions were necessary to obtain a good fit. However, it is clear that the fit could be improved further by assuming more than three fractions or even a continuous distribution.
The analysis above yielded data on the time dependence of two important parameters, the number N(t) of particles observed in a certain small volume and the mean square displacements MSD(t) of these particles. For simple Brownian motion, N(t) decreases nonlinearly, and MSD(t) increases linearly with time (34). In our experiments using U1 snRNPs, we observed a decrease in N(t) (Fig. 4), but not an increase of MSD(t) (Fig. 5). This fact suggests that the observed movements took place within a limited space. Furthermore, we noted transient immobilization of a fraction of U1 snRNPs with a mean duration of 65 ms. It appears that U1 snRNPs attached transiently to sites within regions of highly concentrated binding sites—nuclear speckles—and then either jumped to other binding sites (as often observed in the time-lapse images) or left the speckles when encountering no further sites. Dissociation from the speckles precluded further observations and, thus, prevented an increase of the MSD. Therefore, the sum of the jump-distance distributions of the mobile U1 snRNP fractions reflected the static spatial distribution of accessible binding sites within a speckle rather than a dynamical jump-distance distribution that expanded with time. The greatest jump distances were ≈0.7 μm. Therefore, this distance characterizes the maximum spatial extension of speckles. This number agrees well with electron microscopic investigations of speckles (45) and also with the extensions of the observed U1 snRNP clusters (Fig. 3A).
We suspect that the mobility of U1 snRNPs outside the speckles was so high that noticeable particle signals could not be obtained during the image integration time of 20 ms. This possibility sets a lower limit for the U1 snRNP diffusion coefficient of 2 μm2/s for these nuclear regions (34). In fact, the diffusion coefficient of free U1 snRNPs in the nonspeckled regions of the nucleus may be up to 4–8 μm2/s, as mentioned above.
Altogether, our analysis substantiates the view that splicing factors are highly mobile in the nucleus, and that splicing factor compartments are very dynamic structures (16, 18, 19, 46, 47), concepts based so far solely on mobility measurements by photobleaching techniques. In comparison, single-molecule techniques yield a higher spatial resolution and facilitate the discrimination between different modes of mobility. We observed single U1 snRNPs in a semi-intact cell system. Although we did not perform a quantitative comparison, it can be assumed that splicing is reduced in this system, as compared with living cells. It seems, however, as if the basic structural and mobility features of splicing factors within the nucleus are preserved. Experiments using microinjection of minute amounts of fluorescently labeled U snRNPs into the cytoplasm, which are imaged after their import into the nucleus, should help to understand better the functions of splicing-factor domains and other subnuclear structures in living cells.
Acknowledgments
We gratefully acknowledge Deutsche Forschungsgemeinschaft Grants Ku 975/3–2 (to U.K.) and SFB523/AB (to R.L.), a Volkswagen-Stiftung grant (to U.K. and R.P.), and a postdoctoral fellowship of the Deutsches Krebsforschungszentrum (to A.D.).
Abbreviations
- snRNA
small nuclear RNA
- U snRNP
uridine-rich small nuclear ribonucleoprotein
- MSD
mean square displacements
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Burge C B, Tuschl T, Sharp P A. In: The RNA World. Gesteland R F, Czech T R, Aktins J F, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1999. pp. 525–560. [Google Scholar]
- 2.Mattaj I W, De Robertis E M. Cell. 1985;40:111–118. doi: 10.1016/0092-8674(85)90314-9. [DOI] [PubMed] [Google Scholar]
- 3.Mattaj I W. Cell. 1986;46:905–911. doi: 10.1016/0092-8674(86)90072-3. [DOI] [PubMed] [Google Scholar]
- 4.Plessel G, Fischer U, Lührmann R. Mol Cell Biol. 1994;14:4160–4172. doi: 10.1128/mcb.14.6.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Will C L, Lührmann R. Curr Opin Cell Biol. 2001;13:290–301. doi: 10.1016/s0955-0674(00)00211-8. [DOI] [PubMed] [Google Scholar]
- 6.Neuman de Vegvar H E, Dahlberg J E. Mol Cell Biol. 1990;10:3365–3375. doi: 10.1128/mcb.10.7.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamm J, Mattaj I W. Cell. 1990;63:109–118. doi: 10.1016/0092-8674(90)90292-m. [DOI] [PubMed] [Google Scholar]
- 8.Fischer U, Lührmann R. Science. 1990;249:786–790. doi: 10.1126/science.2143847. [DOI] [PubMed] [Google Scholar]
- 9.Fischer U, Sumpter V, Sekine M, Satoh T, Lührmann R. EMBO J. 1993;12:573–583. doi: 10.1002/j.1460-2075.1993.tb05689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huber J, Cronshagen U, Kadokura M, Marshallsay C, Wada T, Sekine M, Lührmann R. EMBO J. 1998;17:4114–4126. doi: 10.1093/emboj/17.14.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sleeman J E, Lamond A I. Curr Opin Cell Biol. 1999;11:372–377. doi: 10.1016/S0955-0674(99)80052-0. [DOI] [PubMed] [Google Scholar]
