

Abstract
Natural carbohydrate polymers such as starch, cellulose, and chitin provide renewable alternatives to fossil fuels as a source for fuels and materials. As such, there is considerable interest in their conversion for industrial purposes, which is evidenced by the established and emerging markets for products derived from these natural polymers. In many cases, this is achieved via industrial processes that use enzymes to break down carbohydrates to monomer sugars. One of the major challenges facing large-scale industrial applications utilizing natural carbohydrate polymers is rooted in the fact that naturally occurring forms of starch, cellulose, and chitin can have tightly packed organizations of polymer chains with low hydration levels, giving rise to crystalline structures that are highly recalcitrant to enzymatic degradation. The topic of this review is oxidative cleavage of carbohydrate polymers by lytic polysaccharide monooxygenases (LPMOs). LPMOs are copper-dependent enzymes (EC 1.14.99.53–56) that, with glycoside hydrolases, participate in the degradation of recalcitrant carbohydrate polymers. Their activity and structural underpinnings provide insights into biological mechanisms of polysaccharide degradation.
Graphical Abstract
1. INTRODUCTION
Most glycoside hydrolases use general acid/base catalysis to hydrolyze substrates (EC 3.2.1.-) (Figure 1A and 1B), with acidic groups provided by aspartic acid or glutamic acid. For instance, endoglucanases hydrolyze carbohydrate polymers to shorter, soluble oligo-saccharides. Their active sites are often described as grooves that accommodate single, isolated substrate chains. However, isolating a single chain from the bulk (decrystallization) requires considerable energy, resulting in the action of many glycosyl hydrolases on crystalline substrates being orders of magnitude slower than comparable activity on soluble carbohydrate chains.1 In contrast, LPMOs break polysaccharide chains in an oxygen- and electron-dependent process (Figure 1C), apparently without the need for decrystallization, and may contribute to reduced dosing of enzymatic cocktails for the industrial degradation of lignocellulosic biomass substrates. These and other features of LPMOs are discussed in more detail below.
Figure 1.

Catalytic schemes for hydrolytic and oxidative cleavage of glycosidic bonds. (A) Retaining mechanism for hydrolysis of glycosidic bonds by glycoside hydrolases. (B) Inverting mechanism for hydrolysis of glycosidic bonds by glycoside hydrolases (Modified with permission from ref 2. Copyright 1995 Elsevier). (C) General scheme for oxygen and electron-dependent cleavage of glycosidic bonds by LPMOs. Vaaje-Kolstad et al. demonstrated that oxygen atoms from molecular oxygen and solvent water end up in reaction products.3 Ascorbic acid has been used extensively as an electron donor for LPMOs, but various other compounds and enzyme systems can provide electrons as well.4–6
The naming convention for LPMO enzymes was not consistent until recently, and those familiar with LPMO literature will have noted that some LPMOs have historic names that do not reflect their activity. Throughout this review a LPMO enzyme will be referred to by a combination of (i) a two-letter abbreviation of the species it originates from, (ii) “LPMO”, (iii) the number of the Auxiliary Activity family it has been assigned to, and (iv) a one-letter code for the gene designation. Any alternative name(s) for the enzyme will be included between brackets. For instance, chitin-active CBP21 from Serratia marcescens will be referred to as SmLPMO10A (CBP21).
Several aspects of LPMOs have been reviewed in recent years. The role of LPMOs in the breakdown of cellulose was reviewed by Beeson and co-workers.7 Other reviews include those by Vu et al. on starch-active LPMOs, a crystallographer’s perspective on LPMOs provided by Frandsen and Lo Leggio, and an additional structural review by Vaaje-Kolstad et al.8–10 Johansen recently reviewed the use of LPMOs in commercial enzyme cocktails, and other reports on LPMOs have addressed the issue of electron donor promiscuity.11,12 In this review, we aim to provide a focus on three aspects of LPMOs: (i) use of LPMOs in industrial processes, predominantly degradation of lignocellulosic biomass into fermentable sugars, (ii) LPMO protein structures, and (iii) properties of the Cu active site and the catalytic mechanism.
2. BACKGROUND
2.1. Recalcitrant Substrates
The array of LPMO substrate specificities are reflected in the diversity of natural recalcitrant polysaccharides. In this section three main types of polysaccharides are described: (i) starch, (ii) polysaccharides found in lignocellulosic plant cell walls, and (iii) chitin. The descriptions are not intended to be an exhaustive review in themselves, as this has been done elsewhere, as referenced in the sections. Instead, this background provides a foundation to support the following sections in the review.
2.1.1. Starch
Starch is the most abundant storage polysaccharide in plants and is a polymer of α-1,4-linked glucose with α-1,6 side branches (Figure 2A). It is widely used as a source for renewable fuels such as ethanol and renewable materials such as bioplastics, food, feed, and a range of other industrial products and applications. The main sources of starch for these applications are wheat, coarse grains, corn, and rice, the global production of which amounted to a combined 3500 M metric tons in 2014/2015 according to the USDA.13
Figure 2.

Structures of starch. (A) Molecular structure of α-1,4-linked glucan with C1 carbon indicated with blue shading and C4 carbon indicated with yellow shading. α-1,4 linkage results in helical structures in solution. (B) Molecular structures of amylose and amylopectin showing branching patterns and formation of secondary structures. Individual glucose residues are represented by gray circles. (C) Alignment of amylopectin double helices giving rise to crystalline regions (1) interspersed with amorphous regions (2). Adapted with permission from ref 14. Copyright 2016 Springer.
Starch is produced in the chloroplast of green leaves and amyloplast of seeds, pulses, and tubers. Starch is made up of two polymers, amylopectin and amylose, which together are deposited into insoluble, semicrystalline granules (Figure 2B). Amylopectin, which makes up about 75–90% of the starch granule, has a degree of polymerization on the order of 105–106, with 4–5% of its linkages being α-1,6 branching points. By comparison, amylose is smaller, less branched, and has a degree of polymerization of 103. The branch points in amylopectin are clustered at repeating intervals. The α-1,4-linked backbone of amylopectin is helical in solution and lies parallel to two neighboring branch chains that self-associate into a double helix, giving rise to the crystalline regions in starch granules. Together, alternating regions of clustered branch points and extended helical side chains combined with tightly packed starch chains give the granules concentric circles of semicrystalline regions and a low moisture content, making them highly resistant to enzymatic degradation (Figure 2C).14
2.1.2. Lignocellulosic Biomass
Initially, plant cells are surrounded by thin primary cell walls that allow for cell growth. Once cells have reached their final shape a thicker secondary cell wall encapsulates them and serves to provide structural rigidity and pathogen defense to the plant. These are the major sources of carbohydrates in lignocellulosic biomass. Major components of cell walls are polysaccharide cellulose, a collection of predominantly β-1,4-linked polysaccharides collectively called hemicellulose, a family of complex polysaccharides containing β-1,4-linked galacturonic acid called pectin, and the polyaromatic polymer lignin.
2.1.2.1. Cellulose
Cellulose is a polymer of β-1,4-linked glucopyranose (Figure 3A). It is the major structural component of plant cells, and with an estimated half-life of 22 million years, it is the most stable and abundant polymer found in nature.15 Cellulose is synthesized in plants and bacteria by the enzyme cellulase synthase.16 In higher plants, the cellulase synthase complex is located in the plasma membrane of plant cells and consists of a rosette of six cellulase synthase trimers.17 Since each cellulase synthase molecule produces one chain of cellulose, 18 cellulose chains are proposed to form a microfibril, in agreement with previously published spectroscopic studies.18,19 The degree of polymerization of cellulose chains varies among plant species but generally ranges between 1500 and 5000.18 The intra- and interchain hydrogen bonds between the hydroxyl groups and oxygen atoms of neighboring chains result in a compact, rigid structure that is insoluble. To put this into perspective, starch gelatinizes by incubation at 60–70 °C at atmospheric pressure, while cellulose gelatinizes at much higher temperature (320 °C) and pressure (25 MPa).19
Figure 3.
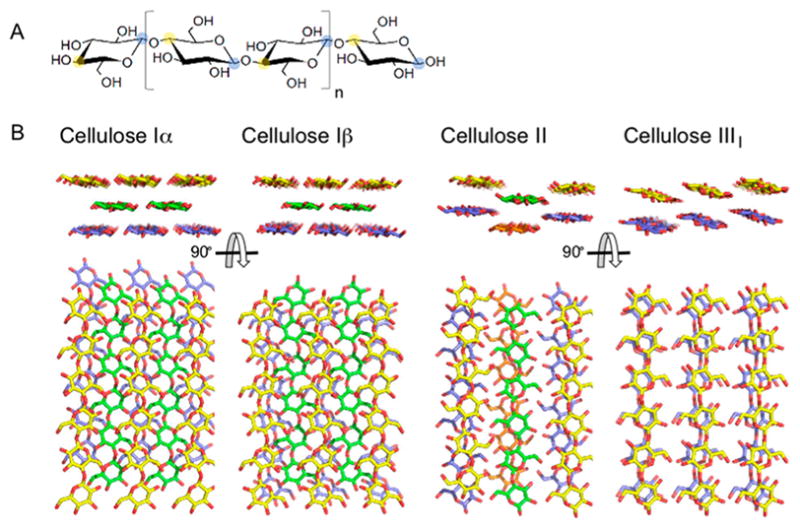
(A) Molecular structure of cellulose with structural cellobiose repeating unit indicated by brackets with C1 carbon indicated with blue shading and C4 carbon indicated with yellow shading. (B) Molecular structures of Cellulose Iα, Iβ, II, and IIII. Hydrogen atoms have been omitted from view for clarity. Crystallites were generated using Cellulose-Builder and visualized using PyMol.20,21
Overall, cellulose fibrils show highly organized or crystalline regions interspersed with less organized, amorphous regions. Various crystalline forms of cellulose have been identified, which differ in chain orientation, chain arrangement, and hydrogen-bonding pattern (Figure 3B). Cellulose I consists of parallel sheets of hydrogen-bonded chains that stack on top of each other via hydrophobic interactions. Subtle changes in crystal packing discriminate Cellulose Iα, found in algae and bacteria, from Cellulose Iβ, found in tunicin and higher plants.22 Chemical treatment can convert Cellulose I into other types, as evidenced by the observation that treatment with alkali or ionic liquids yields a structure with antiparallel chains and intersheet hydrogen bonding, characteristic of Cellulose II.23,24 Treatment of Cellulose I and II with anhydrous ammonia results in Cellulose IIII and IIIII, respectively. Cellulose III is the result of ammonia-induced swelling of the crystal to a metastable phase that returns to Cellulose I upon heating.25,26 A fourth type of crystalline cellulose has been identified,27 but more recent studies indicate Cellulose IVI might be the same as Cellulose Iβ.28 Amorphous cellulose, with a low degree of crystallinity, can be made by dissolution of cellulose in phosphoric acid and subsequent extensive washing with water. The resulting phosphoric acid swollen cellulose, or PASC, is a model substrate for characterization of cellulases and is widely used in LPMO studies.29
2.1.2.2. Hemicelluloses
A second major component of plant cell walls are hemicelluloses (Figure 4), of which the four main types are xyloglycans (xylans), xyloglucans, mannoglycans (mannans), and mixed-linkage β-glucans, which all have (predominantly) β-1,4-linked saccharide backbones (Figure 4).30 Contrary to cellulose, heteroxylans can be made up of multiple saccharide components coupled by a variety of glycosidic linkages.31 Each of these four types will be defined in more detail below.
Figure 4.
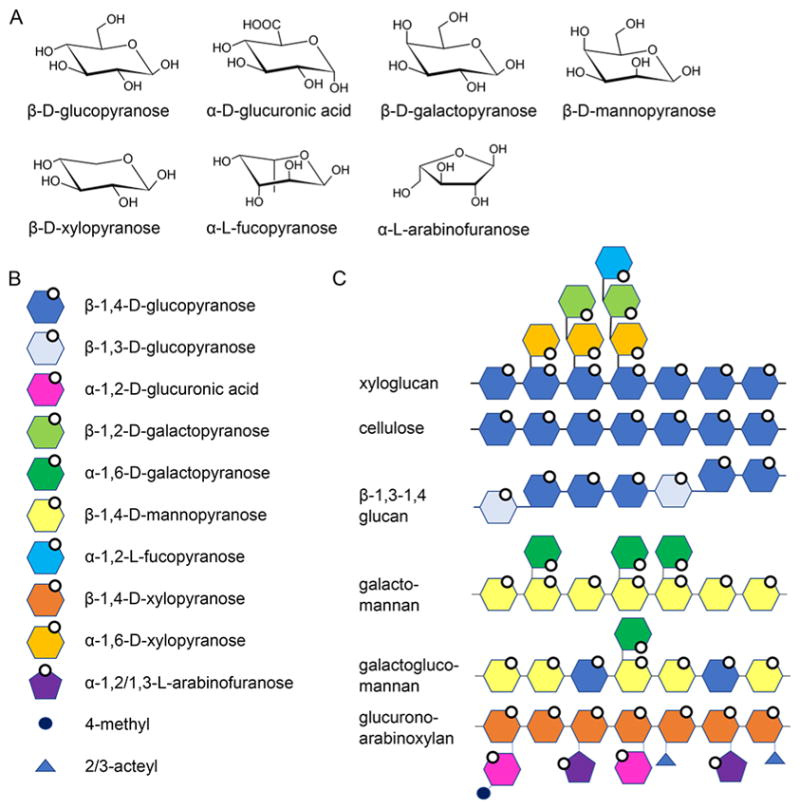
Hemicelluloses. (A) Molecular structures of monosaccharide building blocks. (B) Schematic representations of hemicellulose building blocks. (C) Hemicellulose polymers. of branching and, in many cases, is matched to the role of the polysaccharide in the cell wall.36
Xylans have a β-1,4-linked xylose backbone, which can be decorated with α-1,2 glucuronic acid, or 4-O-methyl glucuronic acid, α-1,2 or α-1,3 arabinofuranose, and 2-O- and 3-O-acetyl groups. In dicots, the xylose backbone is predominantly decorated with glucuronic acid (glucuronoxylan) and can be found in secondary cell walls, where it is the major hemicellulose. Corn and other grasses also have a xylose backbone, although substituted with glucuronic acid and arabinose (glucuronoarabinoxylan), and it is the major hemicellulose in both primary and secondary cell walls. The xylose residues can be either singly (α-1,2 or α-1,3) or doubly (α-1,2–1,3) substituted with arabinose, and the xylan backbone may be acetylated at O2 and O3 to varying degrees, ranging between 40% and 70%.32 In grasses, arabinose residues of xylan can be covalently linked to each other or to lignin through hydroxycinnamic acid esters, such as ferulic acid and p-coumaric acid.33
A second component, xyloglucan, is the major hemicellulose in primary cell walls of dicots. Xyloglucan has a β-(1,4)-linked glucopyranosyl backbone that is branched to varying degrees with xylopyranosyl, acetylated galactopyranosyl, and fucopyranosyl substituents.34 Contrary to xylan, xyloglucan can have a regular structure with repeating structural blocks. Type I has three xylosylated residues followed by a single glucose unit and is found in hardwoods, herbal plants, and some grasses. Type II has two xylosylated units followed by two nonsubstituted glucose residues. In both types the repeating blocks are interspersed with xylosylated units that are substituted with galactose and fucose.30
The third component, mannoglycans (mannans), have a backbone consisting of mannopyranose (mannan) or mannopyranose and glucopyranose (glucomannan). A portion of the mannose residues are branched with α-1,6-galactopyranose in galactomannan and galactoglucomannan. Although mannans are present in varying amounts in all cell walls, glucomannans are the main hemicellulose type in secondary cell walls of softwoods and a minor component in those of hardwoods, herbal plants, and grasses.30
Finally, a fourth type of hemicellulose, β-1,3–1,4 glucans, which are commonly found in cereal, can be thought of as an irregular string of cellotriosyl and cellotetraosyl units coupled by β-1,3 linkages.33 β-1,3–1,4-Glucan is unbranched and kinked due to the irregularly spaced β-1,3 linkages between linear β-1,4-linked stretches.35
The saccharide units in the backbones of cellulose (glucose), xylan (xylose), xyloglucan (glucose), and mannans (mannose and glucose) all have an equatorial C4-hydroxyl group, which, when β-linked, results in a linear backbone conformation. Linear polysaccharides tend to align, form interchain bonds, and aggregate out of solution. This has been well established for cellulose. Cellulose always has the same chemical structure and properties, whereas the aggregation propensity of xylan, xyloglucan, and mannans is modulated by the type and degree
2.1.2.3. Pectin
Pectins are a family of complex polysaccharides containing α-1,4-linked galacturonic acid (GalpA, Figure 5) and are found in primary cell walls during cell growth.37,38 Pectins form a gel-like matrix that can be altered to allow cell elongation, provide protective barriers for the cell, and are virtually absent from secondary cell walls. Three main types of pectin are homogalacturonan, substituted galacturonan, and rhamnogalacturonan. Homogalacturonan consists of an α-1,4-linked GalpA backbone that can be substituted with methyl or acetyl groups. In xylogalacturonan, the GalpA backbone is substituted with β-1,3-linked xylopyranose, while in apiogalacturonan, it is substituted with apiose. In rhamnogalacturonan I, rhamnopyranose (Rhap) and galacturonic acid alternate in an α-1,2-Rhap-α-1,4-GalpA-linked backbone. The rhamnosyl residues can be substituted with galactose or arabinose. Rhamnogalacturonan II are comprised of short stretches of homogalacturonan that have been decorated with exotic saccharide residues, such as apiose, aceric acid (3-C-carboxy-5-deoxy-L-xylose), 3-deoxy-lyxo-2-heptulosaric acid, and 3-deoxy-manno-2-octulosonic acid. Pectins may also be cross-linked by esters.
Figure 5.

Pectins. Schematic representation of five types of pectins, and the molecular structures of the monosaccharide components of pectins.
