

Summary
We performed an extensive immunogenomic analysis of over 10,000 tumors comprising 33 diverse cancer types utilizing data compiled by TCGA. Across cancer types, we identified six immune subtypes: Wound Healing, IFN-γ Dominant, Inflammatory, Lymphocyte Depleted, Immunologically Quiet, and TGF-β Dominant, characterized by differences in macrophage or lymphocyte signatures, Th1:Th2 cell ratio, extent of intratumoral heterogeneity, aneuploidy, extent of neoantigen load, overall cell proliferation, expression of immunomodulatory genes, and prognosis. Specific driver mutations correlated with lower (CTNNB1, NRAS, or IDH1) or higher (BRAF, TP53, or CASP8) leukocyte levels across all cancers. Multiple control modalities of the intracellular and extracellular networks (transcription, microRNAs, copy number and epigenetic processes) were involved in tumor-immune cell interactions, both across and within immune subtypes. Our immunogenomics pipeline to characterize these heterogeneous tumors and the resulting data are intended to serve as a resource for future targeted studies to further advance the field.
Keywords: Immuno-oncology, Cancer Genomics, Immune Subtypes, Tumor immunology, Integrative Network Analysis
eTOC blurb
Thorsson et al. (2018) present immunogenomics analyses of over 10,000 tumors, identifying six immune subtypes that encompass multiple cancer types and are hypothesized to define immune response patterns impacting prognosis. This work provides a resource for understanding tumor-immune interactions, with implications for identifying ways to advance research on immunotherapy.
Introduction
The Cancer Genome Atlas (TCGA) has profoundly illuminated the genomic landscape of human malignancy. Genomic and transcriptomic data derived from bulk tumor samples has been used to study the tumor microenvironment (TME), and measures of immune infiltration define molecular subtypes of ovarian, melanoma, and pancreatic cancer (Bailey et al., 2016; The Cancer Genome Atlas Network, 2015; The Cancer Genome Atlas Research Network, 2011), and immune gene expression in other tumors varies by molecular subtype (Iglesia et al., 2016). Characterization of the immune microenvironment using gene expression signatures, T cell receptor (TCR) and B cell receptor (BCR) repertoire, and analyses to identify neo-antigenic immune targets provide a wealth of information in many cancer types, and have prognostic value (Bindea et al., 2013; Brown et al., 2015; Brown et al., 2014; Charoentong et al., 2017; Gentles et al., 2015; Iglesia et al., 2016; Li et al., 2016; Porta-Pardo and Godzik, 2016; Rooney et al., 2015).
Contemporaneous with the work of TCGA, cancer immunotherapy has revolutionized cancer care. Antibodies against CTLA-4, PD-1, and PD-L1 are effective in treating a variety of malignancies. However, the biology of the immune microenvironment driving these responses is incompletely understood (Hugo et al., 2016; McGranahan et al., 2016), but is critical to the design of immunotherapy treatment strategies.
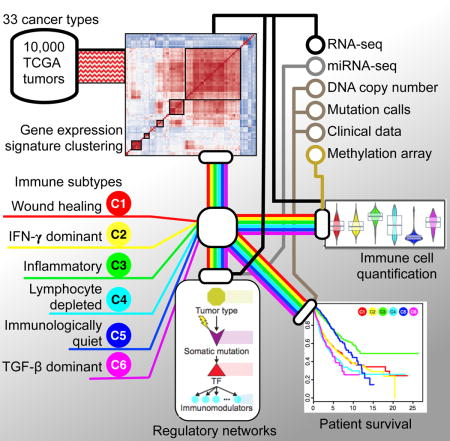
We integrated major immunogenomics methods to characterize the immune tumor microenvironment (TME) across 33 cancers analyzed by TCGA, applying methods for the assessment of total lymphocytic infiltrate (from genomic and H&E image data), immune cell fractions from deconvolution analysis of mRNA-Seq data, immune gene expression signatures, neoantigen prediction, TCR and BCR repertoire inference, viral RNA expression, and somatic DNA alterations (Table S1). Transcriptional regulatory networks and extracellular communication networks that may govern the TME were found, as were possible germline determinants of TME features, and prognostic models were developed.
Through this approach, we identified and characterized 6 immune subtypes spanning multiple tumor types, with potential therapeutic and prognostic implications for cancer management. All data and results are provided in Supplemental Tables, at the NCI Genomic Data Commons (GDC, portal.gdc.cancer.gov) and though the CRI iAtlas portal for interactive exploration and visualization (www.cri-iatlas.org), and are intended to serve as a resource and inspiration for future studies in the field of immunogenomics.
Results
Analytic Pipeline
To characterize the immune response to cancer in all TCGA tumor samples, identify common immune subtypes, and evaluate if tumor extrinsic features can predict outcomes, we analyzed the TME across the landscape of all TCGA tumor samples. First, source datasets from all 33 TCGA cancer types and six molecular platforms (mRNA-, microRNA- and exome-sequencing; DNA methylation-, copy number-, and reverse-phase protein arrays) were harmonized by the PanCanAtlas consortium for uniform quality control, batch effect correction, normalization, mutation calling, and curation of survival data(Ellrott et al., 2018; Liu et al., 2018). We then performed a series of analyses, which we summarize here and describe in detail in the ensuing manuscript sections as noted within parentheses. We first compiled published tumor immune expression signatures and scored these across all non-hematologic TCGA cancer types. Meta-analysis of subsequent cluster analysis identified characteristic immunooncologic gene signatures, which were then used to cluster TCGA tumor types into 6 groups, or subtypes (described in “Immune Subtypes in Cancer”). Leukocyte proportion and cell type were then defined from DNA methylation, mRNA, and image analysis (“Composition of the Tumor Immune Infiltrate”). Survival modeling was performed to assess how immune subtypes associate with patient prognosis (“Prognostic Associations of Tumor Immune Response Measures”). Neoantigen prediction and viral RNA expression (“Survey of Immunogenicity”), TCR and BCR repertoire inference (“The Adaptive Immune Receptor Repertoire in Cancer”), and immunomodulator (IM) expression and regulation (“Regulation of Immunomodulators”) were characterized in the context of TCGA tumor types, TCGA-defined molecular subtypes, and these 6 immune subtypes, so as to assess the relationship between factors affecting immunogenicity and immune infiltrate. In order to assess the degree to which specific underlying somatic alterations (pathways, copy number alterations, and driver mutations) may drive the composition of the TME we identified which alterations correlate with modified immune infiltrate (“Immune Response Correlates of Somatic Variation”). We likewise asked whether gender and ancestry predispose individuals to particular tumor immune responses (“Immune Response Correlates of Demographic and Germline Variation”). Finally, we sought to identify the underlying intracellular regulatory networks governing the immune response to tumors, as well as the extracellular communication networks involved in establishing the particular immune milieu of the TME (“Networks Modulating Tumoral Immune Response.”)
Immune Subtypes in Cancer
To characterize intratumoral immune states, we scored 160 immune expression signatures, and used cluster analysis to identify modules of immune signature sets (Figure 1A, top panel). Five immune expression signatures (macrophages/monocytes (Beck et al., 2009), overall lymphocyte infiltration (dominated by T and B cells) (Calabro et al., 2009), TGF-β response (Teschendorff et al., 2010), IFN-γ response (Wolf et al., 2014), and wound healing (Chang et al., 2004)), which robustly reproduced co-clustering of these immune signature sets (Figures 1A middle panel, S1A), were selected to perform cluster analysis of all 30 non-hematologic cancer types. The six resulting clusters “Immune Subtypes”, C1-C6 (with 2416, 2591, 2397, 1157, 385 and 180 cases, respectively) were characterized by a distinct distribution of scores over the five representative signatures (Figure 1A, bottom panel), and showed distinct immune signatures based on the dominant sample characteristics of their tumor samples (Figure 1B–C). Immune subtypes spanned anatomical location and tumor type, while individual tumor types and TCGA subtypes (Figures 1D, S1B–D) varied substantially in their proportion of immune subtypes.
Figure 1. Immune Subtypes in Cancer.
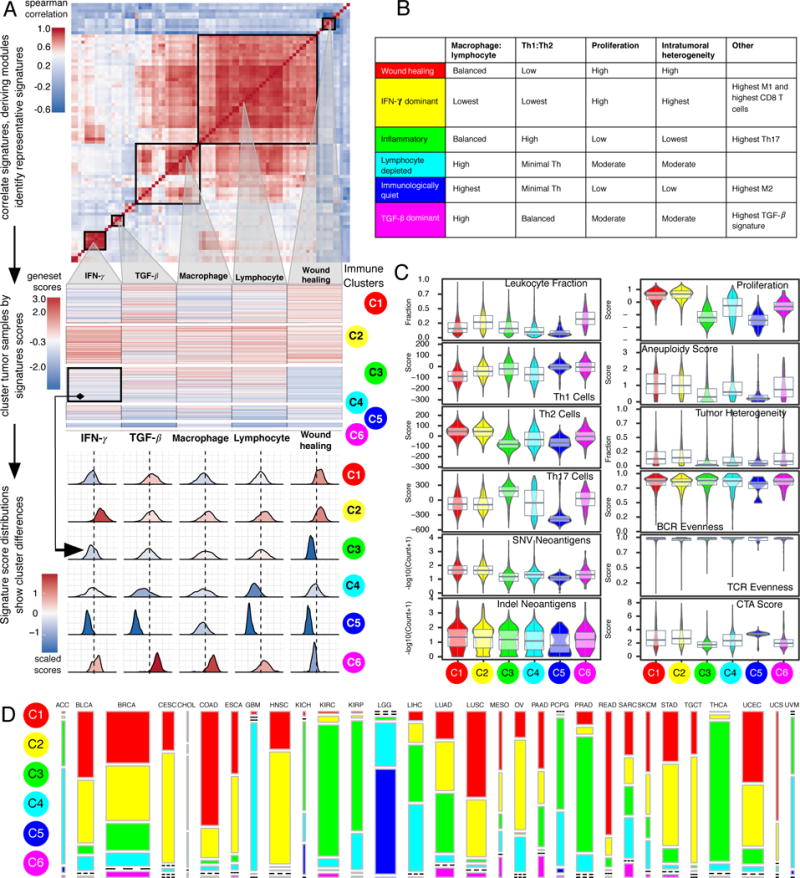
A. Expression signature modules and identification of immune subtypes. Top Consensus clustering of the pairwise correlation of cancer immune gene expression signature scores (rows and columns). Five modules of shared associations are indicated by boxes. Middle Representative gene expression signatures from each module (columns), which robustly reproduced module clustering, were used to cluster TGCA tumor samples (rows), resulting in 6 immune subtypes C1-C6 (colored circles). Bottom Distributions of signature scores within the six subtypes (rows), with dashed line indicating the median. B. Key characteristics of immune subtypes. C. Values of key immune characteristics by immune subtype. D. Distribution of immune subtypes within TCGA tumors. The proportion of samples belonging to each immune subtype is shown, with colors as in A. Bar width reflects the number of tumor samples. See also Figure S1 and Table S1.
C1 (Wound Healing) had elevated expression of angiogenic genes, a high proliferation rate (Figure 1C), and a Th2 cell bias to the adaptive immune infiltrate. Colorectal cancer (COAD, READ) and lung squamous cell carcinoma (LUSC) were rich in C1, as were breast carcinoma (BRCA) luminal A (Figure S1C–D), head and neck squamous cell carcinoma (HNSC) classical, and the chromosomally unstable (CIN) gastrointestinal subtype.
C2 (IFN-γ Dominant) had the highest M1/M2 macrophage polarization (Figure S2A, mean ratio=0.52, p<10−149, Wilcoxon test relative to next-highest), a strong CD8 signal and, together with C6, the greatest TCR diversity. C2 also showed a high proliferation rate, which may override an evolving Type I immune response, and was comprised of highly mutated BRCA, gastric, ovarian (OV), HNSC, and cervical tumors (CESC).
C3 (Inflammatory) was defined by elevated Th17 and Th1 genes (Figure 1C, both p<10−23), low to moderate tumor cell proliferation, and, along with C5, lower levels of aneuploidy and overall somatic copy number alterations than the other subtypes. C3 was enriched in most kidney, prostate (PRAD), pancreatic cancers (PAAD), and papillary thyroid carcinomas (THCA).
C4 (Lymphocyte Depleted) was enriched in particular subtypes of adrenocortical carcinoma (ACC), pheochromocytoma and paraganglioma (PCPG), hepatocellular carcinoma (LIHC), and gliomas, and displayed a more prominent macrophage signature (Figure 2A), with Th1 suppressed and a high M2 response (Figure S2A).
Figure 2. Composition of the Tumor Immune Infiltrate.
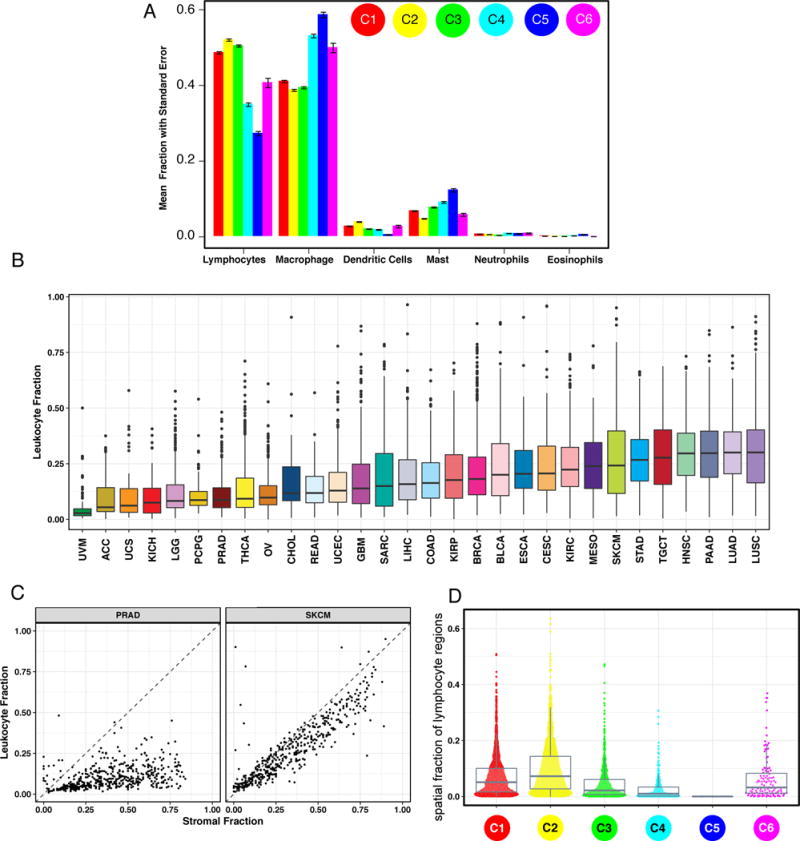
A. The proportion of major classes of immune cells (from CIBERSORT) within the leukocyte compartment for different immune subtypes. Error bars show the standard error of the mean. B. Leukocyte Fraction (LF) within TCGA tumor types, ordered by median. C. LF (y-axis) vs. non-tumor stromal cellular fraction in the TME (x-axes) for two representative TCGA tumor types: PRAD, (low LF relative to stromal content), and SKCM (high leukocyte fraction in the stroma). Dots represent individual tumor samples. D. The spatial fraction of lymphocyte regions in tissue was estimated using machine learning on digital pathology H&E images (see also (Saltz et al, 2018)).
C5 (Immunologically Quiet), consisted mostly of lower grade gliomas (LGG) (Figures 1D, S1B), exhibited the lowest lymphocyte (p<10−17), and highest macrophage (p<10−7) responses (Figure 2A), dominated by M2 macrophages (Figure S2A). Glioma subtypes (Ceccarelli et al., 2016) CpG island methylator phenotype-high (CIMP-H), the 1p/19q codeletion subtype and pilocytic astrocytoma-like (PA-like) were prevalent in C5, with remaining subtypes enriched in C4. IDH mutations were enriched in C5 over C4 (80% of IDH mutations, p<2×10−16, Fisher’s exact test), suggesting an association of IDH-mutations with favorable immune composition. Indeed, IDH-mutations associate with TME composition (Venteicher et al., 2017) and decrease leukocyte chemotaxis, leading to fewer tumor-associated immune cells and better outcome (Amankulor et al., 2017).
Finally, C6 (TGF-β Dominant), which was a small group of mixed tumors not dominant in any one TCGA subtype, displayed the highest TGF-β signature (p<10−34) and a high lymphocytic infiltrate with an even distribution of Type I and Type II T cells.
These six categories represent features of the TME that largely cut across traditional cancer classifications to create groupings and suggest certain treatment approaches may be independent of histologic type.
Composition of the Tumor Immune Infiltrate
Leukocyte fraction (LF) varied substantially across immune subtypes (Figure 1C), and tumor types (Figure 2B). Tumors within the top third LF included cancers most responsive to immune checkpoint inhibitors, such as lung adenocarcinoma (LUAD), LUSC, cutaneous melanoma (SKCM), HNSC, and clear cell renal cell carcinoma (KIRC), and in particular, the LUSC.secretory, LUAD.6, bladder (BLCA.4), papillary renal cell carcinoma (KIRP.C2a), and HNSCC mesenchymal subtypes. Uveal melanoma (UVM) and ACC had very low LF. Glioma subtypes displayed a greater range in LF than other tumors, which may reflect the presence or absence of microglia.
The leukocyte proportion of tumor stromal fraction, ρ, varied across tumor types and immune subtypes (Figure 2C, S2B), ranging from >90% in SKCM to <10% in stroma-rich tumors such as PAAD, PRAD and LGG. Some tumors, e.g. BRCA, showed variation within annotated or immune subtypes. In BRCA, C1 has the lowest ρ, with ρC1 = 0.44, while ρC2 = 0.60 was 37% higher (p<0.001) (Figure S2B); and there were likewise differences between Luminal A and Basal BRCA (ρLumA = 0.45 and ρBasal = 0.67 (p<0.001)). For LGG, ρC5=0.28 (p<0.001), whereas ρC3 = 0.48 and ρC4 = 0.50 (p<0.001) (Figure S2B), and in READ, ρCIN=0.40 and ρHM-indel = 0.78 (p<0.001).
The spatial fraction of tumor regions with tumor infiltrating lymphocytes (TILs), estimated by analysis of digitized TCGA H&E stained slides (Saltz et al, 2018), varied by immune subtype, with C2 the highest (p<10−16, Figure 2D). Image estimates correlated modestly with molecular estimates of lymphocyte proportion (Figures S2C–D), in part because the molecular estimate is more similar to cell count, while TIL fraction is spatially-derived. The relative similarity of the estimates of lymphocytic content between two radically different methodologies reinforces the robustness of individual methods.
Prognostic Associations of Tumor Immune Response Measures
Immune subtypes associated with overall survival (OS) and progression free interval (PFI) (Figures 3A, S3A). C3 had the best prognosis (OS HR 0.628, p=2.34×10−8 relative to C1, adjusted for tumor type), while C2 and C1 had less favorable outcomes despite having a substantial immune component. The more mixed-signature subtypes, C4 and C6, had the least favorable outcome. Functional orientation of the TME for tumor and immune subtypes was measured using the Concordance Index (CI) (Pencina and D’Agostino, 2004) and found to have context-dependent prognostic impact (Figures 3B–C, S3B). Higher lymphocyte signature associated with improved outcome in C1 and C2. An increased value of any of the 5 signatures led to worse outcome in C3 (Figure 3B), perhaps reflecting a balanced immune response. While increased Th17 cells generally led to improved OS, Th1 associated with worse OS across most immune subtypes, and Th2 orientation had mixed effects (Figure 3C). Tumor types displayed two behaviors relative to immune orientation (Figure 3B, OS; S3B, PFI). In the first group including SKCM and CESC, activation of immune pathways was generally associated with better outcome, while in the other, the opposite was seen. The relative abundance of individual immune cell types had complex associations that differed between tumor types (Figure S3C–D). These analyses extend beyond mere determination of lymphocyte presence to suggest testable properties that correlate with patient outcome in different tumor types and immune contexts.
Figure 3. Immune Response and Prognostics.
