

Abstract
Objectives
Type 2 diabetes mellitus (T2DM) and the atherometabolic syndrome exhibit a deadly dyslipoproteinemia that arises in part from impaired hepatic disposal of cholesterol- and triglyceride-rich remnant apoB-lipoproteins (C-TRLs). We previously identified syndecan-1 as a receptor for C-TRLs that directly mediates endocytosis via rafts, independent from coated pits. Caveolins and flotillins form rafts but facilitate distinct endocytotic pathways. We now investigated their participation in syndecan-1-mediated disposal of C-TRLs and their expression in T2DM liver.
Approach and Results
In cultured liver cells and non-diabetic murine livers, we found that syndecan-1 robustly co-immunoprecipitates with flotillin-1 but not with caveolin-1. Binding of C-TRLs to syndecan-1 on liver cells enhanced syndecan-1/flotillin-1 association, and the two molecules then trafficked together into the lysosomes, implying limited if any recycling back to the cell surface. The interaction requires the transmembrane/cytoplasmic region of syndecan-1 and the N-terminal hydrophobic domain of flotillin-1. Knock-down of flotillin-1 in cultured liver cells substantially inhibited syndecan-1 endocytosis. Livers from obese, T2DM KKAy mice exhibited 60-70% less flotillin-1 mRNA and protein than in non-diabetic KK livers. An adenoviral construct to enhance hepatic expression of wild-type flotillin-1 in T2DM mice normalized plasma triglycerides, whereas a mutant flotillin-1 missing its N-terminal hydrophobic domain had no effect. Moreover, the adenoviral vector for wild-type flotillin-1 lowered plasma TG excursions and normalized retinyl excursions in T2DM KKAy mice after corn-oil gavage, without affecting postprandial production of C-TRLs.
Conclusions
Flotillin-1 is a novel participant in the disposal of harmful C-TRLs via syndecan-1. Low expression of flotillin-1 in T2DM liver may contribute to metabolic dyslipoproteinemia.
Keywords: type 2 diabetes mellitus, atherogenic remnant lipoproteins, hypertriglyceridemia, flotillin-1, syndecan-1
Introduction
Currently, the most successful therapies against atherosclerotic cardiovascular disease (ASCVD) act by lowering plasma concentrations of LDL. Statins, ezetimibe, and PCSK9 inhibitors stimulate hepatic expression of LDL receptors that then safely dispose of plasma LDL, reducing ASCVD risk.1–6 But with the rise of overnutrition, obesity, the atherometabolic syndrome, and type 2 diabetes mellitus (T2DM), a second apolipoprotein-B (apoB)-containing lipoprotein has emerged as a major driver of human ASCVD worldwide – namely, cholesterol- and triglyceride-rich remnant apoB-lipoproteins (C-TRLs).7 The characteristic dyslipoproteinemia of the atherometabolic syndrome and T2DM increases plasma levels of C-TRL remnants, not LDL. The major defect is impaired hepatic removal of C-TRLs from plasma.8 Like LDL,9 C-TRL remnants become retained within the arterial wall,5, 10–12 thereby causing ASCVD events in humans.13 Unfortunately, subjects treated with optimal statin therapy14 and even PCSK9 inhibitors3, 4 exhibit considerable residual risk for ASCVD events, which may occur, in large part, because these agents lower fasting or postprandial C-TRL levels by only 0% to 28%.15–17 There is a need to better understand how normal, rapid, safe hepatic removal of C-TRLs from plasma becomes impaired in overnutrition, obesity, and their sequelae.
We previously identified syndecan-1 heparan sulfate proteoglycan (HSPG) as a participant in C-TRL remnant catabolism, based on its abundance along the sinusoidal surface of hepatic parenchymal cells and our finding that it directly mediates endocytosis of model remnant lipoproteins.18–20 These results were extended by the demonstration that syndecan-1 knockout mice exhibit substantially impaired postprandial remnant lipoprotein clearance.21 Thus, the syndecan-1 HSPG is a major hepatic receptor for remnant lipoproteins,18, 19, 21 Moreover, we extensively characterized the endocytic pathway of syndecan-1 that is triggered when it binds large, multivalent lipoproteins, such as C-TRLs.18, 19, 22 A crucial early step is clustering of syndecan-1, which triggers its movement into cholesterol-rich, detergent-insoluble membrane microdomains (rafts), independent of coated pits.19, 22
Two families of proteins – caveolins and flotillins– have been implicated in the formation of specific subsets of rafts.23, 24 Moreover, caveolins and flotillins have both been implicated in membrane internalization and endocytosis.24–26 Caveolar-mediated endocytosis has been linked to the Src kinase,27 and flotillin-mediated endocytosis was reported to involve Fyn.28 Both Src and Fyn are members of the Src family of kinases, a class of enzymes that we identified to participate in syndecan-1-mediated endocytosis.22 Flotillins were implicated in general proteoglycan internalization, without identifying which species of proteoglycan.29 Importantly, caveolins and flotillins segregate into distinct regions of the plasma membrane, with distinct ultrastructural morphologies and different pathways for endocytosis.24
In the current study, we sought to determine if either caveolins or flotillins participate in syndecan-1-mediated endocytosis of C-TRLs. Surprisingly, we found that syndecan-1-mediated endocytosis of multivalent lipoproteins depends specifically on flotillin-1 (FLOT1) and not on caveolin-1 (CAV1). Importantly, flotillin-1 expression is suppressed in the livers of hyperphagic, obese, T2DM KKAy mice, and their diabetic dyslipoproteinemia can be substantially improved by an adenoviral construct to restore hepatic FLOT1 expression to normal levels.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Flotillin-1, but not caveolin-1, associates with syndecan-1 in a dynamic fashion that is sensitive to ligand binding
We began with co-immunoprecipitations. To move syndecan-1 into rafts, we added a model multivalent ligand, LpL-enriched methylated LDL (mLDL+LpL), to cultured McArdle hepatocytes, which express endogenous syndecan-1. The ligand was added 0-5h before harvesting the cells. The upper two immunoblots in Figure 1A show modest, but easily detectable, baseline association of flotillin-1 with syndecan-1 (immunoblot of flotillin-1 at 0h), consistent with the presence of a minority of unclustered syndecan-1 in rafts.19 Incubation of cells with mLDL+LpL caused the association of flotillin-1 with syndecan-1 to became stronger, peaking at 2h after addition of the multivalent ligand. Incubations longer than 2h caused the signals for both flotillin-1 and syndecan-1 to decrease in the immunoprecipitates. This timing is consistent with our previous demonstrations that it takes over an hour for the movement of syndecan-1 ligands into lysosomes and then lysosomal-mediated destruction of these ligands.18 Nevertheless, we had not previously examined if syndecan-1 recycles back to the cell surface or if it becomes destroyed in the lysosomes with its ligands. The lower three immunoblots in Figure 1A show time-dependent loss of flotillin-1 and syndecan-1, but not GAPDH, from whole-cell homogenates from the same experiment. Moreover, natural multivalent ligands for syndecan-1 – namely, human VLDL (Figure 1B) and murine VLDL (Supplemental Figure I), also provoked concurrent loss of these two molecules from McArdle hepatocytes starting after approximately 2h of incubation.
Figure 1. Flotillin-1, but not caveolin-1, associates with syndecan-1 in a dynamic fashion that is sensitive to ligand binding.