2.1.2.4. Lignin
A fourth major component of plant cell walls is the heterogenic polyphenolic polymer lignin, which provides structural strength and acts as a diffusion barrier.33 Lignin is composed of 4-hydroxypropanoids that are connected by both ether and carbon–carbon linkages. The three main monolignol building blocks are p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol (Figure 6A) that are polymerized through a free-radical coupling process mediated by peroxidases.39,40 The degree of methoxylation of the phenyl ring results in hydroxyphenyl (H), guaiacyl (G), and syringyl (S) lignin, respectively (Figure 6B). The predominant monolignol in lignin varies between plant species and even plant tissues. For instance, lignins from softwoods are derived mainly from coniferyl alcohol with some p-coumaryl alcohol, while hardwood lignins mostly contain coniferyl and sinapyl alcohols with smaller amounts of p-coumaryl alcohol.39 Grasses have all three monolignols with a majority of p-coumaryl alcohol-derived lignin.41 The number-average molecular weight of lignin is in the range of 7500 g·mol−1, while the weight-average molecular weight is around 25 000 g·mol−1.42 The free phenolic hydroxyl groups and o-methoxy substitutions in the aromatic ring lend lignin antioxidant properties, which are an active area of research.43–45
Figure 6.
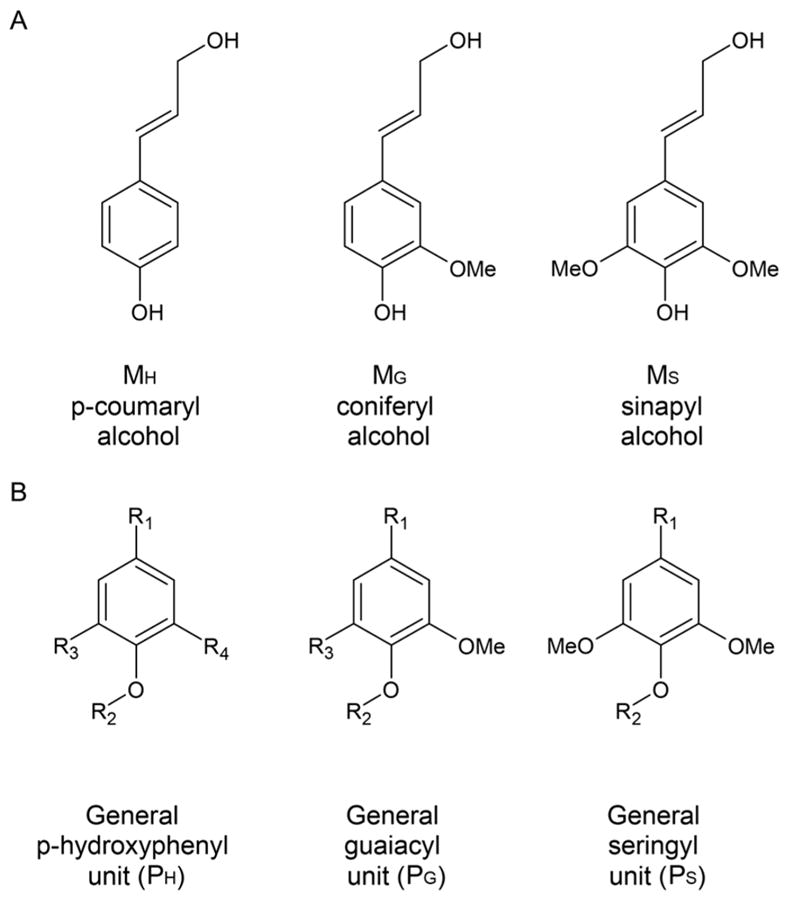
(A) Structures of major monolignol building blocks. (B) Substituted phenyl rings in polymerized lignin. Adapted with permission from ref 40. Copyright 2004 Springer.
2.1.2.5. Plant Cell Wall Synthesis
While the plant cell is growing, the primary cell wall consists mainly of hemicellulose and pectin, with relatively little cellulose and lignin.46 When the cell has reached its final shape and size, the cell wall becomes thicker and the composition shifts to primarily cellulose, xylan, and lignin. Cellulose is synthesized in the plasma membrane from which the fibrils get deposited directly into the matrix.47 Hemicellulose and other matrix components are synthesized in the Golgi and transported in vesicles to the plasma membrane.48 Lignin deposition starts from several well-defined initiation sites within the cell wall region, which expand uniformly until the regions coalesce.39 A primary cell wall consists mainly of polysaccharides placed in water and has a water content of 60–70%. The water content in secondary cell walls with lignin deposition is much lower (about 5%) due to the hydrophobic nature of the aromatic polymer.46 The general polysaccharide compositions of primary and secondary cell walls vary per major plant type (Table 1). Thus, in order to gain further insight into the localization, identification, and quantification of cell wall polysaccharide and lignin components, molecular tools have been developed. For example, in glycome profiling, plant cell walls are subjected to extractions of increasing severity and extracts are screened with monoclonal antibodies and carbohydrate-binding domains with specificities for polysaccharide components.49 Use of these tools as probes in fluorescence microscopy has allowed visualization of the heterogeneity and diversity in cell wall structures.50
Table 1.
Occurrence of Polysaccharides in Plant Cell Wallsa
| polysaccharide | amount of polysaccharide (% w/w) | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| dicots | grasses | conifers | ||||
|
|
|
|
||||
| primary | secondary | primary | secondary | primary | secondary | |
| cellulose | 15–30 | 45–50 | 20–30 | 35–45 | present | present |
| xyloglucan | 20–25 | minor | 2–5 | minor | 10 | minor |
| glucuronoxylan | 20–30 | |||||
| glucuronoarabinoxylan | 5 | 20–40 | 40–50 | 2 | 5–15 | |
| (gluco)mannan | 3–5 | 2–5 | 2 | 0–5 | ||
| galactoglucomannan | absent | 0–3 | present | 10–30 | ||
| β-(1→3,1→4)-glucan | absent | 2–15 | minor | absent | absent | |
2.1.2.6. Industrial Lignocellulosic Substrates
Major lignocellulosic substrates for production of fuels and materials at commodity scale are corn stover in the United States, sugar cane bagasse in Brazil, and wheat straw in Europe. All three belong to the family of Poaceae, also known as grasses. Both corn stover and wheat straw are waste products of grain production and are otherwise left on the field for fertilization and mineralization or harvested for further use. A significant amount of corn is produced in the United States yearly, and therefore, the corn stover supply chain in the United States has received considerable attention. According to reports, corn stover can be harvested in a sustainable manner, providing a stable supply of lignocellulosic substrate for, for instance, biofuel production.52,53 In Brazil, sugar cane is collected routinely and processed for the sugar and ethanol industry. Sugar cane bagasse is the waste product left after crushing the stalks for their juice and is available for energy production (i.e., electricity or biofuels).54,55 The main polysaccharides in the secondary cell walls of Poaceae, which make up the bulk of the biomass, are cellulose and glucuronoarabinoxylan (Table 1).
2.1.2.7. Pretreatment of Lignocellulosic Biomass
The digestibility of polysaccharides in lignocellulosic biomass is hindered by physicochemical, structural, and compositional features. A (thermo)chemical treatment of lignocellulosic biomass can be beneficial for enzymatic conversion to fermentable sugars. Widely used methods include pretreatment with dilute acid, steam, or ammonia, all of which have previously been reviewed.56–59 Acid pretreatment solubilizes hemicellulose and increases the pore size of biomass, thus increasing accessibility. Steam pretreatment generates acetic acid in situ from hydrolysis of acetyl groups from hemicellulose and is comparable to dilute acid pretreatment, although to a lesser extent. Ammonia pretreatment such as ammonia fiber explosion (AFEX) deacetylates hemicellulose and disrupts the biomass structure by, among other possibilities, breaking lignin–carbohydrate bonds which loosens the cellulose crystal structure and increases digestibility. Hemicellulose polymers are converted to oligomers, which stay insoluble. Another option is acid pretreatments, which can degrade amorphous regions of cellulose fibrils and as a result increase the crystallinity of the substrate. Ammonia can loosen up the cellulose crystal structure, increasing digestibility. As mentioned, soaking Cellulose Iβ in anhydrous ammonia results in swelling of the crystal lattice to form Cellulose IIII, which reduces the required amounts of cellulase for hydrolysis. A process that uses this principle to reduce the recalcitrance of lignocellulosic biomass by generating Cellulose IIII was recently described by Da Sousa Costa et al.60 In addition to changing cellulose crystallinity, the process also extracts about one-half of the lignin, which may be beneficial for enzymatic conversion of polysaccharides. Dilute acid pretreatment can solubilize lignin, which is redeposited onto the biomass, hindering enzymatic hydrolysis.61 Ammonia pretreatment has a minimal effect on lignin, although it appears to modify it in such a way that it reduces interference with enzymatic biomass conversion.59
Thus, the type of pretreatment can affect the structure and composition of lignocellulosic biomass, and the choice of pretreatment technologies can influence the composition of the noncarbohydrate part of substrates. It is worth noting here that the noncarbohydrate biomass components can also be involved in the activity of LPMOs, as will be discussed in further sections.
2.1.3. Chitin
Chitin is a polymer of β-1,4-linked N-acetylglucosamine and is found in the cell walls of fungi, the egg shells and gut lining of nematodes, and the exoskeleton of arthropods (including crustaceans, insects, and arachnids) (Figure 7A). Like cellulose, chitin chains are linear, unbranched, and self-associate into fibrils as a result of interchain hydrogen bonding. Crystalline chitin can occur in three polymorphs, α, β, and γ, which differ mainly in the degree of hydration.62 In α-chitin, the polysaccharide chains are antiparallel, which results in a tight packing (Figure 7B). The cuticular structures in arthropods are predominantly of this type. For comparison, the chains in β-chitin are parallel, while sets of two parallel chains aligned with an antiparallel one are observed in γ-chitin.
Figure 7.

Chitin. (A) Molecular structure of chitin with C1 carbon indicated with blue shading and C4 carbon indicated with yellow shading. (B) Crystal structure determined for α-chitin with the crystallographic repeating unit indicated by blue boxes. Reproduced from with permission from ref 63. Copyright 2009 American Chemical Society. (C) Molecular structure of chitosan. Repeating structural disaccharide units in A and C indicated by brackets.
Both β-and γ-chitin are more hydrated than α-chitin resulting in a softer and more flexible structure as found in insect guts. Deacetylated chitin, called chitosan (Figure 7C), is more flexible than chitin and can be solubilized due to the presence of 2-amine, 2-deoxy glucose units that have a pKa of ~6.3.64 The ability to increase the solubility of chitin oligosaccharides by deacetylation (into chitosan oligosaccharides) has facilitated the detection of enzymatic polysaccharide oxidation.3
Chitin is an abundant natural resource and a waste product of fishery and aquaculture industries. In 2014 an estimated 6.9 Mt of crustaceans were caught globally.65 About 35–45% of this is discarded as waste and is available for industrial, agricultural, and pharmaceutical applications.66
2.1.4. Enzymatic Degradation of Recalcitrant Polysaccharides
In the preceding paragraphs, we discussed natural polysaccharides that can be recalcitrant for enzymatic conversion. Starch, lignocellulosic polysaccharides, and chitin can occur in (semi-)crystalline forms that consist of tightly packed saccharide chains and have a low water content. Enzymatic degradation of these substrates by glycoside hydrolases is a large field of research and has been reviewed previously.67–69 There are glycoside hydrolases that have evolved to act on the crystalline polysaccharide forms, and many organisms encode such enzymes in their genome. Upon encountering recalcitrant substrates, enzymes are often excreted for degradation of the substrates. The active sites of cellulases, for instance, have been described as “grooves” or “tunnels”.70 Hydrolysis of glycosidic bonds by these enzymes would require polysaccharide chains to be isolated from the bulk of the crystal, which in turn requires energy.67 The active sites of LPMOs are located on the protein surface, and polysaccharide chains in crystalline substrates could interact with the active site without an apparent need for isolation of individual substrate chains from the bulk (see section 4). As will be discussed below, many organisms contain LPMO-encoding genes in their genomes, which can be upregulated simultaneously with glycoside hydrolases during growth on recalcitrant polysaccharides.
2.2. LPMOs are Abundant in Nature
2.2.1. LPMO-Encoding Genes in Genomes
Many organisms, including cellulolytic organisms, encode one or several LPMOs in their genome. The majority reported to date have been observed in fungi and bacteria. Among fungi, the phyla of Ascomycota and Basidiomycota are a source of LPMO genes. Reported numbers of LPMO-encoding genes in fungal genomes typically range from a few up to a few dozen.71–74 Traditionally, wood-decaying Basidiomycetes have been characterized as white rot fungi or brown-rot fungi based on their ability to degrade lignin (in white rot species) in addition to plant cell wall polysaccharides. The genomes of both types encode LPMOs, with brown rot fungi typically containing a larger number of LPMO genes compared to white rot fungi.75
In the Kingdom of Bacteria, the majority of LPMO-encoding genes have been reported in genomes of species belonging to the phyla of Actinobacteria, Firmicutes, and Proteobacteria with a noted expansion of LPMO genes in the genomes of bacteria belonging to the genera Streptomyces, Bacillus, and Vibrio.76 Bai et al. studied the occurrence of chitinolytic systems in bacterial genomes and reported that AA10 LPMO genes are present in roughly one-third of terrestrial bacterial genomes but are absent in strict anaerobic bacteria.77
While most LPMOs have been identified in fungi and bacteria, they have also been annotated in the genome of the phytoplankton Emiliania huxleyi, as well as in insect virus genomes where they are part of fusolin spindle proteins and facilitate invasion of the host.78–80 The presence of genes corresponding to LPMO profiles have been reported for Drosophila melanogaster, bivalves, stony corals, and sea anemones.71
2.2.2. LPMOs are Induced by Recalcitrant Substrates
Many species that have LPMOs encoded in their genome can grow on recalcitrant polysaccharide substrates. Similar to glycoside hydrolases, production of LPMOs is induced during growth on recalcitrant substrates such as starch,81 plant cell wall polysaccharides,82–86 or chitin.79,87–89 Fungi containing multiple LPMO genes in their genome can display substrate-dependent expression profiles of LPMO genes.86 The observed expressional coregulation with other carbohydrate active enzyme genes suggests involvement of LPMO gene products in microbial breakdown of polysaccharides. Vaaje-Kolstad et al. even reported that the degradation of chitin by chitinases from the soil bacterium Serratia marsescens depended on the presence of SmLPMO10A (CBP21), a chitin-active LPMO.79 Thus, LPMOs are likely to be present in the secretomes of LPMOcontaining organisms grown on recalcitrant polysaccharide substrates.
2.2.3. Oxidative Degradation of Lignocellulosic Substrates
In 1950, Reese and co-workers described a two-component cellulase model for cellulose degradation by fungi in which a first step, termed C1, enables the second hydrolytic step, termed Cx.90 They also noted that several Ascomycete fungi produced Cx enzymes that were able to hydrolyze modified cellulose, but not all were able to degrade crystalline cellulose, where the latter was attributed to the absence of a C1 component.91 It is possible that the C1 component described by Reese et al. is at least in part LPMO activity.
LPMOs are copper-dependent enzymes that bind copper with a 1:1 stoichiometry.92,93 The coordination of the copper atom by two histidine residues is similar to that of CopC, a protein involved in copper resistance in bacteria.93 The copper dependence of LPMOs was further demonstrated by the observation that metal chelators reduced the lignocellulose-degrading efficiency of LPMO-containing enzyme cocktails. 94–96
Many mechanistic proposals to date contend that an electron reduces a LPMO-bound Cu2+ to Cu+, which can then bind molecular oxygen and abstract a proton from the polysaccharide substrate at either the C1 or the C4 position. A second electron is then thought to cause hydroxylation of the substrate and cleavage of the glycosidic bond.93 In many cases, ascorbic acid has been efficient as an electron donor for LPMOs; however, electrons can be acquired from various molecules and enzyme systems.3–5,92,93,96
LPMOs utilize molecular oxygen to oxidize polysaccharide substrates, incorporating one atom of molecular oxygen in the reaction product.3 Further evidence for the oxygen dependence comes from studies showing that the activity of LPMOcontaining enzyme cocktails is reduced under oxygen-limiting conditions.97,98 In the absence of substrate and in the presence of O2 and reductant, LPMOs can generate H2O2.99 This observation has been used to quantify LPMO activity.99,100 Recently, Bisarro et al. reported that in fact H2O2, not oxygen, is the cosubstrate for LPMO activity.101 The presence of H2O2 in the reaction resulted in oxygen-independent formation of oxidized reaction products with an oxygen atom from H2O2 incorporated in the product, even in the presence of molecular oxygen. See section 5 for a discussion of the possible reaction mechanisms of LPMOs.
Brown-rot fungi also use oxidative mechanisms for plant cell wall degradation. However, they do so by generating hydroxyl free radicals via Fenton chemistry to degrade lignocellulosic plant cell walls. For a recent review see Arantes et al.102 The radicals generated by Fenton chemistry are promiscuous and nonspecific. This latter point seems counter to the distinct substrate specificities demonstrated by LPMOs to date (discussed below).
2.3. Nomenclature and Carbohydrate Active Enzyme Database
2.3.1. LPMO Families
In 1991, Henrissat and co-workers initiated an amino acid sequence-based classification of glycosyl hydrolases that resulted in the Carbohydrate Active Enzymes (CAZy) database (http://www.cazy.org).103 Starting with a total of 301 sequences classified into 35 glycosyl hydrolase families, the CAZy database has developed into an authoritative resource for the annotation and classification of sequences of proteins and enzymes that interact with carbohydrates.104 The advent of genome sequencing has caused a large increase in the number of publicly available sequences, and in early 2014 the database held close to 340,000 sequences divided over six major classes (Glycoside Hydrolases, Glycosyl Transferases, Polysaccharide Lyases, Carbohydrate Esterases, Auxiliary Activities, and Carbohydrate-Binding Modules).104 Note that the CAZy database only includes sequences from finished GenBank entries and that additional sequences can be found in unfinished genomes.105
An additional source for protein sequence information that can be complementary to the CAZy database is the Protein Family database (PFAM).78 The number of sequences in a selected enzyme family can differ by an order of magnitude between the two databases, possibly caused by differences in search models and curation.