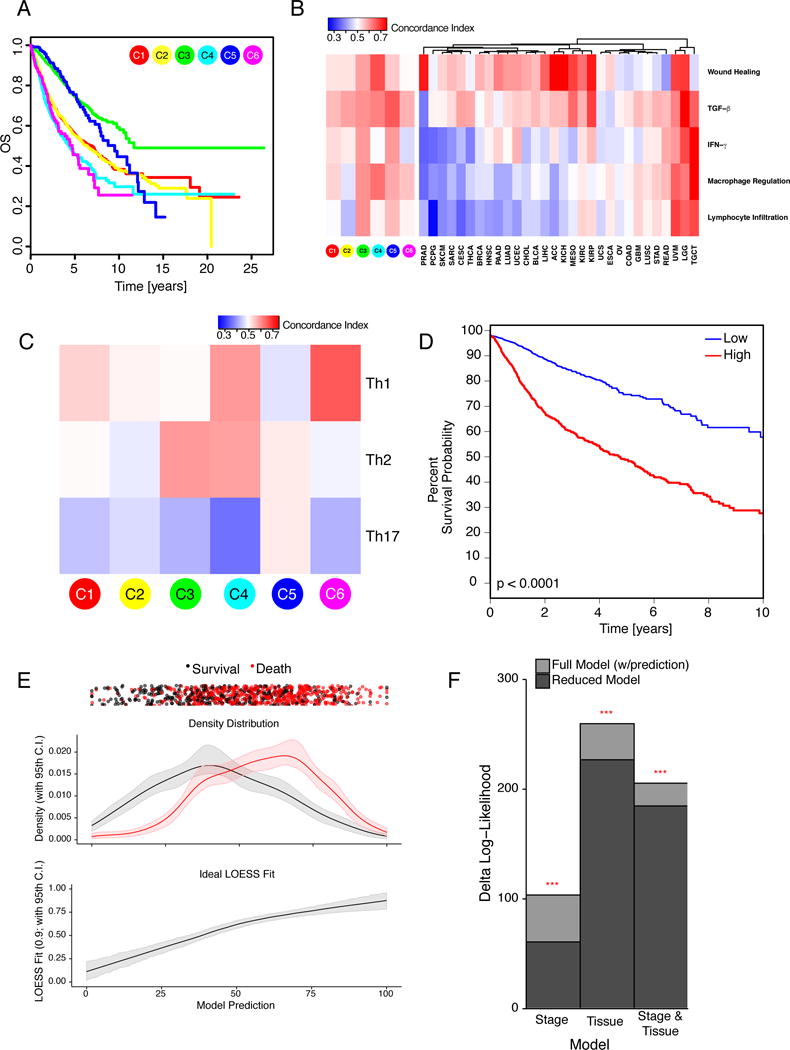
A. Overall survival (OS) by immune subtype. B. Concordance Index (CI) for 5 characteristic immune expression signature scores (Figure 1A) in relation to OS, for immune subtypes and TCGA tumor types. Red denotes higher, and blue lower risk, with an increase in the signature score. C. CI for T-helper scores in relation to OS within immune subtypes. D. Risk stratification from elastic net modeling of immune features. Tumor samples were divided into discovery and validation sets, and an elastic net model was optimized on the discovery set using immune gene signatures, TCR/BCR richness, and neoantigen counts. Kaplan-Meier plot shows the high (red) and low (blue) risk groups from this model as applied to the validation set, p<0.0001 (G-rho family of tests, Harrington and Fleming). E. Prediction vs. outcome from elastic net model in validation set data (from 3D). Top Patient outcomes for each sample (black, survival; red, death) plotted with vertical jitter, along the sample’s model prediction (x-axis). Middle Fractional density of the outcomes plotted against their model predictions. Confidence intervals were generated by bootstrapping with replacement. Bottom LOESS fit of the actual outcomes against the model predictions; narrow confidence bands confirm good prediction accuracy. F. CoxPH models of stage and tumor type (“Tissue”) with (full model) or without (reduced model) the validation set predictions of the elastic net model were compared; the full model significantly outperformed the reduced model in all comparisons (p<0.001; false discovery rate (FDR) BH-corrected). See also Figure S3.
We obtained and validated a survival model using elastic-net Cox proportional hazards (CoxPH) modeling with cross-validation. Low and high-score tumors displayed significant survival differences in the validation set (Figure 3D), with good prediction accuracy (Figure 3E). Incorporating immune features into Cox models fit with tumor type, stage, and tumor type + stage (Figure 3F) improved predictive accuracy, highlighting the importance of the immune TME in determining survival. Lymphocyte expression signature, high number of unique TCR clonotypes, cytokines made by activated and Th1-and Th17 cells, and M1 macrophages most strongly associated with improved OS (Figure S3E), while wound healing, macrophage regulation, and TGF-β associated with worse OS, recapitulating survival associations in immune subtypes. Within tumor types, the prognostic implications of immune subtypes seen in univariate analyses were largely maintained, with C3 correlating with better OS in 6 tumor types, and C4 with poor OS in 3 cancer types (Figure S3F).
Immune Response Correlates of Somatic Variation
The immune infiltrate was related to measures of DNA damage, including copy number variation (CNV) burden (both in terms of number of segments and fraction of genome alterations), aneuploidy, loss of heterozygosity (LOH), homologous recombination deficiency (HRD), and intratumor heterogeneity (ITH) (Figure 4A). LF correlated negatively with CNV segment burden, with strongest correlation in C6 and C2, and positively with aneuploidy, LOH, HRD, and mutation load, particularly in C3. These results suggest a differential effect of multiple smaller, focal copy number events versus larger events on immune infiltration in certain immune subtypes.
Figure 4. Immune Response and Genome State.
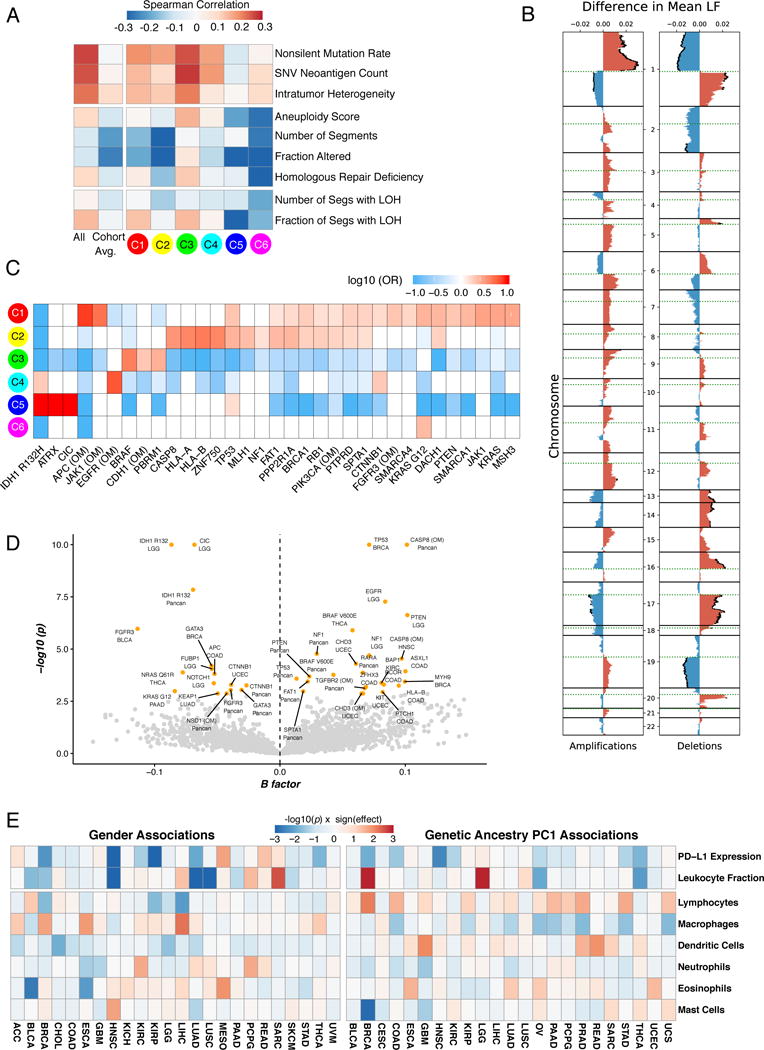
A. Correlation of DNA damage measures (rows) with LF. From left to right: all TCGA tumors; averaged over tumor type; grouped by immune subtype. B. LF association with copy number (CN) alterations. Left Differences between observed and expected mean LF in tumors with amplifications, by genomic region. Significant (FDR < 0.01) differences in mean LF are marked with black caps on the profiles. Right Same, for deletions. C. Enrichment and depletion of mutations in driver genes and oncogenic mutations (OM) within immune subtypes, displayed as fold enrichment. Significance was evaluated by the Cochran-Mantel-Haenszel χ2 test, to account for cancer type (white, no significant association) D. Volcano plot showing driver genes and OMs associated with changes in LF, across all tumors (“Pancan”) and within specific tumor types as indicated. X-axis: Multivariate correlation with LF (B-factor), taking into account tumor type and number of missense mutations. Values >0 represent positive correlation with LF and vice versa; Y-axis: -log10(p). Significant events (FDR < 0.1; p<0.003) are in orange, others in gray. E. Left Degree of association between gender for 8 selected immune characteristics (rows) within TCGA tumor types (columns). Blue denotes a higher value in women than in men, and red the opposite. Right Degree of association between the immune characteristics and the first principal component of genetic ancestry in TCGA participants (PC1), reflecting degree of African ancestry. Blue reflects lower values in individuals of African descent. See also Figure S4 and Table S2.
Specific SCNAs affected LF and immune composition (Figures 4B, S4A). Chromosome 1p (including TNFRS9 and VTCN1) amplification associated with higher LF, while its deletion did the opposite. 19q deletion (including TGFB1) also correlated with lower LF, consistent with the role of TGF-β in immune cell recruitment (Bierie and Moses, 2010). Amplification of chr2, 20q, and 22q (including CTLA4, CD40, and ADORA2 respectively), and deletions of 5q, 9p, and chr19 (including IL13 and IL4, IFNA1 and IFNA2, and ICAM1 respectively) associated with changes in macrophage polarity (Figure S4A). IL-13 influences macrophage polarization (Mantovani et al., 2005), implying a possible basis for our observation that IL13 deletions associated with altered M0 macrophage fractions.
Increased ITH associates with worse clinical outcomes or lower efficacy of immunomodulator (IM) therapy in a number of cancer types (McGranahan et al., 2016; Morris et al., 2016). ITH correlated (Spearman, Benjamini-Hochberg (BH)-adjusted p<0.05) with total LF in 9 tumor types (LUAD, BRCA, KIRC, HNSC, GBM, OV, BLCA, SKCM, and READ (data not shown)), and with individual relative immune cell fractions in many tumor types (Figure S4B). ITH was highest in C1 and C2 (p<10−229 relative to all others), and lowest in C3 (p=3×10−5, Figure 1C), possibly supporting the link between lower ITH and improved survival.
We correlated mutations in 299 cancer driver genes with immune subtypes, and found 33 significant associations (q<0.1) (Figure 4C, Table S2). C1 was enriched in mutations in driver genes, such as TP53, PIK3CA, PTEN or KRAS. C2 was enriched in many of these genes, as well as HLA-A and B and CASP8, which could be immune-evading mechanisms (Rooney et al., 2015). C3 was enriched in BRAF, CDH1 and PBRM1 mutations, a finding of note since patients with PBRM1 mutations respond particularly well to IM therapy (Miao et al., 2018). C4 was enriched in CTNNB1, EGFR, and IDH1 mutations. C5 was enriched in IDH1, ATRX and CIC, consistent with its predominance of LGG samples. C6 only showed an enrichment in KRAS G12 mutations. Mutations in 23 driver genes associated with increased LF either in specific tumor types or across them, including TP53, HLA-B, BRAF, PTEN, NF1, APC and CASP8. Twelve other events were associated with lower LF, including the IDH1 R132H mutation, GATA3, KRAS, NRAS, CTNNB1 and NOTCH1 (Figure 4D).
Since driver mutations in the same pathway had opposing correlations with LF (e.g. BRAF, KRAS, NRAS), we considered the overall effect of somatic alterations (mutations and SCNAs) on 8 oncogenic signaling pathways. PI3K, NOTCH and RTK/RAS pathway disruptions showed variable, tumor type specific effects on immune factors, while TGF-β pathway disruptions more consistently associated with lower LF (most prominently in C2 and C6; Figure S4C), higher eosinophils (C2), and increased macrophages. However, in C3, TGF-β pathway disruption associated with higher LF and M1 macrophages, and lower memory B cells, helper T cells, and M0 macrophages. Thus, TGF-β pathway disruption has context-dependent effects on LF, but may promote increased macrophages, particularly M1. Higher M1/M2 ratio, in turn, may reiterate the local pro-inflammatory state in these patients.
Immune Response Correlates of Demographic and Germline Variation
Immune cell content and expression of PD-L1 varied by gender and genetic ancestry (Figures 4E, S4D). PD-L1 expression was greater (p<0.05, Kruskal-Wallis test, unadjusted) in women than in men in HNSC, KIRC, LUAD, THCA and KIRP (Figure S4E), while mesothelioma (MESO) showed an opposite trend. PD-L1 expression was lower in individuals of predicted African ancestry (overall p=5×10−6). This association was consistent across most cancer types and was significant (p<0.05, unadjusted) in BRCA, COAD, HNSC (Figure S4F), and THCA. No single cis-eQTL significantly correlated with PD-L1 expression, although the SNP rs822337, approximately 1KB upstream of CD274 transcription start, correlated weakly (p=0.074;1.3×10−4 unadjusted; Figure S4G). Lymphocyte fractions tended to be lower in people of Asian ancestry, particularly in UCEC and BLCA (Figure S4H). The significance of these demographic associations remains unclear, but provides hypotheses for the efficacy of checkpoint inhibitor therapy based on genetic ancestry.
Survey of Immunogenicity
Peptides predicted to bind with MHC proteins (pMHCs) and induce antitumor adaptive immunity were identified from SNV and indel mutations. The number of pMHCs (neoantigen load) varied between immune subtypes (Figure 1C), correlated positively with LF in most immune subtypes (Figure S4I), and trended positive in most TCGA tumor subtypes, with some negative correlation seen among GI subtypes, and differential trending seen among individual LUAD, LUSC, OV, and KIRP subtypes (Figure S4J). Neoantigen load also associated with higher content of CD8 T cells, M1 macrophages, and CD4 memory T cells, and lower Tregs, mast, dendritic, and memory B cells in multiple tumor types (Figure S4K).
Most SNV-derived peptides which bind to MHC were each found in the context of a single MHC allele (89.9%). Single mutations generate 99.8% of unique pMHCs while 0.2% result from distinct mutations in different genetic loci yielding identical peptides (Figure 5A). In BRCA and LIHC, worse PFI was associated with higher neoantigen load, while BLCA and UCEC showed the opposite effect (Figure S5A). For most tumors, however, there were no clear associations between predicted pMHC count and survival. Within immune subtypes (Figure S5B), higher neoantigen load was associated with improved PFI in C1 and C2, and worse PFI in C3, C4, and C5. These results suggest that neoantigen load provides more prognostic information within immune subtypes than based on tissue of origin, emphasizing the importance of overall immune signaling in responding to tumor neoantigens
Figure 5. The Tumor-Immune Interface.
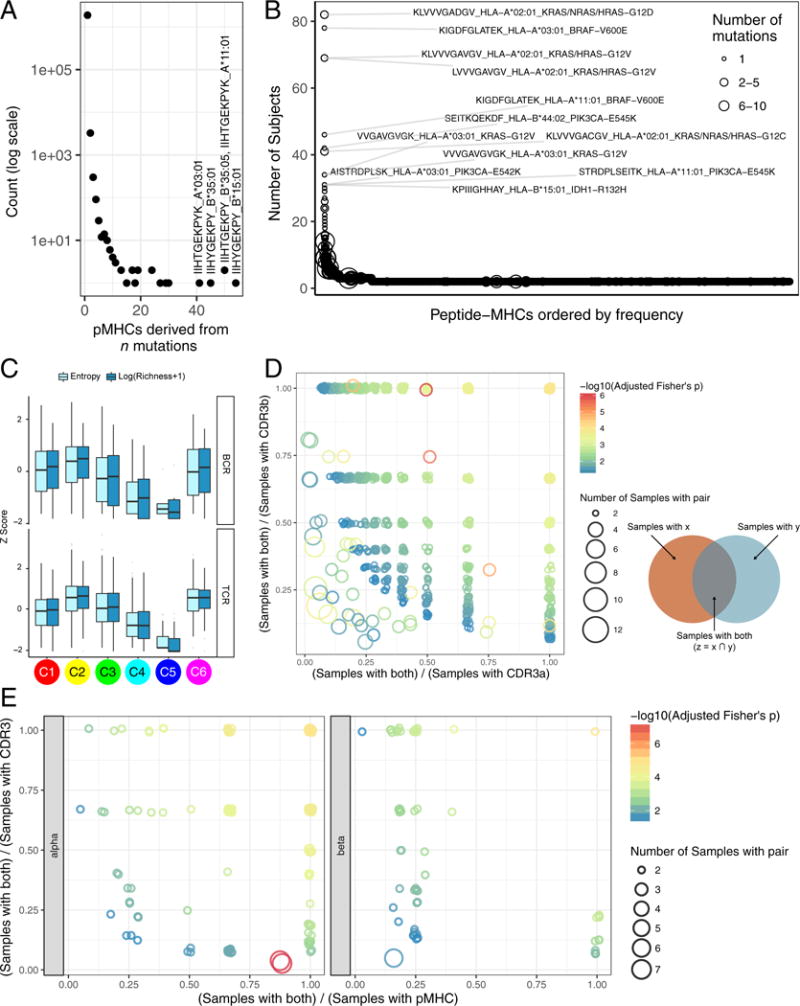
A. Distribution of the number of pMHCs associated with number of mutations; the 4 pMHCs derived from >40 mutations are labeled. B. Numbers of tumors expressing shared pMHCs. The known cancer genes from which the most frequent pMHCs in the population are derived are indicated C. Top BCR and Bottom TCR diversity measured by Shannon entropy and species richness, logarithmically transformed, and expressed as Z-scores, for immune subtypes. D & E. Co-occurrence of CDR3a-CDR3b (D) and pMHC-CDR3 pairs (E) as a surrogate marker for shared T cell responses. Pairs found in at least 2 samples and meeting statistical significance are plotted, with jitter. X and Y axes indicate how exclusive the pair members are: pairs in the top right typically co-occur, whereas along the axes each member is more often found separately. Size of the circle indicates how many samples that pair was found in. See also Figure S5 and Tables S3, S4 and S5.
Cancer testis antigens (CTA) overall expression, and that of individual CTAs, varied by immune subtype with C5 having the highest (p<10−13) and C3 the lowest (p=10−4) expression values (Figure 1C). CEP55, TTK, and PBK were broadly expressed across immune subtypes, with enrichment in C1 and C2. C5 demonstrated high expression of multiple CTAs, illustrating that CTA expression alone is insufficient to elicit an intratumoral immune response.
We found Human Papilloma virus (HPV) in 6.2% of cases, mainly in CESC, GBM, HNSC, and KIRC, whereas Hepatitis B virus (HBV) and Epstein Barr Virus (EBV) were mainly found in LIHC and STAD, respectively. In a regression model of all tumors, high load of each virus type associated with immune features (Figure S5C, cancer-type adjusted). High EBV content associated strongly with high CTLA4 and CD274 expression, and low B cell signatures. High HPV levels associated with increased proliferation and Th2 cells, but low macrophage content. In contrast, high HBV levels associated with Th17 signal and δγ T cell content. These findings highlight the diverse effect of different viruses on the immune response in different cancer types.
Our findings suggest that pMHC burden and viral content impact immune cell composition, while CTAs have inconsistent effects on the immune response. Moreover, the effect of pMHC load on prognosis is disease specific and influenced by immune subtype.
The Adaptive Immune Receptor Repertoire in Cancer
Antigen-specific TCR and BCR repertoires are critical for recognition of pathogens and malignant cells, and may reflect a robust anti-tumor response comprising a large number of antigen-specific adaptive immune cells that have undergone clonal expansion and effector differentiation.