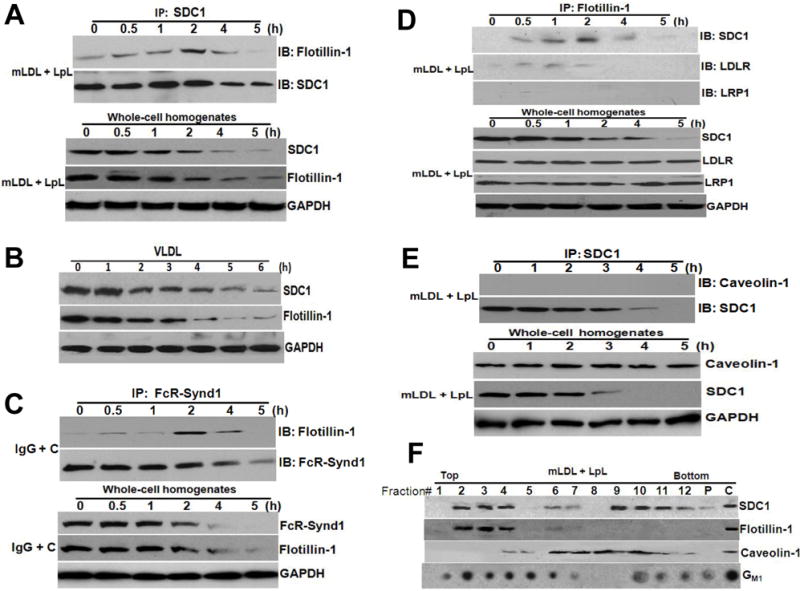
Panel A: Association of flotillin-1 with syndecan-1 in the basal state and after syndecan-1 binds a multivalent ligand. LpL-enriched mLDL was added to untransfected McArdle hepatocytes at the indicated times before harvest. All cells were plated simultaneously and then harvested simultaneously. A portion of the cellular extracts was used for immunopreciptation of syndecan-1 (IP: SDC1) as described in Methods, and then the IP pellets were subjected to electrophoretic separation. The upper two immunoblots were performed to detect flotillin-1 (IB: Flotillin-1), and then to verify immunoprecipitation and evaluate the loaded material, the same membranes were stripped and reprobed to detect syndecan-1 (IB: SDC1). The lower three immunoblots show the contents of flotillin-1, syndecan-1, and GAPDH in whole-cell homogenates from the same experiment, i.e., the starting material for each of the co-immunoprecipitations. Panel B: Human VLDL, a natural multivalent ligand for syndecan-1, drives concurrent loss of both syndecan-1 and flotillin-1 in cultured hepatocytes. Human VLDL was applied at 25 μg protein/ml to untransfected cultured McArdle cells at the indicated times before harvest. All cells were plated simultaneously and harvested simultaneously. Shown are immunoblots for syndecan-1, flotillin-1, and GAPDH in whole-cell extracts. Panel C: Association of flotillin-1 with the FcR-Synd1 chimera in the basal state and after clustering of FcR-Synd1. McArdle hepatocytes stably expressing the FcR-Synd1 chimera were treated with ligand plus our clustering agent (IgG + C), as described in the Methods, and then incubated for the indicated times. A portion of the cellular extracts was used for immunopreciptation of FcR-Synd1 (IP: FcR-Synd1). The upper two immunoblots were performed to detect flotillin-1, and then the same membranes were stripped and reprobed to detect FcR-Synd1. The lower three immunoblots show the contents of flotillin-1, FcR-Synd1, and GAPDH in whole-cell homogenates from the same experiment. Panel D: Robust association of flotillin-1 with syndecan-1 but not with other receptors, as assessed by immunoprecipitation of flotillin-1. Untransfected McArdle hepatocytes were treated as in Panel A, i.e., incubated with LpL-enriched mLDL for the indicated times and then used to prepare cellular homogenates. Here, flotillin-1 rather than syndecan-1 was immunoprecipitated (IP: Flotillin-1), followed by immunoblotting (IB) to detect syndecan-1, the LDL receptor (LDLR), and the LDLR-related protein-1 (LRP1). The lower four immunoblots show the contents of syndecan-1, LDLR, LRP1, and GAPDH in whole-cell homogenates from the same experiment. Panel E: No detectable association of caveolin-1 with syndecan-1 and no loss of caveolin-1 during syndecan-1-mediated endocytosis. Untransfected McArdle hepatocytes were treated as in Panels A and D, i.e., incubated with LpL-enriched mLDL for the indicated times and then used to prepare cellular homogenates. Syndecan-1 was immunoprecipitated (IP: SDC1), followed by immunoblotting to detect caveolin-1 (IB: Caveolin-1), and then the same membranes were stripped and reprobed to detect syndecan-1 (IB: SDC1). The lower three immunoblots show the contents of caveolin-1, syndecan-1 and GAPDH in whole-cell homogenates from the same experiment. Panel F: Co-fractionation of syndecan-1 and flotillin-1 in detergent-insoluble, buoyant membrane domains (rafts). Untransfected McArdle hepatocytes were treated with LpL-enriched mLDL for 1h to cluster syndecan-1, and then lyzed in ice-cold 1% Triton X-100 buffer to disrupt non-raft membranes. Residual detergent-insoluble cellular material was homogenized and then fractionated by ultracentrifugation across a sucrose gradient, as described in the Methods. Shown are immunoblots (proteins) and dot-blots (GM1) of sequential fractions from the top (buoyant lipid rafts) to the bottom of the gradient. The “P “indicates the cell pellet at the bottom of the ultracentrifugation tube, and the “C” indicates a whole-cell homogenate used as a positive control for the immunoblots and dot-blots. Ganglioside GM1 is a marker for lipid rafts. Data in each panel of this figure is representative of three independent experiments.
We next studied our FcR-Synd1 chimera, which we had made by linking the ectodomain of the IgG Fc receptor Ia to the transmembrane and cytoplasmic domains of human syndecan-1.18,19, 22, 30 Figure 1C shows essentially the same pattern for the FcR-Synd1 chimera that we saw with syndecan-1 in Figure 1A – namely, association with flotillin-1 in a dynamic fashion that is sensitive to ligand binding and clustering. These results implicate the transmembrane and cytoplasmic domains of syndecan-1 in the association with flotillin-1 after clustering and movement into rafts. Likewise, after 2h, the signals for the chimera and flotillin-1 fade from immunoprecipitates of FcR-Synd1 and from whole-cell homogenates (Figure 1C).
To assess the specificity of the association of flotillin-1 with syndecan-1, we immunoprecipitated flotillin-1 and then immunoblotted for syndecan-1, the LDL receptor (LDLR), and the LDLR-related protein-1 (LRP1). Figure 1D confirms the association of flotillin-1 with syndecan-1 in a dynamic fashion that is sensitive to multivalent ligand binding. Figure 1D indicates no association of LRP1 with flotillin-1 and a weak association of LDLR with flotillin-1. These results are consistent with our prior finding of an unexpected consensus motif shared by the transmembrane domains of syndecan-1 and the LDL receptor, but absent from the transmembrane domains of LRP1 and the other members of the LDL receptor gene family.31
In contrast to Figures 1A and 1D, Figure 1E shows that caveolin-1 does not co-immunoprecipitate with syndecan-1, even after addition of a multivalent ligand (upper immunoblots). Both caveolin-1 and syndecan-1 are easily detected in whole-cell homogenates, and, importantly, the signal for cellular caveolin-1 is unaffected by the binding of a multivalent ligand to syndecan-1 and the subsequent loss of cellular syndecan-1 (Figure 1E, lower immunoblots).
As an independent method to assess the relationships between syndecan-1, flotillin-1, and caveolin-1, we incubated McArdle hepatocytes with mLDL+LpL for 1h to cluster syndecan-1, lysed the cells in cold 1% Triton X-100 to disrupt non-raft membranes, and then fractionated the remaining detergent-insoluble membranes by ultracentrifugation across a sucrose gradient, as described in the Methods. Figure 1F shows that this technique almost entirely separates FLOT1-containing rafts (fractions 2-4) from CAV1-containing rafts (predominantly fractions 6-10). Syndecan-1 appears in two regions of the gradient: fractions 2-4, i.e., co-purifiying with flotillin-1, and fractions 9-12, i.e., only partially overlapping with the major caveolin-1-containing fractions.
Flotillin-1, but not caveolin-1, participates in endocytosis and lysosomal targeting of syndecan-1 with its multivalent ligands
Next, using siRNAs to suppress the expression of flotillin-1 or caveolin-1, we examined the roles of these molecules in syndecan-1-mediated endocytosis. Figure 2A shows that knock-down of flotillin-1 had no effect on cell-surface binding of VLDL, a physiologic, multivalent ligand for syndecan-1, but impaired the internalization, degradation, and hence total cellular catabolism of this ligand. Figure 2B shows the same effects for LpL-enriched 125I-labeled mLDL, a multivalent model remnant lipoprotein. Supplemental Figure II shows the same data as in Figure 2A,B, but plotted in absolute units (ng of lipoprotein protein catabolized per mg cell protein). Similar to Figure 1 and Supplemental Figure I, Figure 2C shows a decrease in the signal for syndecan-1 in control cells by 5h after the addition of a multivalent ligand for syndecan-1. This effect was abolished by knock-down of flotillin-1, consistent with impaired endocytosis and impaired movement into lysosomes for destruction.
Figure 2. Flotillin-1, but not caveolin-1, participates in endocytosis and lysosomal targeting of syndecan-1 with its multivalent ligands.