LPMOs have been assigned to the Auxiliary Activities class in the CAZy database, which is made up of a number of redox enzyme families involved in the degradation of lignocellulosic substrates.73 Within this class are the LPMO families AA9 (formerly GH61), AA10 (formerly CBM33), AA11, and AA13. (Family AA12 contains the non-LPMO auxiliary activity pyrroloquinoline quinone-dependent oxidoreductase.) LPMOs display low sequence homology between families but share a common β-sandwich immunoglobulin-like fold.105–107 A recent bioinformatics study has presented evidence for a new LPMO family, tentatively named LPMO14.71
AA9 is a family with about 2500 annotated sequences that display low overall sequence homology.108 Phylogenetic analyses of these classifications have resulted in further division into three subfamilies within the AA9 family.76,109 For some CAZy families, the separation into subfamilies indicates differences in substrate specificity,104 but for AA9 LPMOs, subfamily classification is based on differences in reaction specificity, i.e., oxidation of either C1 or C4 of the glucopyranose ring, discussed above.110 Sequence homology studies reveal that while most sequences consist of a single AA9 domain, about 20% contain a C-terminal linker and CBM1 module, the latter of which are fungal cellulose-binding domains.
In contrast to the AA9 family, the AA10 family (formerly CBM33) contains two phylogenetic clades that are distinguished by their substrate specificity for chitin or for cellulose.76 This is in agreement with the remark by Vu and Marletta that distinct active-site differences between AA10 enzymes with different substrate specificities support the further separation into two subfamilies.8
The known AA11 sequences have a mix of active-site features found in AA9 and AA10 LPMOs and can be further separated into two subfamilies through bioinformatics analysis.111 However, only the specificity for chitin of the larger subfamily has been determined.107
The PFAM database listed fungal CBM33 sequences with C-terminal starch-binding CBM20 domains, providing a hint of LPMO substrate reactivity beyond straight polysaccharides like cellulose and chitin.78 Today, these LPMOs are classified in a separate family, referred to as AA13 LPMOs, and three phylogenetic subfamilies have been identified within this family.105
An additional LPMO family, named LPMO14 by the authors, has been identified by Voshol et al. after constructing sensitive and specific Hidden Markov Models and subsequent genome mining.71 Members of this family are found predominantly in fungi as well as in a plant pathogenic protist, bivalves, sea anemones, and stony corals. In addition, the search models found significant hits for the presence of LPMOs in the genome of D. melanogaster.
2.3.2. Substrate Specificities
AA9 enzymes have reported activities on substrates with a β-1,4-linked glucose backbone found in plant cell walls, such as cellulose, xyloglucan, and glucomannan.92,93,100,112–117 Another AA9 member, MtLPMO9A, was also shown to oxidize xylan oligosaccharides when xylan was associated with cellulose (oxidized cellulose products were most abundant).118,119
Reported substrate affinities for AA10 LPMOs include α- and β-chitin.3,87,120–123 Others are active on both β-chitin and cellulose and yet others are reported to be cellulose specific.124,125 Chitin- and cellulose-specific AA10 enzymes fall into separate phylogenetic clades.76 Horn et al. commented on the diversity of modular topologies observed for AA10-containing enzymes126 and suggested that the associated modules indicate that AA10 activity can be associated with glycosyl hydrolases or targeted to a wide range of substrates including cellulose, chitin, xylan, and mannan.
The single characterized AA11 LPMO from Aspergillus oryzae was active on squid pen chitin.107 Known AA11 sequences can be separated into two subfamilies based on bioinformatic analysis.111 Chitin activity has only been demonstrated for the larger subfamily.107 AA13 starch-active LPMOs have reported activities on polysaccharides containing an α-1,4-linked glucose backbone such as amylose, amylopectin, and starches.105,127 For the putative LPMO14 family, the authors draw from genomic clustering and transcriptomic data and propose involvement of its members in the degradation of glucans and pectin.71
2.3.3. Reaction Specificities
The structural repeating unit of both cellulose and chitin is a disaccharide of glucose or acetylglucosamine, which are shown in Figures 2A and 6A, respectively. The pyranose rings of the disaccharide are rotated by 180° around the axis of the cellulose or chitin chain. Glycosidic oxygens are flanked by C1 and C4 atoms of adjacent sugar residues. In the case that cellulose or chitin chains are on top or bottom of a polysaccharide fibril, the glycosidic oxygens alternate between exposure to the solvent and burial in the cellulose crystal. When the oxygen atom is buried, both C1 and C4 atoms are exposed for oxidative cleavage of the glycosidic bond (Figure 8).
Figure 8.

Exposure of C1 and C4 atoms in cellulose chains. (A) Stick and (B) space-filling representations of three cellohexaose molecules of a Cellulose Iβ crystal as presented in Figure 3. Hydrogen atoms are not shown for clarity. C1 and C4 atoms have been colored in blue and yellow, respectively. Due to the opposite orientation of glucose molecules in the cellobiose repeating unit, every other C1–C4 atom pair is exposed to the surface in cellulose chains that are at the top or bottom of the cellulose fibril. Type 1 LPMOs specifically attack C1 atoms (blue), type 2 enzymes specifically attack C4 atoms (yellow), and type 3 enzymes attack both C1 and C4 atoms.110
In principle, oxidation could occur at either the C1 or the C4 position, leading to different products as illustrated in Scheme 1. Analysis of the oxidized products of LPMO activity has been reported for several LPMOs from different families, including SmLPMO10A (CBP21), a chitin-active AA10 enzyme.3 The oxidative activity of SmLPMO10A was apparent from formation of the C1 lactone of glucosamine that would hydrate to an aldonic acid at pH above 3.5. Phillips et al. studied AA9 LPMOs from N. crassa and communicated that some LPMOs preferentially formed aldonic acids while others produced keto sugars.129 Two LPMO reactivities were proposed: PMO1 enzymes preferentially oxidized at the C1 position, while PMO2 enzymes selectively oxidized at the C4 position. Beeson et al. proposed that the keto sugars formed by PMO2 enzymes were indeed the result of oxidation at C4 (Scheme 1).128 They describe the reduction of isolated reaction products of AA9 PMO2 NcLPMO9D (NCU01050, PMO2, GH61–4) with sodium borohydride and hydrolysis by trifluoroacetic acid and reported that upon reduction of the C4-keto sugar a racemic mixture of glucose and galactose was formed. Eventually, three classifications were proposed based on reaction specificities within family AA9.110 PMO1 enzymes act on C1, PMO2s are specific for C4, and PMO3 enzymes oxidize both C1 and C4. Oxidation at the C6 position by LPMO action has been proposed based on mass spectrometry data of oxidized products.92,130 However, products of hypoiodite treatment of LPMO oxidation products were not consistent with the assignment of C6 oxidation.128 The capacity of LPMOs to oxidize the C6 position has therefore not been definitively proven.
Scheme 1. Regioselectivity of LPMOsa.
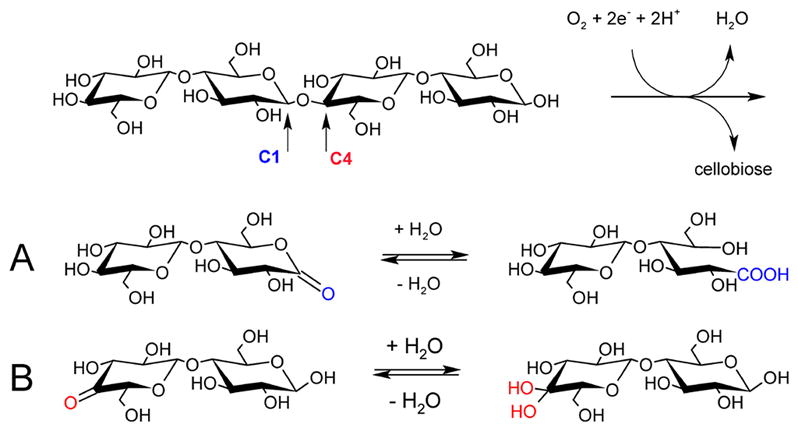
a(A) Oxidative cleavage at C1 generates a lactone, which hydrates to a reducing-end aldonic acid. (B) Oxidation of C4 results in a 4-ketoaldose, which hydrates to a gemdiol. Adapted with permission from ref 128. Copyright 2012 American Chemical Society.
Forsberg et al. described that, in all cases, AA10 enzymes oxidized at either C1 or both C1 and C4.125 Similar information is also available for the other AA families. It has been reported that AA11 LPMO from A. oryzae reportedly oxidizes the C1 position of acetyl-glucosamine and perhaps also C4, although the latter has not been conclusively determined. 107 Last, to date, all reported AA13 enzymes oxidize the substrate C1 position.105,127
2.4. Industrial Enzymes
LPMO genes are regularly found in the genomes of Ascomycete and Basidomycete fungi, with the number of LPMO-encoding genes in fungal genomes varying from a few to several dozen. The large diversity and frequent occurrence of these enzymes may reflect the array of possible naturally occurring carbohydrate structures outlined above. Fungal enzymes have been studied for commercial use for over a century. Takamine studied A. oryzae amylase for starch processing and was granted a U.S. patent in 1894.131 In 1964, U.S. patents on the use of fungal cellulases, including those from Trichoderma and Aspergillus, were granted.132,133 Nowadays, the use of enzymes in industrial processes is well established and polysaccharide-degrading enzymes find applications in a number of industries, including paper and pulp, textile processing, baking, animal feed, beverage, and biofuels.134–136 Table 2 highlights a few species of particular industrial relevance, especially with regard to saccharification of lignocellulosic biomass.
Table 2.
Fungal Species of Industrial Relevance with a Focus on Cellulosic Biofuels
| species | source | LPMO genes | details | example commercial products |
|---|---|---|---|---|
| Trichoderma reesei | canvas bivouac tents WWII137 | 3 | established enzyme production host138 | Spezyme CP (DuPont) Celluclast (Novozymes) |
| Aspergillus niger | soil | 8 | used for commercial citric acid production | E19 (Rohm and Haas) C6105, C1184 (Sigma-Aldrich) Rapidase Press (DSM) |
| Myceliophthora thermophila | compost piles139 | 23–30 | thermophilic, established enzyme production host140,141 | Fibrezyme G4 (DuPont) |
| Thermoascus aurantiacus | self-heating hay142 | 3 | thermophilic, proposed as enzyme production host143 | |
| Rasamsonia emersonii (Talaromyces emersonii) | self-heating wood chip pile144 | 2 | thermophilic | Filtrase NL (DSM) |
Many industrially relevant fungi that have been studied for their capability to degrade lignocellulosic biomass substrates encode LPMOs in their genomes. Furthermore, when grown on biomass substrates, LPMO enzymes will be present in secretomes. In the next section, we review aspects relating to LPMO activity for use in applications.
3. INDUSTRIAL USE OF LPMOS
3.1. Industrial Significance
Lignocellulosic biomass represents an alternative source for fuels and materials and has the potential to replace fossil fuels as we strive to become a sustainable and carbon-neutral society. Cellulose is the major structural polysaccharide of plant cell walls and is the most abundant and readily accessible source of renewable organic carbon on the planet. The enzymatic conversion of cellulose to glucose provides a carbon source for the biomanufacturing of fuels and chemicals. To this end, some LPMOs have been used in concert with cellulases to degrade of lignocellulosic biomass substrates, such as acid-pretreated corn stover95 and milled birch wood,97 and have been reported as components of certain commercial enzyme cocktails for the production of ethanol from lignocellulosic biomass.11 In the following paragraphs we will address the electron requirement for LPMO activity as well as the fundamental and applied protein engineering studies to investigate LPMO function.
3.2. Reported Electron Donors
3.2.1. Overview
The catalytic mechanism of LPMOs remains unclear and, as such, has yet to be described in detail. Current mechanistic proposals (described in section 5) require either stoichiometric single-electron reduction for each catalytic turnover with oxygen as the cosubstrate or a single priming reduction of the active-site copper with peroxide as the cosubstrate. Several possible donors that may act as the source(s) of these electrons are outlined below (Figure 9).
Figure 9.
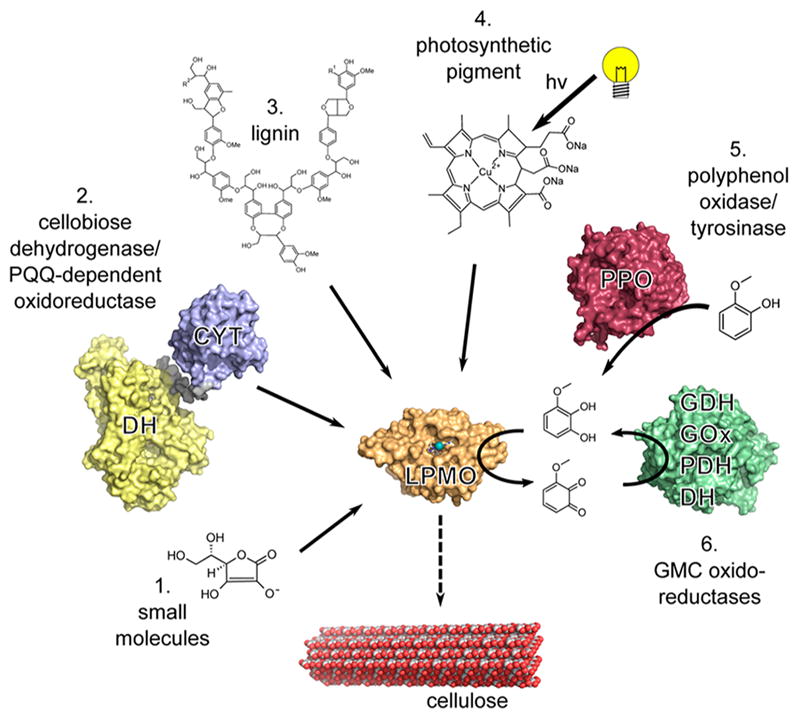
Examples of electron sources that can reduce the LPMO (orange) active-site copper (teal). (1) Small molecules such as ascorbate can donate electrons directly.3 (2) Enzymatic electron donors such as cellobiose dehydrogenase (CDH) can transfer electrons from the catalytic dehydrogenase domain (DH, yellow) via the cytochrome heme domain (CYT, blue) to a LPMO.145 Pyrroloquinoline quinone (PQQ)-dependent pyranose dehydrogenase has been postulated to transfer electrons in a similar fashion.146 (3) Insoluble high-molecular-weight lignin can serve as a reservoir for electrons facilitating LPMO activity.6 (4) Excited photosynthetic pigments can provide electrons for LPMO activity.147 (5) Polyphenol oxidase (red) can generate small molecule electron donors for LPMO activity from lignin building blocks.148 (6) Principle of regeneration of an oxidized quinoid form of electron donor by glucose-methanolcholine (PMC) oxidoreductases (green) such as glucose dehydrogenase (GDH), glucose oxidase (GOx), pyranose dehydrogenase (PDH), and the dehydrogenase domain of CDH (DH).4 Adapted with permission from ref 4. Copyright 2016 The American Association for the Advancement of Science.
3.2.2. Small Molecules
Several small molecule reductants have been used in vitro. In 2010, Vaaje-Kolstad and co-workers used ascorbic acid and glutathione to provide electrons to SmLPMO10A (CBP21) in a description of LPMO activity.3 Many studies have employed ascorbic acid as the reducing agent. Other small organic compounds such as hydroquinone, catechin, and gallic acid have also been shown to provide electrons to LPMOs.92,119,149 However, Frommhagen et al. demonstrated that LPMOs may have not only a carbohydrate substrate specificity but also a reducing agent specificity (see section 5.4).5 This is an important consideration for industrial applications as the ideal reducing equivalents in industrial processes are bulk chemicals or already present in the substrate.
Lignin-derived compounds, abundant in many biomass feedstocks, have been identified as additional electron sources for LPMOs. This point was confirmed by Dimarogona et al., who reported the enhanced activity of a heterologously expressed LPMO from M. thermophila upon addition of lignin from several sources.150 Westereng and colleagues showed that high molecular weight lignin can function as a reservoir of electrons and that reducing equivalents can be donated to LPMOs via long-range electron transfer mediated by soluble low molecular weight lignins present in plant cell walls.6
A chemical treatment of lignocellulosic biomass may be used to reduce biomass recalcitrance before enzymatic treatment.59 Pretreatment of carbohydrate materials can affect the presence of potential reducing agents in the substrate. Acid-or base-dependent pretreatment methods will not remove lignin and lignin breakdown products that could act as electron sources. Alternatively, delignifying and extractive techniques such as organosolv and ionic liquid pretreatment will result in a cleaner cellulose substrate, potentially making it necessary to add an external electron donor for LPMO activity.151,152 Rodríguez-Zúñiga et al. measured the impact of LPMO addition to cellulase mixtures in the degradation of hydrothermally pretreated corn stover, sugar cane bagasse, and wheat straw, as well as on alkaline and organosolv-pretreated sugar cane bagasse.151 Interestingly, the greatest impact from LPMO (and the most oxidized glucose detected) was with hydrothermally pretreated biomasses. We discuss the alternative enzymatically produced reducing equivalents that are able to generate electron donors in situ below.