We evaluated TCR α and β immunoglobulin heavy and light chain repertoires from RNA-seq. Mean TCR diversity values differed by immune subtype, with the highest diversity in C6 and C2 (p<10−183, Wilcoxon, relative to all other subtypes; Figure 5C), and by tumor type (Figure S5D, lower panel). We saw recurrent TCR sequences across multiple samples (Figure S5E, Table S5), suggesting a common, but not necessarily cancer-related, antigen (the top recurrent TCRs include known mucosal associated invariant T cell sequences). We assessed co-occurrence of complementarity determining region 3 (CDR3) α and β chains, in order to determine the frequency of patients with identical TCRs (a surrogate marker for shared T cell responses). We identified 2812 α-β pairs present in at least 2 tumors (p≤0.05, Fisher’s Exact Test with Bonferroni correction; Figure 5D and Table S54). Likewise, testing for co-occurrence of specific SNV pMHC-CDR3 pairs across all patients identified 206 pMHC-CDR3 α pairs, and 196 pMHC-CDR3 β pairs (Figure 5E, Table S5). Thus, a minority of these patients appear to share T cell responses, possibly mediated by public antigens. That said, there is relatively little pMHC and TCR sharing among tumors, highlighting the large degree of diversity in TILs.
Higher TCR diversity only correlated with improved PFI in a few tumor types (BLCA, COAD, LIHC, and UCEC)(Figure S5F). Therefore, it may be more important for the immune system to mount a robust response against only a few antigens, than a diverse response against many different antigens.
The pattern of immunoglobulin heavy chain diversity was similar to that of TCR diversity (Figures 5C & S5D), with tumors showing significant variance in IgH repertoire diversity, suggesting differential B cell recruitment and/or clonal expansion within the tumor types.
Regulation of Immunomodulators
IMs are critical for cancer immunotherapy with numerous IM agonists and antagonists being evaluated in clinical oncology (Tang et al., 2018). To advance this research, understanding of their expression and modes of control in different states of the TME is needed. We examined IM gene expression, SCNAs, and expression control via epigenetic and miRNA mechanisms.
Gene expression of IMs (Table S6, Figure 6A) varied across immune subtypes, and IM expression largely segregated tumors by immune subtypes (Figure S6A), perhaps indicative of their role in shaping the TME. Genes with the greatest differences between subtypes (Figures 6B, S6B) included CXCL10 (BH-adjusted p<10−5), most highly expressed in C2 (consistent with its known interferon-inducibility) and EDNRB (BH adjusted p<10−5), most highly expressed in the immunologically quiet C5. DNA methylation of many IM genes, e.g. CD40 (Figure 6C), IL10 and IDO1, inversely correlated with gene expression, suggesting epigenetic silencing. 294 miRNAs were implicated as possible regulators of IM gene expression; among these, several associated with IMs in multiple subtypes (Figure S6C) including immune inhibitors (EDNRB, PD-L1 or VEGFA) and activators (CD28 or TNFRSF9). The immune activator BTN3A was one of the most commonly co-regulated IMs from the SYGNAL-PanImmune network (below). Negative correlations between miR-17 and BTN3A1, PDCD1LG2 and CD274 may relate to the role of this miRNA in maturation and activation of cells into effector or memory subsets (Liang et al., 2015).
Figure 6. Regulation of Immunomodulators.
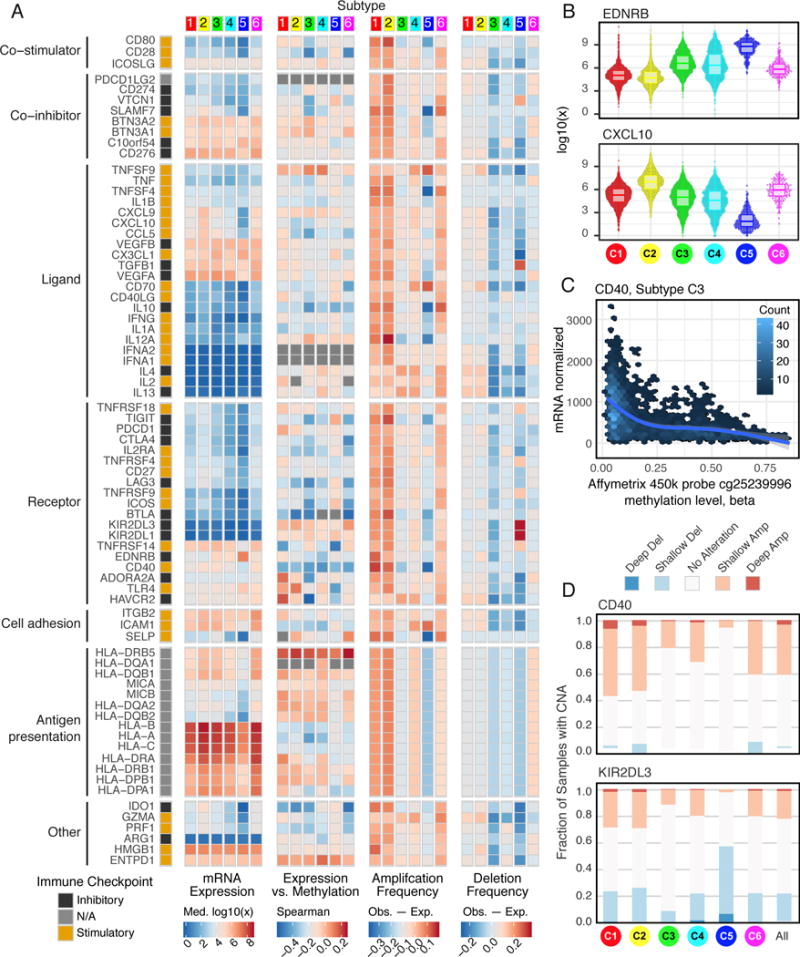
A. From left to right: mRNA expression (median normalized expression levels); expression vs. methylation (gene expression correlation with DNA-methylation beta-value); amplification frequency (the difference between the fraction of samples in which an IM is amplified in a particular subtype and the amplification fraction in all samples); and the deletion frequency (as amplifications) for 75 IM genes by immune subtype. B. Distribution of log-transformed expression levels for IM genes with largest differences across subtypes (by Kruskal-Wallis test). C. CD40 expression is inversely correlated to methylation levels (Affymetrix 450K probe cg25239996, 125 bases upstream of CD40 TSS) in C3. Each point represents a tumor sample, and color indicates point density. D. Proportion of samples in each immune subtype with copy number alterations in CD40 (top) and KIR2DL3 (bottom). The “All” column shows the overall proportion (8461 tumors). See also Figure S6 and Table S6.
Copy-number alterations affected multiple IMs and varied across immune subtypes. C1 and C2 showed both frequent amplification and deletion of IM genes, consistent with their greater genomic instability, while subtypes C3 and C5 generally showed fewer alterations in IM genes. In particular, IMs SLAMF7, SELP, TNFSF4 (OX40L), IL10, and CD40 were amplified less frequently in C5 relative to all samples, while TGFB1, KIR2DL1, and KIR2DL3 deletions were enriched in C5 (Figure 6D), consistent with our observation of lower immune infiltration with TGFB1 deletion (Figure S4A). CD40 was most frequently amplified in C1 (Figure 6D) (Fisher’s exact p<10−10 for all comparisons mentioned). Overall, these marked differences in IM copy number may be reflective of more direct modulation of the TME by cancer cells.
Among IMs under investigation for cancer therapy, expression of VISTA is relatively high in all tumor types, and highest in MESO; BTLA expression is high in C4, C5; HAVCR2 (TIM-3) shows evidence of differential silencing among immune subtypes, and IDO1 is amplified, mostly in C1. The observed differences in regulation of IMs might have implications for therapeutic development and combination immune therapies, and the multiple mechanisms at play in evoking them further highlights their biological importance.
Networks Modulating Tumoral Immune Response
The immune response is determined by the collective states of intracellular molecular networks in tumor, immune and other stromal cells and the extracellular network encompassing direct interaction among cells and communication via soluble proteins such as cytokines to mediate interactions among those cells.
Beginning with a large network of extracellular interactions known from other sources, we identified which of those met a specified precondition for interaction, namely that both interaction partners are consistently present within samples in an immune subtype, according to our TME estimates. We focused the network on IMs. Networks in C2 and C3 had abundant CD8 T cells, while C3, C4, and C6 were enriched in CD4 T cells.
A small sub-network (Figure 7A) focused around IFN-γ, illustrates some subtype-specific associations. In both C2 and C3, CD4 T cells, CD8 T cells, and NK cells correlated with expression of IFN-γ and CCL5, a potent chemoattractant. A second sub-network (Figure 7B), centered on TGFB1, was found in the C2, C3, and C6 networks. Across subtypes, different cell types were associated with abundant expression of TGFB1: CD4 T cells and mast cells in C3 and C6, macrophages in C6, neutrophils and eosinophils in C2 and C6, and B cells, NK cells and CD8 T cells in C2 and C3. The receptors known to bind TGFB1 were likewise were subtype specific and may help mediate the TGFB1 driven infiltrates, with TGFBR1, 2, & 3 found only in the C3 and C6 networks. These results largely echo findings seen in our TGF-β pathway analysis (Figure S4C) which examined the effects of intracellular, rather than extracellular, signaling disruption on immune TME composition across immune subtypes. Finally, a third cytokine subnetwork illustrates variation in T cell ligands and receptors across immune subtypes (Figure 7C). CD4 and CD8 receptors fell into two groups, those found in C2, C3, and C6 networks, such as PDCD1, and those absent in C3, such as IL2RA and LAG3. Some T cell associated ligands were subtype specific, such as CD276 (C2, C6), IL1B (C6), and VEGFB (C4).
Figure 7. Predicted Networks Modulating the Immune Response to Tumors.
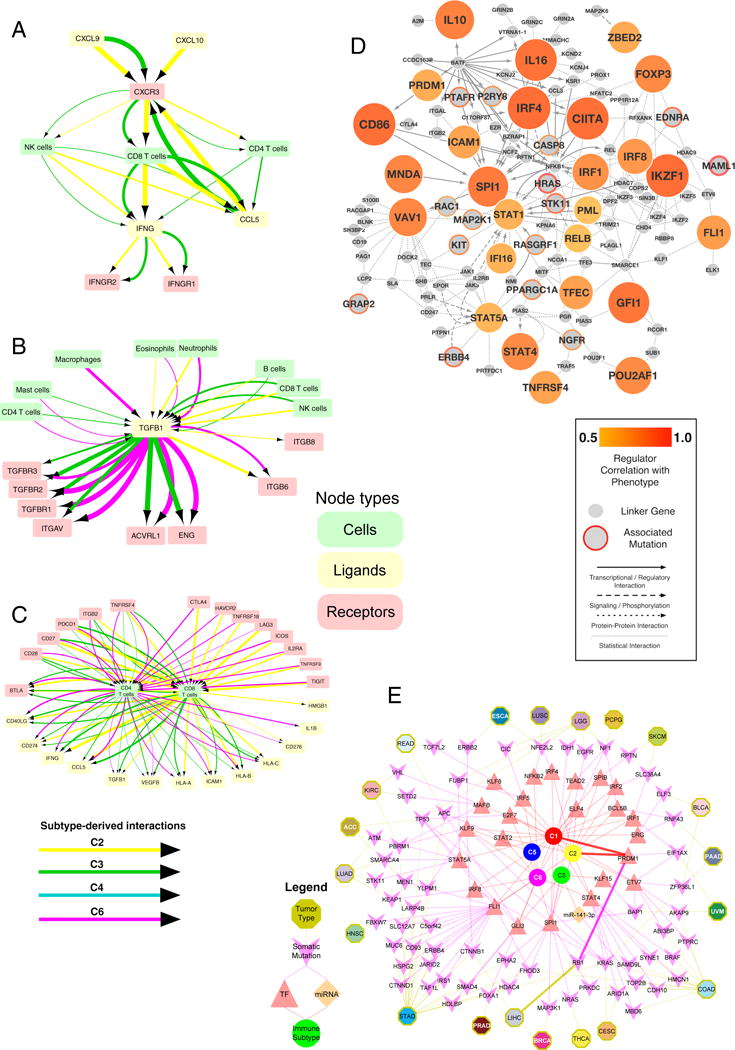
TME estimates and tumor cell characteristics were combined with available data on possible physical, signaling and regulatory interactions to predict cellular and molecular interactions involved in tumoral immune responses A. Immune subtype-specific extracellular communication network involving IFN-γ (IFNG, bottom of the diagram), whose expression is concordant with that of its cognate receptors IFNGR1 and IFNGR2 (bottom right and left, respectively), in C2 and C3 (yellow and green arrows, respectively; line thickness indicates strength of association). NK cells (left), which are known to secrete IFN-γ, could be producing IFN-γ in C2 and C3, as the NK cellular fraction is concordant with IFNG expression in both. CXCR3 is known to be expressed on NK cells, and has concordant levels, but only in C3 (green arrow). This is a subnetwork within a larger network constructed by similarly combining annotations of known interactions between ligands, receptors, and particular immune cells types, with evidence for concordance of those components. B. TGF-β subnetwork. Magenta: C6 C. T cell subnetwork. D. Master Regulator (MR) Pan-Immune Network. The network diagram shows 26 MRs “hubs” (filled orange) significantly associated with 15 upstream driver events (orange rings), along with proteins linking the two. The lineage factor VAV1 (on left) is inferred to be a MR by combining predicted protein activity with data on gene expression, protein interactions and somatic alterations. VAV1 activity correlates with LF (degree of correlation depicted as degree of orange). Mutations in HRAS (center of network), are statistically associated with changes in LF. The HRAS and VAV1 proteins are in close proximity on a large network of known protein-protein interactions (not shown), as both can lead to activation of protein MAP2K1, (as shown connecting with dotted lines). Mutations in HRAS are associated (p<0.05) with VAV1 activity, and their link through documented protein interactions implies that HRAS could directly modulate the activity of VAV1. In the diagram, the size of MR nodes represents their ranked activity. Smaller nodes with red borders represent mutated and/or copy-number altered genes statistically associated with one or more MR and LF, with the thickness of the border representing the number of associated MRs; small grey nodes are ‘linker’ proteins. E. Regulators of immune subtypes from SYGNAL-PanImmune Network. Tumor types (octagons) linked through mutations (purple chevrons) to transcription factors (TFs, red triangles) and miRNAs (orange diamonds) that actively regulate the expression of IMs in biclusters associated with a single immune subtype (circles). The network describes predicted causal and mechanistic regulatory relationships linking tumor types through their somatic mutations (yellow edges) which causally modulate the activity of TFs and/or miRNAs (purple edges), which in turn regulate genes (not shown) whose expression is associated with an immune subtype (red edges). For example, RB1 mutations in LIHC (5% of patients) have significant evidence for causally modulating the activity of PRDM1 which in turn regulates genes associated (causal model at least 3 times as likely as alternative models and p-value < 0.05) with C1 and C2. Interactions for this path are bolded.
The derived extracellular networks reflect the properties of immune subtypes in terms of cellular propensities and immune pathway activation noted earlier (Figures 1B,1C, 2A, S2A), but also place those properties in the context of possible interactions in the TME that may play a role in sculpting those same properties. The particular associations observed among IMs within distinct subtypes may be important for identifying directions for therapy.
We next used two complementary approaches, Master Regulators (MRs) and SYGNAL to synthesize a pan-cancer transcriptional regulatory network describing the interactions linking genomic events to transcriptional regulators to downstream target genes, and finally to immune infiltration and patient survival. In both approaches, somatic alterations were used as anchors to infer regulatory relationships, in that they can act as a root cause of the “downstream” transcriptional changes mediated through transcription factors (TFs) and miRNAs.
This resulted in two transcriptional networks. The first one, MR-PanImmune, consisted of 26 MRs that acted as hubs associated with observed gene expression and LF, connected with 15 putative upstream driver events (Figure 7D). The second one, SYGNAL-Panimmune, comprised 171 biclusters enriched in IMs and associated with LF.
Seven TFs were shared between the MR- and SYGNAL-PanImmune networks, a significant overlap (p = 4.8 × 10−10, Fisher’s exact test): PRDM1, SPI1, FLI1, IRF4, IRF8, STAT4 and STAT5A. Additional MRs included the hematopoietic lineage specific factor IKZF1, which may reflect variation in immune cell content, and known IMs, such as IFN-γ, IL16, CD86, and TNFRSF4. The regulators in SYGNAL-PanImmune were inferred to regulate a total of 27 IM genes (Figure S7C). The top two most commonly co-regulated IMs from SYGNAL-PanImmune, BTN3A1 and BTN3A2, are of particular interest as they modulate the activation of T cells (Cubillos-Ruiz et al., 2010) and have antibody-based immunotherapies (Benyamine et al., 2016; Legut et al., 2015).
Somatic alterations in AKAP9, HRAS, KRAS and PREX2 were inferred to modulate the activity of IMs according to both the MR- and SYGNAL-PanImmune, a significant overlap (p=1.6×10−7, Fisher’s exact test). In MR-PanImmune, MAML1 and HRAS had the highest number of statistical interactions with 26 MRs. This analysis identified complex roles for the RAS-signaling pathway (Figure 7D) specifically through connections to lineage factor VAV1 (implicated in multiple human cancers), potentially mediated by MAP2K1. Similarly, MAML1, hypothesized to mediate cross-talk across pathways in cancer (McElhinny et al., 2008), was associated (p≤0.05) with multiple MRs, including STAT1, STAT4, CIITA, SPI1, TNFRSF4, CD86, VAV1, IKZF1 and IL16.
In SYGNAL-PanImmune some regulators of IMs, but not upstream somatic mutations were shared between tumor types, including STAT4, which regulated BTN3A1 and BTN3A2 in both LUSC and UCEC, secondary to implied causal mutations TP53 and ARHGAP35, respectively. Conversely, causal mutations shared across tumor types may associate with different tumor-specific downstream regulators. TP53 was a causal mutation in UCEC acting through IRF7 to regulate many of the same IMs as was seen in LUSC. These differences in causal relationships arise because the different cell types giving rise to each tumor type affect oncogenic paths.
We identified the putative regulators of immune gene expression within immune subtypes (Figure 7E). In these predictions, C1-associated biclusters were regulated by ERG, KLF8, MAFB, STAT5A and TEAD2. C1 and C2 shared regulation by BCL5B, ETV7, IRF1, IRF2, IRF4, PRDM1 and SPIB, consistent with IFN-γ signaling predominance in these subtypes. C3 was regulated by KLF15 and miR-141-3p. C6 associated biclusters were regulated by NFKB2. C1, C2 and C6 shared regulation by STAT2 and STAT4, implying shared regulation by important immune TF families, such as STAT and IRF, but also differential employment of subunits and family members by the immune milieu.
In SYGNAL-PanImmune, the increased expression of biclusters enriched with IMs from KIRC, LGG, LUSC, and READ was associated with worse patient survival (CoxPH BH adjusted p-value ≤ 0.05). Conversely, the increased expression of biclusters enriched with IMs from SKCM, containing CCL5, CXCL9, CXCL10, HAVCR2, PRF1 and MHC class II genes, were associated with improved patient survival (BH-adjusted p≤ 0.05).
Discussion
We report an extensive evaluation of immunogenomic features in over 10,000 tumors from 33 cancer types. Data and results are available as Supplemental Tables, at NCI GDC and interactively at the CRI iAtlas portal, which is configured to accept new immunogenomics datasets and feature calculations as they come available, including those from derived from immunotherapy clinical trials, to develop as a “living resource” for the immunogenomics community. Meta-analysis of consensus expression clustering revealed immune subtypes spanning multiple tumor types, and characterized by a dominance of either macrophage or lymphocyte signatures, T-helper phenotype, extent of intratumoral heterogeneity, and proliferative activity. All tumor samples were assessed for immune content by multiple methods. These include the estimation of immune cell fractions from deconvolution of gene expression and DNA methylation data, prediction of neoantigen-MHC pairs from mutations and HLA-typing, and evaluation of BCR and TCR repertoire from RNA-sequencing data. Immune content was compared among immune and cancer subtypes, and somatic alterations were identified that correlate with changes in the TME. Finally, predictions were made of regulatory networks that could influence the TME, and intracellular communication networks in the TME, based on integrating known interactions and observed associations. Immunogenomic features were predictive of outcome, with OS and PFS differing between immune subtypes both within and across cancer types.