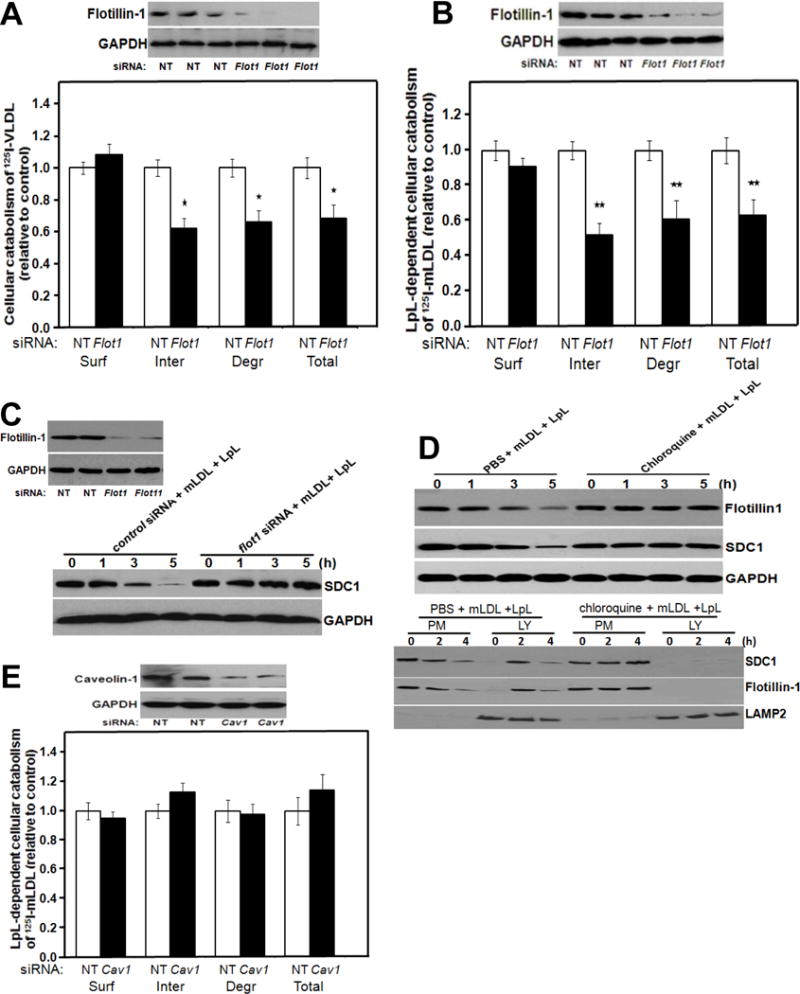
Panels A, B: Participation of flotillin-1 in syndecan-1-mediated catabolism of VLDL and model remnant lipoproteins. McArdle hepatocytes were incubated at 37°C for 4 hours with 100 nM nontarget (NT) control siRNA or Flot1 siRNA, as indicated. Cells were rinsed to remove unincorporated siRNA, and then incubated at 37°C in serum-containing medium for an additional 44 hours. Cells were switched to serum-free medium and incubated at 37°C for 4 hours with labeled lipoproteins. The upper images in Panels A, B show immunoblots of whole-cell homogenates, and the column graphs display values for cell-surface binding (Surf), internalization (Inter, calculated as the sum of intracellular accumulation plus degradation), degradation (Degr), and total cellular catabolism (Total, calculated as surface binding plus internalization) of the labeled lipoproteins, normalized to control values from cells treated with the nontarget siRNA (mean ± SEM, n=3). Panel A: Cellular catabolism of 125I-labelled human VLDL; nonnormalized control values were 117± 4.1, 421 ± 10.2, 219 ± 6.5, and total 539 ± 16.2 ng/mg, respectively. *p<0.05 between nontarget control and flotillin-1 knock-down (two-tailed Student’s t-test). Panel B: LpL-dependent cellular catabolism of LpL-enriched 125I-labelled mLDL; nonnormalized control values were 156 ± 4.2, 301 ± 8.6, 110 ± 3.1, and total 457 ± 10.6 ng/mg, respectively. **p<0.01 between nontarget control and flotillin-1 knock-down (two-tailed Student’s t-test). Panel C: Participation of flotillin-1 in the loss of cellular syndecan-1 during ligand catabolism. McArdle hepatocytes were treated with nontarget and Flot1 siRNAs as in Panels A, B and then LpL-enriched mLDL was added at the indicated times before harvest. Displayed are immunoblots of whole-cell homogenates to detect flotillin-1 at t=0h to verify the knock-down (left) and then syndecan-1 during the time course from 0 to 5h (SDC1; right). The protein GAPDH was used as a loading control. Panel D: Lysosomal targeting of flotillin-1 with syndecan-1 during catabolism of model remnant lipoproteins. McArdle hepatocytes were treated with PBS or chloroquine, as indicated, and then incubated at 37°C for the indicated times with unlabeled model remnant lipoproteins (LpL-enriched mLDL). The upper three immunoblots were performed to detect flotillin-1, syndecan-1 (SDC1), and, as a loading control, GAPDH in whole-cell homogenates. The lower three immunoblots show the contents of syndecan-1, flotillin-1, and LAMP2 (a lysosomal marker) in plasma membrane (PM) and lysosomal (LY) fractions that we isolated from these hepatocytes, as described in the Methods. Panel E: No detectable role for caveolin-1 in syndecan-1-mediated catabolism of model remnant lipoproteins. McArdle hepatocytes were treated exactly as in Panel B, except that the target siRNA was directed against caveolin-1 (Cav1) and, accordingly, the immunoblot verified caveolin-1 knock-down. Nonnormalized control values for surface binding, internalization, degradation, and total cellular catabolism of LpL-enriched 125I-labeled mLDL were 224 ± 6.2, 457 ± 10.6, 217 ± 4.1, and total 681 ± 15.2 ng/mg, respectively (mean ± SEM, n=3). p﹥0.5 between nontarget control and caveolin-1 knock-down for each of the four catabolic parameters (two-tailed Student’s t-test). All data in this figure are representatives of three independent experiments.
The upper immunoblots in Figure 2D show that chloroquine, which inhibits lysosomal acidification and endosomal-lysosomal fusion, completely blocks cellular loss of both flotillin-1 and syndecan-1 during catabolism of a multivalent ligand. The lower immunoblots in Figure 2D show that, in the absence of chloroquine, mLDL+LpL causes syndecan-1 and flotillin-1 to move from their predominant localization in the plasma membrane (PM) fraction at 0h, to substantial lysosomal (LY) localization at 2h. By 4h, total signals for both proteins have attenuated. The addition of chloroquine blocked ligand-induced movement of syndecan-1 and flotillin-1 into lysosomes, as well as ligand-induced loss of the two proteins.
Figure 2E indicates that caveolin-1 knock-down does not affect cell-surface binding, internalization, degradation, or total cellular catabolism of LpL-enriched 125I-labeled mLDL, in contrast to the effects of flotillin-1 knock-down in Figure 2B. Supplemental Figure III shows that individual knockdowns of flotillin-1 and caveolin-1 substantially and similarly disrupted overall raft structure, but only the knockdown of flotillin-1 excluded syndecan-1 from the most buoyant raft fractions. Two additional molecules – namely, Fyn, a member of the Src family of kinases, and the glycosylphosphatidylinositol-anchored form of CD55, another raft marker, also depend on FLOT1 to associate with raft fractions, with no apparent dependence on CAV1 (Supplementary Figure III).
Type 2 diabetes in KKAy mice suppresses hepatic levels of flotillin-1 in association with hypertriglyceridemia
In previous studies, we identified the sulfatase-2 (SULF2) as an endogenous inhibitor of the binding of C-TRLs to syndecan-1,32 and we implicated abnormal hepatic overexpression of SULF2 in postprandial dyslipoproteinemia in T2DM db/db mice.33 Moreover, we and others identified a human SULF2 gene polymorphism that associates with improved postprandial fat tolerance in T2DM patients34 and in healthy subjects.35 Surprisingly, we now found that hepatic expression levels of SULF2 protein in another widely used model of hyperphagia, T2DM, and metabolic dyslipoproteinemia – the KKAy mouse7, 36–38 – are not elevated compared to levels in non-diabetic KK littermate controls (Supplemental Figure IV). To explore the molecular basis for metabolic dyslipidemia in KKAy mice,36, 37 we examined the expression of flotillin-1 in their livers.
Figure 3A shows a striking deficiency of flotillin-1 protein in livers from T2DM KKAy mice compared to non-diabetic KK controls. Supplemental Figure V extends this finding to db/db mice, while also confirming their hepatic overexpression of SULF2. Figure 3B indicates a marked deficiency of Flot1 mRNA in KKAy livers. The T2DM KKAy mice have normal hepatic levels of syndecan-1 (Figure 3C) and no deficiency of the mRNA for the syndecan-1 core protein (Figure 3D). Similar to previous data in obese Zucker rats39 and T2DM db/db mice,32 hepatic Sdc1 mRNA levels in T2DM KKAy livers showed a trend to higher values than those in KK controls, although here the difference was not statistically significant. Importantly, co-immunoprecipitations reveal that far less flotillin-1 protein associates with syndecan-1 in the livers of KKAy mice than does in control KK livers (Figure 3E).