3.2.3. Cellobiose Dehydrogenase
Bao et al. reported that cellobiose dehydrogenase (CDH) from Phanaerochete chrysosporium enhances the hydrolysis of Sigmacell type 50 microcrystalline cellulose by Trichoderma cellulases.153 CDHs have been linked to lignin degradation and proposed providing electrons for Fenton chemistry.154 A link between CDHs and LPMOs in vivo has been demonstrated.93 CDHs are flavocytochromes containing a heme b-binding cytochrome domain (CYT) connected by a flexible linker to a flavin adenine dinucleotide (FAD)-binding dehydrogenase domain.155 CDHs have been divided into two main classes. Class I CDHs are produced by basidiomycetes and lack additional domains, whereas class II CDHs occur in ascomycetes with class IIA having a type 1 carbohydrate-binding module (CBM) and class IIB that lacks a binding domain.156 CDHs catalyze the oxidation of the disaccharide cellobiose (Figure 10) as well as other oligosaccharides and transfer electrons to external electron acceptors via interdomain electron transfer from the reduced FAD to CYT heme b. This process presumably occurs by single-electron-transfer (ET) events, followed by ET from CYT to the external electron acceptor. Langston et al. showed that addition of Humicola insolens CDH boosted T. terrestris LPMO activity and that the combination of an LPMO with a CDH from T. terrestris increased the performance of various cellulases by approximately 2-fold on cellulose substrates.157 Active combinations of CDHs and LPMOs from different species have been reported, and in some cases CDHs from several fungi have been shown to provide electrons for LPMOs.93,110,119,130 More recently, Loose et al. demonstrated activation of a bacterial AA10 LPMO by a CDH from the fungus Myriococcum thermophilum.158 The interactions of CDH with LPMOs are discussed further in sections 4 and 5.
Figure 10.

Summary of the reactions catalyzed by cellobiose dehydrogenase (CDH) and β-glucosidase. (A) Oxidation of cellobiose to cellobionolactone by CDH. Hydrolysis of (B) cellobiose to glucose and (C) cellobionolactone to glucose and gluconolactone by β-glucosidase (BGL). Adapted with permission from ref 159. Copyright 2012 Springer.
Although CDHs have the potential to serve as electron donors, their activity results in the formation and accumulation of cellobionolactone. Fortunately, there are β-glucosidases (BGLs) present that can hydrolyze cellobiose and cellobionolactone, the latter of which hydrates to gluconic acid (Scheme 1) and results in accumulation of gluconolactone or gluconic acid (Figure 10).159
Although the cellobionolactone and gluconolactone have relatively short lifetimes in solution, these compounds are strong inhibitors of some cellulases, potentially reducing the efficiency of the saccharification process.160 Alternatively, to circumvent the accumulation of lactones, some organisms produce aldonolactonase to drive the reaction to the acid and avoid accumulation of lactones.161,162
CDHs can be beneficial for saccharification efficiency under certain conditions; however, the cellobiose oxidized by CDH is formed at the expense of one unit of glucose and will impact the final titer of reducing sugars.163 The economics of biorefinery applications that utilize CDH might benefit from a fermenting organism that, in addition to reducing sugars, is able to utilize the CDH-derived product gluconic acid.
In addition to the inhibitory effect of the oxidized products and the reduction in glucose yield, gluconic acid accumulation may also acidify the hydrolysate that will have to be neutralized by base addition, adding to the cost of the process. Therefore, the level of CDH activity for use in industrial saccharification requires careful consideration.163
3.2.4. PQQ-Dependent Oxidoreductases
Matsumura described an extracellular Pyrroloquinoline quinone (PQQ)-dependent oxidoreductase in the basidiomycete Coprinopsis cinerea.164 Similar to some CDHs, the PQQ-dependent dehydrogenase contains a type 1 CBM domain for adsorption on cellulose as well as a cytochrome domain. However, in PQQ-dependent oxidoreductases, the flavin domain is replaced by a PQQ-dependent sugar dehydrogenase domain. Takeda concluded that the cytochrome domain of CcPDH possesses similar biophysical properties to those of CDH. However, unlike CDH, CcPDH prefers monosaccharide substrates. This finding combined with binding studies reveals a high binding affinity of CcPDH for cellulose, suggesting that CcPDH function is related to the enzymatic degradation of plant cell walls.146
3.2.5. Cytochrome Domains
Yoshida identified a single cytochrome domain secreted by Phanerochaete chrysosporium. 165 The hemoprotein, similar to the cytochrome domain of CDH, consists of an N-terminal cytochrome domain and a C-terminal family 1 carbohydrate-binding module (CBM1). The heterologously expressed carbohydrate-binding cytochrome b562 (CBCyt. b562) was found to be redox active with a redox potential (measured by cyclic voltammetry) similar to that of the cytochrome domain of CDH, suggesting that the protein may have an electron-transfer function. In a binding study with various carbohydrates, CBCyt. b562 was adsorbed with high affinity on both cellulose and chitin. Although the protein is redox active and the presence of the CBM1 suggests a role in cellulosic degradation, a direct interaction with LPMO has not been reported.
3.2.6. GMC Oxidases and Polyphenol Oxidases
Kracher et al. reported that plant-derived diphenols, can be regenerated by glucose-methanol-choline (GMC) oxidoreductases and that these diphenols are efficient reducing agents for LPMO activity.4 Similarly, Garajova et al. reported that single-domain glucose dehydrogenase and aryl-alcohol quinone oxidoreductases are catalytically efficient electron donors for LPMOs. These single-domain flavoenzymes display redox potentials compatible with electron transfer between partners. 166 More recently, Frommhagen et al. demonstrated that tyrosinase-like polyphenol oxidases are able to generate reducing agents from methoxy-phenolic compounds in a similar mechanism.148 By enzymatically converting small organic compounds, possibly derived from lignin present in the biomass, reducing equivalents may be generated without the oxidation of sugars as with enzymes such as CDH.
3.2.7. Light
Studies by Canella et al. report that photosynthetic pigments can be used to provide electrons to LPMOs. The authors show that when LPMOs from Thielavia terrestris and T. aurantiacus as well as a bacterial LPMOs from Thermobifida fusca were exposed to light in the presence of pigments and reducing agents, activity was enhanced and substrate specificity was broadened.147 More recently, others have challenged this interpretation and have instead proposed that photosynthetic pigments generate peroxide under the conditions tested and that peroxide, not O2, is an efficient substrate for LPMOs (see section 5).167 However, work by Möllers et al. showing that catalase addition (to reduce peroxide) does not reduce the effect of the photosynthetic pigment system seems to contradict this argument as it relates to peroxide formation by photosynthetic pigments.168 Although more research is needed to establish a mechanism for light-induced reduction of LPMOs, the use of light as an energy source in an industrial saccharification would have significant consequences for the design and operation of a reactor.
3.3. LPMO Activity Determination
Accurate determination of the activity of LPMOs allows for greater understanding of enzyme function and for improvements in enzyme efficiency through protein engineering. Unfortunately, the direct measurement of LPMO activity is challenging because of the insolubility of the substrates and products and the specialized analytical tools required to measure oxidized products formed and to distinguish them from product formation by hydrolytic enzymes.169
3.3.1. Reports of Hydrolytic Activity
Saloheimo reported the direct detection of endoglucanase activity for T. reesei LPMO9A (HjLPMO9A, EG4) expressed in Saccharomyces cerevisiae,115 which was also reported after expression in T. reesei113 and Pichia pastoris.170 These reports served as the basis for its classification as endoglucanase 4 (EG4) and Cel61A, placing the enzyme in a cellulosic glycosyl hydrolase family 61. Hydrolytic activity on carboxymethyl cellulose (CMC) has been reported for an Aspergillus nidulans LPMO9 (AN1602.2) expressed in P. pastoris,114 an Aspergillus kawachii LPMO9 with and without the cellulose-binding module,112 and for HjLPMO9A expressed in P. pastoris, the latter of which also showed cellooligosaccharide hydrolytic activity.170,171 Oxidative cleavage by LPMO was confirmed by a combination of chromatographic methods and mass spectrometry,3 and oxidative activity for HjLPMO9A has been observed.172
3.3.2. Indirect Measurement of Activity
The activity of LPMOs has often been measured by the enhancement of other cellulolytic enzymes with known hydrolytic activity in lignocellulosic biomass, where lignin components act as external electron donors.95
Using a method that coupled peroxide formation by N. crassa LPMOs to horseradish peroxidase and Amplex Red, Kittl et al. were able to quantitatively assay LPMO activity along with large oligosaccharides which were detected by HPLC.99 However, Marinai reported that formation of peroxide is largely dependent on the background copper available in the assay.173 Loose et al. observed a tight coupling of the cellobiose dehydrogenase as an external electron donor and the activity of a chitin active LPMO and proposed the indirect measurement of LPMO activity via the consumption of cellobiose or lactose.158 Similarly, efforts by Yu et al. to measure the activity of chitin-active LPMOs rely on monitoring the consumption of ascorbic acid.174 All of these methods are dependent on a tight coupling to other activities and require that those other activities are not limiting. Although not readily available, a direct measure of activity will not have these limitations.
3.3.3. Direct Measurement of Activity
Span et al. used an oxygen electrode to directly measure dioxygen consumption during the oxidation of cellulose by MtLPMO9.175 This same study also reported peroxide formation in the absence of cellulose and showed that formation of peroxide was not a significant contributor to dioxygen consumption. While measuring direct consumption of a reactant may seem simple, a level of care was taken in this study given the diverse reactivity of dioxygen.
Quinlan et al. and Westereng et al. reported measurement of LPMO activity by monitoring the enhancement of glucose release by a β-glucosidase using MALDI mass spectrometry as well as by direct detection of oxidized cello-oligosaccharides via chromatographic methods.92,176 In both cases, these methods are efficient but require appropriate instrumentation. Hansson et al. use high-performance anion exchange chromatography (HPAEC) to detect products formed from phosphoric acid -swollen cellulose (PASC) by HjLPMO9A and present quantitation of HjLPMO9A activity based on measuring the reduction of light scattering as PASC is depolymerized.177
The insolubility of substrates and products make activity measurement analytically challenging. Researchers have turned to atomic force microscopy, solid state NMR, high-performance size exclusion chromatography coupled with light scattering and refractive index detection (Villares et al.), and confocal microscopy (Eibinger et al.) to directly measure changes to cellulose fibers after treatment with Podospora anserina LPMO9H (PaLPMO9H).178,179
Frandsen et al. measured substrate saturation of Lentinulis similis LPMO9A with a cellotetraose FRET-based substrate.180 The turnover rate for reaction with this substrate is ~1 s−1, similar to those reported for other LPMOs and hydrolytic cellulases acting on crystalline cellulose.3,169,181,182 However, it is important to note that this substrate is only useful for LPMOs capable of interacting with soluble cello-oligosaccharides.
3.4. LPMO Protein Engineering
Protein engineering of LPMOs has been carried out by both academic and industrial research groups. Some of these studies were carried out to map substrate-binding behavior and active-site properties to features observed in protein crystal structures. Protein engineering was also used to aid understanding of the function of modular enzyme domains as they pertain to the activity and substrate interaction of LPMOs.
3.4.1. Identification of Catalytic Sites
The structural features of LPMOs have been characterized (see sections 4 and 5), and protein engineering has helped correlate structural features with function. Vaaje-Kolstad et al. identified six amino acid sites important for substrate binding to the chitin-active SmLPMO10A (CBP21) from S. marcescens.183
Harris and co-workers used protein crystallography to identify metal-binding ligands and active-site residues in T. terrestris LPMO9E (TtLPMO9E).95 In order to gain insight into the roles of these residues in enzyme function and catalysis, site-selective mutagenesis studies were employed. These studies revealed that substitution of either of the metal-coordinating histidines (His-1 or His-68) abolished activity as tested in a biomass saccharification assay. Likewise, the H1A substitution in T. fusca LPMO10A eliminates activity and cellulose binding.184
The substitution of the AA9 family conserved tyrosine at position 153 to phenylalanine near the metal-binding site of TtLPMO9E resulted in an 85% reduction in performance relative to wild type.95 Analogous variants Y177F Tf LPMO10A and Y190F Neurospora crassa LPMO9E (NcLPMO9E, NCU08760) exhibited 71% and 50% reductions in activity relative to wild-type enzyme when acting on bacterial microcrystalline cellulose or phosphoric acid swollen cellulose, respectively.184 Phenylalanine is largely conserved at the analogous position in the family AA10. Substitution of F219 to A or Y (numbering starting with the first histidine at position 35) in Streptomyces coelicolor LPMO10C (CelS2) resulted in low or no detectable activity on phosphoric acid swollen cellulose.125
Interestingly, substitution of the second-sphere residue glutamine 151 to glutamate (or asparagine or leucine) that interacts with tyrosine at position 153 was reported to eliminate performance completely in TtLPMO9E.95 Marletta et al. studied the role of the analogous glutamine residue in NcLPMO9E and reported a large reduction in activity on phosphoric acid swollen cellulose for the Q188A NcLPMO9E variant.185 The importance of these residues in the hydrogen-bonding network surrounding the metal ion is discussed in more detail in section 5.1.
3.4.2. Modules and Mini-Cellulosomes
Carbohydrate-binding modules (CBMs) can modulate the activity of catalytic domains by altering the binding affinity for substrate. Crouch et al. genetically deleted the family 2 CBMs from Cellulomonas fimi LPMO10 (Cf LPMO10) and Thermobispora bispora LPMO10 (TbLPMO10) or replaced the CBMs with CBMs from other proteins.186 The effect of removing the CBM or the replacement of the native CBM with a non-native one was dependent on the catalytic domain and the form of the cellulose substrate (i.e., phosphoric acid swollen cellulose, Avicel, or bacterial microcrystalline cellulose). Hansson et al. show that the removal of the CBM1 domain from HjLPMO9A does not alter the active site but does reduce the activity and cellulose binding relative the full-length protein.177 Borisova and co-workers removed the CBM1 from N. crassa LPMO9C (NcLPMO9C) which resulted in reduced activity on tamarind xyloglucan by 50% with no change in the activity on phosphoric acid swollen cellulose.182 The binding affinity of the variant without the CBM1, determined by isothermal titration calorimetry, was decreased for both substrates relative to the wild-type NcLPMO9C. These studies indicate that CBMs can have a significant effect on LPMO catalysis and that their roles are not the same for all catalytic domains or all substrates.
Kruer-Zerhusen analyzed the significance of the domains of T. fusca LPMO10B (Tf LPMO10B, also called TfAA10B or E8).184 The cellulose-active Tf LPMO10B is composed of an N-terminal catalytic domain, a central X1 (Fn3) domain, and a CBM2 domain. The removal of the central X1 domain does not alter the activity or cellulose binding of the resulting Tf LPMO10B variant. Variants with the CBM2 domain removed or both the X1 and the CBM2 domains removed displayed 10–20% residual binding and 50–60% residual activity compared to that of wild type, respectively. The activity of this enzyme seems to be completely independent of the X1 domain, the role of which has not yet been identified.
Arfi et al. engineered artificial cellulosomes to contain two LPMOs from T. fusca.187 A variety of engineered scaffoldins were used to assemble a variety of chimeric enzymes. These chimeric enzymes included engineered hydrolases (GH5 and GH48 families) and T. fusca LPMO10A (Tf LPMO10A, also called TfAA10A or E7) and T. fusca LPMO10B (TfAA10B or E8). The dockerin domains are attached at the C-termini of TfLPMO10A and Tf LPMO10B with or without a linker and in the case of Tf LPMO10B with and without X1 (Fn3 domain). The inclusion of Tf LPMO10A or Tf LPMO10B in assembled artificial cellulosomes improved degradation of cellulose by 70%. Assembly of the enzymes into artificial cellulosomes resulted in a 1.7-fold increase of soluble sugar relative to the free enzymes.
Liang et al. constructed an artificial mini-cellulosome with the LPMO from T. aurantiacus and the CDH (see section 3.2.3) from H. insolens attached with other cellulases to the surface of yeast as an engineered complex for consolidated bioprocessing. 188 The artificial cellulosome is assembled by engineering cellulases, CDH, and LPMO to contain dockerin domains. These proteins are then allowed to assemble into specific cohesin scaffolds that are linked together and to an Aga2 protein that tethers the complex to the surface of yeast via an interaction with the α-agglutinin mating adhesion receptor. Liang et al. mention the potential difficulty of providing enough oxygen to the CDH/LPMO enzyme system while sustaining fermentation conditions that allow yeast to produce ethanol efficiently. However, yeast expressing the mini-cellulosome are able to grow on phosphoric acid swollen cellulose as a carbon source.
4. TERTIARY PROTEIN STRUCTURES
4.1. Overall LPMO Architecture
The multiple structures of LPMO enzymes available show that the enzyme families share a core immunoglobulin-like β-sandwich topology in which certain individual β-strands are connected by loops of variable length and structure. 95,105–107,183 Also common to LPMOs with known structures is a relatively flat surface containing the catalytic site with a mononuclear copper center. In LPMOs active on noncrystalline substrates, this flat surface is less pronounced. The copper atom of LPMOs is coordinated by two conserved histidine residues, one of which is the N-terminal residue and binds bidentate through the imidazole side chain and the amine of the N-terminus.92,93,120,189 This motif is referred to as the histidine brace (discussed in more detail in section 5).92
An early structure of an LPMO, at the time known as CBM33 (AA10), was that of SmLPMO10A (CBP21) from the bacterium S. marcescens (Figure 11A) and was published in 2005. The fold was described as a “budded” fibronectin type III fold (fnIII), which is named after the third β-sandwich domain of the glycoprotein fibronectin. The “bud” referred to a three-helix insert between β1 and β2, which comprise one of the β-sheets within the β-sandwich (Figure 11A).183
Figure 11.