C4 and C6 subtypes conferred the worst prognosis on their constituent tumors, and displayed composite signatures reflecting a macrophage dominated, low lymphocytic infiltrate, with high M2 macrophage content, consistent with an immunosuppressed TME for which a poor outcome would be expected. In contrast, tumors included in the two subtypes displaying a Type I immune response, C2 and C3, had the most favorable prognosis, consistent with studies suggesting a dominant Type I immune response is needed for cancer control (Galon et al., 2013). In addition, C3 demonstrated the most pronounced Th17 signature, in agreement with a recent systematic review suggesting that Th17 expression is generally associated with improved cancer survival (Punt et al., 2015). C2 was IFN-γ dominant, and showed a less favorable survival despite having the highest lymphocytic infiltrate, a CD8 T cell associated signature, and highest M1 content, suggesting a robust anti-tumor immune response. One explanation for this discrepancy is the aggressiveness of both the tumor types and specific cases within C2 relative to C3. C2 showed the highest proliferation signature and ITH while C3 was the lowest in both those categories. It may be that the immune response simply could not control the rapid growth of tumors comprising C2. A second hypothesis is that tumors in C2 are those that have already been remodeled by the existing robust Type I infiltrate and have escaped immune recognition. While signatures biased towards interferon-mediated viral sensing and antigen presentation genes were often associated with higher survival, interferon signatures without increased antigen presentation showed an opposite association. Loss of genes associated with antigen processing and presentation is often found in tumors that have been immune edited. In contrast to the potential immune editing of C2, C3 may represent immunologic control of disease, that is, immune equilibrium.
Possible impact of somatic alterations on immune response was seen. For example, KRAS mutations were enriched in C1 and but infrequent in C5, suggesting that mutations in driver oncogenes alter pathways that affect immune cells. Driver mutations such as TP53, by inducing genomic instability, may alter the immune landscape via the generation of neoantigens. Our findings confirmed previous work showing that mutations in BRAF (Ilieva et al., 2014) enhance the immune infiltrate while those in IDH1 diminish it (Amankulor et al., 2017). Further work is needed to determine the functional aspects of these associations.
Tumor-specific neoantigens are thought to be key targets of anti-tumor immunity, and are associated with improved OS and response to immune checkpoint inhibition in multiple tumor types (Brown et al., 2014). We found OS correlated with pMHC number in only a limited number of tumors, with no clear association in most tumors, including several responsive to immune checkpoint inhibitor therapy. There are some caveats to this finding. The current predictors are highly sensitive, but poorly specific for neoantigen identification, and our approach did not include neoantigens from introns or spliced variants. Moreover, it is not possible to fully determine the ability to process and present an epitope or the specific T cell repertoire in each tumor, which impacts the ability to generate a neoantigen response. It is also possible that the role of neoantigens may vary with tumor type, as supported by our per-tumor results.
Integrative methods predicted tumor-intrinsic and tumor-extrinsic regulation in, of, and by the TME, and yielded information on specific modes of intracellular and extracellular control, the latter reflecting the network of cellular communication among immune cells in the TME. The resulting network was rich in structure, with mast cells, neutrophils, CD4 T cells, NK cells, B cells, eosinophils, macrophages, and CD8 T cells figuring prominently. The cellular communication network highlighted the role of key receptor and ligands such as TGFB1, CXCL10 and CXCR3, and receptor-ligand pairs, such as the CCL5-CCR5 axis, and illustrated how immune cell interactions may differ depending on the immune system context, manifested in the immune subtype.
Predicted intracellular networks implied that seven immune related TFs(including interferon and STAT-family transcription factors) may play an active role in transcriptional events related to leukocyte infiltration, and that mutations in six genes (including Ras-family proteins) may influence immune infiltration. Across tumor types the TFs and miRNAs regulating the expression of IMs tended to be shared, while somatic mutations modulating those regulatory factors tended to differ. This suggests that therapies targeting regulatory factors upstream of IMs should be considered, and that they may have a broader impact across tumor types than therapies focusing on somatic mutations. Of note, in these approaches, it is not always possible to fully ascertain if some particular interactions act in the tumor, immune, or stromal cell compartments, but this could be improved on by incorporating additional cell-type specific knowledge. Shared elements of intra- and extracellular network models should also be explored, with particular regard to the IMs and cytokines in both.
There are important caveats to using TCGA data. First, survival event rates and follow-up durations differ across the tumor types. Second, for most tumor types, samples with less than 60% tumor cell nuclei by pathologist review were excluded from study, thus potentially removing the most immune-infiltrated tumors from analysis. The degree to which this biases the results, relative to the general population of cancer patients, is difficult to ascertain. Our analyses were also limited by restriction to data from genome-wide molecular assays, in absence of targeted classical cellular immunology assays for confirming cell phenotype distribution, as those types of data have not been collected from TCGA patients.
In summary, six stable and reproducible immune subtypes were found to encompass nearly all human malignancies. These subtypes were associated with prognosis, genetic, and immune modulatory alterations that may shape the specific types of immune environments we have observed. With our increasing understanding that the tumor immune environment plays an important role in prognosis as well as response to therapy, the definition of the immune subtype of a tumor may play a critical role in the predicting disease outcome as opposed to relying solely on features specific to individual cancer types.
STAR ★ Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Vesteinn Thorsson (Vesteinn.Thorsson@systemsbiology.org)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
A total of 11,180 participants were included in this study. This study contained both males and females, with inclusions of genders dependent on tumor types. There were 5,621 females, 5,138 males and 321 with missing information about gender. TCGA’s goal was to characterize adult human tumors; therefore, the vast majority of participants were over the age of 18. However, 20 participants under the age of 18 had tissue submitted prior to clinical data. Age was missing for 188 participants. The range of ages was 10–90 (maximum set to 90 for protection of human subjects) with a median age of diagnosis of 60 years of age. Institutional review boards at each tissue source site reviewed protocols and consent documentation and approved submission of cases to TCGA. Detailed clinical, pathologic and molecular characterization of these participants, as well as inclusion criteria and quality control procedures have been previously published for each of the individual TGCA cancer types.
Sample Inclusion Criteria
Surgical resection of biopsy biospecimens were collected from patients that had not received prior treatment for their disease (ablation, chemotherapy, or radiotherapy). Cases were staged according to the American Joint Committee on Cancer (AJCC). Each frozen primary tumor specimen had a companion normal tissue specimen (blood or blood components, including DNA extracted at the tissue source site). Adjacent tissue was submitted for some cases. Specimens were shipped overnight using a cryoport that maintained an average temperature of less than −180°C.
Pathology quality control was performed on each tumor and normal tissue (if available) specimen from either a frozen section slide prepared by the TCGA Biospecimen Core Resource (BCR) or from a frozen section slide prepared by the Tissue Source Site (TSS). Hematoxylin and eosin (H&E) stained sections from each sample were subjected to independent pathology review to confirm that the tumor specimen was histologically consistent with the submitted diagnosis; as required, tumor reclassification and/or exclusion was performed by expert pathology review. Pathology review also confirmed that the adjacent non-neoplastic “normal” tissue specimen contained no tumor cells. For cases of LIHC, adjacent tissue with cirrhotic changes was not acceptable as a germline control, but was characterized if accompanied by DNA from a patient-matched blood specimen. The percent tumor nuclei, percent necrosis, and other pathology annotations were also assessed. Tumor samples with ≥ 60% tumor nuclei and ≤ 20% necrosis were submitted for nucleic acid extraction.
METHOD DETAILS
Clinical and Molecular Data
The standardized, normalized, batch corrected and platform-corrected data matrices and mutation data generated by the PanCancer Atlas consortium, available at the publication page (https://gdc.cancer.gov/about-data/publications/pancanatlas), were used in this study. Gene expression, protein, and miRNA expression, DNA methylation, copy number variation, and gene mutations were obtained for this study for 11,080 participants. TCGA aliquot barcodes flagged as “do not use” or excluded by pathology review by the PanCancer Atlas Consortium, and annotated according to the Merged Sample Quality Annotation file were removed from the study. For somatic mutations FILTER values were required to be one of PASS, wga, or native_wga_mix, and only protein coding mutations retained (Variant_Classification one of Frame_Shift_Del, Frame_Shift_Ins,In_Frame_Del,In_Frame_Ins,Missense_Mutation, Nonsense_Mutation,Nonstop_Mutation,Splice_Site, and Translation_Start_Site). Mutations calls were required to be made by two or more mutations callers (NCALLERS>1). Where both normal tissue and blood was available as reference, the blood reference sample was used. The values of OS, OS.time, PFI, and PFI.time were used from obtained from (Liu et al., 2018)
Immune-related tumor sample characteristics and selected base data values such as demographic information, survival data and expression of key immumodulators for the 11,080 participants were collected into a per participant summary matrix (Table S1). For the molecular data matrices above, a single representative aliquot was selected per participant for cases where more than one aliquot was available, as follows. When data on more than one tumor sample was available, a choice of primary tumor sample was favored, and in remaining cases metastatic were selected over “additional metastatic”. For gene expression, a handful of cases were not resolved by these rules and the following aliquots were adopted TCGA-23-1023: TCGA-23-1023-01A-02R-1564-13; TCGA-06-0156-01:TCGA-06-0156-01A-02R-1849-01; TCGA-06-0211-01:TCGA-06-0211-01B-01R-1849-01; TCGA-21-1076-01:TCGA-21-1076-01A-01R-0692-07 based on BCR annotations. Each primary data file was loaded into a Google BigQuery table on the ISB Cancer Genomics Cloud, annotated with uniform TCGA barcode information, permitting integration of heterogeneous sources into a single matrix through cloud queries.
Contributors: Vesteinn Thorsson, David L. Gibbs,Tai-Hsien Ou Yang, Dante Bortone, Katherine Hoadley, Tathiane Malta, Houtan Noushmehr
TCGA Molecular Subtypes
Previously published TCGA molecular subtypes from multiple tumor types were collected and compiled into a single matrix. A total of 7,734 TCGA samples were annotated with with molecular subtypes based on TCGA Research Network tumor-specific publications for the following tumor types: ACC, AML, BLCA, BRCA, LGG/GBM, Pan-GI (ESCA/STAD/COAD/READ), HNSC, KICH, KIRC, KIRP, LIHC, LUAD, LUSC, OVCA, PCPG, PRAD, SKCM, THCA, UCEC, and UCS, with publication sources detailed on http://bioinformaticsfmrp.github.io/TCGAbiolinks/subtypes.html. The unified patient-centric matrix contains a comprehensive collection of the subtypes by molecular platform. Each column contains subtype assignments of a particular molecular platform (e.g., mRNA, DNA methylation, protein). We selected the most prominent subtype classification of a particular tumor type based on the corresponding paper recommendation and stored this information in column named “Subtype_Selected”. The subtype collection matrix and the bibliography associated with them are available within TCGAbiolinks on R/Bioconductor (http://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html) (Colaprico et al., 2016) and using the TCGAbiolinksGUI (Silva et al., 2016). The function “PanCancerAtlas_subtypes()” provides full access to the curated matrix used for this study. The “Subtype_Selected” column was used for molecular subtypes in this study.
Immune Subtype Identification
Immune Signature Compilation
We undertook an extensive literature search and assembled a collection of 160 immune expression signatures utilizing diverse resources which were considered to be reliable and comprehensive, based on the expert opinions of immune-oncologists in the group. Of these signatures, 83 were derived in the context of studies of the immune response in cancer and the remaining 77 are of general validity for immunity. The 83 signatures that are known to be associated with immune activity in tumor tissue consisted of 68 gene sets collected from earlier studies (Wolf et al., 2014), 9 co-expression signatures derived from computational analysis of all TCGA gene expression data sets (immune metagene attractors), (Cheng et al., 2013a, b), 3 signatures representing the functional orientation of the immune contexture (or Immunologic Constant of Rejection, ICR) (Bedognetti et al., 2016; Galon et al., 2013; Hendrickx et al., 2017), and 3 signatures from a recent study characterizing the immune microenvironment of clear cell renal cell carcinoma (Senbabaoglu et al., 2016). The 77 more general signatures comprised scores of 45 signatures representing individual cell types from two sources (20 from (Gentles et al., 2015) and 25 from (Bindea et al., 2013)) and 32 scores encompassing the dominant modes of scores derived from the ImmuneSigDB (Godec et al., 2016)(Collection C7 of MSigDB, Broad Institute). The modes were determined as the first 32 principal components of 1888 Immune C7 human gene sets, and were used as the full set was overwhelmingly large and complex. Gene sets were scored using single-sample gene set enrichment (ssGSEA) analysis (Barbie et al., 2009), as implemented in the GSVA R package.
Immune Signature Cluster Modeling
All available TCGA tumor samples (n=9126) were scored for each of the 160 identified gene expression signatures. Prior to model-based clustering, we began by identifying a limited set of distinct and representative gene signatures to use for the model-based clustering analysis based on consensus clustering of all available gene signature scores over all available samples. Initial data exploration using all 160 gene sets implied that including the 77 more general immune signatures did not affect the identified signature clusters, and we performed the final analysis with the 83 signatures derived in the cancer immune response context. Representative clusters were identified as follows: two independent analysts used weighted gene correlation network analysis (WGCNA) to produce clusters of signatures (Langfelder and Horvath, 2008). First, using gene set scores (ssGSEA) (Barbie et al., 2009) over all samples, Spearman correlations were computed between signatures creating a correlation matrix. Then, the correlation matrix was scaled by taking each element to a specified power and clustered using the WGCNA R package. Various WGCNA parameters were explored, but good results were found with TOMType=“signed”, power=18, pamStage=F, minModuleSize=3. Each identified module contained an ‘eigen-signature’ which is used to identify possible “most representative” gene expression signatures from those contained in the cluster module by computing a distance from each signature to the ‘eigen-signature’. Signatures having short distances to the eigen-signature would be considered to be more representative of the signature-module.
Representative Gene Signature Identification
Results from the 2 independent WGCNA analyses yielded 9 potential signatures considered representative of identified module eigen-signatures. We then evaluated each of the potential representative signatures using the strategy put forth in “cluster validation by predictive strength” (Tibshirani and Walther, 2005). This strategy involves building cluster-models using random subsets of samples, and making cluster predictions on the remaining unclustered samples. The predicted cluster labels are compared across models built from random sample subsets. For sets of features that produce strong clustering models, the labels will be consistent.
To do this work, model based clustering, performed with the mclust R package (Scrucca et al., 2016), which uses finite normal mixture modelling, was in part selected as it can readily handle the large set of scores from the Pancancer Atlas (9,129 samples). This approach identified 3 of the potential signatures as lacking robustness and they were excluded from further analysis.
Finally, the actual genes contained in each of the potential signatures were examined by an expert in the immuno-oncology field for validity (Nora Disis), and one of two highly similar IFN signatures was excluded for redundancy. This left five final representative gene signatures, each standing in for one of five signature-similarity modules (Figure 1A, top). The five identified representative signatures are: “CSF1_response” for activation of macrophages/monocytes (Beck et al., 2009)(referred to throughout text and figures as “Macrophage”, “LIexpression_score” representing overall lymphocyte infiltration, and dominated by B and T cell signatures (Calabro et al., 2009) (referred to throughout text and figures as “Lymphocyte”), TGF-β response “TGFB_score_21050467” (Teschendorff et al., 2010)(“ TGF-β” in text and figures), “Module3_IFN_score” representing IFN-γ response (Wolf et al., 2014)(“IFN-γ” in text and figures), and wound healing “CHANG_CORE_SERUM_RESPONSE_UP”(Chang et al., 2004) (“Wound healing” in text and figures).
Using the final five signatures to cluster TCGA tumor samples, the number of clusters, K, was determined using scores that were median centered and scaled by median-absolute-deviation (MAD). Possible values for K (the number of clusters) ranged from 2 to 32. Then, 21 random subsets, each representing 50% of 9,129 TCGA aliquots, (from 9,126 participants) were selected and mclust models were fit to each subset, resulting in 21 clustering models. In each model, the parameter K was selected that maximized the Bayesian Information Criterion (BIC), and an average K was computed. Maximal BIC was found to occur with a six cluster solution, thus 6 clusters were used for the remainder of analyses.
An ensemble approach was used to improve predictability and increase robustness. To produce the final clustering, 256 sub-samples were taken (each representing a random 50% of 9,129 samples), and a model was fit to each sub-sample, setting K=6. Then, the “GV1” method in the R package ‘clue’ (CLUster Ensembles) was used to call the consensus clusters (Hornik, 2005). This method takes the list of 256 clusterings, each containing a subset of the samples, and produces a consensus cluster by minimizing an objective function. The entire process was performed twice to ensure reproducibility.
Contributors: David L. Gibbs, Denise Wolf, Vesteinn Thorsson, Benjamin Vincent, Ilya Shmulevich
Validation of Model-based Clustering
To determine the robustness of model-based clustering, we performed an analysis in which the samples were partitioned into training and test sets in varying proportions that ranged from 0.5% to 30%. The training set was used to build the ensemble model, which in turn is used to predict cluster labels on the test set (the held-out samples). The clustering of the training and test sets was compared to results from the full model using all samples. 20 repetitions were performed. Cluster purity (CP, not to be confused with tumor purity) and Normalized mutual information (NMI) were used to evaluate the training and test results. Cluster-purity describes the fraction of the most common label within a cluster. So, if 9/10 members of a cluster (from the reported clustering) share a label, then the purity is 90%. Secondly, the NMI describes the mutual information between our new clusters and reported clusters, normalizing by average entropy which puts it on a scale of zero to one. Considering both the test set and training set, when the proportion of samples removed was less than 16%, the NMI averaged greater than 0.9, which indicates an excellent level of similarity to the full model. When 32% of samples were removed, the NMI was 0.81 and 0.82 respectively, still indicating very good concordance. In both above cases (training and test) when 16% of samples were held-out, cluster purity (CP) levels were greater than 95%. Overall, there is very good NMI and CP scores found when removing even up to 32% of samples (2,921samples held out).
Of note, using cluster purity (CP), the training set maintained levels above 89% even when 32% of the samples were missing. The exception being C6, which is noisy and had a purity level of 72% when 32% of samples were removed. The test set prediction results showed slightly better CP, with 32% missing samples, purity levels for all subtypes were greater than 90%, the exception being C6 which had purity 71%. In addition, we explored the extent to which clustering results vary when different, but correlated, signatures are used. In clustering, the results (the cluster labels assigned to samples) are always dependent on the inputs, or in this case, the signatures. It is often the case that by using different signatures, the clustering structure will change. The question we aim to answer with this is: if one uses related signatures, how different is the clustering structure? In each iteration, either one or two signatures was randomly selected from the 5 main signatures. The selected signature was then replaced with a signature(s) that was sampled with probability proportional to the correlation structure (as seen in the heatmap of gene set signatures). After the replacement of a signature (one or two), the complete ensemble clustering model was constructed, and new clusters called. Again, cluster purity and normalized mutual information were used to evaluate the clustering results.
In total, using the full set of available signatures, 363 new cluster models were constructed, and across clusters (C1-C6) we found that as new replacement signatures have greater correlation with the original signatures, the NMI gradually increases. Starting from ~0.4 for single replacements and ~0.35 for double replacements. As the replacement signature correlation increases past 0.95, we saw NMIs of 0.7 to 0.8 which indicate between 8-15% of cluster labels changing. Using cluster purity we found a similar effect where increasing levels of correlation with the replacement signatures produced higher levels of purity. There are several exceptions. The C5 cluster is very robust regardless of the replacement signature with purity levels above 90%. The C6 cluster is (as above) very noisy with purity levels around 50-60%. Among the remainder of the clusters (C1-C4), the C3 cluster shows the lowest levels of purity with an average of 0.80 when the signature correlation is greater than 0.95. When the correlation drops to 0.9, the purity level for C3 drops to 70%. Overall, while the purity levels gradual increase with signature correlation, the exception is C3 where the variance in purity values was relatively strong, indicating that the cluster was splitting. As the field moves forward, it is likely that we will see a more detailed classification of samples found in C3.
Contributor: David L. Gibbs
Biclustering of Immune-Expression Signatures
As another measure of the robustness of the above model based sample clustering, we applied an entirely different clustering method, iterative binary biclustering using iBBiG (Gusenleitner et al., 2012). The iterative biclustering identifies similarity blocks within the matrix of signature scores, but with tumor sample groups (clusters) that are to allowed to overlap, unlike the model-based clustering. We analyzed the total 160 gene signature score sets using iBBiG, which and this yielded 15 biclusters. Model-based clustering and biclustering have commonalities both in terms of shared tumor sample groupings and in the association of clusters to phenotypes, as evidenced by 13 significant overlaps between the biclusters and the six immune subtypes according to a hypergeometric test. Comparing functional annotations of these clusters, we found that overlap to be reflected in the concordant distribution of mean scores of IFN-γ, TGF-β, mutation load and overall leukocyte infiltrate among the overlapping clusters.