Figure 3. Type 2 diabetes in KKAy mice suppresses hepatic levels of flotillin-1 in association with hypertriglyceridemia.
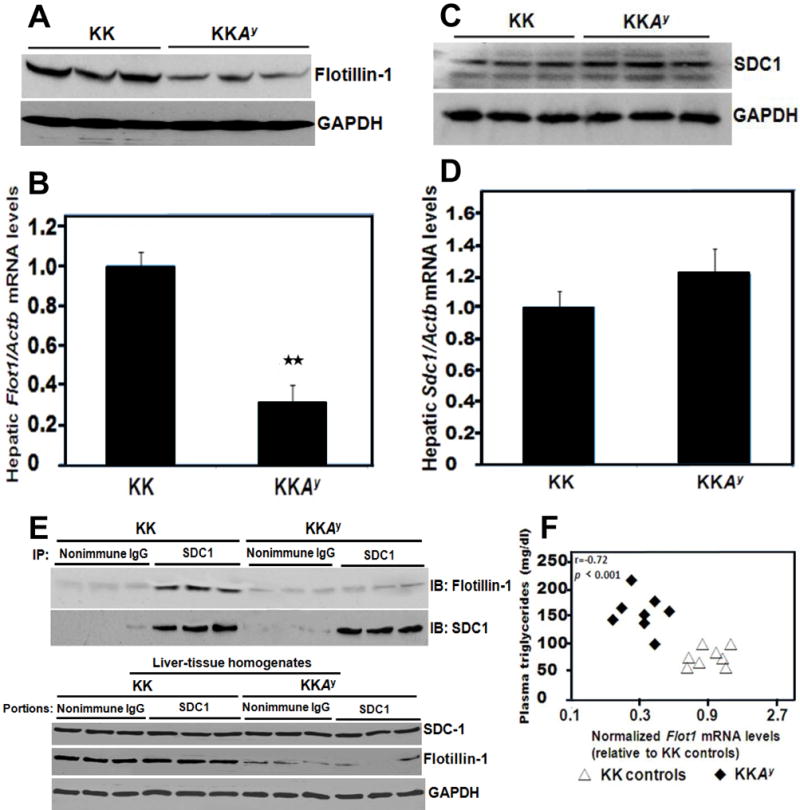
Protein and RNA were extracted from liver tissues of 12-week-old phenotypically lean KK mice (controls) and their hyperphagic, centrally obese, T2DM KKAy littermates. Panel A: Immunoblots in triplicate for hepatic flotillin-1 protein and, as a loading control, GAPDH. Each lane represents a sample from a different animal. Panel B: Hepatic flotillin-1 mRNA levels normalized to β-actin (Actb) mRNA levels, as assessed by quantitative reverse transcription polymerase chain reactions (qRT-PCR). Shown are means ± SEMs, n=6-8 per group, **p< 0.01 (two-tailed Student’s t-test). Panels C,D: The same liver samples used in Panels A,B were analyzed for their contents of the syndecan-1 HSPG by immunoblot (Panel C) and the syndecan-1 core protein mRNA by qRT-PCR (Panel D, p﹥0.5 by the two-tailed Student’s t-test). Panel E: Syndecan-1 was immunoprecipitated from mouse liver extracts (IP: SDC1, with nonimmune IgG as the negative control), and the IP pellets were subjected to electrophoretic separation. The upper two immunoblots were performed to detect flotillin-1 (IB: Flotillin-1) in the immunoprecipitate, and then the same membranes were stripped and reprobed to detect syndecan-1 (IB: SDC1). The lower three immunoblots show the contents of syndecan-1, flotillin-1, and GAPDH in liver tissue homogenates from the same experiment, i.e., the starting material for each of the co-immunoprecipitations, arranged in the same order as in the upper immunoblots. Panel F: Plot of plasma triglyceride concentrations versus hepatic Flot1 mRNA levels for all control KK mice and all T2DM KKAy mice. All data in this figure are representatives of three independent experiments.
To assess possible relationships between plasma lipoprotein concentrations and hepatic Flot1 mRNA levels, we examined statistical correlations in T2DM KKAy and control KK mice. A graph of fasting plasma triglyceride concentrations versus hepatic Flot1 mRNA levels showed complete segregation of T2DM KKAy mice, in the upper left-hand corner, from control KK mice, which are all in the lower right-hand sector (Figure 3F).
The first hydrophobic hairpin of flotillin-1 is required for association with syndecan-1 in cultured liver cells
To identify molecular determinants within flotillin-1 responsible for its interaction with syndecan-1, as well as the functional importance of this interaction, we sought to make two plasmid constructs, one encoding wild-type (WT, unmutated) mouse flotillin-1, and the other a site-directed mutant (Mut) of mouse flotillin-1 that would no longer associate with syndecan-1. Figure 4A shows schematics of two constructs in the pcDNA3(-) expression plasmid that we prepared to test in cultured cells. The upper schematic summarizes the structure of WT flotillin-1, which includes an N-terminal prohibitin homology (PHB) domain [also known as the stomatin, prohibitin, flotillin and HflK/C (SPFH) domain]. The PHB domain is thought to mediate membrane association via a single palmitoylation site at Cys34 and two putative hydrophobic hairpins that are thought to insert into the inner leaflet of the plasma membrane.40, 41 The C-terminus of flotillin-1 contains a conserved flotillin domain involved in oligomerization and raft formation.41, 42 The lower schematic in Figure 4A summarizes our site-directed flotillin-1 mutant, in which we converted the first hydrophobic hairpin, including the palmitoylation site, into a run of 27 consecutive alanines (the first residue of the unmutated hairpin is already an alanine). A truncated epitope-tagged mouse flotillin-1 construct missing the first 35 residues was previously shown to localize to the plasma membrane in non-raft domains.42 Each of our expression constructs, pcDNA3-Flot1(WT)-Flag and pcDNA3-Flot1(Mut)-Flag, was linked at the 3′ end of the coding region (C-terminus of the protein) to a sequence encoding the Flag epitope tag (DYKDDDDK).43 A control plasmid encoded only the Flag epitope tag (pcDNA3-Flag, not shown schematically).
Figure 4. The first hydrophobic hairpin of flotillin-1 is required for association with syndecan-1 in cultured liver cells.
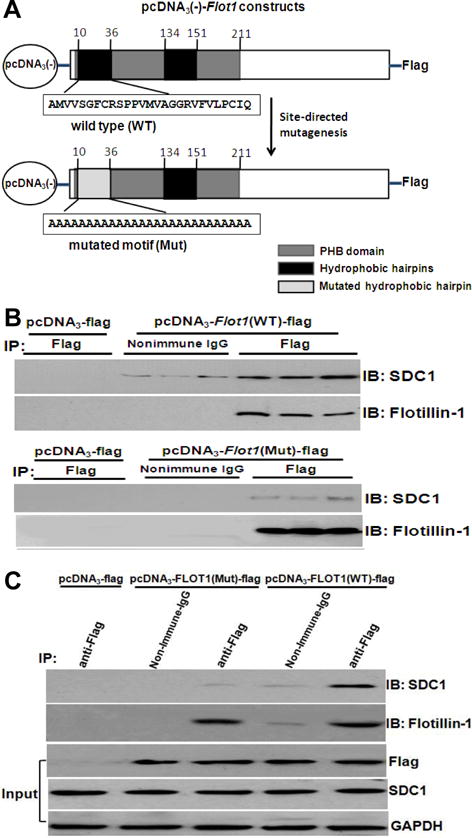
Panel A: Schematics of expression plasmids for Flag-tagged wild-type (WT, unmutated) flotillin-1 and a site-directed mutant (Mut) of flotillin-1. The upper schematic summarizes the structure of wild-type flotillin-1, which includes an N-terminal prohibitin homology (PHB) domain. The PHB domain contains a single palmitoylation site at Cys34 and two putative hydrophobic hairpins that are thought to insert into the inner leaflet of the plasma membrane. The lower schematic summarizes our site-directed flotillin-1 mutant (Mut), in which we converted the first hydrophobic hairpin, including the palmitoylation site, into a run of 27 consecutive alanines (the first residue of the unmutated hairpin is already an alanine). Each of these expression constructs was linked at the 3′ end of the coding region (C-terminus of the protein) to a sequencing encoding the Flag epitope tag (DYKDDDDK). These coding sequences were placed into the pcDNA3-minus expression vector to produce pcDNA3-Flot1(WT)-Flag and pcDNA3-Flot1(Mut)-Flag, respectively. A control plasmid encoded only the Flag epitope tag (pcDNA3-Flag, not shown schematically). Panels B,C: Flag-tagged wild-type flotillin-1 protein, but not the Flag-tagged flotillin-1 mutant, associates with clustered syndecan-1. McArdle cells were transiently transfected for 24h at 37°C with the indicated pcDNA3 expression constructs. To maximize the association of Flag-tagged WT and Mut flotillin-1 with endogenous syndecan-1, a multivalent ligand for syndecan-1 was added 2h before harvest (LpL-enriched mLDL). Cells were extracted into Nonidet P-40, immunoprecipitations were performed using anti-Flag (IP: Flag) or nonimmune IgG (IP: Nonimmune IgG as control), and then the IP pellets were subjected to electrophoretic separation. Shown are immunoblots that were performed to detect syndecan-1 (IB: SDC1), and then to verify specific immunoprecipitation and loading, the same membranes were stripped and reprobed to detect flotillin-1 (IB: Flotillin-1). Data in panel B and C are representatives of three independent experiments.