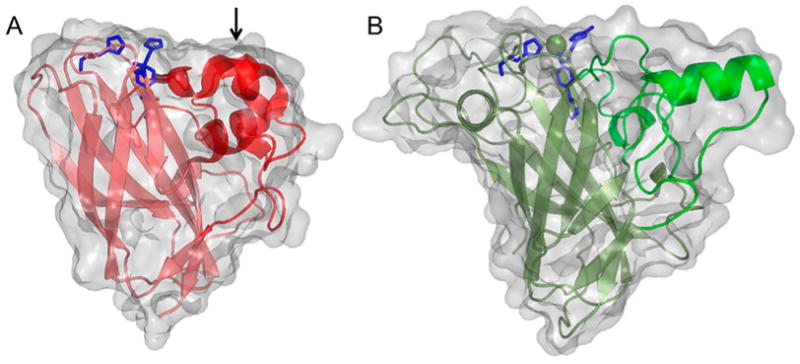
Illustration of early published LPMO structures. (A) S. marcescens LPMO10A (CBP21). (B) T. reesei (H. jecorina) LPMO9B (Cel61B, EG7) presented with the backbone as cartoon and with the molecular surface in transparent gray.106,183 Three-helix insert of SmLPMO10A or “bud” (as described in the text) is marked with an arrow and shown in a more intense red color. Corresponding region in HjLPMO9B is shown as a more intense green color. LPMO characteristic metal-coordinating histidines and active-site phenylalanine/tyrosine are shown as blue colored sticks.
With the first publication of structures of fungal LPMOs (at the time known as GH61 proteins, see Figure 11B), a functional link between the SmLPMO10A and the fungal LPMOs was suggested based on the fold similarities.95,106 In addition to the observed structural similarities, Harris and coworkers95 reported enhancing effects in cellulose degradation similar to what had previously been shown for SmLPMO10A in chitin degradation.79 In their review, Frandsen and LoLeggio pointed out that the β-sandwich by itself is not a unique fold.9 They further remark that the observed backbone fits of the β-sandwiches of the SmLPMO10A enzyme and of another AA10 enzyme, Jonesia denitrificans LPMO10A (JdLPMO10A) to a non-LPMO (the MG2 domain of human 2-macroglobulin) were “remarkable despite the absence of an obvious functional relationship”.123,183,1909 This may be explained, in part, by the fact that the fnIII fold is a variant of the immunoglobulin-like β-sandwich fold, which is one of the most commonly occurring protein folds and have multiple examples in the Structural Classification of Proteins (SCOP) protein fold classification database.191
4.1.1. LPMO AA Families
4.1.1.1. AA9
According to the Protein Data Bank, the structures of 11 different AA9 LPMOs from seven different fungal species have been reported to date (Table 3).92,95,106,172,177,180,182,189,192,193
Table 3.
AA9 LPMO Structures Available in the Protein Data Bank
| source, name, and alt. name | PDB code | resolution [Å] | metal ion | type192 and brief highlights | ref |
|---|---|---|---|---|---|
| T. reesei (Hypocrea jecorina) HjLPMO9B, Cel61B, EG7 | 2VTC | 1.60 | Ni2+ | type 3: first AA9 and fungal LPMO structure reported | 106 |
| T. terrestris TtLPMO9E, GH61E | 3EII | 2.25 | Zn2+ | type 1: Zn-bound GH61 structure; noted similarities with aromatics on surface to Family 1 CBMs | 95 |
| T. terrestris TtLPMO9E, GH61E | 3EJA | 1.90 | Mg2+ | type 1: Mg-bound GH61 structure | 95 |
| T. aurantiacus TaLPMO9A, GH61A | 2YET | 1.50 | Cu2+ | type 3: observed N-terminal histidine N-methylation | 92 |
| T. aurantiacus TaLPMO9A, GH61A | 3ZUD | 1.25 | Cu2+ | type 3 | 92 |
| P. chrysosporium PcLPMO9D, GH61D | 45BQ | 1.75 | Cu2+ | type 1: first basidiomycete GH61 structure reported | 189 |
| N. crassa NcLPMO9D, PMO-2 | 4EIR | 1.10 | Cu2+ | first type 2: LPMO structure reported with activity for the C4 carbon | 192 |
| N. crassa NcLPMO9M, PMO-3 | 4EIS | 1.37 | Cu2+ | first type 3: LPMO structure reported with activity for the C1 and C4 carbons | 192 |
| N. crassa NcLPMO9C | 4D7U | 1.56 | Cu2+ | type 2: LPMO active on cellulose, cellodextrins and xyloglucan | 182 |
| N. crassa NcLPMO9C | 4D7V | 1.90 | Zn2+ | type 2: LPMO active on cellulose, cellodextrins and xyloglucan | 182 |
| N. crassa NcLPMO9F | 4QI8 | 1.10 | Cu2+ | type 1: high resolution | 172 |
| L. similis LsLPMO9A, LsAA9A | 5ACF | 1.80 | Cu2+ | type 2: LPMO with substrate bound, G3 | 180 |
| L. similis LsLPMO9A, LsAA9A | 5ACI | 1.75 | Cu2+ | type 2: LPMO with substrate bound, G6 | 180 |
| L. similis LsLPMO9A, LsAA9A | 5ACJ | 1.70 | Cu2+ | type 2: LPMO with substrate bound, G3 | 180 |
| L. similis LsLPMO9A, LsAA9A | 5ACG | 1.91 | Cu2+ | type 2 | 180 |
| L. similis LsLPMO9A, LsAA9A | 5ACH | 1.28 | Cu2+ | type 2 | 180 |
| L. similis LsLPMO9A, LsAA9A | 5N04 | 1.76 | Cu2+ | type 2 | 194 |
| L. similis LsLPMO9A, LsAA9A | 5N05 | 1.58 | Cu2+ | type 2: with cellohexaose bound | 194 |
| M. thermophila (Thermothelomyces thermophila) MtLPMO9, MtPMO-3 | 5UFV | 2.45 | Cu2+ | type 3: LPMO from a thermotolerant fungus | 175 |
| T. reesei (H. jecorina) HjLPMO9A, Cel61A, EG4 | 5O2X | 0.95 | Cu2+ | type 3: extended LPMO domain containing linker residues as an integral part, biochemical removal of CBM | 177 |
| T. reesei (H. jecorina) HjLPMO9A, Cel61A, EG4 | 5O2W | 2.00 | Cu2+ | type 3: extended LPMO domain containing linker residues as an integral part, genetic removal of CBM | 177 |
Li and co-workers suggested that AA9 LPMOs can be divided into three types: type 1, type 2, and type 3.192 This classification was based on a phylogenetic analysis, for which sequence divergences between the three suggested types were shown to reflect distinct structural differences between T. terrestris LPMO9E (type 1), N. crassa LPMO9D (type 2), and LPMO9M (type 3).95,192 The three types are illustrated in Figure 12. This classification was refined by Vu et al., and the differences in regioselectivity of AA9 LPMOs were found to be related to the three types.110 This classification was further refined by Moses et al. in a bioinformatic study and was included as three separate clusters (6, 7, and 8) in the larger structural classification of all LPMOs defined by Vaaje-Kolstad and co-workers in a review on the structural diversity of LPMOs.109,10 Although there are large structural variations among AA9 LPMOs, these differences are localized to two regions, the L2 and L3 regions (colored yellow and red, respectively, in Figure 12). In type 1 AA9s, both the L2 and the L3 regions are shorter than those observed in type 2 and type 3 LPMOs. Additionally, the L2 region commonly lacks aromatic residues, while there are solvent-exposed aromatic residues in the L3 region and also in a loop region close to the C-terminus (LC region; colored green in Figure 12). Harris and co-workers noted that the orientations of the surface-exposed tyrosines in T. terrestris LPMO9E were structurally analogous to those of family 1 cellulose-binding module (CBM1) from T. reesei CBH1. Substitution of Tyr-192 in the LC region of of TtLPMO9E (analogous to Y31 of TrCBH1) reduced performance to about 30% that of wild type, supporting the proposal that cellulose substrates bind at the surface of TtLPMO9E. Type 1 is usually described as having a CBM1-like arrangement of altogether three aromatic residues on the flat surface containing the active site (see section 4.2.1 for examples of the arrangement of aromatic residues in CBM modules).95 The type 2 LPMOs have a longer loop insert in the L3 region, forming a more pronounced broad surface on one side of the active site. Aromatic residues suggested to be involved in substrate recognition are present on three sides of the catalytic site, in loops L2 and L3 and in the LC region. At present, AA9 LPMOs known to be active on soluble substrates, such as cellooligosaccharides, are of this type. Type 3 LPMOs are characterized by a longer flat surface formed by a large L2 region, with aromatic residues at each end of the flat surface in the L2 and the LC regions. As reported by Vu and co-workers, the classification also reflects the regioselectivity of AA9 enzymes, so that type 1 enzymes mainly oxidize the C1 position of the cellulose polymer, type 2 enzymes mainly oxidize the C4 position, and type 3 enzymes showed mixed behavior by being able to oxidize both C1 and C4 positions; however, there are exceptions. For example, M. thermophila LPMO9, which was reported as a strict C1 oxidizer, may form a subgroup of the type 3 LPMOs.110 Interestingly, the same study showed that a mutant type 3 LPMO from N. crassa, lacking key parts of the L2 region loses much of its ability to oxidize the C4 position. Similarly, Danneels et al. observed that substitutions of two surface-exposed aromatic residues located near the L2 region of HjLPMO9A resulted in variants that produce more C1-oxidized products of cellulose, whereas substitution of Y211 to alanine resulted in a variant that yielded more C4-oxidized products.195 Both of these observations indicate that there are determinants for regioselectivity within the L2 region.
Figure 12.

Illustration of the three main types of AA9 LPMOs. (Left and right) Top-down view of a transparent protein surface with the backbone represented as a cartoon and side view of the backbone, respectively. (A) Type 1: T. terrestris LPMO9E (PDB ID 3EJA).95 (B) Type 2: N. crassa LPMO9D (PMO-2, PDB ID 4EIR).192 (C) Type 3: N. crassa LPMO9M (PMO-3, PDB ID 4EIS).192 Catalytic site metal atom is shown as a sphere (Mg in 3EJA), active-site residues are colored blue, varying parts of the L2 region are colored yellow, varying parts of the L3 region are colored red, partially conserved loop on the opposite side of the catalytic site is colored cyan, and surface parts of the LC region are colored green.
4.1.1.2. AA10
Fourteen different structures of proteins from the AA10 family (listed in Table 4) have been published in the Protein Data Bank, both with X-ray crystallographic and with NMR methods. Eleven of these are of bacterial origin and are functional in the oxidation of biomass.120,121,123,124,183,196–201 There are also three proteins of viral origin classified as AA10.80 These are all from insect poxviruses, in which the LPMO domain plays a role as part of the spindle protein fusolin, essential for the virus to infect its target insect and has been suggested to cause disruption of the chitin-rich peritrophic matrix in the insect gut (Figure 13D).80,202
Table 4.
AA10 LPMO Structures Available in the Protein Data Bank
| source, name and alt. name | PDB code | resolution [Å] | metal ion | cluster10 and brief highlights | ref |
|---|---|---|---|---|---|
| S. marcescens LPMO10A, CPB21 | 2BEM | 1.55 | N/A | cluster 1: first reported bacterial LPMO structure | 183 |
| S. marcescens LPMO10A, CPB21 | 2BEN | 1.80 | N/A | cluster 1: Y54A variant of SmLPMO10A | 183 |
| S. marcescens LPMO10A, CPB21 | 2LHS | NMR | N/A | cluster 1: first reported NMR structure of a LPMO | 196 |
| Enterococcus faecalis V583 LPMO10A, CBM33 | 4A02 | 0.95 | N/A | cluster 1: high-resolution apo structure of an AA10 LPMO | 120 |
| E. faecalis V583 LPMO10A, CBM33 | 4ALC | 1.49 | Cu2+ | cluster 1: six structures in one study of the Cu(II) to Cu(I) transition by X-ray photoreduction | 199 |
| E. faecalis V583 LPMO10A, CBM33 | 4ALE | 1.48 | cluster 1: six structures in one study of the Cu(II) to Cu(I) transition by X-ray photoreduction. | 199 | |
| E. faecalis V583 LPMO10A, CBM33 | 4ALQ | 1.48 | cluster 1: six structures in one study of the Cu(II) to Cu(I) transition by X-ray photoreduction | 199 | |
| E. faecalis V583 LPMO10A, CBM33 | 4ALR | 1.49 | cluster 1: six structures in one study of the Cu(II) to Cu(I) transition by X-ray photoreduction | 199 | |
| E. faecalis V583 LPMO10A, CBM33 | 4ALS | 1.47 | cluster 1: six structures in one study of the Cu(II) to Cu(I) transition by X-ray photoreduction | 199 | |
| E. faecalis V583 LPMO10A, CBM33 | 4ALT | 1.49 | Cu1+ | cluster 1: six structures in one study of the Cu(II) to Cu(I) transition by X-ray photoreduction | 199 |
| Bacillus amyloliquefaciens LPMO10A, CBM33 | 2YOW | 1.80 | N/A | cluster 1 | 198 |
| B. amyloliquefaciens LPMO10A, CBM33 | 2YOX | 1.90 | Cu1+ | cluster 1: metal center (X-ray) photoreduced to Cu(I) | 198 |
| B. amyloliquefaciens LPMO10A, CBM33 | 2YOY | 1.70 | Cu1+ | cluster 1: metal center chemically (ascorbate) reduced to Cu(I) | 198 |
| B. amyloliquefaciens LPMO10A, CBM33 | 5IJU | 1.70 | Cu2+ | cluster 1 | 201 |
| S. coelicolor LPMO10B | 4OY6 | 1.29 | Cu2+ | cluster 2: one of two first cellulose active bacterial (AA10) LPMO structures reported. | 124 |
| S. coelicolor LPMO10B | 4OY8 | 1.40 | Zn2+ | cluster 2: one of two first cellulose active bacterial (AA10) LPMO structures reported | 124 |
| S. coelicolor LPMO10C, CelS2 | 4OY7 | 1.50 | Cu1+ | cluster 2: one of two first cellulose active bacterial (AA10) LPMO structures described | 124 |
| T. fusca LPMO10A, TfAA10A, E7 | 4GBO | 2.00 | Cu2+ | cluster 2: cellulose active LPMO, C1, and C4 oxidizer | 203 |
| Vibrio cholerae LPMO10B, GbpA, AA10B | 2XWX | 1.80 | N/A | cluster 1: AA10 domain from the V. cholerae colonization factor GbpA | 197 |
| Bacillus thuringiensis LPMO10A | 5WSZ | 2.57 | Cu2+ | 204 | |
| Burkholderia pseudomallei LPMO10A, BpAA10A | 3UAM | 2.00 | N/A | cluster 1 | 205 |
| Cellvibrio japonicus LPMO10A, Cbp33A | 5FJQ | 1.85 | Cu2+ | cluster 2 | 121 |
| J. denitrificans LPMO10A | 5AA7 | 1.66 | Cu1+ | cluster 3 | 123 |
| Streptomyces lividans LPMO10E | 3FTZ | 1.38 | Cu2+ | 200 | |
| Anomala cuprea entomopoxvirus CV6M fusolin | 4YN1 | 1.90 | N/A | cluster 4 | 80 |
| unidentified entomopoxvirus fusolin | 4YN2 | 2.02 | Zn2+ | cluster 4 | 80 |
| Melolontha melolontha entomopoxvirus (MMEV) fusolin | 4OW5 | 1.90 | N/A | cluster 4 | 80 |
| M. melolontha entomopoxvirus (MMEV) fusolin | 4X27 | 2.40 | Cu2+ | cluster 4 | 80 |
| M. melolontha entomopoxvirus (MMEV) fusolin | 4X29 | 2.41 | Zn2+ | cluster 4 | 80 |
Figure 13.
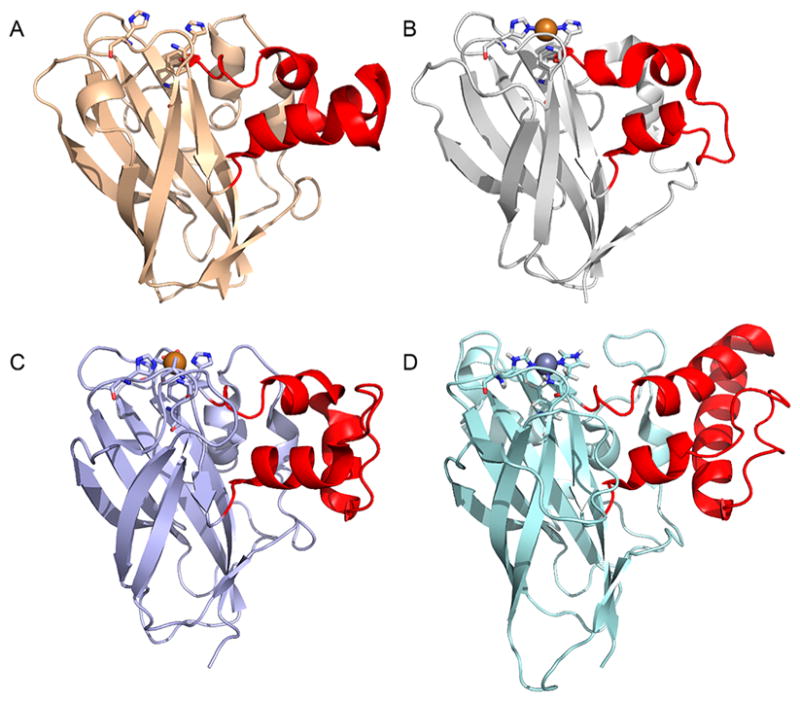
Four different AA10 LPMOs, one from each cluster 1–4 defined by Vaaje-Kolstad and co-workers with the L2 region/loops marked in red.10 (A) Chitin-active BaLPMO10A (PDB ID 2OYX).198 (B) Cellulose-active ScLPMO10C (PDB ID 4OY7)).124 (C) Chitin-active JdLPMO10A (PDB ID 5AA7).123 (D) AA10 LPMO domain of the entomopoxvirus fusolin protein (PDB ID 4YN2).80
Despite sharing a common topology and copper active site, sequence identities between bacterial (AA10) and fungal (AA9) LPMOs are often low, e.g., 16.5% between SmLPMO10A and HjLPMO9B.106 While AA9s contain cellulose- and hemicellulose-active LPMOs, the AA10 family contains LPMOs active on both chitin and cellulose. Thus far, the chitin-active AA10 LPMOs have been shown to produce C1-oxidized products. AA10 LPMOs active on cellulose have been characterized to be either mixed C1/C4 oxidizers or strict C1 oxidizers, though two AA10 LPMOs active on both chitin and cellulose produce a mixed C1/C4 product from cellulose while only C1-oxidized products from chitin. This is an empirical correlation for regioselectivity, and more data may alter this picture. In a review on the structural diversity of LPMOs, Vaaje-Kolstad and co-workers suggested a structure-based classification of all LPMOs into 9 clusters, of which the AA10 enzymes were divided into four clusters.10 While chitin-activity seems a more general theme for AA10 LPMOs, activity has been shown for enzymes belonging to only three of the clusters. Cellulose-active AA10 LPMOs have only been reported for one of the clusters. Most of the variation between AA10 enzymes (Figure 13) occurs in the region/loops connecting the first β-strand to the third β-strand, referred to as L2 (for loop 2). Cluster 1 contains only chitin-active LPMOs, while cluster 2 contains both chitin-active and cellulose-active LPMOs. Cluster 3 contains only one known member, which is active on chitin. The AA10 domains found in the spindle proteins from insect poxviruses fall into cluster 4, for which both the substrate and the regioselectivity are unknown.