Contributors: Aedin Culhane, Azfar Basunia
Leukocyte and Stromal Fractions
Methylation Analysis
Overall leukocyte content in 10,817 TCGA tumor aliquots was assessed by identifying DNA methylation probes with the greatest differences between pure leukocyte cells and normal tissue, then estimating leukocyte content using a mixture model. From Illumina Infinium DNA methylation platform arrays HumanMethylation450, 2000 loci were identified (200 for HumanMethylation27) that were the most differentially methylated between leukocyte and normal tissues, 1000 in each direction. For each locus i, assuming two populations (j), for each sample we have
Using the tumor with the least evidence of leukocyte methylation as a surrogate for the beta value (β) for each locus in the pure tumor, 2000 estimates were made, solving for π. We took the mode of 200 estimates to avoid loci that violate the assumptions. Using the estimated π and the measured β for tumor and leukocyte, with the same linear model, solved for β (deconvoluted value) extracting the leukocyte fraction (LF). Estimates for DLBC, THYM, LAML were masked as their tissues of origin are expected to be related to leukocytes, and therefore there were not enough tissue-specific DNA methylation loci to distinguish the two.
Stromal fraction (SF) was defined as the total non-tumor cellular component, obtained by subtracting tumor purity from unity, with the leukocyte proportion of stromal content R=LF/SF. Tumor purity was generated using ABSOLUTE (Carter et al., 2012) by the TGCA Research Network,(Taylor et al., 2018). R was estimated by the Pearson correlation coefficient between SF and LF, ρ, assessed for individual sample groups (TCGA tumor types, subtypes, and immune subtypes).
Contributors: Hui Shen, Vesteinn Thorsson
Whole-Slide Image Analysis
Characterization of tumor-infiltrating lymphocytes (TILs) from TCGA H&E images was carried out using deep learning-based lymphocyte classification with Convolutional Neural Networks (CNNs) (TCGA Research Network, “Spatial Organization And Molecular Correlation Of Tumor Infiltrating Lymphocytes Using Deep Learning On Pathology Images,” unpublished data). TIL infiltrated regions are presented as heatmaps overlaying H&E diagnostic images, allowing pathologists to curate those heatmaps to create final lymphocyte distribution maps. The tool was trained by experts to delineate lymphocyte-infiltrated tumor regions for each slide. In a whole slide image, a given small region of 50×50 microns is considered lymphocyte infiltrated if and only if 1) the predicted probability of lymphocyte infiltration is above a threshold and 2) the patch is not classified as necrotic tissue. The associated software provides a visual interface for threshold selection but due to the large number of whole slide images, we developed the following semi-automatic method for setting thresholds. We select ten patches for each whole slide image stratified by predicted probability. The whole slide images are then grouped into a small number of categories (seven) based on the agreement between predicted probabilities and pathologist labels. We sample eight slides per category and select thresholds visually based on the heatmap overlaying images. The averaged threshold is used for all slides in the same category. TCGA tumor types analyzed were LUAD, BRCA, PAAD, COAD, LUSC, PRAD, UCEC, READ, BLCA, STAD, CESC, SKCM and UVM. We began with generating 48K labeled patches to train our model for LUAD and incrementally added additional patches as necessary to train the model for BRCA, PAAD, COAD, LUSC, PRAD, UCED, READ, BLCA, STAD, CESC (in that order). For each new cancer type, we first applied the trained deep learning model. Pathologists then reviewed the results on a set of sample whole slide images. If the pathologists judged that the lymphocyte classification was inadequate, we retrained the model with additional training patches extracted from the new given cancer type, repeating this process until adequate accuracy was obtained. The deep learning model for the two melanoma types – SKCM and UVM was trained separately. The TIL regional fraction was estimated obtained as the number of TIL positive 50×50 micron regions over the total number of those 50×50 micron regions on the tissue image.
Contributors: Joel Saltz, Arvind U.K. Rao, Alexander J. Lazar, Ashish Sharma
Immune Cellular Fraction Estimates
The relative fraction of 22 immune cell types within the leukocyte compartment were estimated using CIBERSORT (Newman et al., 2015). These proportions were multiplied by LF to yield corresponding estimates in terms of overall fraction in tissue. Further, values were aggregated in various combinations to yield abundance of more comprehensive cellular classes, such as lymphocytes, macrophages and CD4 T cells. More specifically, we applied CIBERSORT to TCGA RNASeq data. CIBERSORT (cell-type identification by estimating relative subsets of RNA transcripts) uses a set of 22 immune cell reference profiles to derive a base (signature) matrix which can be applied to mixed samples to determine relative proportions of immune cells. As several key immune genes used in the signatures are absent from TCGA GAF (Generic Annotation File) Version 3.0, we applied CIBERSORT to a re-quantification of the TCGA data using Kallisto (Bray et al., 2016) and the Gencode GTF (Harrow et al., 2012)(available from https://www.gencodegenes.org/), which includes the missing genes. A version of the entire TCGA RNA-seq data normalized to Gencode with Kallisto was computed on the ISB Cancer Genomics Cloud by Steve Piccolo’s group at BYU (https://osf.io/gqrz9/wiki/home/) (Tatlow and Piccolo, 2016).
In order to relate to results to other estimates in this study, three aggregation schemes were defined as follows
Aggregate 1
(6 classes; Used in Figure 2A, e.g.)
Lymphocytes=B.cells.naive+B.cells.memory+T.cells.CD4.naive+T.cells.CD4.memory.resting+T.cells.CD4.memory.activated+T.cells.follicular.helper+T.cells.regulatory..Tregs+T.cells.gamma.delta+T.cells.CD8+NK.cells.resting+NK.cells.activated+Plasma.cells,
Macrophages=Monocytes + Macrophages.M0 + Macrophages.M1 + Macrophages.M2
Dendritic.cells=Dendritic.cells.resting + Dendritic.cells.activated,
Mast.cells=Mast.cells.resting + Mast.cells.activated,
Neutrophils=Neutrophils,
Eosinophils=Eosinophils,
Aggregate 2
(9 classes; used for cytokine network, including Figure 7A,B,C)
T.cells.CD8=T.cells.CD8,
T.cells.CD4=T.cells.CD4.naive+T.cells.CD4.memory.resting+T.cells.CD4.memory.activated,
B.cells=B.cells.naive + B.cells.memory,
NK.cells=NK.cells.resting+NK.cells.activated,
Macrophage=Macrophages.M0 + Macrophages.M1 + Macrophages.M2,
Dendritic.cells=Dendritic.cells.resting + Dendritic.cells.activated,
Mast.cells=Mast.cells.resting + Mast.cells.activated,
Neutrophils=Neutrophils,
Eosinophils=Eosinophils
Aggregate 3
(11 classes)
T.cells.CD8=T.cells.CD8,
T.cells.CD4=T.cells.CD4.naive+T.cells.CD4.memory.resting+T.cells.CD4.memory.activated+T.cells.follicular.helper+T.cells.regulatory..Tregs,
T.cells.gamma.delta=T.cells.gamma.delta,
B.cells=B.cells.naive + B.cells.memory,
NK.cells=NK.cells.resting+NK.cells.activated,
Plasma.cells=Plasma.cells,
Macrophage=Monocytes + Macrophages.M0 + Macrophages.M1 + Macrophages.M2,
Dendritic.cells=Dendritic.cells.resting + Dendritic.cells.activated,
Mast.cells=Mast.cells.resting + Mast.cells.activated,
Neutrophils=Neutrophils,
Eosinophils=Eosinophils
Contributors: Andrew Gentles, Vesteinn Thorsson, Alex J. Lazar, David L. Gibbs
Prognostic Correlations of Immune Phenotypes
Univariate Analysis
We first estimated the prognostic impact of immune subtypes on OS and PFI using Kaplan-Meier analysis and computed hazard ratios for each immune subtype relative to C1 in unadjusted models and in CoxPH models adjusted for tumor type. To further dissect the prognostic impact of individual gene expression signatures or immune cell types within immune subtypes and tumor types, we used the concordance index (CI) (Pencina and D’Agostino, 2004) to correlate the immune signatures and the cellular fractions with the outcomes (OS and PFI). The concordance index is defined by the relative frequency of accurate pairwise predictions of survival over all pairs of patients for which such a meaningful determination can be achieved. Samples with missing values in the features of interest or the outcomes were excluded from the analysis. Heatmaps were generated in R using the heatmap.2 function from the gplots package.
Contributors: Tai-Hsien Ou Yang, Dimitris Anastassiou
Multivariate Analysis
Elastic net regression was performed on primary tumor data to predict overall survival using glmnet in R (Friedman et al., 2010). Features tested included subtype scores, CIBERSORT data, immune gene signatures, TCR/BCR richness, neoantigen counts (Indel and SNV), lymphocyte fraction and average cancer testis antigen expression. Data were divided into discovery and validation sets (2/3 and 1/3 of the samples, respectively), which were balanced for survival events. The discovery set was further divided into test and training sets over 50 cross validation cycles across 20 alpha values to select optimal alpha and lambda values for the final model. Optimal parameters (alpha = 0.0022, lambda = 0.0066) were selected on model performance by taking the combination that produced the highest average C-Index. LOESS fit of the actual outcomes was plotted against the model predictions. The span for the LOESS fit was optimized by k-fold cross validation, using randomized training sets to fit the LOESS and testing the root mean square (RMS) of the residual in a test set. The LOESS span producing the smallest RMS was selected for the final fit. Confidence intervals were generated using bootstrapping with replacement using the optimized span.
For each immune subtype, Cox Proportional Hazards (CoxPH) modeling was done to determine whether belonging to that subtype predicts patient survival. These data were divided according to cancer tissue type. Bars were colored according to whether there was a negative or positive association with survival (blue or red outlines, respectively). A False Discovery Rate (FDR) correction using the BH method was applied to p-values for the addition of stars. If data were significant after FDR correction red stars were added to show significance with 1, 2 and 3 stars indicating FDR corrected values below 0.05, 0.01 and 0.001, respectively. Black stars indicate data that were only significant prior to FDR correction.
Contributors: Dante Bortone, Benjamin Vincent
Copy Number and DNA Damage Scores
All purity, ploidy, LOH and CNV calls used to generate the DNA damage scores used in this study and summarized below were generated by the TCGA Aneuploidy AWG using ABSOLUTE (Carter et al., 2012; Taylor et al., 2018). In brief, ABSOLUTE was run, using default parameters, on segmentation data generated from Affymetrix genome-wide human SNP6.0 arrays by hapseg and on SNV and indel calls from the MC3 variant file. All clonality calls for quantifying intratumoral heterogeneity (ITH) were also determined by ABSOLUTE, which models tumor copy number alterations and mutations as mixtures of subclonal and clonal components of varying ploidy. Specifically, for these analyses, ITH score was defined as the subclonal genome fraction (which measures the fraction of tumor genome that is not part of the “plurality” clone), as determined from ABSOLUTE.
Scores for copy number burden, aneuploidy, loss of heterozygosity, and homologous recombination deficiency (HRD) were derived (Knijnenburg et al., 2018). Copy number burden scores frac_altered and n_segs (“fraction altered”, and “number of segments”, respectively) represent the fraction of bases deviating from baseline ploidy (defined as above 0.1 or below - 0.1 in log2 relative copy number (CN) space), and the total number of segments in each sample’s copy number profile, respectively. LOH_n_seg and LOH_frac_altered are the number of segments with LOH events and fraction of bases with LOH events, respectively. HRD score is a measure quantifying defects in homologous recombination that sums 3 separate metrics of genomic scarring: large (>15 Mb) non-arm-level regions with LOH, large-scale state transitions (breaks between adjacent segments of >10 Mb), and subtelomeric regions with allelic imbalance.
Aneuploidy scores were calculated as the sum total of amplified or deleted (collectively “altered”) arms (Taylor et al., 2018). To call arm alterations, sample chromosome arms were first stratified by sample tumor type, type of alteration being tested (amplification or deletion), and chromosome arm (1p, 1q, etc.). The samples are then clustered using an n-component Gaussian Mixture Model fitted on that particular arm’s start coordinate, end coordinate, and percentage length of longest joined segment in that arm for each sample (segments were joined until the joined segment either encompassed the entire chromosome or achieved >20% contamination by segments not of that alteration type) for each sample. For each clustering, number of clusters n was chosen from 2-9 based on lowest Bayesian Information Criterion. Arms were designated as as altered if they belonged to a cluster of arms with mean fraction altered >=80%. Each segment was designated amplified, deleted, or neutral based on its copy number relative to the sample’s rounded ploidy.
Contributors: Galen Gao, Andrew Cherniack
Genomic Correlations with Immune Phenotypes
DNA Damage Scores
For each TCGA subtype containing at least 10 tumors, Spearman correlations were calculated between leukocyte fraction and measures of DNA alteration. Cohort-averaged correlation between DNA damage scores and leukocyte fraction was computed as the arithmetic mean of the Spearman correlation coefficients for each TCGA disease type considered individually.
Contributors: Galen Gao, Vesteinn Thorsson
Copy Number Variation
Amplification and deletion were defined as follows using a PanCan GISTIC2.0 run on the samples after performing In Silico Admixture Removal (ISAR) (Zack et al., 2013) on the relative copy number values using the ABSOLUTE-estimated purity and ploidy values of each sample (Mermel et al., 2011). For each tumor sample, the median copy-ratio for each chromosome arm is calculated. For each locus, a sample is called deep amplification if the value is +2 (i.e. higher than the maximum of these arm values), while a −2 (deep deletion) is a value less than the minimum of these values. Shallow (+/− 1) amplifications and deletions correspond to alterations between 0.1 relative copy number and the thresholds for deep alterations.
To determine correlations between gene amplification (GISTIC2.0 CN=1 or CN=2 as described above) and LF, expected mean leukocyte fraction for each gene was computed as the average of the mean leukocyte fractions for each individual TCGA disease type weighted by the number of “amplified” samples present in each disease type. One-sample t-tests were then used with BH multiple hypothesis correction to assess the significance of the difference between the observed mean LF among “amplified” samples and this expected mean LF. We report both this difference and its significance. This analysis was then repeated for “deleted” genes (GISTIC2.0 CN=−1 or CN=−2 as described above). Furthermore, for each gene, we similarly computed significances of differences of CIBERSORT-estimated relative immune cell subtype levels from their expected levels first in “amplified” and then in “deleted” samples in order to identify the effects of copy number amplification and deletion respectively on immune infiltrate composition while controlling for cancer disease type. Genes localized on the X chromosome were disregarded for all analyses.
Contributors: Galen Gao, Andrew Cherniack
Driver Gene Mutations
We focused our analysis on genes identified as drivers by the TCGA PanCancer Atlas Driver Mutation Working Group (the CGAT list; TCGA Research Network, “Comprehensive Discovery and Characterization of Driver Genes and Mutations in Human Cancers”, unpublished data) that were identified as 1) having 10 or more mutations overall and 2) mutated in two or more tissues. For each gene that fit these criteria, we created a three-dimensional matrix contingency table using the mutation status of each sample, its immune subtype and its cancer type. We next used the Cochran-Mantel-Haenszel Chi-Squared test function from the R statistical package to test whether the immune subtype and the genotype are independent. We kept all the associations that had a FDR below 0.1 after BH correction. Finally, we used Fisher’s test to find which pairs of driver mutations and immune subtypes were statistically significant and their associated odds ratio. We repeated the analysis using only the subset of mutations in each driver gene that are predicted to be oncogenic according to the above source to ensure that we would not miss associations that might be weaker due to the presence of passenger mutations in driver genes.
Contributor: Eduard Porta Pardo
We used domainXplorer to identify driver genes and mutations that correlate with the leukocyte fraction of the tumor sample. The algorithm uses a linear model that takes into account potential biases caused by differences in the immune responses between the tissues of origin of the tumors, the gender of the patient, the total number of missense mutations in the sample or the patient’s age as covariates. The model is:
where LF is the leukocyte fraction of each sample, T is the tissue of origin, N the total number of immunogenic mutations in the sample and D is a binary variable showing whether the sample has a mutation in the driver gene. To correct for multiple testing, the BH method is applied to p-values of the D factor from the ANOVA test of each driver event. We repeated the analysis using only the subset of mutations in each driver gene that are predicted to be oncogenic according to the TCGA Driver Genes Analysis Working Group to ensure that we would not miss associations that might be weaker due to the presence of passenger mutations in driver genes.
Contributors: Eduard Porta-Pardo and Adam Godzik
Genomic Alterations in Signaling Pathways
To study correlation of pathway aberrations with the leukocyte fraction and other immune composition scores, we used membership of the eight signaling pathways curated by the TCGA PanCancer Atlas Pathway subgroup (Sanchez-Vega et al., 2018). The eight pathways are PI3K signaling, RTK/RAS signaling, WNT signaling, TGF-β signaling, NOTCH signaling, HIPPO signaling, MYC signaling, and Mismatch Repair machinery (MMR). For each pathway, samples from each of 30 tumor types were divided into two groups of altered and intact cases based on acquisition of non-silent or frameshift mutations, heterozygous or homozygous deletions, or amplifications, in at least one member of the pathway. The association of the genomically-altered pathways in each tumor type or patients subgroup with each CIBERSORT immune estimated score was calculated by a two-sided Student t-Test, assuming unequal variances (Welch’s t-test). Associations were assumed significant if their BH p-value, adjusted for multiple comparisons, were below 0.05. Tumor types with less than 5 samples in each of the comparison arms were excluded from association studies. To ascertain whether the observed associations are derived by specific molecular subtypes, we repeated this analysis using the molecular subtypes previously identified by the TCGA tumor-specific studies instead of tumor tissue of origin. The same approach was used to discover the association of tumor types or immune subgroups with 6 aggregated CIBERSORT estimates (using Aggregate 1 above).
Contributor: Farshad Farshidfar
Genetic Ancestry
Principal Components Analysis
We evaluated the relationship between genetic ancestry and immune signatures in 9003 samples from which genome wide array genotype data from normal blood and immune phenotypes were available. To infer genetic ancestry, we used the germline genetic data (Affymetrix 6.0 normal). We downloaded the cel files from the TCGA datasets and used Affymetrix software to make genotype calls. Genotype calls were made to human genome Build37, forward strand. We used EIGENSOFT (Price et al., 2006) to perform principal components analysis on the genotype data. We inferred how the principal components related to continental ancestry by comparing self report of race/ethnicity to the principal components. High values of principal component 1 (PC1) were found among African Americans, high values of PC2 were found among Asians, high values of PC3 were found among Hispanics and Native Americans, and low values for PC1, PC2 and PC3 were found among Whites. We clustered genetic ancestry into 4 ancestry clusters (AC1-AC4) by performing K means clustering on genotype principal components PC1, PC2 and PC3.
Correlation with Immune Phenotypes
We then tested the association between PC1, PC2 and PC3 and phenotypes: Leukocyte Fraction, log transformed PD-L1 expression, and CIBERSORT immune cell proportions by combined using Aggregate1 (see “Immune cellular fraction estimates” above) using linear regression models. In models which included all cancers, we adjusted for cancer type as a categorical model in the regression model.
Correlation with SNPs
To perform association analyses with single nucleotide polymorphisms (SNPs) at the PDL1 locus, we imputed the genotype data using the Haplotype Reference Consortium as a reference (McCarthy et al., 2016). We defined the region in cis as 1 megabase (500 kilobases upstream and 500 kilobases downstream) around the transcriptional start side of PDL1. We tested the association of all SNPs that had imputation quality R2>0.5 and allele frequency >0.01 using linear regression. Each SNP was tested using an additive model and we adjusted for genetic ancestry using PC1-PC10 and also adjusted for cancer subtype as a categorical variable in the model. To determine significance level for SNP associations we used a method which calculated the effective number of independent SNPs at the locus (Li et al., 2012) and derived a threshold of 9.3×10−5.