- 12.Misteli T. J Cell Sci. 2000;113:1841–1849. doi: 10.1242/jcs.113.11.1841. [DOI] [PubMed] [Google Scholar]
- 13.Lewis J D, Tollervey D. Science. 2000;288:1385–1389. doi: 10.1126/science.288.5470.1385. [DOI] [PubMed] [Google Scholar]
- 14.Misteli T, Spector D L. Curr Opin Cell Biol. 1998;10:323–331. doi: 10.1016/s0955-0674(98)80007-0. [DOI] [PubMed] [Google Scholar]
- 15.Misteli T, Caceres J F, Spector D L. Nature (London) 1997;387:523–527. doi: 10.1038/387523a0. [DOI] [PubMed] [Google Scholar]
- 16.Kruhlak M J, Lever M A, Fischle W, Verdin E, Bazett-Jones D P, Hendzel M J. J Cell Biol. 2000;150:41–51. doi: 10.1083/jcb.150.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phair R D, Misteli T. Nature (London) 2000;404:604–609. doi: 10.1038/35007077. [DOI] [PubMed] [Google Scholar]
- 18.Melcak I I, Melcakova S, Kopsky V V, Vecerova J, Raska I I. Mol Biol Cell. 2001;12:393–406. doi: 10.1091/mbc.12.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Misteli T. Science. 2001;291:843–847. doi: 10.1126/science.291.5505.843. [DOI] [PubMed] [Google Scholar]
- 20.Staley J P, Guthrie C. Cell. 1998;92:315–326. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- 21.Politz J C, Browne E S, Wolf D E, Pederson T. Proc Natl Acad Sci USA. 1998;95:6043–6048. doi: 10.1073/pnas.95.11.6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sleeman J E, Lamond A I. Curr Biol. 1999;9:1065–1074. doi: 10.1016/s0960-9822(99)80475-8. [DOI] [PubMed] [Google Scholar]
- 23.Eils R, Gerlich D, Tvarusko W, Spector D L, Misteli T. Mol Biol Cell. 2000;11:413–418. doi: 10.1091/mbc.11.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters R. EMBO J. 1984;3:1831–1836. doi: 10.1002/j.1460-2075.1984.tb02055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lang I, Scholz M, Peters R. J Cell Biol. 1986;102:1183–1190. doi: 10.1083/jcb.102.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seksek O, Biwersi J, Verkman A S. J Cell Biol. 1997;138:131–142. doi: 10.1083/jcb.138.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wachsmuth M, Waldeck W, Langowski J. J Mol Biol. 2000;298:677–689. doi: 10.1006/jmbi.2000.3692. [DOI] [PubMed] [Google Scholar]
- 28.Saxton M J, Jacobson K. Annu Rev Biophys Biomol Struct. 1997;26:373–399. doi: 10.1146/annurev.biophys.26.1.373. [DOI] [PubMed] [Google Scholar]
- 29.Nie S, Zare R N. Annu Rev Biophys Biomol Struct. 1997;26:567–596. doi: 10.1146/annurev.biophys.26.1.567. [DOI] [PubMed] [Google Scholar]
- 30.Weiss S. Science. 1999;283:1676–1683. doi: 10.1126/science.283.5408.1676. [DOI] [PubMed] [Google Scholar]
- 31.Schütz G J, Sonnleitner M, Hinterdorfer P, Schindler H. Mol Membr Biol. 2000;17:17–29. doi: 10.1080/096876800294452. [DOI] [PubMed] [Google Scholar]
- 32.Kubitscheck U, Kückmann O, Kues T, Peters R. Biophys J. 2000;78:2170–2179. doi: 10.1016/S0006-3495(00)76764-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goulian M, Simon S M. Biophys J. 2000;79:2188–2198. doi: 10.1016/S0006-3495(00)76467-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kues T, Peters R, Kubitscheck U. Biophys J. 2001;80:2954–2967. doi: 10.1016/S0006-3495(01)76261-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newmeyer D D, Wilson K L. Methods Cell Biol. 1991;36:607–634. doi: 10.1016/s0091-679x(08)60299-x. [DOI] [PubMed] [Google Scholar]
- 36.Marshallsay C, Lührmann R. EMBO J. 1994;13:222–231. doi: 10.1002/j.1460-2075.1994.tb06252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmidt T, Schütz G J, Baumgartner W, Gruber H J, Schindler H. J Chem Phys. 1995;99:17662–17668. [Google Scholar]
- 38.Kubitscheck U, Kues T, Peters R. Methods Enzymol. 1999;307:207–230. doi: 10.1016/s0076-6879(99)07015-9. [DOI] [PubMed] [Google Scholar]
- 39.Born M, Wolf E. Principles of Optics. Cambridge, U.K.: Cambridge Univ. Press; 1980. [Google Scholar]
- 40.Crank J. The Mathematics of Diffusion. Oxford: Clarendon; 1975. [Google Scholar]
- 41.Stark H, Dube P, Lührmann R, Kastner B. Nature (London) 2001;409:539–542. doi: 10.1038/35054102. [DOI] [PubMed] [Google Scholar]
- 42.Berg H C. Random Walks in Biology. Princeton: Princeton Univ. Press; 1993. [Google Scholar]
- 43.Schütz G J, Schindler H, Schmidt T. Biophys J. 1997;73:1073–1080. doi: 10.1016/S0006-3495(97)78139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith P R, Morrison I E, Wilson K M, Fernandez N, Cherry R J. Biophys J. 1999;76:3331–3344. doi: 10.1016/S0006-3495(99)77486-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spector D L. Exp Cell Res. 1996;229:189–197. doi: 10.1006/excr.1996.0358. [DOI] [PubMed] [Google Scholar]
- 46.Shopland L S, Lawrence J B. J Cell Biol. 2000;150:F1–F4. doi: 10.1083/jcb.150.1.f1. [DOI] [PubMed] [Google Scholar]
- 47.Pederson T. Cell. 2001;104:635–638. doi: 10.1016/s0092-8674(01)00258-6. [DOI] [PubMed] [Google Scholar]