4.1.1.3. AA11
AA11 LPMOs comprise a family of chitin-active LPMOs of fungal origin that share an overall fold similar to that of AA9 LPMOs (Figure 14). Less than 20 members are currently classified in this family, and of these, those with published crystal structures are listed in Table 5. This LPMO family was identified using what Hemsworth et al. referred to as “module walking” where they pointed out a sequence motif, “X278”, among AA9-like sequences and found that this motif also occurs among modular chitinase sequences that have a GH18 catalytic domain.107 They expressed and determined the structure of one such protein from A. oryzae and demonstrated copper binding.107 They also made the observation that the proposed binding surface lacked aromatic residues which, while similar to AA10 LPMOs active on chitin substrates, is in contrast to AA9 LPMOs.107 Furthermore, all AA11 LPMOs characterized thus far have been shown to produce C1-oxidized products, another feature similar to AA10 LPMOs active on chitin.
Figure 14.
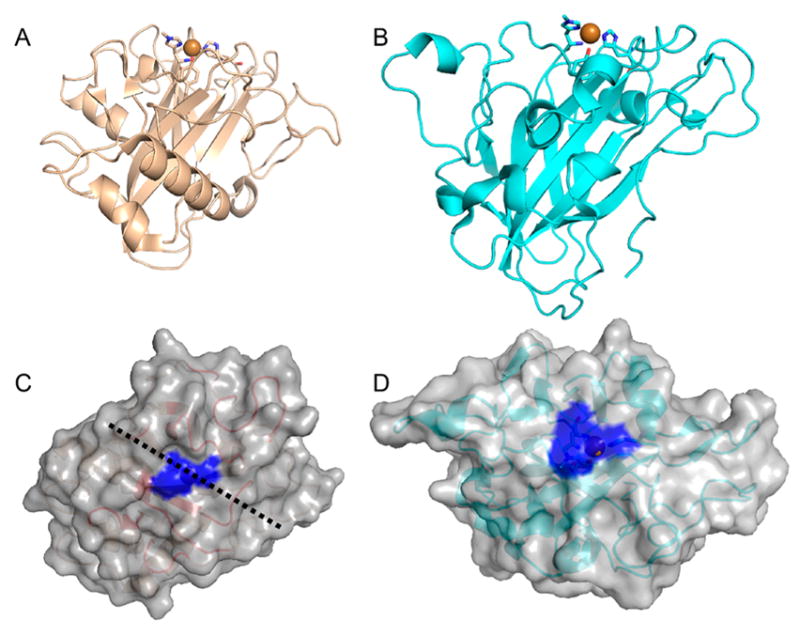
Side-by-side comparison of AA11, represented by A. oryzae LPMO11 (AoAA11; PDB ID 4MAI) and AA9, represented by T. aurentiacus LPMO9A (PDB ID 3ZUD).92,107 (A) Fold of AoLPMO11 as a cartoon representation with the histidine brace at the top. (B) Same view of TaLPMO9A. (C) AoLPMO11 presumed chitin-interacting surface (active-site histidines colored blue). (D) For comparison, the cellulose-interacting surface of TaLPMO9A (active-site histidines colored blue).
Table 5.
AA11 LPMO Structures Available in the Protein Data Bank
4.1.1.4. AA13
Vu and co-workers showed that N. crassa secretes a protein with a starch-binding CBM20 motif C-terminally connected to an unknown domain, which had the LPMO sequence signatures, e.g., the N-terminal histidine. They also showed that this was an enzyme that could degrade starch by oxidizing α-glycosidic linkages at the C1 position, analogous to LPMOs from AA9, AA10, and AA11. Together these observations formed a base for the AA13 family of the CAZy database.127 The published structure of A. oryzae AA13 (PDB ID 4OPB) showed both the β-sandwich and the active-site topology of a typical LPMO (Figure 15A and 15C).105 This protein also bound a copper atom in the active site and had the methylated N-terminal histidine similar to that previously found in AA9 LPMOs (see section 5.1). Though the AoLPMO13 itself did not have oxidative activity on any starch substrates, one AA13 from A. nidulans with ~70% sequence identity to AoLPMO13 within the AA13 domain was shown to have oxidative activity on starch substrates. These two enzymes are reported to have the same affinity for copper as well as similar EPR spectra, thereby verifying that the AoLPMO13 structure is that of the catalytic domain.105 Much of the topological and structural divergences from the other LPMOs are localized to the residues between the first and the second β-strand. The protein face around the copper atom, presumed to be the starch-interacting surface, was less flat than in cellulose-active LPMOs and described rather as a groove (illustrated by the black dashed line in Figure 15), postulated to accommodate a helical α-1,4-linked glucan substrate (Figure 2; Table 6).
Figure 15.
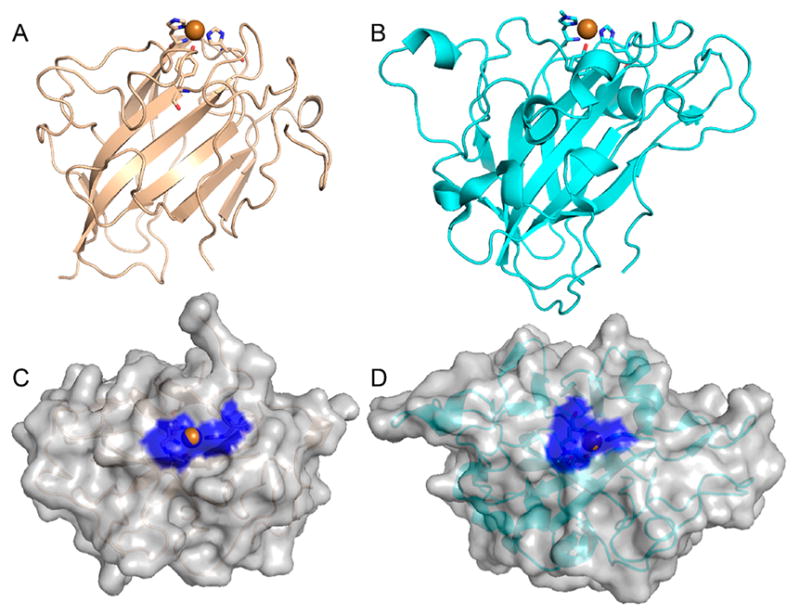
Side-by-side comparison of AA13, represented by A. oryzae LPMO13 (PDB ID 4OPD) and AA9, represented by TaLPMO9A (PDB ID 3ZUD).92,105 (A) AoLPMO13 fold as a cartoon representation with the histidine brace at the top. (B) Same view of TaLPMO9A. (C) AoLPMO13 presumed starch-interacting surface (active-site histidines colored blue; groove indicated by a black dotted line). (D) For comparison, the cellulose-interacting surface of TaLPMO9A (active-site histidines shaded blue).
Table 6.
AA13 LPMO Structures Available in the Protein Data Bank
| source, name and alt. name | PDB code | resolution [Å] | metal ion | brief highlights | ref |
|---|---|---|---|---|---|
| A. oryzae LPMO13, AoAA13 | 4OPB | 1.50 | Cu2+ | first reported AA13 structure; Cu(II) cation in the active site | 105 |
| A. oryzae LPMO13, AoAA13 | 5LSV | 1.10 | Zn2+ | in complex with ligand (maltose) though not in active site | 206 |
| A. oryzae LPMO13, AoAA13 | 5T7J | 1.65 | Zn2+ | 206 | |
| A. oryzae LPMO13, AoAA13 | 5T7N | 1.60 | Zn2+ | in complex with ligand though not in active site | 206 |
4.2. LPMO Modularity
Most LPMOs with solved 3D structures are single-domain enzymes. The major exception is the AA10 V. cholerae colonization factor GlcNAc-binding protein A (GbpA), which has been suggested to mediate cell adhesion and has been shown to bind both chitin and mucin.207 This protein consists natively of four domains, but the structural model (PDB ID 2XWX) includes only three: one AA10 domain (VcLPMO10B), one resembling a flagellin domain, and a third resembling the FimC-chaperone from the Usher pathway of bacterial pili-assembly.197 The fourth domain, not included in the structure model, is a chitin-binding module. Among fungal enzymes there are also a few other exceptions, namely, N. crassa LPMO9A and LPMO9C, T.reesei HjLPMO9A, and also A. oryzae LPMO11, all of which contain a carbohydrate-binding module (CBM) in their native forms that has been removed genetically and/or enzymatically prior to structure determination.107,177,182,193 On the basis of annotated sequences deposited in sequence databases, the dominant architecture of an LPMO is that of a single-domain enzyme. According to Horn and co-workers, 30% of the AA10 sequences in the manually curated CAZy database contain one or more modules in addition to the AA10 LPMO module.104,126,208 In their survey, AA9 LPMOs were found to show little variation in modularity despite constituting a family of enzymes with high sequence diversity. Of 534 analyzed sequences, 409 were single-domain enzymes while 100 (19%) had the fungal-specific CBM1 module attached. More recently, Book and co-workers made a phylogenetic analysis based on sequences in the CAZy database and found that 31% of 184 AA9 sequences contained a known CBM and that the percentage was the same among AA10 gene sequences.76 The present data from Pfam suggest that approximately 24% of the AA10 sequences (annotated as PF03067, LPMO_10) and 18% of the AA9 sequences (annotated as PF03443, Glyco_hydro_61) contain one or more carbohydrate-binding modules.78 This percentage is in the same range as the survey by Horn and co-workers as well as the occurrence of CBM1 in cellulases annotated as GH family 7 (PF00840), which resemble the AA9s of fungal origin.126 Harris and co-workers pointed out that figures based on less curated sequence data may be underestimated due to methodological sequencing limitations that create incomplete gene models especially at the 3′ end where these domains would be linked to the catalytic domain.95
Horn and co-workers also observed much larger modular variation for AA10 LPMOs. The variations in modularity were not restricted to carbohydrate-binding modules but were also observed for genes with other kinds of modules as well, including modules with sequence motifs related to other enzyme activities, e.g., glycoside hydrolases.126 These observations were further investigated by Book and co-workers in their phylogenetic analysis of AA9 and AA10 sequences.76
4.2.1. Carbohydrate Binding Modules
Carbohydrate-binding modules (CBMs) are structurally discrete modules that form part of a larger multimodular enzyme and can act to direct the enzyme to its substrate (for reviews see refs 67 and 211–215). At present, 79 families of CBMs have been characterized according to the CAZy database.104,208 In a recent modified classification, these different families of CBMs can be categorized as belonging to one of three types (A, B, or C) based on the shape and degree of polymerization of the target ligand.211,212 Type A (Figure 16) have flat glycan-binding sites and recognize crystalline surfaces of cellulose and chitin. Type B has glycan-binding sites in clefts or grooves to accommodate longer single-glycan chains, e.g., in hemicellulose and amorphous cellulose and chitin, and binds in an “endolike” fashion. Type C binds “exolike” at the ends of glycan chains.212 For some types of CBMs, disruptive and adhesive roles have been shown, though the primary functional roles of CBMs have been suggested to include targeting enzymes to the correct part of the substrate and increasing the enzyme concentration adjacent to the substrate.216–223
Figure 16.
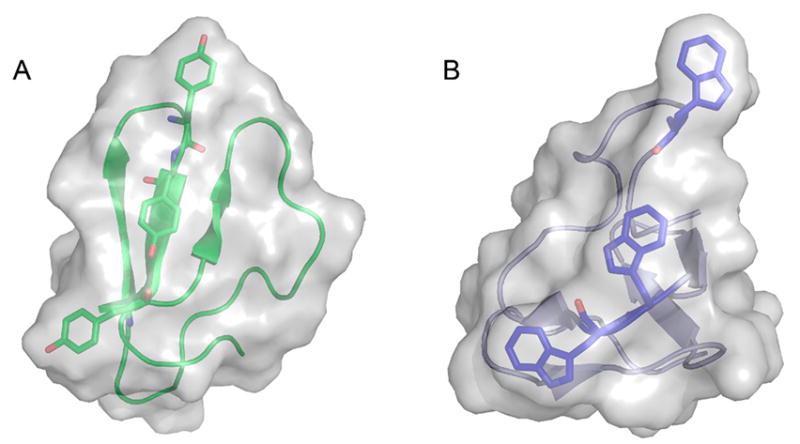
Type A carbohydrate-binding modules CBM1 (cellulose) and CBM5 (chitin) with the aromatic residues important for substrate binding shown as sticks. (A) CBM1 domain of T. reesei Cel7A cellobiohydrolase, PDB ID 2MWK.209 (B) CBM5 domain of Moritella marina chi60 Chitinase, PDB ID 4MB4.210
The dominant CBM among AA9 LPMO sequences (Pfam PF03443) is that of CBM1. CBM1 is a fungal-specific module of type A known to interact with crystalline cellulose (for reviews see refs 67, 224, and 225). Among AA10 LPMOs (Pfam PF03067) there exist both chitin-specific CBMs (e.g., CBM5, CBM12, CBM14) and cellulose-specific CBMs (e.g., CBM1, CBM2a), which can recognize crystalline substrates. 211,224–228 Recently, a new family of CBMs, CBM73, was identified as specific for chitin binding by studies of the AA10 LPMO10A from C. japonicus.121 Although there are few publicly available annotated sequences for AA11 and AA13 LPMOs, the occurrence of genes containing chitin-specific (AA11) and starch-specific (AA13) CBMs together with the observation that they contained characteristics of LPMOs ultimately led to the categorization of these two families of LPMOs.105,107,127
For H. jecorina cellobiohydrolase Cel7A it was reported as early as 1986 that removing the CBM lowered the affinity for crystalline cellulose.229 Reduction in activity and substrate binding in comparisons of the same enzyme with and without CBMs can thus be attributed to a reduced local enzyme concentration adjacent to the substrate. Such roles of CBMs in LPMOs have been investigated and are described in section 3.3.3. In a study to introduce LPMO activities in designer cellulosomes, control experiments showed that removal of the CBMs from two T. fusca AA10 LPMOs reduced activity on Avicel relative to the full-length enzyme.187 In another study, a pair of AA10 LPMOs from Streptococcus coelicolor, LPMO10B and the CBM containing LPMO10C (CelS2), were structurally and functionally characterized. Comparison revealed that LPMO10C without its native CBM showed 25–30% of the activity of the full-length enzyme when measured on PASC, though the product profile seemed not to be affected by the presence or absence of a CBM.125 While cellulose binding by LPMO10C was greatly diminished without its native CBM, the enzyme bound to both α- and β-chitin despite low activity toward those substrates.125 On the basis of these observations, the authors conclude that chitin binding by the catalytic domain is nonproductive and that the ability to bind the substrate is not necessarily connected to substrate activity and/or specificity. For the chitin-active C. japonicus LPMO10A it was shown that the two CBMs, one CBM5 and the newly described CBM73, were responsible for substrate binding by the enzyme and that the LPMO domain alone bound very weakly to chitin.121 Similar to ScLPMO10C, the product profile was the same for both the CjLPMO10A catalytic domain alone and the fulllength enzyme.121,124 Crouch and co-workers further showed that removal of the CBM from C. fimi reduced the activity at least 2-fold on PASC and bacterial microcrystalline cellulose (BMCC).186 However, when they removed the CBM from the similar T. bispora LPMO, most of its activity on PASC and Avicel was lost, while activity on BMCC was unaffected. The creation of hybrid enzymes by swapping the CBMs between the two LPMOs gave different effects for the two enzymes. This was detrimental for the activity on PASC for the T. bispora LPMO but less so for the activity on BMCC. For the C. fimi LPMO there was an increase in activity on PASC but a reduction in the activity on BMCC.186 While the product profile was not significantly affected by the removal of the CBM, module swapping had larger effects that resulted in higher amounts of nonoxidized released cello-oligosaccharides. On the basis of these findings and the observation that introduction of other non-native CBMs as hybrid enzymes affected the product profile, the study authors concluded that the substrate recognition by CBMs to some extent can be LPMO specific and that the CBMs may target subtle differences on the cellulose surfaces.186
N. crassa LPMO9C has been shown to be active on both soluble cellodextrins and xyloglucan.116,119 A recent study looked at a model of NcLPMO9C where the native CBM1 module had been removed.182 This study used ITC experiments to determine the affinities for cellulose and xyloglucan and showed that substrate affinities are higher for the full-length enzyme than for the LPMO domain alone. The binding isotherm for cellulose was also different, indicating two binding sites for the full-length enzyme, one site with higher affinity and one with lower affinity, which seemed to be the site present in the CBM1 deletion model. The product profile and the activity on cellulose, represented by PASC, and on cellodextrin did not show any dependence on the CBM, while the activity on xyloglucan was much lower for the LPMO domain alone than for the full-length NcLPMO9C. To complement these studies, EPR spectra of the full-length NcLPMO9C and the CBM1 deletion were collected and did not exhibit a dependence on the presence or absence of the CBM1 module.182
4.2.2. Linker Region
Multimodular enzymes contain linker regions of various lengths that connect the modules/domains to each other. While hydrophobic residues are prevalent in the core of proteins, linker regions usually lack hydrophobic residues, resulting in a bias in the amino acid composition. For this reason, linker regions are often referred to as low-complexity regions (LCRs).230 Linker sequences, such as those present in fungal cellulases, have been reviewed elsewhere.67 In some carbohydrate-active enzymes of bacterial origin, short linkers have been thought to create a longer carbohydrate-binding surface by allowing intimate contact between the catalytic domain and the CBM.231–233 Longer linker regions between catalytic glycoside hydrolase domains and CBMs have been proposed to create flexibility for the enzyme to bind and act on its substrate. Among fungal cellulases and also within AA9 LPMOs the linkers are rich in serine and threonine residues that are targets for O-glycosylations, as observed in both cellobiohydrolase I and II (Cel7A and Cel6A) from T. reesei. Early studies of these two enzymes using small-angle X-ray scattering (SAXS) experiments indicated elongated structures and structural changes upon substrate binding.234,235 In a more recent study, linkers of both bacterial and fungal origin were compared for GH families 6 and 7.236 In this study, O-glycosylation sites were observed to be uniformly distributed and were determined to be more common in fungal than in bacterial linker sequences. In general, serine, threonine, and proline were more common in linker sequences, though proline was twice as common in bacterial linkers as in fungal linkers, and O-glycosylation sites were more common in fungal linkers. For both fungal and bacterial linkers, the amino acid content of serine and threonine was found to increase with linker length. The same trend was also observed for proline content in bacterial linkers.236
While there are some studies that have addressed the CBM in LPMOs (see previous section), the linker region has not been addressed per se. Recently, the structure of HjLPMO9A (EG4) of T. reesei (H. jecorina) was published.177 This structure included 21 residues beyond the acknowledged AA9 LPMO catalytic core, which formed an extended loop that wrapped around the molecule placing the C-terminus on the opposite side of the LPMO domain (Figure 17, magenta). Though the removal of the CBM1 was performed enzymatically, genetically truncated variants were also produced with different linker lengths. A variant that was similar in length to the enzymatically produced variant was expressed and structurally characterized. Studies have concluded that this part of the linker should be regarded as an integrated part of the LPMO domain.177 In the case of Lentinus similis LPMO9A, however, extra residues are present despite the fact that this this enzyme does not have a native CBM (Figure 17B).180
Figure 17.