Contributors: Elad Ziv, Donglei Hu, Karen Wong
Identification of Neoantigens
HLA typing with OptiType
HLA class I typing of samples (raw RNA-Seq from 8872 samples and aligned reads from 715 samples) was performed on the Seven Bridges Cancer Genomics Cloud using a Common Workflow Language (CWL) description of the OptiType tool (version 1.2) (Szolek et al., 2014). The aligned RNA-Seq samples were first converted to raw sequences using a CWL description of the Picard SamtoFastq tool (version 1.140). The reads from each raw RNA-Seq sample were first aligned to the HLA class I database using a CWL description of the yara aligner (version 0.9.9) (Siragusa et al., 2013) with its error rate parameter set to 3%. Next, the CWL description of OptiType was used to compute the HLA class I types for the sample. OptiType was run under its default parameters for RNA sequencing data using the GLPK linear programming solver and the CBC linear programming solver in samples where the GLPK solver failed. In order to validate the typing results from OptiType, we compared the HLA class I four-digit types obtained from the software PolySolver on TCGA Whole Exome Sequencing data samples (Shukla et al., 2015). For the 5222 patient cases shared by the two studies, approximately 90% of the typing calls were completely concordant for all HLA-A, HLA-B or HLA-C alleles, whereas completely discordant calls were found in less than 1.5% of cases for each of the genes. The HLA typing results are available at https://portal.gdc.cancer.gov/.
Contributors: Raunaq Malhotra, Alexander Krasnitz
Neoantigen Prediction from SNVs
Potential neoantigenic peptides were identified using NetMHCpan v3.0 (Nielsen and Andreatta, 2016), based on HLA types derived from RNA-seq using OptiType as above. In brief, using the HLA calls from OptiType, for each sample, all pairs of MHC and minimal mutant peptide were input into NetMHCpan v3.0 using default settings. NetMHCpan will automatically extract all 8-11mer peptides from a minimal peptide sequence and predict binding for each peptide-MHC pair. After computation, the results were parsed to only retain peptides which included the mutated position. Peptides containing amino acid mutations were identified as potential antigens on the basis of a predicted binding to autologous MHC (IC50 < 500 nM) and detectable gene expression meeting an empirically determined threshold of 1.6 transcripts-per-million (TPM). This threshold was selected in order to divide the bimodal distribution in the expression data.
Specifically, somatic nonsynonymous coding single nucleotide variants were extracted from the MC3 variant file (mc3.v0.2.8.CONTROLLED.maf) with the following filters: FILTER in “PASS”, “wga”, “native_wga_mix”; NCALLERS > 1; barcode in whitelist where do_not_use = False; Variant_Classification = “Missense_Mutation”; and Variant_Type = “SNP”. For each SNV, the Ensembl protein reference sequence was obtained, and the minimal peptide encompassing the mutation site plus 10 amino acids up and downstream of the mutation site was extracted (21 aa long peptide). If the mutation occurred within 10 amino acids of the N- or C-terminal end of the protein, all available sequence between the mutation and start/end of the protein was taken, resulting in a minimal peptide shorter than 21 aa. The variant position within the minimal peptide was recorded, and the mutation was applied to the minimal peptide, resulting in a mutant minimal peptide. Variation in sequencing coverage and tumor purity require careful consideration in order to mitigate the risk of impacting mutation calls and on pMHC, and prior to pMHC calling, sequencing data was subjected to rigorous harmonization efforts, performed by the PanCancer MC3 Consortium(Ellrott et al., 2018).
Contributors: Scott D. Brown, Robert A. Holt
Neoantigen Prediction from Indels
Somatic indel variants were extracted from the MC3 variant file (mc3.v0.2.8.CONTROLLED.maf) with the following filters: FILTER in “PASS”, “wga”, “native_wga_mix” (with no combination with other tags); NCALLERS > 1; barcode in whitelist where do_not_use = False; Variant_Classification = “Frame_Shift_Ins”, “Frame_Shift_Del”, “In_Frame_Ins”, “In_Frame_Del”, “Missense_Mutation”, “Nonsense_Mutation”; and Variant_Type = “INS”, “DEL”. For each Indel, the downstream protein sequence was obtained using VEP v87 (Ensembl Variant Effect Predictor) (McLaren et al., 2016) using default settings.
Using 9-mer peptides extracted from VEP downstream protein sequences and the HLA calls from OptiType, for each sample, binding for each pair of mutant peptide-MHC were predicted using pVAC-Seq v4.0.8 pipeline (Hundal et al., 2016) with NetMHCpan v3.0 using default settings, of which an IC50 binding score threshold 500 nM was used to report the predicted binding epitopes as neoantigens.
Contributors: Nam Sy Vo, Ken Chen
Prognostic Associations
Cox models with predicted neoantigen number (including SNV and indel neoantigens) binned into high and low groups across all possible neoantigen count thresholds and including as covariates patient age, gender, leukocyte fraction, and tumor type (if applicable) were used to evaluate PFI for each tumor type or immune subtype, and HR for each predicted neoantigen count threshold calculated.
Contributor: Scott D. Brown
Genomic Viral Content Analysis
Viral Read Counts
Viral sequence libraries (filter sets) were constructed for known tumor viruses EBV, HBV, and HPV. Scans were performed using BioBloom Tools (Chu et al., 2014) on the ISB Cancer Genomics Cloud, reporting the number of hits and misses per filter set as well as shared and unique reads. For each virus and each sample, a score of normalized reads per million (NRPM) was defined as 106 times the number of hits over the total reads in the sample. NRPM Thresholds HPV: 10, EBV: 5, HBV; 5. The NRPM values are provided in Table S1.
Correlation with Immune Response
Viral read counts were correlated with expression signatures see (“Immune-Expression Signatures”),CIBERSORT fractions (both original and aggregated), expression of key immunotherapy targets (PD-L1,CTLA4,PD-1), Th1/Th2/Th17 signatures, DNA damage scores (AS,LOH), ITH, TCR/BCR diversity, stromal fraction and LF. Regression of read counts with these immune characterizations was performed, using immune subtype as a covariate, and resulting p-values were corrected for multiple testing using the BH method. For HPV, tumor types STAD, ESCA, LAML, and OV were excluded, due to evidence of possible false positives.
Contributors: Sheila M. Reynolds, Varsha Dhankani, Margaret Gulley, Reanne Bowlby, Yusanne Ma, Payal Sipahimalani, Karen Mungall, Chandra Sekhar Pedamallu, Susan Bullman, Akinyemi I. Ojesina, Denise Wolf, Vesteinn Thorsson
T- and B- Cell Receptor Analysis
TCR Inference from Tumor RNA-Seq Data
Identification of TCR CDR3 sequences from T cells present in the sequenced tumor sections was performed using MiTCR v1.0.3, and previously described parameters to optimize extraction from RNA-seq datasets (Brown et al., 2015). Briefly, paired-end fastq files were concatenated into a single file and run through MiTCR using the appropriate parameter set for the sequence read length as described in Brown et al. Runs were performed on the ISB Cancer Genomics Cloud. TCR diversity scores (Shannon Entropy, Evenness, and Richness) are provided in Table S1.
Contributors: Scott D. Brown, Sheila M. Reynolds
Prognostic Impact of TCR Diversity Scores
Cox models for TCR diversity within each TCGA tumor type were generated with Shannon entropy scores binned into high and low groups across all possible thresholds and including as covariates patient age, gender, leukocyte fraction, and used to evaluate PFI for each tumor type, and HR for each predicted neoantigen count threshold calculated. Due to the effect of read length on TCR extraction, 76 bp datasets were used for each TCGA tumor type or immune subtype if available, otherwise 50 bp datasets were used.
Contributor: Scott D. Brown
BCR Inference from Tumor RNA-Seq Data
We used the VDJer tool (Mose et al., 2016), running on the ISB Cancer Genomics Cloud, to reconstruct the immunoglobulin heavy chain for all tumor samples. Paired end mRNASeq FASTQ data were aligned to human reference genome hg38 using STAR version 2.4.2a (Dobin et al., 2013). FASTQ files containing more than one read length were truncated to the shorter length. STAR was configured to emit unmapped reads within the output BAM files and samtools was used to generate BAM indices. An estimated insert size for each sample was calculated by using bwa version 0.7.12 (Li and Durbin, 2009) to align the first 1,000,000 read pairs of each sample to a reference human transcriptome and identifying the median bwa computed insert length. BCR heavy chain contigs and read alignments were generated using V’DJer version 0.12 run in standard mode. RSEM version 1.2.21 (Li and Dewey, 2011) was then used to quantify the BCR contigs. The RSEM reference was generated by running rsem-prepare-reference against the BCR contig fasta file and quantification was performed using rsem-calculate-expression. Expression counts were normalized to the total mRNASeq count for each sample. Isotypes for each contig were identified by mapping the trailing 48 bases to the hg38 reference and using the resultant alignment coordinates to call the isotype. IMGT/HighV-Quest (Lefranc et al., 2009) (http://www.imgt.org/IMGTindex/IMGTHighV-QUEST.php)was used to identify V and J gene segments, CDR3 sequence and V region identity for each contig. IgH diversity scores (Shannon Entropy, Evenness, and Richness) are provided in Table S1. Contributors: Joel Parker, Lisle E. Mose, Sheila M. Reynolds, Benjamin Vincent
Immunomodulator Identification and Analysis
Immunomodulator Compilation
A list of immunomodulatory genes (Table S6) was curated from a literature review performed by immuno-oncology experts within the TCGA immune response working group, who reviewed each entry and confirmed the immunomodulatory function of each gene, resulting in a list of 78 immunomodulators (IMs).
IM Gene Expression
Corresponding mRNA expression was unavailable for 3 of these IMs (HLA-DRB3, HLA-DRB4, KIR2DL2), which were excluded from subsequent analysis. Median expression levels (used to summarize expression in each subtype) were computed only using samples with non-missing values.
Prior to differential expression and miRNA correlation analysis for IMs, any genes with missing expression values in at least one sample were removed; any samples for which LF or subtype designation were unavailable were also excluded. The resulting expression data included 67 genes and 9,058 samples. PCA of all normalized expression values (log10(expression + 1)) was performed to check for batch or confounding effects.
To examine differences in IM expression across subtypes, we performed a Kruskal-Wallis test for each gene expression level with respect to subtype; p-values were adjusted for for multiple testing based on the BH method. Based on the observation from PCA that IM gene expression is correlated with LF within subtypes, we controlled for differences in LF by calculating residuals for expression with respect to LF. We recomputed Kruskal-Wallis results for expression residuals and found all genes to remain significant.
Expression Correlation with DNA Methylation
To study the relationship between gene expression and DNA methylation of immunomodulators, we mapped DNA methylation probes to genes using bioconductor packages IlluminaHumanMethylation450kanno.ilmn12.hg19 and IlluminaHumanMethylation27kanno.ilmn12.hg19, containing manifests and annotation for Illumina’s 450k and 27k arrays. For a given IM gene, Spearman correlation between gene expression and each corresponding gene-associated probe was evaluated, within each immune subtype. Results were then filtered to retain sets of probes with similarly signed correlations, to reduce noise and increase robustness of signal. The filter produces probe-clusters, where probes are uniquely assigned a cluster, are within 10KB and have the same correlation sign. Single correlation values per probe-cluster were found by averaging probes. In cases where multiple probe clusters were associated with a single gene, the corresponding correlation value were averaged to yield the single correlation value reported in Figure 6A.
IM Copy Number
Using output from a PanCan GISTIC2.0 run on ISAR-corrected Affymetrix genome-wide human SNP6.0 array data, deep amplifications, shallow amplifications, non-alterations, shallow deletions, and deep deletions of each immunomodulator gene were called as described in “Genomic Correlations with Immune Phenotype” above for 8461 tumors that both were immune subtyped and had ABSOLUTE purity and ploidy calls. Proportions of samples with each type of copy number alteration were then compared across immune subtypes. We also report the difference between observed and expected frequencies of amplification for each immunomodulator gene in each immune subtype, where the expected frequency is the overall frequency of amplification among all 8461 tumors. This difference calculation was then repeated for immunomodulator deletions.
IM Gene Expression Correlation with miRNA
We examined the association of microRNA (miRNA) expression with immune populations and signatures across all immune subtypes. The normalized, batch corrected expression levels of 743 miRNA genes were tested for significant correlation (Spearman, BH corrected p-value < 0.05) within each subtype against mRNA expression of IM genes. Predicted binding targets for miRNA genes were obtained from version 5.0 of the miRDB database (http://www.mirdb.org/) and mapped to IMs based on HGNC gene symbol.
Contributors: Christopher Plaisier, Benjamin Vincent, Galen Gao, David L. Gibbs, James A. Eddy
Immune Phenotype Correlation with miRNA & IMs
We examined the association of microRNA (miRNA) expression with immune populations and signatures across all tumor types. The normalized, batch corrected expression levels of 743 miRNA genes were tested for significant correlation (Spearman, BH corrected p-value < 0.05) within each tumor group against 95 different features from several other working group datasets and observations: total leukocyte fraction (based on DNA methylation assays); immune infiltrate subpopulations estimated by CIBERSORT (9 adaptive immune cell types, 13 innate immune cell types); and mRNA expression of immune-related genes (22 checkpoint stimulator genes, 34 checkpoint inhibitor genes, 5 MHC class I genes, 9 MHC class II genes, and 2 cytolytic markers). Hematologic (LAML, THYM) and lymphatic (LAML) cancers were excluded from all correlations.
Contributor: James Eddy
The Cell-to-Cell Communication Network
A network of documented ligand-receptor, cell-receptor, and cell-ligand pairs (Ramilowski et al., 2015) was retrieved from the FANTOM5 resource at (http://fantom.gsc.riken.jp/5/suppl/Ramilowski_et_al_2015/). CIBERSORT cell types are more granular than the immune cells in FANTOM5 and were therefore summed to yield estimates for FANTOM5 immune cell abundances, as defined above in “Immune cellular fraction estimates” Aggregate 2. For example, FANTOM5 CD19 B cell estimates are the combination of CIBERSORT naive and memory B cells. This network was augmented with additional known interactions of immumodulators, and only ligand-receptor edges that contained at least one cell or one immune modulator were retained, yielding a ‘scaffold’ of possible interactions.
From the scaffold of possible interactions, interactions were identified that could be playing a role within the TME in each subtype as follows. Cellular fractions were binned into tertiles (low, medium, high), as were gene expression values for ligands and receptors, yielding ternary values for all ‘nodes’ in the network. The binning was performed over all TCGA samples. In subsequent processing, nodes and edges were treated uniformly in processing, without regard to type (cell,ligand,receptor). From the scaffold, interactions predicted to take place in the TME were identified first by a criterion for the nodes to be included (‘present’ in the network), then by a criterion for inclusion of edges, potential interactions. For nodes, if at least 66% of samples within a subtype map to mid or high value bins, the node is entered into the subtype-network. An edge present in the scaffold network between any two nodes is then evaluated for inclusion. A contingency table is populated for the ternary values of the two nodes, over all samples in the subtype, and a concordance vs discordance ratio (“concordance score”) is calculated for the edge in terms of the values of ((high,high)+(low,low))/((low,high)+(high,low)). Edges were retained with concordance score > 2.9, set based on evaluation of quantile distributions.
Contributors: David L. Gibbs, Vesteinn Thorsson, Ilya Shmulevich
Master Regulators of Immune Genes
The Master Regulators (MRs) are identified by first inferring protein activity of candidate MRs as transcriptional influence on groups of co-expressed genes using the VIPER algorithm (Alvarez et al., 2016), then using the DIGGIT algorithm (Chen et al., 2014) to find somatically altered proteins significantly associated with the MRs, and finally linking the two through a method called TieDIE (Drake et al., 2016; Paull et al., 2013), which finds connecting “paths” through a network of known and predicted interactions. MRs that correlate with leukocyte fraction (LF) are prioritized, as are somatic alterations seen by domainXplorer.
We applied the VIPER algorithm (Alvarez et al., 2016) across all samples, using tissue-matched ARACNE (Margolin et al., 2006) interactomes, to infer protein-activity for 2506 potential transcription factor and co-factor candidate “master regulators” (cMRs) from the expression of their downstream targets. Pearson correlation of the inferred protein activity with LF was calculated. Samples were clustered into an optimal number of 67 clusters based on inferred cMR activity, using a modified silhouette score based on the native distance metric defined by VIPER. We then integrated the p-values of the mean activity in each cluster to rank overall cMR activity across the PanCancer dataset.
Similarly, we used the DIGGIT algorithm (Chen et al., 2014) to find mutation and copy-number events significantly associated with each cMR. Briefly: for each tumor type, we computed the aREA (Alvarez et al., 2016) enrichment of the sample set with non-silent coding mutations in a given gene, against the ranked protein-activity signature inferred by VIPER for a given MR. This was performed for each cMR / mutated gene pair with at least 4 samples with a non-silent alteration. Similarly SNP6 copy number profiles were downloaded from the Broad Institute and thresholded at a value of 0.5. We then ranked the cMRs by combining the p-values of all significant DIGGIT interactions (p < 0.05; uncorrected) across all tumor types using Stouffer’s method. Similarly, we overlapped predicted protein-protein interactions taken from the PrePPI 1.2.0 database (Zhang et al., 2012)(https://bhapp.c2b2.columbia.edu/PrePPI/) with DIGGIT interactions generated in the previous step to generate a second ranking of cMRs based on structural data. These (2) separate rankings were integrated in a Bayesian context with the ranks derived from VIPER clustering to produce a single PanCancer ranking of cMR activity. In the top decile, we found 32 candidate MRs that also had a positive correlation of 0.5 or greater with LF.
Mutation or copy-number events identified by the domainXplorer algorithm were tested for statistical association with the 32 cMRs identified, using the DIGGIT algorithm (above), and retained if associated with one or more of the 32 cMRs in at least one tumor-specific context. In addition we considered genomic events with broad statistical association with leukocyte fraction across the PanCancer dataset that were not identified by domainXplorer (< 0.15 FDR; BH correction), resulting in 44 total genomic events significantly associated with both the phenotype and the cMRs identified in the first step.
To elucidate functional and molecular relationships between these genomic events and the 32 cMRs, we applied the TieDIE algorithm (Drake et al., 2016; Paull et al., 2013) with a database consisting of literature-based regulatory and signaling interactions as well as high-confidence predicted protein-protein interactions (Khurana et al., 2013). TieDIE found the 44 genomic events were significantly “close” to the 32 MRs in pathway space (p-value < 0.021) and identified a network MR-PanImmune connecting 15 of these altered genes to 26 MRs across 222 database interaction containing 60 transcriptional regulatory, 8 signaling, 3 phosphorylation and 151 protein-protein interactions.
Contributors: Evan O. Paull, Mariano Alvarez, Federico Giorgi, Jing He and Andrea Califano
SYstems Genetics Network AnaLysis
The SYstems Genetics Network AnaLysis (SYGNAL) pipeline is composed of 4 steps (Plaisier et al., 2016). Command line parameters for all programs in SYGNAL pipeline can be found in Plaisier et al., 2016 (Plaisier et al., 2016). Each tumor type was run separately through the pipeline to reduce the confounding from tissue of origin differences. Highly expressed genes were discovered for each tumor type by requiring that genes have greater than or equal to the median expression of all genes across all conditions in ≥50% of patients (Plaisier et al., 2016). These gene sets were then used as input to SYGNAL.
Mechanistic Regulatory Network Inference
In the first step, the cMonkey2 biclustering algorithm (Reiss et al., 2015) was used to reduce the genes expression profiles from each tumor type into co-regulated biclusters. The number of biclusters was determined using two times the number of genes divided by the expectation of 30 genes on average per cluster. The training configuration for cMonkey2 included co-expression, GeneMania gene-gene interaction network, and enrichment of either TF or miRNA target genes using the set-enrichment module (Reiss et al., 2015). In total, cMonkey2 was run three times for each tumor type and we discovered 43,000 biclusters. The first run used the TF-target gene interaction database as input to the set-enrichment module to discover TF mediated regulation. The second and third runs used PITA (Kertesz et al., 2007) and TargetScan (Agarwal et al., 2015) as input to the set-enrichment module to discover miRNA mediated regulation.