The upper panel of Figure 4B shows that transient transfection of the pcDNA3-Flot1(WT)-Flag construct in cultured McArdle hepatocytes results in expression of Flag-tagged flotillin-1 protein that can be immunoprecipitated with anti-Flag antibodies and, importantly, also associates with endogenous syndecan-1 after clustering. The lower panel of Figure 4B shows that the mutated flotillin-1-Flag construct, pcDNA3-Flot1(Mut)-Flag, drives abundant expression of immunoreactive flotillin-1 protein, but the mutated protein does not associate with clustered syndecan-1. Figure 4C shows all experimental conditions analyzed together on single immunoblots. The upper two immunoblots of Figure 4C allow the direct comparison of different associations of WT flotillin-1 versus mutated flotillin-1 with syndecan-1. Of the five experimental conditions shown, only immunoprecipitation of the Flag epitope in cells expressing the pcDNA3- Flot1(WT)-Flag construct brought down significant amounts of syndecan-1. The lower three immunoblots of Figure 4C show the contents of the Flag epitope, syndecan-1, and GAPDH in whole-cell homogenates from the same experiment, i.e., the starting material for the co-immunoprecipitations in the upper immunoblots of Figure 4C.
Restoration of wild-type flotillin-1 expression in livers of T2DM KKAy mice corrects their fasting hypertriglyceridemia
Figure 5A shows schematics of adenoviral constructs analogous to the plasmid constructs of Figure 4A, to drive expression of WT or mutated flotillin-1 in liver in vivo [AdV-flot1(WT), AdV-flot1(Mut); not shown schematically is our control adenovirus, AdV-gfp, that drives expression of green fluorescent protein]. Administration of 109 plaque-forming units (pfu) per injection of either of the two flotillin-1 adenoviral constructs to individual KKAy mice, followed by four weeks to allow expression, was able to normalize hepatic content of immunoreactive flotillin-1 protein (Figure 5B). Expression of WT flotillin-1, but not the mutant, caused a moderate decrease in hepatic syndecan-1 (Figure 5C), presumably reflecting enhanced trafficking to lysosomes. As in cultured hepatocytes, only WT flotillin-1 in the liver co-immunoprecipitated with syndecan-1; the mutated flotillin-1 did not (Figure 5D).
Figure 5. Restoration of wild-type flotillin-1 expression in livers of T2DM KKAy mice in vivo corrects their random, fasting hypertriglyceridemia.
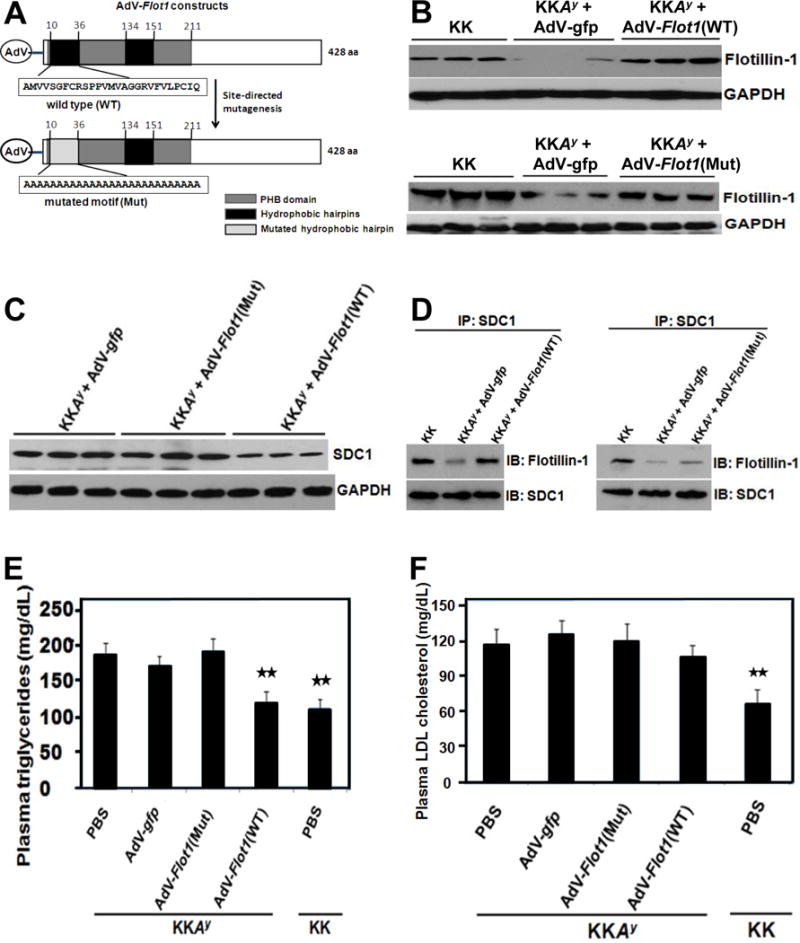
Panel A: Schematics of adenoviral (AdV) expression vectors for wild-type flotillin-1 [AdV-flot1(WT)] and a site-directed mutant of flotillin-1 in which the first hydrophobic hairpin has been replaced with a run of alanine residues [AdV-flot1(Mut)]. A control adenovirus encoded green fluorescent protein (AdV-gfp, not shown schematically). Other abbreviations follow Figure 5A. Panels B-F: Adenoviral particles (109 pfu per mouse) or phosphate-buffered saline (PBS) were administrated via caudal vein to 12-week-old phenotypically lean KK mice and their centrally obese, T2DM KKAy littermates, as indicated. Four weeks after injection, livers and blood plasma were collected. Immunoblots of liver extracts were performed in triplicate to assess contents of immunoreactive flotillin-1 protein (Panel B), syndecan-1 HSPG (SDC1, Panel C), and, as a loading control, GAPDH (Panels B,C). Each lane represents a sample from a different animal. In Panel D, syndecan-1 was immunoprecipitated from mouse liver extracts (IP: SDC1), and the IP pellets were subjected to electrophoretic separation. Shown are immunoblots to detect immunoreactive flotillin-1 (IB: Flotillin-1), and then the same membranes were stripped and reprobed to detect syndecan-1 (IB: SDC1). Panels E,F show triglyceride and LDL cholesterol concentrations, respectively, in fasting plasma samples four weeks after administration of adenoviruses or saline. Displayed are means ± SEMs, n=6 per group. p< 0.001 by ANOVA; **p<0.01 versus saline-treated KKAy (Dunnett q’ test). Data in panels B, C, D, E, F are representative of three independent experiments.
Next, we examined metabolic effects of the flotillin-1 adenoviral constructs in T2DM KKAy mice. Controls included PBS- or AdV-gfp-treated T2DM KKAy mice and PBS-treated non-diabetic KK mice. These treatments did not affect body weights, hyperglycemia, or HOMA-IR (Table 1). There was no significant difference in plasma insulin concentrations in KKAy mice treated with the two AdV-flot1 constructs (Table 1). Figure 5E shows the effects of these treatments on fasting plasma triglyceride concentrations. The KKAy mice that we treated with PBS, Adv-gfp, or AdV-flot1(Mut) showed statistically indistinguishable levels of hypertriglyceridemia. In contrast, the AdV-flot1(WT) construct lowered fasting plasma triglyceride levels in KKAy mice by 35%, to achieve values indistinguishable from those in nondiabetic KK mice. Similarly, the AdV-flot1(WT) construct, but not the AdV-flot1(Mut) adenovirus, corrected plasma non-esterified fatty acid concentrations in KKAy mice to normal levels (Table 1). The AdV-flot1(WT) construct did not significantly affect fasting plasma LDL-cholesterol in KKAy mice (Figure 5F).
Table 1.