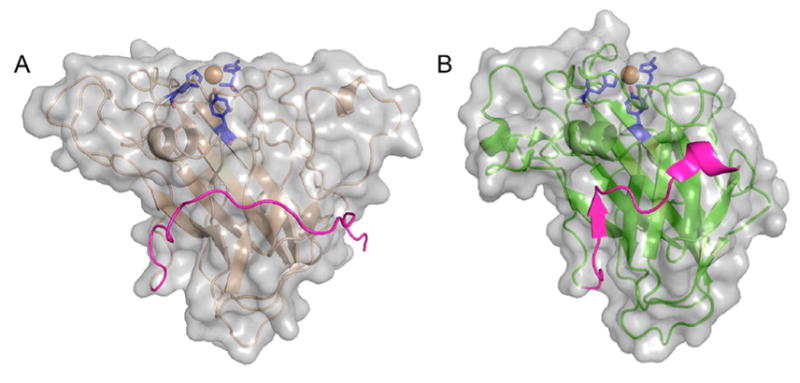
Extended C-terminal linker in T. reesei (H. jeocorina) LPMO9A and the extra residues in L. similis LPMO9A. Surface and cartoon representations of (A) HjLPMO9A (PDB ID 5O2X) and (B) LsLPMO9A (PDB ID 5ACJ).180 HjLPMO9A, active on crystalline substrates, has a flat surface, while Ls(AA9)A, active on both cellulose and soluble substrates, has a less pronounced binding surface also containing a “ridge-like” topology. Orientations of the two molecules are the same with the binding surfaces on top of the molecules. Catalytic sites with two histidines: one tyrosine (blue) and the copper atom (orange) are shown with sticks/sphere. Both structure models contain 15–20 extra C-terminal residues (magenta). While the native full-length HjLPMO9A has a CBM1 module, the native LsLPMO9A does not (see section 4.2).
4.3. Glycosylation
Glycosylations are post-translational modifications of proteins and can be of two types: N-linked, when a glycan is attached to the Nδ2 nitrogen atom of an asparagine residue, or O-linked, where a glycan is attached to the Oγ of a serine or threonine residue. In secreted LPMOs from fungi, N-linked glycan chains begin with a β-linked N-acetylglucosamine (GlcNac) residue and O-linked glycans begin with an α-linked mannose. Both types of glycosylations also exist in some bacteria.237,238 More glycosidic residues may be attached to the protein-linked glycan, but both the type of glycan and the kind of glyocsidic linkage may differ depending on the organism.237,239,240 Studies on cellulases have shown that while N-linked glycosylations do not seem to be enriched in any particular part of multidomain enzymes, O-linked glycosylations are enriched in linker regions.241 There are several suggested functions of glycosylations, though for secreted biomass-acting enzymes the main functions are thought to be structural, protective, and/or stabilizing. For the glucoamylase from A. niger, removal of glycosylation lead to a less thermostable enzyme.237,242 This observation supports the proposal that glycosylation improves protein rigidity, especially in linker regions, and reduces surface hydrophobicity that may prevent aggregation.243 This was complemented by another study on the same enzyme that showed that the presence of glycosylation resulted in a more efficient enzyme.244 Similarly, glycosylation was correlated with improved activity of the cellobiohydrolases of GH families 5, 6, and 7.245–248
In many structural studies of (fungal) enzymes, the preparation for crystallization involves a deglycosylation step to make the enzyme sample homogeneous, which usually reduces glycosylation to one glycan at each site. Work in this area has shown that the expression host organism used to produce the protein for structural studies affects the glycosylation status of the enzyme. Furthermore, since the commonly used bacterial expression systems do not introduce glycosylations, it may be expected that none of the available bacterial AA10 LPMO structure models contain glycosylation. However, both N-linked and O-linked glycosylation sites are present in several of the available AA9 LPMO structure models as well as AoAA13 and two of the viral AA10s that contain N-linked glycans. Each of these contain one or more glycosylation sites with the exception of HjLPMO9A, which contains two N-linked glycosylation sites as well as 17 O-glycosylation sites (mainly in a linker-like region), 15 of which have one mannose in the high-resolution structure model.177 These glycosylations are distributed around the molecule with the exception of the flat surface suggested to interact with crystalline cellulose.
4.4. Substrate Binding
LPMO substrates are polymeric, and many are insoluble, which present significant challenges for studing enzyme–substrate interactions. Much of what is known about the LPMO–ligand interaction stems from studies on LPMOs that are active on soluble substrates, though there is a study of insoluble β-chitin binding to CBP21/SmAA10A using NMR and an indirect method utilizing 2H/1H exchange.180,196,249 There are also site-directed mutagenesis studies in which the effect of selected amino acid residues on the catalytic activity and/or substrate binding have been probed.95,110,183 In an early publication on SmLPMO10A (CBP21), Vaaje-Kolstad and co-workers showed that the single aromatic residue on the proposed binding surface is important for chitin binding.183 In this study, sites for substitution were identified by inspection of the crystal structure and sequence alignment with related proteins. The importance of these residues in substrate binding was demonstrated by generating variants where several acidic or aromatic residues along the binding surface were mutated (Y54A, E55A, E60A, H114A, D182A, and N185A; numbering from the first methionine though histidine at position 28 is the first amino acid observed in the structure) and measuring changes in affinity to chitin using a noncatalytic binding assay. Marinai later extended the analysis of the substrate-binding surface of SmLPMO10A by constructing the S58A, T111A, A112G, and T183A variants.173 In the 2H/1H exchange by Aachmann et al., some of these residues appeared to be protected by chitin, indicating that they are likely close to the substrate upon chitin binding.196 Similarly, surface variants W46A and N47A of TfAA10A demonstrated reduced cellulose binding to 40% and 19% of that of wild type and were found to retain only 6% and 48% the activity of wild type, respectively.184 Li et al. provided a crude model of LPMO interaction with cellulose via manual docking of the NcLPMO9M (PMO-3) to a model of a flat surface of cellulose Iβ. Comparison with similarly constructed models of CBM1, TtLPMO9E and TaLPMO9A interacting with cellulose Iβ suggested that the NcLPMO9M bound differently to cellulose than the other three, i.e., across the cellulose chains rather than along a cellulose chain.192 On the basis of the interaction of CBMs with cellulose and the apparent similarities between CBM1 and LPMO, Wu and co-workers modeled the P. chrysosporium LPMO9D interaction with the hydrophobic face of cellulose using MD simulation.189 In this study, they examined the role of three tyrosine residues on the proposed binding surface in the interaction with cellulose and showed that these also had the highest interaction energies. Two of the tyrosine residues interacted with pyranoses on the same central cellulose chain, while the third tyrosine interacted with a pyranose on an adjacent parallel cellulose chain. The position of the copper active site above the central cellulose chain was stable during the 100 ns MD simulation. A detailed study originates from the X-ray crystallography of protein–ligand structure complex of Lentinula similis LPMO9A. This C4-oxidizing LPMO is active on soluble cello-oligo sacccharides. Frandsen and co-workers obtained crystals in the presence of high salt concentration in which cellotriose (G3) and cellohexaose (G6) were soaked and shown to bind to the protein.180 In the general definition of subsites also used for glycoside hydrolases, the subsites for pyranoses are numbered beginning on each side of the cleavage site and with integers of –1 toward the nonreducing end and +1 toward the reducing end of the glycan, respectively.250 According to this nomenclature, the G3 bound –1 to +2 subsites with glycosyl linkage above the copper atom and an empty putative O2 binding site (illustrated by the black dashed line in Figure 18). It is worth noting here that several hydrogen bonds contribute to the protein–ligand interaction via both direct and water-bridged hydrogen bonds; however, many of these are specific to LsLPMO9A and are not conserved. While the G3 binds in a flat conformation resembling that of cellulose, the G6 deviates slightly from the flat conformation. Interestingly, the glycosyl unit in the 3 position seems to have an aromatic ring interaction with a (AA9) tyrosine residue (Tyr 203 in Figure 18). This tyrosine residue has previously been identified as a possible cellulose-interacting residue, and of the AA9 LPMOs with known structures, all but NcLPMO9F (PDB ID 4QI8) have tyrosine in a similar position.189,192 This indicates some degree of conservation and suggests that this aromatic-ring interaction should be important for substrate interaction among AA9 LPMOs. In an NMR study of N. crassa LPMO9C, the 15N-amide nitrogen chemical shifts for the corresponding tyrosine residue changed upon addition of xyloglucan and, to a lesser extent, upon addition of cellohexaose.249 The loop region on which this tyrosine is located, denoted the LC loop in the study, did not show any significant changes in the 15N-amide nitrogen chemical shift. In the same study, the authors performed an autodocking procedure using NMR data as restraints to make a model of cellohexaose bound to NcLPMO9C. It has been suggested that differences in the NcLPMO9C interactions with cellohexaose in this model and those observed in the LsLPMO9A structure models are mainly due to variability in loop lengths between the two enzymes including the LC loop. On the basis of structural alignments of LPMOs from other AA families with AA9 LPMOs, there were no regions corresponding to the LC loop in AA10, AA11, or AA13s. Larger differences in chemical shifts upon substrate interaction were noted for the residues around the copper atom, in particular, for the N-terminal histidine, and may be due in part to the fact that in the structure of LsAA9 the glucosyl unit in +1 subsite is stacked immediately above the N-terminal histidine (Figure 18). In this structure, the substrate ring oxygen is facing and interacting with the aromatic histidine ring via an unusual noncovalent bond referred to as the lone-pair-to-π* interaction.180 The electronic nature of the interaction may explain the large effect on 15N chemical shift reported for His-1 of NcLPMO9C.249 Beneath the –1 subsite in LsAA9A structure complexes is a conserved glutamine residue interacting with a chloride ion that is localized near the substrate. This glutamine is conserved not only in AA9 but also in AA13 LPMOs. A glutamine residue is also present in some AA10 enzymes that are active on cellulose, with one exception being CelS2/ScLPMO10C, which is active on both chitin and cellulose.124 In the chitin-active AA10 and AA11 LPMOs, the corresponding residue is a glutamate which, in addition to glutamine, has been suggested to be involved in a conserved hydrogen-bonding motif.107,124 Additionally, mutation studies have shown it to be important for enzyme function.95,175,183 The structural features responsible for determining the LPMO substrate specificity for chitin vs cellulose are not fully understood, though a conserved cavity in chitin-active AA10 LPMOs could accommodate the N-acetyl group of the chitin polymer.124
Figure 18.
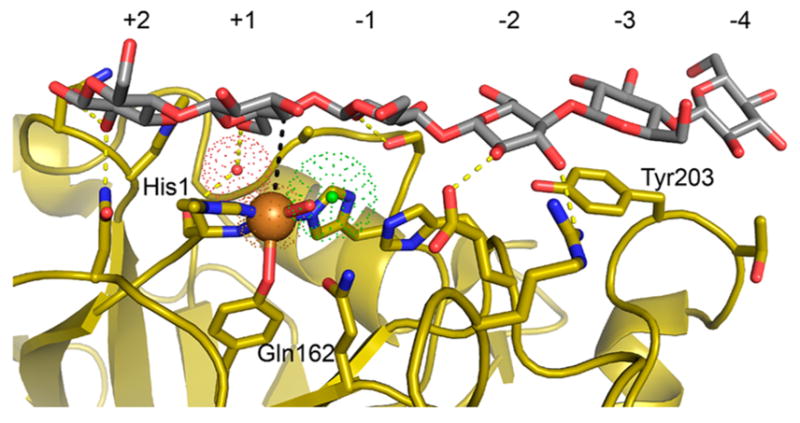
Cellohexaose, in gray and red, bound to L. similis LPMO9A (PDB ID 1ACI) in yellow.180 Copper atom is shown as an orange sphere, while a hydrogen-bond bridging water molecule (red) and a chloride ion (green) are shown with their vdW radius as “dots”. Hydrogen bonds are shown as yellow dotted lines. Copper atom and C4 atom of the glucosyl unit in +1 subsite is connected with a black dotted line in the space corresponding to the presumed O2-binding site.
4.5. Potential CDH-Binding Surface
As described in the section on the Industrial use of LPMOs, one potential source of electrons for the oxidation by LPMOs is the enzyme cellobiose dehydrogenase (CDH). Langston and co-workers demonstrated this with the binary combination of H. insolens CDH and T. aurentiacus LPMO that produced soluble oxidized oligosaccharides.157 They also reported that the CDH/LPMO likely acted in concert with cellulases and hydrolases in the breakdown of cellulose by the fungus T. terrestris. The combined action of CDH/LPMO has also been shown for N. crassa enzymes for which an interaction model of a CDH/LPMO pair was proposed between the CDH and a conserved part of NcLPMO9M (PMO-3).93,192 This hydrophilic patch was located on a different side of the LPMO relative to the copper-containing active site, along with a putative electron-transfer chain across the enzyme. Two separate studies have since suggested a direct interaction between the cytochrome domain and the copper atom in the active site.172,249 In the first study, Tan et al. determined the structures of full-length CDH and the LPMO9F from N. crassa as well as the structure of the CDH from M. thermophilum.172 By measuring the kinetics of the electron transfer from the NcCDH cytochrome domain to NcLPMO9F and comparing with stopped-flow experiments where the flavin domain was included, they concluded that the interaction is between the NcCDH cytochrome domain and NcLPMO9F. Automatic docking of NcCDH to NcLPMO9F, performed in the absence of any cellulosic substrate, resulted in a direct interaction between the heme of the cytochrome domain and the copper center of the LPMO.172 In the second study, direct evidence for an interaction between the CDH and the copper center was provided by monitoring chemical shifts of mixtures of NcCDH enzyme (or the NcCDH cytochrome domain only) with 15N13C-labeled NcLPMO9C.249 Interestingly, the chemical shifts of NcLPMO9C were perturbed for residues clustered around the copper center, the same residues that were affected by substrate binding. Furthermore, increasing substrate concentration seemed also to hamper the interaction with CDH, further indicating that the interaction surfaces are the same.249
5. ACTIVE SITE GEOMETRIC AND ELECTRONIC STRUCTURE AND MOLECULAR MECHANISM
5.1. Cu Active Site Structure
The first coordination sphere of the LPMO copper active site includes three nitrogen atoms from two histidine residues, with one binding bidentate through the N-terminal amine in what has been called a His-brace motif. X-ray crystallography has revealed that reduced LPMOs exhibit three-coordinate T-shaped Cu(I) ligand geometries (shown in Figure 19A) with Cu–N distances ranging from 1.9 to 2.4 Å.123,182,189,198 Additional water-derived ligand(s) are resolved in the Cu(II) structures, which show some variability in water number and position (Figure 19B).92,95,105,107,121,125,180,183,192,199
Figure 19.