Post-Processing and Filtering of Biclusters
Biclusters were considered significantly co-expressed if the variance explained by first principal component was greater than or equal to 0.3 and was significantly larger than random samples (empirical p-value ≤ 0.05). Each of the 43,000 cMonkey2 biclusters were then post-processed to discover: (i) co-expression quality via variance explained by first principal component (empirical p-value < 0.05 and variance explained ≥ 0.3), (ii) putative TF regulators via de novo motif detection with MEME or WEEDER (Bailey et al., 2009; Pavesi and Pesole, 2006) and comparison of motif to known DNA recognition motifs (TOMTOM q-value ≤ 0.05), and enrichment of TF target genes (Bonferroni corrected p-value ≤ 0.05 and percent target genes ≥ 10%); (iii) TF family expansion using the TFClass database (Wingender et al., 2013); (iv) putative miRNA regulators via the FIRM pipeline (Plaisier et al., 2012), (v) correlation of TF and miRNA regulators with bicluster eigengenes (Langfelder and Horvath, 2007)(TFs: R ≥ 0.3 or ≤ −0.3 and p-value ≤ 0.05; miRNAs: R ≤ −0.3 and p-value ≤ 0.05); (vi) enrichment of IM genes (p-value ≤ 0.05); (vii) association of total leukocyte fraction bicluster eigengenes (p-value ≤ 0.05); (viii) functional enrichment with GO biological process terms (BH-corrected p-value ≤ 0.05) (Plaisier et al., 2012); and (ix) association with hallmarks of cancer (Jiang-Conrath Semantic Similarity Score ≥ 0.8, permuted p-value ≤ 5.1 × 10−4) (Hanahan and Weinberg, 2011; Plaisier et al., 2012). The biclusters were filtered by validating co-expression and ensuring disease relevance. A bicluster was considered significantly co-expressed if the variance explained by first principal component was greater than or equal to 0.3 and was significantly larger than random samples (empirical p-value ≤ 0.05). A bicluster was considered immune-related if the genes were significantly enriched with immunomodulators (p-value ≤ 0.05) and conditional elevated and decreased regulation was significantly associated with total leukocyte fraction (p-value ≤ 0.05) or associated with either evading immune detection or tumor promoting inflammation (the two immune hallmarks of cancer (Plaisier et al., 2016).
In all 6,667 biclusters were significantly associated with total leukocyte fraction (p-value ≤ 0.05). Additionally, 197 biclusters were significantly enriched with a curated set of immunomodulatory genes (Bonferroni corrected p-value ≤ 0.05) There was a significant overlap of 171 biclusters (87%) that were enriched with immunomodulators and associated with total leukocyte infiltration (p-value = 1.4 × 10−110).
Causal regulatory network inference
In the third step of the SYGNAL pipeline, the single.marker.analysis function from the network edge orienting (NEO) package in R (Aten et al., 2008; Plaisier et al., 2009; Plaisier et al., 2016) was applied to infer causal flows of information anchored on a somatically mutated gene or pathway to expression of a TF or miRNA to a bicluster eigengenes. The single.marker.analysis function compares five different causal graphical models to test for significant evidence of causal flow across the variables tested. The model of interest for these studies was the causal graph anchored on a somatically mutated gene or pathway (M) which affects the expression of a TF or miRNA (R) that in turn alters the expression of a bicluster eigengene (B), i.e. the causal graph M→R→B. The fit of this model was assessed using the local structural equation modeling (SEM) based, edge orienting, next best single marker (LEO.NB.SingleMarker) score, which is the log10 probability of this model divided by the log10 probability of the next best fitting alternative model (Aten et al., 2008). A causal flow was inferred when the LEO.NB.SingleMarker score was positive and three times more likely than the next best alternative model (LEO.NB.SingleMarker score ≥ 0.5)(Plaisier et al., 2009). For miRNAs, we imposed the additional requirement that the regulation of the miRNA on the bicluster eigengene must be repressive (ZPathAB < 0). Thus any LEO.NB.SingleMarker score greater than or equal to 0.5 was considered sufficient evidence to infer causal flow through the causal graph M→R→B. To reduce the overall number of tests, only TFs and miRNAs that were significantly associated with a somatic mutation were evaluated (Student’s T-test p-value ≤ 0.05 and FC ≥ 1.25).
Integration of Mechanistic & Causal Networks
In the fourth and final step of the SYGNAL pipeline we integrate the regulatory influences by either taking the intersection for transcription factors and union for miRNAs. For the intersection of TF mediated regulation it was also required that the causal and mechanistic predictions must be for regulation of the same bicluster.
Contributor: Christopher Plaisier
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical details of all experiments are reported in the text, figure legends and figures, including statistical analysis performed, statistical significance and exact n numbers.
SOFTWARE AND DATA AVAILABILITY
The raw data, processed data and clinical data can be found at the legacy archive of the GDC (https://portal.gdc.cancer.gov/legacy-archive/search/f) and the Pancancer Atlas publication page (https://gdc.cancer.gov/about-data/publications/pancanatlas). The mutation data can be found here (https://gdc.cancer.gov/about-data/publications/mc3-2017). TCGA data can also be explored through the Broad Institute FireBrowse portal (http://gdac.broadinstitute.org) and the Memorial Sloan Kettering Cancer Center cBioPortal (http://www.cbioportal.org). Details for software availability are in the Key Resource Table. Interactive exploration and visualization of data and results in this manuscript is available at the CRI iAtlas portal (www.cri-iatlas.org).
Software used for the analyses for each of the data platforms and integrated analyses are described and referenced in the individual Method Details subsections and are listed in the Key Resource Table.
KEY RESOURCES TABLE
Supplementary Material
Table S1 PanImmune Feature matrix of immune characteristics. Related to Figure 1.
Spreadsheet containing all tumor-sample characterizations used in, and generated by this study
Table S2 Driver mutations enriched in immune subtypes. Related to Figure 4.
Table S3 Recurrent pMHCs. Related to Figure 5.
Table S4 Recurrent Indels. Related to Figure 5.
Table S5 Recurrent TCRs and co-occurrence of TCRs and pMHC. Related to Figure 5.
Table S6 Immunomodulators. Related to Figure 6
HIGHLIGHTS.
Six identified immune subtypes span cancer tissue types and molecular subtypes
Immune subtypes differ by somatic aberrations, microenvironment and survival
Multiple control modalities of molecular networks affect tumor-immune interactions
These analyses serve as a resource for exploring immunogenicity across cancer types
Acknowledgments
We are grateful to all the patients and families who contributed to this study. We also thank the Office of Cancer Genomics at the NCI for organizational and logistical support of this study. The high throughput analyses in this study were performed on the Institute for Systems Biology-Cancer Genomics Cloud (ISB-CGC), under contract number HHSN261201400007C and on the Seven Bridges Cancer Genomics Cloud under contract HHSN261201400008C, cloud pilot projects of the National Cancer Institute. Funding from the Cancer Research Institute is gratefully acknowledged, as is support from NCI through U54 HG003273, U54 HG003067, U54 HG003079, U24 CA143799, U24 CA143835, U24 CA143840, U24 CA143843, U24 CA143845,U24 CA143848, U24 CA143858, U24 CA143866, U24 CA143867, U24 CA143882, U24 CA143883, U24 CA144025, P30 CA016672. The study was supported by W81XWH-12-2-0050, HU0001-16-2-0004 from the US Department of Defense through the Henry M. Jackson Foundation for the Advancement of Military Medicine. We thank Peter Hammerman and Yasin Şenbabaoğlu for contributions in early phases of this work.
Abbreviations of the TCGA Tumor Types
- ACC
Adrenocortical carcinoma
- BLCA
Bladder urothelial carcinoma
- BRCA
Breast invasive carcinoma
- CESC
Cervical squamous cell carcinoma and endocervical adenocarcinoma
- CHOL
Cholangiocarcinoma
- COAD
Colon adenocarcinoma
- DLBC
Lymphoid neoplasm diffuse large B-cell lymphoma
- ESCA
Esophageal carcinoma
- GBM
Glioblastoma multiforme
- HNSC
Head and neck squamous cell carcinoma
- KICH
Kidney chromophobe
- KIRC
Kidney renal clear cell carcinoma
- KIRP
Kidney renal papillary cell carcinoma
- LAML
Acute myeloid leukemia
- LGG
Brain lower grade glioma
- LIHC
Liver hepatocellular carcinoma
- LUAD
Lung adenocarcinoma
- LUSC
Lung squamous cell carcinoma
- MESO
Mesothelioma
- OV
Ovarian serous cystadenocarcinoma
- PAAD
Pancreatic adenocarcinoma
- PCPG
Pheochromocytoma and paraganglioma
- PRAD
Prostate adenocarcinoma
- READ
Rectum adenocarcinoma
- SARC
Sarcoma SKCM Skin Cutaneous Melanoma
- STAD
Stomach adenocarcinoma
- TGCT
Testicular Germ Cell Tumors
- THCA
Thyroid carcinoma
- THYM
Thymoma
- UCEC
Uterine Corpus Endometrial Carcinoma
- UCS
Uterine Carcinosarcoma
- UVM
Uveal Melanoma
Secondary author list
Samantha J. Caesar-Johnson, John A. Demchok, Ina Felau, Melpomeni Kasapi, Martin L. Ferguson, Carolyn M. Hutter, Heidi J. Sofia, Roy Tarnuzzer, Zhining Wang, Liming Yang, Jean C. Zenklusen, Jiashan (Julia) Zhang, Sudha Chudamani, Jia Liu, Laxmi Lolla, Rashi Naresh, Todd Pihl, Qiang Sun, Yunhu Wan, Ye Wu, Juok Cho, Timothy DeFreitas, Scott Frazer, Nils Gehlenborg, Gad Getz, David I. Heiman, Jaegil Kim, Michael S. Lawrence, Pei Lin, Sam Meier, Michael S. Noble, Gordon Saksena, Doug Voet, Hailei Zhang, Brady Bernard, Nyasha Chambwe, Varsha Dhankani, Theo Knijnenburg, Roger Kramer, Kalle Leinonen, Yuexin Liu, Michael Miller, Sheila Reynolds, Ilya Shmulevich, Vesteinn Thorsson, Wei Zhang, Rehan Akbani, Bradley M. Broom, Apurva M. Hegde, Zhenlin Ju, Rupa S. Kanchi, Anil Korkut, Jun Li, Han Liang, Shiyun Ling, Wenbin Liu, Yiling Lu, Gordon B. Mills, Kwok-Shing Ng, Arvind Rao, Michael Ryan, Jing Wang, John N. Weinstein, Jiexin Zhang, Adam Abeshouse, Joshua Armenia, Debyani Chakravarty, Walid K. Chatila, Ino de Bruijn, Jianjiong Gao, Benjamin E. Gross, Zachary J. Heins, Ritika Kundra, Konnor La, Marc Ladanyi, Augustin Luna, Moriah G. Nissan, Angelica Ochoa, Sarah M. Phillips, Ed Reznik, Francisco Sanchez-Vega, Chris Sander, Nikolaus Schultz, Robert Sheridan, S. Onur Sumer, Yichao Sun, Barry S. Taylor, Jioajiao Wang, Hongxin Zhang, Pavana Anur, Myron Peto, Paul Spellman, Christopher Benz, Joshua M. Stuart, Christopher K. Wong, Christina Yau, D. Neil Hayes, Joel S. Parker, Matthew D. Wilkerson, Adrian Ally, Miruna Balasundaram, Reanne Bowlby, Denise Brooks, Rebecca Carlsen, Eric Chuah, Noreen Dhalla, Robert Holt, Steven J.M. Jones, Katayoon Kasaian, Darlene Lee, Yussanne Ma, Marco A. Marra, Michael Mayo, Richard A. Moore, Andrew J. Mungall, Karen Mungall, A. Gordon Robertson, Sara Sadeghi, Jacqueline E. Schein, Payal Sipahimalani, Angela Tam, Nina Thiessen, Kane Tse, Tina Wong, Ashton C. Berger, Rameen Beroukhim, Andrew D. Cherniack, Carrie Cibulskis, Stacey B. Gabriel, Galen F. Gao, Gavin Ha, Matthew Meyerson, Steven E. Schumacher, Juliann Shih, Melanie H. Kucherlapati, Raju S. Kucherlapati, Stephen Baylin, Leslie Cope, Ludmila Danilova, Moiz S. Bootwalla, Phillip H. Lai, Dennis T. Maglinte, David J. Van Den Berg, Daniel J. Weisenberger, J. Todd Auman, Saianand Balu, Tom Bodenheimer, Cheng Fan, Katherine A. Hoadley, Alan P. Hoyle, Stuart R. Jefferys, Corbin D. Jones, Shaowu Meng, Piotr A. Mieczkowski, Lisle E. Mose, Amy H. Perou, Charles M. Perou, Jeffrey Roach, Yan Shi, Janae V. Simons, Tara Skelly, Matthew G. Soloway, Donghui Tan, Umadevi Veluvolu, Huihui Fan, Toshinori Hinoue, Peter W. Laird, Hui Shen, Wanding Zhou, Michelle Bellair, Kyle Chang, Kyle Covington, Chad J. Creighton, Huyen Dinh, HarshaVardhan Doddapaneni, Lawrence A. Donehower, Jennifer Drummond, Richard A. Gibbs, Robert Glenn, Walker Hale, Yi Han, Jianhong Hu, Viktoriya Korchina, Sandra Lee, Lora Lewis, Wei Li, Xiuping Liu, Margaret Morgan, Donna Morton, Donna Muzny, Jireh Santibanez, Margi Sheth, Eve Shinbrot, Linghua Wang, Min Wang, David A. Wheeler, Liu Xi, Fengmei Zhao, Julian Hess, Elizabeth L. Appelbaum, Matthew Bailey, Matthew G. Cordes, Li Ding, Catrina C. Fronick, Lucinda A. Fulton, Robert S. Fulton, Cyriac Kandoth, Elaine R. Mardis, Michael D. McLellan, Christopher A. Miller, Heather K. Schmidt, Richard K. Wilson, Daniel Crain, Erin Curley, Johanna Gardner, Kevin Lau, David Mallery, Scott Morris, Joseph Paulauskis, Robert Penny, Candace Shelton, Troy Shelton, Mark Sherman, Eric Thompson, Peggy Yena, Jay Bowen, Julie M. Gastier-Foster, Mark Gerken, Kristen M. Leraas, Tara M. Lichtenberg, Nilsa C. Ramirez, Lisa Wise, Erik Zmuda, Niall Corcoran, Tony Costello, Christopher Hovens, Andre L. Carvalho, Ana C. de Carvalho, José H. Fregnani, Adhemar Longatto-Filho, Rui M. Reis, Cristovam Scapulatempo-Neto, Henrique C.S. Silveira, Daniel O. Vidal, Andrew Burnette, Jennifer Eschbacher, Beth Hermes, Ardene Noss, Rosy Singh, Matthew L. Anderson, Patricia D. Castro, Michael Ittmann, David Huntsman, Bernard Kohl, Xuan Le, Richard Thorp, Chris Andry, Elizabeth R. Duffy, Vladimir Lyadov, Oxana Paklina, Galiya Setdikova, Alexey Shabunin, Mikhail Tavobilov, Christopher McPherson, Ronald Warnick, Ross Berkowitz, Daniel Cramer, Colleen Feltmate, Neil Horowitz, Adam Kibel, Michael Muto, Chandrajit P. Raut, Andrei Malykh, Jill S. Barnholtz-Sloan, Wendi Barrett, Karen Devine, Jordonna Fulop, Quinn T. Ostrom, Kristen Shimmel, Yingli Wolinsky, Andrew E. Sloan, Agostino De Rose, Felice Giuliante, Marc Goodman, Beth Y. Karlan, Curt H. Hagedorn, John Eckman, Jodi Harr, Jerome Myers, Kelinda Tucker, Leigh Anne Zach, Brenda Deyarmin, Hai Hu, Leonid Kvecher, Caroline Larson, Richard J. Mural, Stella Somiari, Ales Vicha, Tomas Zelinka, Joseph Bennett, Mary Iacocca, Brenda Rabeno, Patricia Swanson, Mathieu Latour, Louis Lacombe, Bernard Têtu, Alain Bergeron, Mary McGraw, Susan M. Staugaitis, John Chabot, Hanina Hibshoosh, Antonia Sepulveda, Tao Su, Timothy Wang, Olga Potapova, Olga Voronina, Laurence Desjardins, Odette Mariani, Sergio Roman-Roman, Xavier Sastre, Marc-Henri Stern, Feixiong Cheng, Sabina Signoretti, Andrew Berchuck, Darell Bigner, Eric Lipp, Jeffrey Marks, Shannon McCall, Roger McLendon, Angeles Secord, Alexis Sharp, Madhusmita Behera, Daniel J. Brat, Amy Chen, Keith Delman, Seth Force, Fadlo Khuri, Kelly Magliocca, Shishir Maithel, Jeffrey J. Olson, Taofeek Owonikoko, Alan Pickens, Suresh Ramalingam, Dong M. Shin, Gabriel Sica, Erwin G. Van Meir, Hongzheng Zhang, Wil Eijckenboom, Ad Gillis, Esther Korpershoek, Leendert Looijenga, Wolter Oosterhuis, Hans Stoop, Kim E. van Kessel, Ellen C. Zwarthoff, Chiara Calatozzolo, Lucia Cuppini, Stefania Cuzzubbo, Francesco DiMeco, Gaetano Finocchiaro, Luca Mattei, Alessandro Perin, Bianca Pollo, Chu Chen, John Houck, Pawadee Lohavanichbutr, Arndt Hartmann, Christine Stoehr, Robert Stoehr, Helge Taubert, Sven Wach, Bernd Wullich, Witold Kycler, Dawid Murawa, Maciej Wiznerowicz, Ki Chung, W. Jeffrey Edenfield, Julie Martin, Eric Baudin, Glenn Bubley, Raphael Bueno, Assunta De Rienzo, William G. Richards, Steven Kalkanis, Tom Mikkelsen, Houtan Noushmehr, Lisa Scarpace, Nicolas Girard, Marta Aymerich, Elias Campo, Eva Giné, Armando López Guillermo, Nguyen Van Bang, Phan Thi Hanh, Bui Duc Phu, Yufang Tang, Howard Colman, Kimberley Evason, Peter R. Dottino, John A. Martignetti, Hani Gabra, Hartmut Juhl, Teniola Akeredolu, Serghei Stepa, Dave Hoon, Keunsoo Ahn, Koo Jeong Kang, Felix Beuschlein, Anne Breggia, Michael Birrer, Debra Bell, Mitesh Borad, Alan H. Bryce, Erik Castle, Vishal Chandan, John Cheville, John A. Copland, Michael Farnell, Thomas Flotte, Nasra Giama, Thai Ho, Michael Kendrick, Jean-Pierre Kocher, Karla Kopp, Catherine Moser, David Nagorney, Daniel O’Brien, Brian Patrick O’Neill, Tushar Patel, Gloria Petersen, Florencia Que, Michael Rivera, Lewis Roberts, Robert Smallridge, Thomas Smyrk, Melissa Stanton, R. Houston Thompson, Michael Torbenson, Ju Dong Yang, Lizhi Zhang, Fadi Brimo, Jaffer A. Ajani, Ana Maria Angulo Gonzalez, Carmen Behrens, Jolanta Bondaruk, Russell Broaddus, Bogdan Czerniak, Bita Esmaeli, Junya Fujimoto, Jeffrey Gershenwald, Charles Guo, Alexander J. Lazar, Christopher Logothetis, Funda Meric-Bernstam, Cesar Moran, Lois Ramondetta, David Rice, Anil Sood, Pheroze Tamboli, Timothy Thompson, Patricia Troncoso, Anne Tsao, Ignacio Wistuba, Candace Carter, Lauren Haydu, Peter Hersey, Valerie Jakrot, Hojabr Kakavand, Richard Kefford, Kenneth Lee, Georgina Long, Graham Mann, Michael Quinn, Robyn Saw, Richard Scolyer, Kerwin Shannon, Andrew Spillane, Jonathan Stretch, Maria Synott, John Thompson, James Wilmott, Hikmat Al-Ahmadie, Timothy A. Chan, Ronald Ghossein, Anuradha Gopalan, Douglas A. Levine, Victor Reuter, Samuel Singer, Bhuvanesh Singh, Nguyen Viet Tien, Thomas Broudy, Cyrus Mirsaidi, Praveen Nair, Paul Drwiega, Judy Miller, Jennifer Smith, Howard Zaren, Joong-Won Park, Nguyen Phi Hung, Electron Kebebew, W. Marston Linehan, Adam R. Metwalli, Karel Pacak, Peter A. Pinto, Mark Schiffman, Laura S. Schmidt, Cathy D. Vocke, Nicolas Wentzensen, Robert Worrell, Hannah Yang, Marc Moncrieff, Chandra Goparaju, Jonathan Melamed, Harvey Pass, Natalia Botnariuc, Irina Caraman, Mircea Cernat, Inga Chemencedji, Adrian Clipca, Serghei Doruc, Ghenadie Gorincioi, Sergiu Mura, Maria Pirtac, Irina Stancul, Diana Tcaciuc, Monique Albert, Iakovina Alexopoulou, Angel Arnaout, John Bartlett, Jay Engel, Sebastien Gilbert, Jeremy Parfitt, Harman Sekhon, George Thomas, Doris M. Rassl, Robert C. Rintoul, Carlo Bifulco, Raina Tamakawa, Walter Urba, Nicholas Hayward, Henri Timmers, Anna Antenucci, Francesco Facciolo, Gianluca Grazi, Mirella Marino, Roberta Merola, Ronald de Krijger, Anne-Paule Gimenez-Roqueplo, Alain Piché, Simone Chevalier, Ginette McKercher, Kivanc Birsoy, Gene Barnett, Cathy Brewer, Carol Farver, Theresa Naska, Nathan A. Pennell, Daniel Raymond, Cathy Schilero, Kathy Smolenski, Felicia Williams, Carl Morrison, Jeffrey A. Borgia, Michael J. Liptay, Mark Pool, Christopher W. Seder, Kerstin Junker, Larsson Omberg, Mikhail Dinkin, George Manikhas, Domenico Alvaro, Maria Consiglia Bragazzi, Vincenzo Cardinale, Guido Carpino, Eugenio Gaudio, David Chesla, Sandra Cottingham, Michael Dubina, Fedor Moiseenko, Renumathy Dhanasekaran, Karl-Friedrich Becker, Klaus-Peter Janssen, Julia Slotta-Huspenina, Mohamed H. Abdel-Rahman, Dina Aziz, Sue Bell, Colleen M. Cebulla, Amy Davis, Rebecca Duell, J. Bradley Elder, Joe Hilty, Bahavna Kumar, James Lang, Norman L. Lehman, Randy Mandt, Phuong Nguyen, Robert Pilarski, Karan Rai, Lynn Schoenfield, Kelly Senecal, Paul Wakely, Paul Hansen, Ronald Lechan, James Powers, Arthur Tischler, William E. Grizzle, Katherine C. Sexton, Alison Kastl, Joel Henderson, Sima Porten, Jens Waldmann, Martin Fassnacht, Sylvia L. Asa, Dirk Schadendorf, Marta Couce, Markus Graefen, Hartwig Huland, Guido Sauter, Thorsten Schlomm, Ronald Simon, Pierre Tennstedt, Oluwole Olabode, Mark Nelson, Oliver Bathe, Peter R. Carroll, June M. Chan, Philip Disaia, Pat Glenn, Robin K. Kelley, Charles N. Landen, Joanna Phillips, Michael Prados, Jeffry Simko, Karen Smith-McCune, Scott VandenBerg, Kevin Roggin, Ashley Fehrenbach, Ady Kendler, Suzanne Sifri, Ruth Steele, Antonio Jimeno, Francis Carey, Ian Forgie, Massimo Mannelli, Michael Carney, Brenda Hernandez, Benito Campos, Christel Herold-Mende, Christin Jungk, Andreas Unterberg, Andreas von Deimling, Aaron Bossler, Joseph Galbraith, Laura Jacobus, Michael Knudson, Tina Knutson, Deqin Ma, Mohammed Milhem, Rita Sigmund, Andrew K. Godwin, Rashna Madan, Howard G. Rosenthal, Clement Adebamowo, Sally N. Adebamowo, Alex Boussioutas, David Beer, Thomas Giordano, Anne-Marie Mes-Masson, Fred Saad, Therese Bocklage, Lisa Landrum, Robert Mannel, Kathleen Moore, Katherine Moxley, Russel Postier, Joan Walker, Rosemary Zuna, Michael Feldman, Federico Valdivieso, Rajiv Dhir, James Luketich, Edna M. Mora Pinero, Mario Quintero-Aguilo, Carlos Gilberto Carlotti, Jr., Jose Sebastião Dos Santos, Rafael Kemp, Ajith Sankarankuty, Daniela Tirapelli, James Catto, Kathy Agnew, Elizabeth Swisher, Jenette Creaney, Bruce Robinson, Carl Simon Shelley, Eryn M. Godwin, Sara Kendall, Cassaundra Shipman, Carol Bradford, Thomas Carey, Andrea Haddad, Jeffey Moyer, Lisa Peterson, Mark Prince, Laura Rozek, Gregory Wolf, Rayleen Bowman, Kwun M. Fong, Ian Yang, Robert Korst, W. Kimryn Rathmell, J. Leigh Fantacone-Campbell, Jeffrey A. Hooke, Albert J. Kovatich, Craig D. Shriver, John DiPersio, Bettina Drake, Ramaswamy Govindan, Sharon Heath, Timothy Ley, Brian Van Tine, Peter Westervelt, Mark A. Rubin, Jung Il Lee, Natália D. Aredes, and Armaz Mariamidze
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Analysis, Computation, and Software: VT, DLG, SDB, DW, DSB, TOY, EP, GG, CLP, JAE, EZ, ACC, EOP, IKAS, AJG, RM, FF, AC, JSP, LEM, NSV, JFL, YL, VD, SMR, RB, ADC, DB, AR, AK, HH, TMM, HN, CSP, SB, AIO, AL, WZ, JG, JS, BGV. Supervision: JR, AC, DA, KC, HS, TKC, JNW, JG, RAH, BGV, IS. Writing: VT, DLG, SDB, DW, DSB, TOY, EP, GG, CLP, EZ, ACC, EOP, IKAS, AJG, RM, FF, AC, NSV, HH, TMM, HN, JS, CER, AJL, JSS, EGD, MLD, BGV, IS.