Characteristics of KKAy mice after treatment with AdV-Flot1
| Characteristics | KKAy | KK | p value | |||
|---|---|---|---|---|---|---|
| PBS | AdV-gfp | AdV-Flot1(Mut) | AdV-Flot1(WT) | PBS | ||
| Body weights (g) | 42.0 ± 1.0a | 43.6 ± 1.0a | 40.2 ± 0.8a | 38.7 ± 1.2a | 32.8 ± 1.0b | <0.002 |
| HbA1c (%) | 7.2 ± 0.6a | 7.1 ± 0.5a | 6.8 ± 0.8a | 6.6 ± 0.2a | 5.1 ± 0.1b | <0.001 |
| Glucose (mmol/L) | 9.6 ± 0.2a | 9.2 ± 0.1a | 8.9 ± 0.5a | 8.7 ± 0.6a | 6.5 ± 0.7b | <0.01 |
| Insulin (pmol/L) | 168 ± 41a | 172 ± 23a | 208 ± 56b | 198 ± 31a,b | 64 ± 7.3c | <0.05 |
| HOMA-IR | 10.3 ± 1.7a | 10.1 ± 1.5a | 11.8 ± 2a | 11.0 ± 1.8a | 2.7 ± 0.4b | <0.001 |
| Non-esterified Fatty Acid (mEq/L) | 0.89 ± 0.07a | 0.83 ± 0.09a | 0.91 ± 0.1a | 0.68 ± 0.05b | 0.71 ± 0.1b | <0.01 |
All data are presented as the mean ±SEM (ANOVA), n=6-8 mice/group. For each characteristic, any two numbers labeled with the same lowercase letter (a, b, c) are statistically indistinguishable. If two numbers do not share a lowercase letter, they are statistically different (P <0.01 [Student-Newman-Keuls post hoc pairwise test]).
Restoration of wild-type flotillin-1 expression in livers of T2DM KKAy mice corrects their postprandial dyslipidemia
Similar to other hyperphagic animals33, 44 and obese type 2 diabetic humans,8 T2DM KKAy mice exhibited a substantial impairment in postprandial fat tolerance, indicated here by abnormally large, prolonged excursions in plasma concentrations of triglyceride (Figure 6A,B) and tritium (Figure 6C,D) after a single gavage of corn oil enriched with [3H] retinol. Restoration of normal levels of hepatic flotillin-1 protein in KKAy mice given that the AdV-flot1(Mut) construct failed to significantly improve the postprandial triglyceride or tritium excursions. The AdV-flot1(WT) construct markedly lowered both measures of postprandial fat tolerance, to values statistically indistinguishable from those in non-diabetic KK mice (Figure 6B,D). The AdV-flot1(WT) construct also brought fasting (0h) and persistent postprandial (4.5h) levels of apoB48 in plasma down to normal levels (Supplemental Figure VI).
Figure 6. Restoration of wild-type flotillin-1 expression in livers of T2DMKKAy mice in vivo corrects their postprandial dyslipidemia.
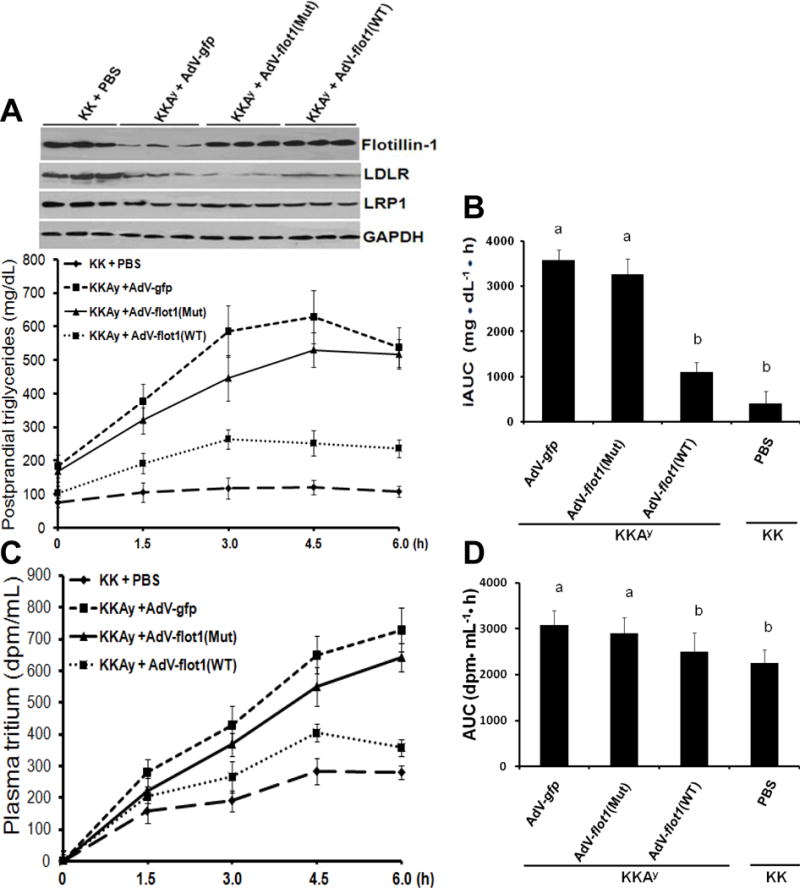
Adenoviral particles or saline (PBS) were administrated via caudal vein to lean KK mice and their centrally obese, T2DM KKAy littermates, as indicated. Four weeks after injection, mice were fasted for 5h, then given a gastric gavage of corn oil enriched with [3H] retinol (10μl of corn oil per gram of body weight). The upper images in Panel A show immunoblots of liver homogenates. The graph in Panel A displays postprandial excursions of plasma triglyceride concentrations. Panel B shows the incremental areas under these triglyceride excursion curves (iAUC). Panels C,D display postprandial excursions of plasma [3H] concentrations and the corresponding AUCs. Panels A (graph) and C display means ± SEMs, n=6 per group. In Panels B and D, p< 0.001 by ANOVA. Any two columns labeled with the same lowercase letter (a, b) are statistically indistinguishable. If two columns do not share a lowercase letter, they are statistically different (p <0.01, Student-Newman-Keuls test). All data in this figure are representatives of three independent experiments.
Based on our demonstration of a role for flotillin-1 in internalization and degradation of lipoproteins by cultured hepatocytes (Figure 2), the most likely explanation for improved postprandial fat tolerance in T2DM KKAy mice after treatment with our AdV-flot1(WT) construct (Figure 6) is enhanced hepatic lipoprotein clearance. Nevertheless, it is theoretically possible that restoration of WT FLOT1 expression in the liver might influence postprandial C-TRL secretion. To investigate this possibility, we performed another fat tolerance test, but in this experiment, some of the KKAy mice that had received Adv-flot1(WT) or Adv-flot1(Mut) were given an intravenous injection of Triton WR1339 to block lipoprotein clearance. Thus, the slope of their plasma triglyceride concentration curves from 0-4h after a gastric gavage of corn oil can be used to calculate the production rates of lipoprotein TG. The results in Supplemental Figure VII confirm the finding from Figure 6AB that Adv-flot1(WT), but not Adv-flot1(Mut), substantially corrects postprandial fat tolerance in T2DM KKAy mice that had not received Triton WR1339. Supplemental Figure VII also shows that this correction occurs without any difference in the postprandial production rate of lipoprotein TG.
Discussion
In the present study, we identified a novel factor, flotillin-1, that physically interacts with syndecan-1 and thereby facilitates normal syndecan-1-mediated endocytosis and then lysosomal disposal of C-TRLs. Binding of C-TRLs to syndecan-1 on liver cells enhances syndecan-1/flotillin-1 association, and the two molecules then traffic together into the lysosomes, implying limited if any recycling back to the cell surface. The interaction requires the transmembrane/cytoplasmic region of syndecan-1 and the N-terminal hydrophobic domain of flotillin-1.