DECLARATION OF INTERESTS
The authors declare no competing interests
References
- Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4 doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, Califano A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet. 2016;48:838–847. doi: 10.1038/ng.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, Margineantu DH, Rao A, Bolouri H, Delrow J, et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017;31:774–786. doi: 10.1101/gad.294991.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aten JE, Fuller TF, Lusis AJ, Horvath S. Using genetic markers to orient the edges in quantitative trait networks: the NEO software. BMC Syst Biol. 2008;2:34. doi: 10.1186/1752-0509-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck AH, Espinosa I, Edris B, Li R, Montgomery K, Zhu S, Varma S, Marinelli RJ, van de Rijn M, West RB. The macrophage colony-stimulating factor 1 response signature in breast carcinoma. Clin Cancer Res. 2009;15:778–787. doi: 10.1158/1078-0432.CCR-08-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedognetti D, Hendrickx W, Ceccarelli M, Miller LD, Seliger B. Disentangling the relationship between tumor genetic programs and immune responsiveness. Curr Opin Immunol. 2016;39:150–158. doi: 10.1016/j.coi.2016.02.001. [DOI] [PubMed] [Google Scholar]
- Benyamine A, Le Roy A, Mamessier E, Gertner-Dardenne J, Castanier C, Orlanducci F, Pouyet L, Goubard A, Collette Y, Vey N, et al. BTN3A molecules considerably improve Vgamma9Vdelta2T cells-based immunotherapy in acute myeloid leukemia. Oncoimmunology. 2016;5:e1146843. doi: 10.1080/2162402X.2016.1146843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierie B, Moses HL. Transforming growth factor beta (TGF-beta) and inflammation in cancer. Cytokine Growth Factor Rev. 2010;21:49–59. doi: 10.1016/j.cytogfr.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Brown SD, Raeburn LA, Holt RA. Profiling tissue-resident T cell repertoires by RNA sequencing. Genome Med. 2015;7:125. doi: 10.1186/s13073-015-0248-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24:743–750. doi: 10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabro A, Beissbarth T, Kuner R, Stojanov M, Benner A, Asslaber M, Ploner F, Zatloukal K, Samonigg H, Poustka A, et al. Effects of infiltrating lymphocytes and estrogen receptor on gene expression and prognosis in breast cancer. Breast Cancer Res Treat. 2009;116:69–77. doi: 10.1007/s10549-008-0105-3. [DOI] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016;164:550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, van de Rijn M, Botstein D, Brown PO. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, Trajanoski Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- Chen JC, Alvarez MJ, Talos F, Dhruv H, Rieckhof GE, Iyer A, Diefes KL, Aldape K, Berens M, Shen MM, et al. Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell. 2014;159:402–414. doi: 10.1016/j.cell.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng WY, Ou Yang TH, Anastassiou D. Biomolecular events in cancer revealed by attractor metagenes. PLoS Comput Biol. 2013a;9:e1002920. doi: 10.1371/journal.pcbi.1002920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng WY, Ou Yang TH, Anastassiou D. Development of a prognostic model for breast cancer survival in an open challenge environment. Sci Transl Med. 2013b;5:181ra150. doi: 10.1126/scitranslmed.3005974. [DOI] [PubMed] [Google Scholar]
- Chu J, Sadeghi S, Raymond A, Jackman SD, Nip KM, Mar R, Mohamadi H, Butterfield YS, Robertson AG, Birol I. BioBloom tools: fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics. 2014;30:3402–3404. doi: 10.1093/bioinformatics/btu558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44:e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos-Ruiz JR, Martinez D, Scarlett UK, Rutkowski MR, Nesbeth YC, Camposeco-Jacobs AL, Conejo-Garcia JR. CD277 is a negative co-stimulatory molecule universally expressed by ovarian cancer microenvironmental cells. Oncotarget. 2010;1:329–338. doi: 10.18632/oncotarget.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JM, Paull EO, Graham NA, Lee JK, Smith BA, Titz B, Stoyanova T, Faltermeier CM, Uzunangelov V, Carlin DE, et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell. 2016;166:1041–1054. doi: 10.1016/j.cell.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellrott K, Covington KR, Kandoth C, Saksena G, McLellan MD, Bailey MH, Sofia H, Hutter C, The Cancer Genome Atlas Research Network Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018 doi: 10.1016/j.cels.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39:11–26. doi: 10.1016/j.immuni.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godec J, Tan Y, Liberzon A, Tamayo P, Bhattacharya S, Butte AJ, Mesirov JP, Haining WN. Compendium of Immune Signatures Identifies Conserved and Species-Specific Biology in Response to Inflammation. Immunity. 2016;44:194–206. doi: 10.1016/j.immuni.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusenleitner D, Howe EA, Bentink S, Quackenbush J, Culhane AC. iBBiG: iterative binary bi-clustering of gene sets. Bioinformatics. 2012;28:2484–2492. doi: 10.1093/bioinformatics/bts438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickx W, Simeone I, Anjum S, Mokrab Y, Bertucci F, Finetti P, Curigliano G, Seliger B, Cerulo L, Tomei S, et al. Identification of genetic determinants of breast cancer immune phenotypes by integrative genome-scale analysis. Oncoimmunology. 2017;6:e1253654. doi: 10.1080/2162402X.2016.1253654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornik K. A CLUE for CLUster ensembles. J Stat Softw. 2005;14:1–25. [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, Mardis ER, Griffith M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016;8:11. doi: 10.1186/s13073-016-0264-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesia MD, Parker JS, Hoadley KA, Serody JS, Perou CM, Vincent BG. Genomic Analysis of Immune Cell Infiltrates Across 11 Tumor Types. J Natl Cancer Inst. 2016;108 doi: 10.1093/jnci/djw144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva KM, Correa I, Josephs DH, Karagiannis P, Egbuniwe IU, Cafferkey MJ, Spicer JF, Harries M, Nestle FO, Lacy KE, et al. Effects of BRAF mutations and BRAF inhibition on immune responses to melanoma. Mol Cancer Ther. 2014;13:2769–2783. doi: 10.1158/1535-7163.MCT-14-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- Khurana E, Fu Y, Chen J, Gerstein M. Interpretation of genomic variants using a unified biological network approach. PLoS Comput Biol. 2013;9:1002886. doi: 10.1371/journal.pcbi.1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijnenburg TA, Wang L, Zimmerman MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, et al. Genomic and molecular landscape of DNA damage repair deficiency across The Cancer Genome Atlas. Cell Rep. 2018 doi: 10.1016/j.celrep.2018.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc MP, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane J, et al. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2009;37:D1006–1012. doi: 10.1093/nar/gkn838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legut M, Cole DK, Sewell AK. The promise of gammadelta T cells and the gammadelta T cell receptor for cancer immunotherapy. Cell Mol Immunol. 2015;12:656–668. doi: 10.1038/cmi.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, Jiang P, Shen H, Aster JC, Rodig S, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17:174. doi: 10.1186/s13059-016-1028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MX, Yeung JM, Cherny SS, Sham PC. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet. 2012;131:747–756. doi: 10.1007/s00439-011-1118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Pan HF, Ye DQ. microRNAs function in CD8+T cell biology. J Leukoc Biol. 2015;97:487–497. doi: 10.1189/jlb.1RU0814-369R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson L, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. An integrated TCGA pan-cancer clinical data resource to drive high quality survival outcome analytics. Cell. 2018 doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–346. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7 Suppl. 2006;1:S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinny AS, Li JL, Wu L. Mastermind-like transcriptional co-activators: emerging roles in regulating cross talk among multiple signaling pathways. Oncogene. 2008;27:5138–5147. doi: 10.1038/onc.2008.228. [DOI] [PubMed] [Google Scholar]
- McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bosse D, Wankowicz SM, Cullen D, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359:801–806. doi: 10.1126/science.aan5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris LG, Riaz N, Desrichard A, Senbabaoglu Y, Hakimi AA, Makarov V, Reis-Filho JS, Chan TA. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget. 2016;7:10051–10063. doi: 10.18632/oncotarget.7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mose LE, Selitsky SR, Bixby LM, Marron DL, Iglesia MD, Serody JS, Perou CM, Vincent BG, Parker JS. Assembly-based inference of B-cell receptor repertoires from short read RNA sequencing data with V’DJer. Bioinformatics. 2016;32:3729–3734. doi: 10.1093/bioinformatics/btw526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8:33. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull EO, Carlin DE, Niepel M, Sorger PK, Haussler D, Stuart JM. Discovering causal pathways linking genomic events to transcriptional states using Tied Diffusion Through Interacting Events (TieDIE) Bioinformatics. 2013;29:2757–2764. doi: 10.1093/bioinformatics/btt471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavesi G, Pesole G. Using Weeder for the discovery of conserved transcription factor binding sites. Curr Protoc Bioinformatics. 2006:11. doi: 10.1002/0471250953.bi0211s15. Chapter 2, Unit 2. [DOI] [PubMed] [Google Scholar]
- Pencina MJ, D’Agostino RB. Overall C as a measure of discrimination in survival analysis: model specific population value and confidence interval estimation. Stat Med. 2004;23:2109–2123. doi: 10.1002/sim.1802. [DOI] [PubMed] [Google Scholar]
- Plaisier CL, Horvath S, Huertas-Vazquez A, Cruz-Bautista I, Herrera MF, Tusie-Luna T, Aguilar-Salinas C, Pajukanta P. A systems genetics approach implicates USF1, FADS3, and other causal candidate genes for familial combined hyperlipidemia. PLoS Genet. 2009;5:e1000642. doi: 10.1371/journal.pgen.1000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisier CL, O’Brien S, Bernard B, Reynolds S, Simon Z, Toledo CM, Ding Y, Reiss DJ, Paddison PJ, Baliga NS. Causal Mechanistic Regulatory Network for Glioblastoma Deciphered Using Systems Genetics Network Analysis. Cell Syst. 2016;3:172–186. doi: 10.1016/j.cels.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisier CL, Pan M, Baliga NS. A miRNA-regulatory network explains how dysregulated miRNAs perturb oncogenic processes across diverse cancers. Genome Res. 2012;22:2302–2314. doi: 10.1101/gr.133991.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta-Pardo E, Godzik A. Mutation Drivers of Immunological Responses to Cancer. Cancer Immunol Res. 2016;4:789–798. doi: 10.1158/2326-6066.CIR-15-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Punt S, Langenhoff JM, Putter H, Fleuren GJ, Gorter A, Jordanova ES. The correlations between IL-17 vs. Th17 cells and cancer patient survival: a systematic review. Oncoimmunology. 2015;4:e984547. doi: 10.4161/2162402X.2014.984547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramilowski JA, Goldberg T, Harshbarger J, Kloppmann E, Lizio M, Satagopam VP, Itoh M, Kawaji H, Carninci P, Rost B, et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat Commun. 2015;6:7866. doi: 10.1038/ncomms8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss DJ, Plaisier CL, Wu WJ, Baliga NS. cMonkey2: Automated, systematic, integrated detection of co-regulated gene modules for any organism. Nucleic Acids Res. 2015;43:e87. doi: 10.1093/nar/gkv300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltz JH, Gupta R, Hou L, Kurc T, Singh P, Nguyen V, Samaras D, Shroyer KR, Zhao T, Batiste R, et al. Spatial Organization And Molecular Correlation Of Tumor Infiltrating Lymphocytes Using Deep Learning On Pathology Images. Cell Reports. 2018 doi: 10.1016/j.celrep.2018.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila W, Luna A, La K, Dimitriadoy S, Liu DL, Kantheti HS, Heins Z, et al. Oncogenic signaling pathways in The Cancer Genome Atlas. 2018 doi: 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrucca L, Fop M, Murphy TB, Raftery AE. mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J. 2016;8:289–317. [PMC free article] [PubMed] [Google Scholar]
- Senbabaoglu Y, Gejman RS, Winer AG, Liu M, Van Allen EM, de Velasco G, Miao D, Ostrovnaya I, Drill E, Luna A, et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016;17:231. doi: 10.1186/s13059-016-1092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33:1152–1158. doi: 10.1038/nbt.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva TC, Colaprico A, Olsen C, D’Angelo F, Bontempi G, Ceccarelli M, Noushmehr H. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Res. 2016;5:1542. doi: 10.12688/f1000research.8923.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siragusa E, Weese D, Reinert K. Fast and accurate read mapping with approximate seeds and multiple backtracking. Nucleic Acids Res. 2013;41:e78. doi: 10.1093/nar/gkt005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30:3310–3316. doi: 10.1093/bioinformatics/btu548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Shalabi A, Hubbard-Lucey VM. Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol. 2018;29:84–91. doi: 10.1093/annonc/mdx755. [DOI] [PubMed] [Google Scholar]
- Tatlow PJ, Piccolo SR. A cloud-based workflow to quantify transcript-expression levels in public cancer compendia. Sci Rep. 2016;6:39259. doi: 10.1038/srep39259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A, Shih J, Ha G, Gao GF, Zhang X, Berger AS, Schumacher S, Wang C, Hu H, Liu J, et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018 doi: 10.1016/j.ccell.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Gomez S, Arenas A, El-Ashry D, Schmidt M, Gehrmann M, Caldas C. Improved prognostic classification of breast cancer defined by antagonistic activation patterns of immune response pathway modules. BMC Cancer. 2010;10:604. doi: 10.1186/1471-2407-10-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R, Walther G. Cluster validation by prediction strength. J Comput Graph Stat. 2005;14:511–528. [Google Scholar]
- Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355 doi: 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E, Schoeps T, Donitz J. TFClass: an expandable hierarchical classification of human transcription factors. Nucleic Acids Res. 2013;41:D165–170. doi: 10.1093/nar/gks1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DM, Lenburg ME, Yau C, Boudreau A, van ‘t Veer LJ. Gene co-expression modules as clinically relevant hallmarks of breast cancer diversity. PLoS One. 2014;9:e88309. doi: 10.1371/journal.pone.0088309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T, et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature. 2012;490:556–560. doi: 10.1038/nature11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 PanImmune Feature matrix of immune characteristics. Related to Figure 1.
Spreadsheet containing all tumor-sample characterizations used in, and generated by this study
Table S2 Driver mutations enriched in immune subtypes. Related to Figure 4.
Table S3 Recurrent pMHCs. Related to Figure 5.
Table S4 Recurrent Indels. Related to Figure 5.
Table S5 Recurrent TCRs and co-occurrence of TCRs and pMHC. Related to Figure 5.
Table S6 Immunomodulators. Related to Figure 6