Syndecan-1-mediated endocytosis occurs in cholesterol-rich membrane microdomains (rafts),19, 22 and flotillin-1 had been identified as a structural component of specific types of rafts.23–25 Moreover, flotillin-1 interacts with a number of elements that were previously showed to participate in syndecan-1-mediated endocytosis,22 such as Src family kinases,41 cortactin,45 and the actin cytoskeleton.41
Consistent with the model of “flotillin-assisted endocytosis”, in which flotillins facilitate but do not necessarily mediate endocytosis,46 our knock-downs of flotillin-1 slowed, but did not stop, internalization and lysosomal delivery of VLDL or model C-TRLs (Figures 2AB). Low amounts of residual flotillin-1 after knock-down may have also contributed. Significant roles for LDL receptor gene family members in these knock-down experiments seem unlikely, based on our previous demonstrations that LpL-dependent cellular internalization of lipoproteins is nearly entirely abolished by digesting cells with heparitinase, an enzyme that removes the heparan sulfate side-chains of syndecan-1 and other HSPGs but does not affect the LDLR or LRP1,47 as well as other distinctive features of ligand internalization via the syndecan-1 pathway versus coated-pit receptors, such as slower kinetics and an unusual pattern of responses to several inhibitors.18, 19, 22
Atherogenic dyslipoproteinemia in syndromes of chronic overnutrition has resisted molecular explanation for many years.12 Our previous systematic survey of proteoglycan-related gene transcripts on the GLYCOv3 microarray revealed a single hepatic mRNA, Sulf2, that was affected by obesity and T2DM in mice in a way that would impair syndecan-1 function as remnant lipoprotein receptors.32 We34 and others35 extended this work by implicating a role for SULF2 in plasma C-TRL accumulation in humans. Our current data add flotillin-1 to the short list of molecules that affect syndecan-1-mediated catabolism of C-TRLs in vivo and are dysregulated in T2DM liver to impair syndecan-1 function. Moreover, restoring hepatic flotillin-1 expression to normal levels in centrally obese, T2DM KKAy mice significantly corrected their fasting and postprandial dyslipoproteinemia (Figure 5E, Figure 6, and Supplemental Figure VII). Of note, our prior work correcting hepatic Sulf2 overexpression in hyperphagic T2DM db/db mice flattened their plasma TG excursions after corn-oil gavage,33 despite our current finding of a dual defect in these mice, i.e., hepatic SULF2 overexpression combined with FLOT1 underexpression (Supplemental Figure V). To resolve the roles of these two proteins, the possible involvement of flotillin-1 dysregulation in human metabolic dyslipoproteinemia will be a high priority to examine. Interestingly, despite correction of plasma NEFA levels after WT flotillin-1 expression in KKAy mice (Table 1), there was no improvement in HOMA-IR, which is consistent with a considerable and growing body of literature contradicting the hypothesis of a central role for NEFAs in causing insulin resistance for handling glucose.7, 48, 49
In addition, our detergent injection study revealed that in vivo restoration of wild type flotillin-1 did not affect VLDL secretion into plasma (Figure VII), while the in vivo rescue of flotillin-1 in diabetic animals improved the hepatic clearance of remnant lipoproteins, for both apoB48 and apoB100 (Figure VI).
The molecular basis for flotillin-1 suppression in the livers of centrally obese, T2DM KKAy mice will be an important topic for future study. Taken together, we have identified a new role for flotillin-1 in syndecan-1-mediated lipoprotein catabolism via raft-dependent endocytosis. Low expression of flotillin-1 in T2DM liver may contribute to metabolic dyslipoproteinemia.
Supplementary Material
Highlights.
Safe hepatic disposal of harmful cholesterol- and triglyceride-rich remnant lipoproteins (C-TRLs) normally occurs via syndecan-1, a receptor that directly mediates endocytosis via rafts, independent from coated pits.
Although both caveolins and flotillins form rafts, we found that syndecan-1-mediated endocytosis involves flotillin-1 but not caveolin-1.
Binding of C-TRLs to syndecan-1 on liver cells enhanced syndecan-1/flotillin-1 association, and the two molecules then trafficked together into the lysosomes, implying limited if any recycling back to the cell surface.
Livers from obese, T2DM KKAy mice exhibited 60-70% less flotillin-1 mRNA and protein than in non-diabetic KK livers, and an adenoviral construct to enhance hepatic expression of wild-type flotillin-1 in T2DM KKAy mice substantially improved their postprandial dyslipoproteinemia.
Flotillin-1 is a novel participant in the disposal of harmful C-TRLs via syndecan-1. Low expression of flotillin-1 in T2DM liver may contribute to metabolic dyslipoproteinemia.
Acknowledgments
None
Sources of funding
This work was supported by national natural science foundation of China (NSFC) grant 81570786; natural science fund of Anhui province of China 1508085MH162 and intramural fund of Anhui Medical University (to K. C); and National Institutes of Health-USA Grant DK100851, the Ruth and Yonatan Ben-Avraham Fund, and the Swedish Heart-Lung Foundation (Hjärt-Lungfonden) (to K.J.W).
Disclosure
KJW reports ownership interest in Hygieia, Inc., and in Gemphire Therapeutics, Inc., and serves on the Medical and Scientific Advisory Board of Gemphire Therapeutics, Inc.
Nonstandarded Abbreviations and Acronyms
- ANOVA
analysis of variance
- apoB
apolipoprotein-B
- ASCVD
atherosclerotic cardiovascular diseases
- BSA
bovine serum albumin
- CAV1
caveolin-1
- CD55
cluster of differentiation 55, also known as complement decay-accelerating factor (DAF)
- C-TRLs
cholesterol- and triglyceride-rich apoB-lipoproteins
- FcR-Synd1
a chimeric receptor consisting of the ectodomain of the IgG Fc receptor Ia linked to the transmembrane and cytoplasmic domains of human syndecan-1
- FLOT1
flotillin-1
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HSPG
heparan sulfate proteoglycan
- LDL
low-density lipoprotein
- LpL
lipoprotein lipase
- mLDL
LDL that we had methylated to an extent that blocks its binding to LDL receptors
- mRNA
messenger RNA
- qRT-PCR
quantitative real-time reverse-transcription polymerase chain reaction
- SDC1
syndecan-1
- siRNA
small interfering RNA
- SULF2
heparan sulfate glucosamine-6-O-endosulfatase-2
- T2DM
type 2 diabetes mellitus
- VLDL
very low-density lipoprotein
Footnotes
Supplemental materials
Refer to Web version on PubMed Central for supplementary materials.
Author contributions
Chen K for experimental design, some key experiments, funding support, and writing the manuscript. Wu Q, Yang C, Wu X and Cheung P for experimental data collection and analysis. Hu K for some animal work. Wlilliams KJ for partial funding support, experimental design, and writing the manuscript.
References
- 1.Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C, Cholesterol Treatment Trialists’ (CTT) Collaborators The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–90. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cannon CP, Blazing MA, Giugliano RP, et al. for the IMPROVE-IT Investigators Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–97. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 3.Robinson JG, Farnier M, Krempf M, et al. for the ODYSSEY LONG TERM Investigators Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99. doi: 10.1056/NEJMoa1501031. [DOI] [PubMed] [Google Scholar]
- 4.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR, for the FOURIER Steering Committee and Investigators Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 5.Williams KJ, Tabas I, Fisher EA. How an artery heals. Circ Res. 2015;117:909–913. doi: 10.1161/CIRCRESAHA.115.307609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borén J, Williams KJ. The central role of arterial retention of cholesterol-rich apoB-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27:473–83. doi: 10.1097/MOL.0000000000000330. [DOI] [PubMed] [Google Scholar]
- 7.Williams KJ, Wu X. Imbalanced insulin action in chronic over nutrition: clinical harm, molecular mechanisms, and a way forward. Atherosclerosis. 2016;247:225–282. doi: 10.1016/j.atherosclerosis.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 8.Taskinen MR, Borén J. New insights into the pathophysiology of dyslipidemia in type 2 diabetes. Atherosclerosis. 2015;239:483–95. doi: 10.1016/j.atherosclerosis.2015.01.039. [DOI] [PubMed] [Google Scholar]
- 9.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.ATV.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flood C, Gustafsson M, Richardson PE, Harvey SC, Segrest JP, Borén J. Identification of the proteoglycan binding site in apolipoprotein B48. J Biol Chem. 2002;277:32228–32233. doi: 10.1074/jbc.M204053200. [DOI] [PubMed] [Google Scholar]
- 11.Williams KJ, Tabas I. Lipoprotein retention—and clues for atheroma regression. Arterioscler Thromb Vasc Biol. 2005;25:1536–1540. doi: 10.1161/01.ATV.0000174795.62387.d3. [DOI] [PubMed] [Google Scholar]
- 12.Williams KJ, Chen K. Recent insights into factors affecting remnant lipoprotein uptake. Curr Opin Lipidol. 2010;21:218–28. doi: 10.1097/MOL.0b013e328338cabc. [DOI] [PubMed] [Google Scholar]
- 13.Varbo A, Benn M, Tybjærg-Hansen A, Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61:427–36. doi: 10.1016/j.jacc.2012.08.1026. [DOI] [PubMed] [Google Scholar]
- 14.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM, Jr, Kastelein JJP, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ, for the JUPITER Study Group Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 15.Deedwania PC, Hunninghake DB, Bays HE, Jones PH, Cain VA, Blasetto JW, for the STELLAR Study Group Effects of rosuvastatin, atorvastatin, simvastatin, and pravastatin on atherogenic dyslipidemia in patients with characteristics of the metabolic syndrome. Am J Cardiol. 2005;95:360–6. doi: 10.1016/j.amjcard.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 16.Blom DJ, Hala T, Bolognese M, et al. for the DESCARTES Investigators: A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370:1809–19. doi: 10.1056/NEJMoa1316222. [DOI] [PubMed] [Google Scholar]
- 17.Reyes-Soffer G, Pavlyha M, Ngai C, et al. Effects of PCSK9 inhibition with alirocumab on lipoprotein metabolism in healthy humans. Circulation. 2017;135:352–362. doi: 10.1161/CIRCULATIONAHA.116.025253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuki IV, Kuhn KM, Lomazov IR, Rothman VL, Tuszynski GP, Iozzo RV, Swenson TL, Fisher EA, Williams KJ. The syndecan family of proteoglycans: novel receptors mediating internalization of atherogenic lipoproteins in vitro. J Clin Invest. 1997;100:1611–1622. doi: 10.1172/JCI119685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuki IV, Meyer ME, Williams KJ. Transmembrane and cytoplasmic domains of syndecan mediate a multi-step endocytic pathway involving detergent-insoluble membrane rafts. Biochem J. 2000;351:607–612. doi: 10.1042/bj3510607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams KJ, Fuki IV. Cell-surface heparan sulfate proteoglycans: dynamic molecules mediating ligand catabolism. Curr Opin Lipidol. 1997;8:253–262. doi: 10.1097/00041433-199710000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Stanford KI, Bishop JR, Foley EM, Gonzales JC, Niesman IR, Witztum JL, Esko JD. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J Clin Invest. 2009;119:3236–45. doi: 10.1172/JCI38251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen K, Williams KJ. Molecular mediators for raft-dependent endocytosis of syndecan-1, a highly conserved, multifunctional receptor. J Biol Chem. 2013;288:13988–13999. doi: 10.1074/jbc.M112.444737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bickel PE, Scherer PE, Schnitzer JE, Oh P, Lisanti MP, Lodish HF. Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J Biol Chem. 1997;272:13793–13802. doi: 10.1074/jbc.272.21.13793. [DOI] [PubMed] [Google Scholar]
- 24.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 25.Glebov OO, Bright NA, Nichols BJ. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat Cell Biol. 2006;8:46–54. doi: 10.1038/ncb1342. [DOI] [PubMed] [Google Scholar]
- 26.Ge L, Qi W, Wang LJ, Miao HH, Qu YX, Li BL, Song BL. Flotillins play an essential role in Niemann-Pick C1-like 1-mediated cholesterol uptake. Proc Natl Acad Sci U S A. 2011;108:551–6. doi: 10.1073/pnas.1014434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pelkmans L, Zerial M. Kinase-regulated quantal assemblies and kiss-and-run recycling of caveolae. Nature. 2005;436:128–33. doi: 10.1038/nature03866. [DOI] [PubMed] [Google Scholar]
- 28.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–94. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 29.Payne CK, Jones SA, Chen C, Zhuang X. Internalization and trafficking of cell surface proteoglycans and proteoglycan-binding ligands. Traffic. 2007;8:389–401. doi: 10.1111/j.1600-0854.2007.00540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams KJ. Interactions of lipoproteins with proteoglycans. Methods Mol Biol. 2001;171:457–477. doi: 10.1385/1-59259-209-0:457. [DOI] [PubMed] [Google Scholar]
- 31.Liu M-L, Davidson WR, Meyer ME, Friedman RA, Williams KJ. An unexpected consensus motif shared by the LDL receptor and syndecan transmembrane domains directs movement into rafts upon clustering. Arterioscler Thromb Vasc Biol. 2004;24(5):E-53. [Google Scholar]
- 32.Chen K, Liu M-L, Schaffer L, Li M, Boden G, Wu X, Williams KJ. Type 2 diabetes in mice induces hepatic overexpression of sulfatase 2, a novel factor that suppresses uptake of remnant lipoproteins. Hepatology. 2010;52:1957–1967. doi: 10.1002/hep.23916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hassing HC, Mooij H, Guo S, Monia BP, Chen K, Kulik W, Dallinga-Thie GM, Nieuwdorp M, Stroes ESG, Williams KJ. Inhibition of hepatic sulfatase-2 in vivo: A novel strategy to correct diabetic dyslipidemia. Hepatology. 2012;55:1746–1753. doi: 10.1002/hep.25580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hassing HC, Surendran RP, Derudas B, Verrijken A, Francque SM, Mooij HL, Bernelot Moens SJ, ’t Hart LM, Nijpels G, Dekker JM, Williams KJ, Stroes ES, Van Gaal LF, Staels B, Nieuwdorp M, Dallinga-Thie GM. SULF2 strongly predisposes to fasting and postprandial triglycerides in patients with obesity and type 2 diabetes mellitus. Obesity (Silver Spring) 2014;22:1309–1316. doi: 10.1002/oby.20682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matikainen N, Burza MA, Romeo S, Hakkarainen A, Adiels M, Folkersen L, Eriksson P, Lundbom N, Ehrenborg E, Orho-Melander M, Taskinen MR, Borén J. Genetic variation in SULF2 is associated with postprandial clearance of triglyceride-rich remnant particles and triglyceride levels in healthy subjects. PLoS One. 2013;8:e79473. doi: 10.1371/journal.pone.0079473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taketomi S. KK and KKAy mice: Models of type 2 diabetes with obesity Chapter 16. In: Shafrir E, editor. Animal Models of Diabetes: Frontiers in Research. CRC Press; Boca Raton: 2007. pp. 335–347. [Google Scholar]
- 37.Shafrir E, Ziv E. A useful list of spontaneously arising animal models of obesity and diabetes. Am J Physiol Endocrinol Metab. 2009;296:E1450–2. doi: 10.1152/ajpendo.00113.2009. [DOI] [PubMed] [Google Scholar]
- 38.Wu X, Williams KJ. A novel insulin signaling cascade that selectively drives hepatic lipogenesis, despite persistently active PTEN. Diabetes; Presented at the 73rd Scientific Sessions of the American Diabetes Association; Chicago, IL, USA. 22 June 2013.2013. p. A467. [Google Scholar]
- 39.Olsson U, Egnell A-C, Lee MR, Lundén GO, Lorentzon M, Salmivirta M, Bondjers G, Camejo G. Changes in matrix proteoglycans induced by insulin and fatty acids in hepatic cells may contribute to dyslipidemia of insulin resistance. Diabetes. 2001;50:2126–2132. doi: 10.2337/diabetes.50.9.2126. [DOI] [PubMed] [Google Scholar]
- 40.Babuke T, Tikkanen R. Dissecting the molecular function of reggie/flotillin proteins. Eur J Cell Biol. 2007;86:525–32. doi: 10.1016/j.ejcb.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 41.Otto GP, Nichols BJ. The roles of flotillin microdomains – endocytosis and beyond. J Cell Sci. 2011;124:3933–40. doi: 10.1242/jcs.092015. [DOI] [PubMed] [Google Scholar]
- 42.Solis GP, Hoegg M, Munderloh C, Schrock Y, Malaga-Trillo E, Rivera-Milla E, Stuermer CAO. Reggie/flotillin proteins are organized into stable tetramers in membrane microdomains. Biochem J. 2007;403:313–22. doi: 10.1042/BJ20061686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hopp TP, Prickett KS, Price VL, Libby RT, March CJ, Cerretti DP, Urdal DL, Conlon PJ. A short polypeptide marker sequence useful for recombinant protein identification and purification. Nat Biotech. 1988;6:1204–1210. doi: 10.1038/Nbt1088-1204. [DOI] [Google Scholar]
- 44.Redgrave TG. Catabolism of chylomicron triacylglycerol and cholesteryl ester in genetically obese rats. J Lipid Res. 1977;18:604–612. [PubMed] [Google Scholar]
- 45.Munderloh C, Solis GP, Bodrikov V, Jaeger FA, Wiechers M, Málaga-Trillo E, Stuermer CAO. Reggies/flotillins regulate retinal axon regeneration in the zebrafish optic nerve and differentiation of hippocampal and N2a neurons. J Neurosci. 2009;29:6607–15. doi: 10.1523/JNEUROSCI.0870-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meister M, Tikkanen R. Endocytic trafficking of membrane-bound cargo: a flotillin point of view. Membranes (Basel) 2014;4:356–71. doi: 10.3390/membranes4030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams KJ, Fless GM, Petrie KA, Snyder ML, Brocia RW, Swenson TL. Mechanisms by which lipoprotein lipase alters cellular metabolism of lipoprotein(a), low density lipoprotein, and nascent lipoproteins. Roles for low density lipoprotein receptors and heparan sulfate proteoglycans. J Biol Chem. 1992;267:13284–13292. [PubMed] [Google Scholar]
- 48.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–23. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011;60:2441–9. doi: 10.2337/db11-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.