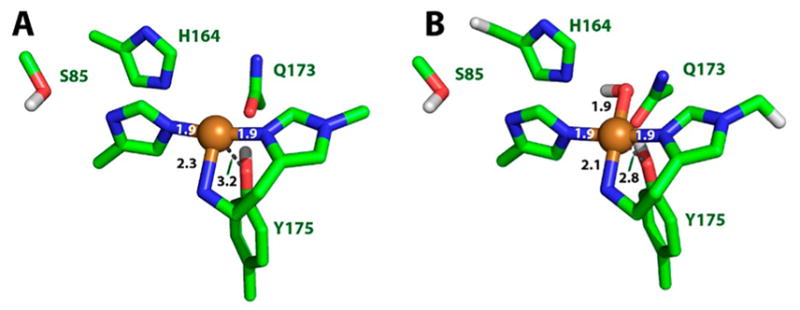
Active site models of the three-coordinate Cu(I)–TaLPMO9A (A) and four-coordinate + H2O Cu(II)–TaLPMO9A (B). Second-sphere residues are labeled, and bond distances to noncoordinating residues are indicated by gray dashed lines. Adapted with permission from ref 251. Copyright 2014 National Academy of Sciences of the United States of America.
In families AA9, 11, and 13, one water/hydroxide ligand is bound in the plane of the three protein-derived nitrogen atoms resulting in a square planar geometry. EXAFS, Cu(I)-XANES, and DFT computations of the cellulose-active fungal T. aurantiacus TaLPMO9A (shown in Figure 19) have established the three- and four-coordinate natures of the reduced and oxidized active sites, respectively.251 These results are in agreement with Cu–O/N ligand distances obtained via crystallography, and the decrease in coordination number is consistent with the coordination preferences for Cu(I) and Cu(II) complexes. Though they share the same T-shaped geometry in the Cu(I) form, the active sites of Cu(II)–AA10 structures are resolved with two water molecules at distances of 2.0–2.7 Å.199 Figure 20 shows a representative crystal structure of the Cu(II) active site of E. faecalis AA10A in which the amine nitrogen of the His-brace and the two water ligands form the equatorial plane of a trigonal bipyramidal structure. Note, however, that EPR data indicate the ground state is close to d(x2–y2) that reflects a more tetragonal electronic structure (vide infra).
Figure 20.
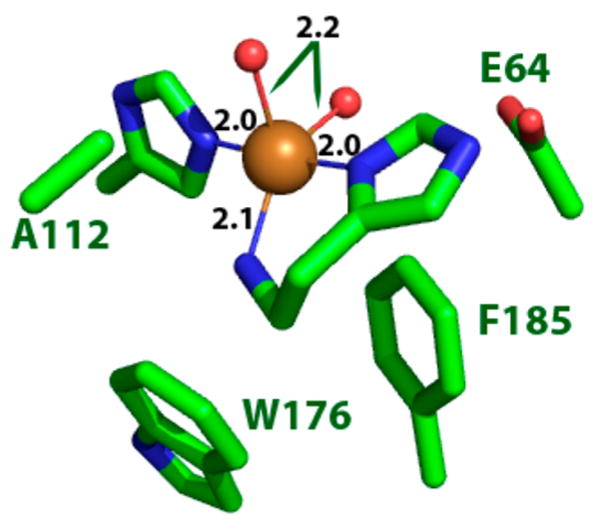
Rendering of the active site of Ef LPMO10A in stick representation showing the trigonal bipyramidal structure and conserved AA10 residues (PDB ID 4ALC).
In AA9s there is often an additional water molecule in an axial position at a distance of ~3 Å and a Tyr residue in the other axial direction that is conserved across the AA9, 11, and 13 LPMO families.105,107,189 The structures published to date show Cu–O(Tyr) distances varying between 2.5 and 3.0 Å. Although not directly bound to the copper and likely protonated, the conserved tyrosine residue is part of a hydrogen-bonding network near the active site that also consists of His-164, Gln-173 (labeled in Figure 19), and several active-site waters.92 The AA10 LPMO families have a conserved Phe (Phe-185) in place of the axial Tyr residue of AA9s as shown in Figure 20.199 Crystal structures show that the side-chain positions in cellulose-active bacterial LPMOs resemble the His-164, Gln-173, and Tyr-175 pattern (2YET numbering, Figure 19) observed for their fungal counterparts, though H-bonding networks are not prevalent in chitin-active bacterial LPMOs.125 The differences in second-sphere environments (e.g., conserved tyrosine residues in AA9s versus conserved phenylalanine residues in AA10s) have been proposed to contribute to differences in Cu(II) affinity in fungal LPMOs compared to their bacterial counterparts.92,196
Due to the low sequence identity across LPMOs, there are no residues apart from the His brace that are strictly conserved among the members of any one LPMO family. However, there are some residues that are generally conserved, including the Tyr/Phe in AA9/10s, mentioned above. Other conserved residues include a Gln (Gln-173 in Figure 19) involved in the hydrogen-bonding network of water molecules near the active site in AA9s. In chitin-active AA10 LPMOs, this Gln residue is replaced by a Glu (Glu-64 in Figure 20). Figure 20 also includes an alanine residue (A112) that is largely conserved in AA10s and is thought to restrict solvent access to the axial position of the Cu.121,252
Cu(II)-bound AA9, 11, and 13 LPMOs exhibit EPR spectra characteristic of type 2, normal copper sites with Jahn–Teller-distorted, axially elongated tetragonal geometries,92,182,251 giving a g-value pattern with gz > gx ≈ gy > 2.00 (Figure 21; gz ≈ 2.27, gy ≈ 2.07, and gx ≈ 2.05). EPR spectra of AA10 LPMOs show approximately axial Cu(II) features similar to AA9s, though with substantial broadening and more rhombicity than other LPMOs (Figure 22) with gz ≈ 2.26, gy ≈ 2.10, and gx ≈ 2.02, consistent with the two bound water-derived ligands more distorted toward a trigonal bipyramidal structure.125,198
Figure 21.
EPR spectra of Cu-loaded (red) and apo (blue) AA9 species from T. aurantiacus. Reprinted with permission from ref 251. Copyright 2014 National Academy of Sciences of the United States of America.
Figure 22.
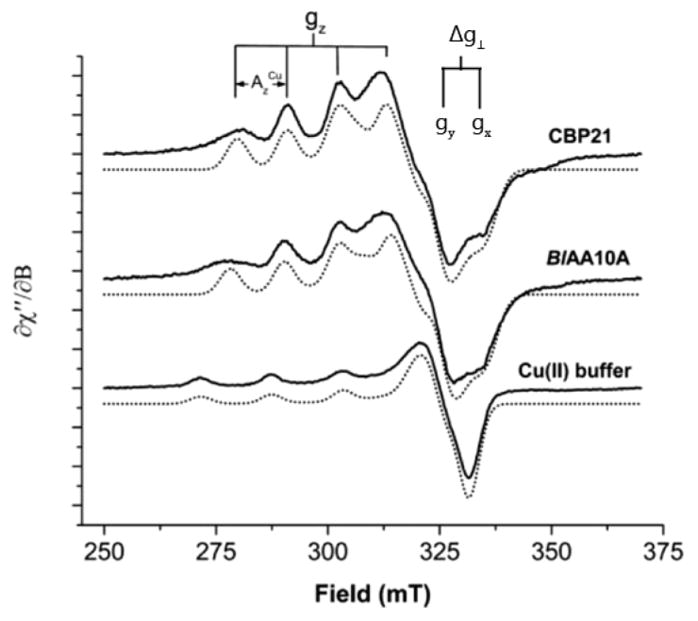
X-band EPR spectra recorded at 30 K for chitin-oxidizing Cu(II)–AA10 LPMOs. Experimental spectra are shown as solid lines, and simulations are shown as dotted lines. These spectra show additional superhyperfine splitting centered around 325 mT. Reprinted in part with permission from ref 125. Copyright 2014 American Chemical Society.
In the study by Forsberg et al.,125 mutation of the axial Phe residue of the cellulose-active AA10, CelS2-N, to either a Tyr or an Ala resulted in a decrease in rhombicity of the EPR signal from Δg⊥ ≈ 0.09 in the wild-type enzyme to Δg⊥ ≈ 0.01 in the variants, resulting in EPR spectra reminiscent of AA9 LPMOs (Figure 23). Spectral perturbations coupled with diminished activity of these variants (0% and 15% of the activity of wild type for the Ala and Tyr variant, respectively) suggest that second-sphere residues have a significant impact on the electronic and geometric structure of the copper active site.
Figure 23.
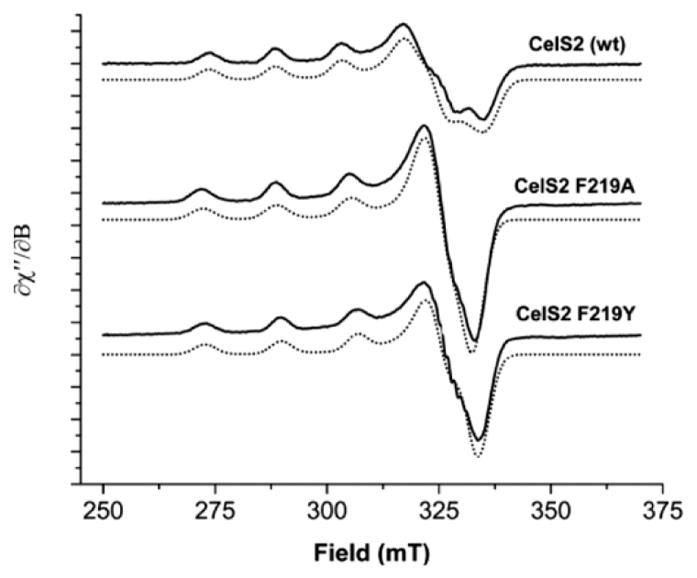
X-band EPR spectra (solid lines) and simulations (dotted lines) of wild-type CelS2 (top), F219A variant (middle), and F219Y variant (bottom). Reprinted in part with permission from ref 125. Copyright 2014 American Chemical Society.
Possible functions of the His-brace, other than equatorial binding, are unknown. This structural feature has also been observed in pMMO253 (thought to use a binuclear copper site to monooxygenate methane) and CopC254 (part of the cop operon involved in copper resistance in bacteria), though the latter has not been reported to have dioxygen activity. Although the active sites found in noncoupled binuclear Cu enzymes (tyramine β-monooxygenase and dopamine β-monooxygenase, TβM and DβM),255 copper amine oxidase,256 and nitrite reductase257 exhibit type 2 or axial spectroscopic features similar to LPMOs, the bidentate ligation of the His-brace is unique. The His-brace of LPMOs has been proposed to enforce critical geometric and steric constraints on the Cu active site using the bidentate ligand to lock the active site in a T-shaped geometry. Computational studies251 suggest that these steric constraints are critical for the associative displacement of superoxide by H2O (see Oxygen Reaction Mechanism section) as well as for maintaining low reorganization energies for enzymes to transition between oxidized and reduced states. Citek et al.258 used model complexes capable of C–H oxidation to suggest that since primary amines are strong ligands they may help to stabilize the higher valence Cu(III) oxidation state as compared to tertiary or peralkylated amines. A recent neutron diffraction crystal structure showed evidence of deprotonation at the amine of the His brace, which if confirmed could also stabilize Cu(III). These are features of the His-brace that can directly impact reactivity. Furthermore, although the factors governing substrate selectivity remain to be determined, some have suggested180 that the His-brace motif may play a role in facilitating the enzyme–substrate interaction.
Another interesting structural feature reported for some fungal LPMOs is N-terminal His-methylation.92 The N-terminal histidine of many AA9 structures and the available AA13 structure show distinct features in electron density maps consistent with methylation, modeled as a 3-methylhistidine (τ-methyl-histidine) residue. This methylation was verified by mass spectrometry.92,105 AoAA11 lacks methylation on the N-terminal histidine, which may be due to the expression of the protein in E. coli.105 Tan and co-workers145 reported that LPMO9F of N. crassa is expressed in P. pastoris as a 214 residue nonglycosylated protein with a nonmethylated N-terminus. Lack of methylation at the N-terminal histidine was also observed for the fungal LPMO GH61D from Phanerochaete chryosporium. These reports show that the enzymes studied retain activity even without this post-translational modification reported for other N. crassa LPMOs expressed in their native fungal hosts. While the function and/or role of this modification is unclear, Aachman et al.196 and others suggested that N-terminal methylation may alter the basicity of the active site and may increase metal-binding affinity. Methylation may also prime the N-terminal His to accommodate the copper ion, keep the active site open to bind dioxygen, and tune reactivity by enforcing a particular binding geometry of the O2 substrate. Crystallographic reports180 have led to proposals that alkyl groups on aromatic ring N atoms may enhance the electrostatic interaction between the substrate and the imidazole ring of His 1. Calculations by Paiva et al.259 and Kim et al.260 challenge these proposals and instead argue that N-terminal methylation has only a minor effect on the active-site structure and reactivity of LPMOs.
Protein modification by methylation is well known in other enzyme systems. In a study of modified histidines in amyloid-β peptide, Tickler et al. studied methylation at either the ε- or the δ-nitrogen of imidazole side chains bound to a copper center and identified a correlation to H2O2 production. They reported that methylation not only affected metal binding but also inhibited surface membrane interactions. Tickler et al. attributed these observations to the fact that N-methylation locks the imidazole ring into a single tautomeric conformation. 261 It is interesting to consider if the N-methylation of the His ligand could impact in situ generation of H2O2 as this has been proposed as a cosubstrate of the LPMOs (vide infra).
As mentioned earlier, another feature observed in a subset of LPMOs is the presence of noncatalytic CBM domains (see section 4.2.1). While the quantitative contribution of the CBM domain remains unclear, understanding this feature has attracted a great deal of attention in the LPMO research community. Studies by Crouch et al. have shown that in some cases deletion of the family 2a CBM from LPMOs natively expressing CBMs resulted in significant loss of activity.186 Crystallographic studies of HjLPMO9A showed that while proteolytic cleavage of the CBM from the enzyme generated a CBM-free form, a portion of the long, flexible linker that connects the CBM to the catalytic core of the enzyme remained. Inclusion of this linker resulted in proper expression and folding of a CBM-free genetic construct. The CBM-deletion variants were found to retain activity, albeit lower activity as compared to wild type, as well as poorer binding to cellulose.177,182 Interestingly, comparison of absorbance, CD, MCD, and X- and Q-band EPR spectra (which are probes of the Cu active site) of the wild-type and CBM-deletion variants revealed no significant changes in spectral features between the two forms. Figure 24 shows overlays of the CD, MCD, and EPR (X- and Q-band) spectra of the full-length and CBM-deletion variant. Comparison of these spectra established that the absence of the noncatalytic domain does not significantly affect the active-site structure of the LPMO, and so the difference in reactivity likely reflects a difference in substrate binding by the forms lacking a CBM.
Figure 24.
Room-temperature CD (A), low-temperature, 5 K MCD (B), 77 K X-band EPR (C), and 77 K Q-band EPR (D) spectra of wild-type HjLPMO9A (blue) and Cu(II)-HjLPMO9A-ΔCBM (black). CD spectra shown have the corresponding spectra of the apo enzyme removed subtracted. Low-temperature MCD spectrum prepared by subtracting the 0 T spectrum from the 7 T data. Reproduced with permission from ref 177. Copyright 2017 The American Society for Biochemistry and Molecular Biology.
5.2. Substrate Interactions with the Cu Active Site
Detailed mechanistic understanding of polysaccharide substrate binding has been inhibited by the insolubility of the majority of known LPMO substrates. Most LPMOs reported to date are only active on recalcitrant polysaccharide substrates,3,92,149 which are insoluble when the degree of polymerization (DP) is greater than 15–20.262 However, a subset of AA9 LPMOs has been reported to have activity on select soluble cellooligosaccharides119,180 as well as larger soluble polymers such as xyloglucan and glucomannan and suspensions of amorphous polysaccharides such as PASC149,263 or starch.105,127
Substrate binding to several AA9s has been monitored via optical measurements and ITC.121,182,249 Crystalline substrates with larger areas exhibit lower dissociation constants that vary across LPMOs. In a study by Kracher et al. on four LPMOs from N. crassa, the reported KD values measured using microcrystalline cellulose varied between 5.7 and 16.5 μM and of PASC between 11.2 and 60.8 μM.4 Similar measurements by Borisova et al. on NcLPMO9C using PASC compared substrate binding of the full-length enzyme with the CBM-deletion variant. They observed two binding modes for the full-length enzyme with KD values of 0.013 and 0.64 μM but only a single binding mode for the CBM-deletion variant with a KD value of 0.54 μM.182 These results indicate substrate binding for LPMOs even in the absence of CBMs, but there appears to be a higher affinity with the CBM. Smaller substrates such as the 14-mer of xyloglucan and cellohexaose were studied by ITC and found to have KD values between 0.33 and 0.81 mM.249 The KD values dropped to 0.13–0.14 mM in the presence of CN−, which is known to bind to Cu(II). Other methods, such as changes in protein stability, have indirectly shown substrate binding in the case of BaLPMO10A.201 In general, the study of substrate-bound LPMOs is hindered by the limited solubility of some polysaccharide substrates of interest.
Despite the inherent limitations of these experiments, EPR180,182 and NMR196,249 spectroscopies have been used to probe the effects of binding of polysaccharide substrates on the Cu(II) site of AA9s. The changes in the EPR spectra include a decrease in g|| (Δg ≈ –0.04), an increase in |A||| (ΔA ≈ 20 × 10−4 cm−1), and the appearance of superhyperfine structure arising from slight alterations in the nitrogen ligation in the presence of substrate near the active site (Figure 25). The axial envelope of the Cu(II) signal is largely unchanged, indicating retention of a d(x2 – y2) ground state and suggesting that substrate does not directly bind to the Cu but near enough to perturb its structure. Both larger crystalline substrates and smaller soluble substrates produce the same perturbations in the EPR spectra, indicating very similar modes of substrate binding with respect to the copper active site. As of this writing, there appear to be no published data on the effects of polysaccharide substrate binding to the EPR spectra of the active sites of the other three classes of LPMOs. It should be noted, however, that the lack of spectroscopic perturbation does not imply a lack of substrate binding. Unpublished results from our lab do not show any perturbation upon addition of substrate to HjLPMO9A.
Figure 25.
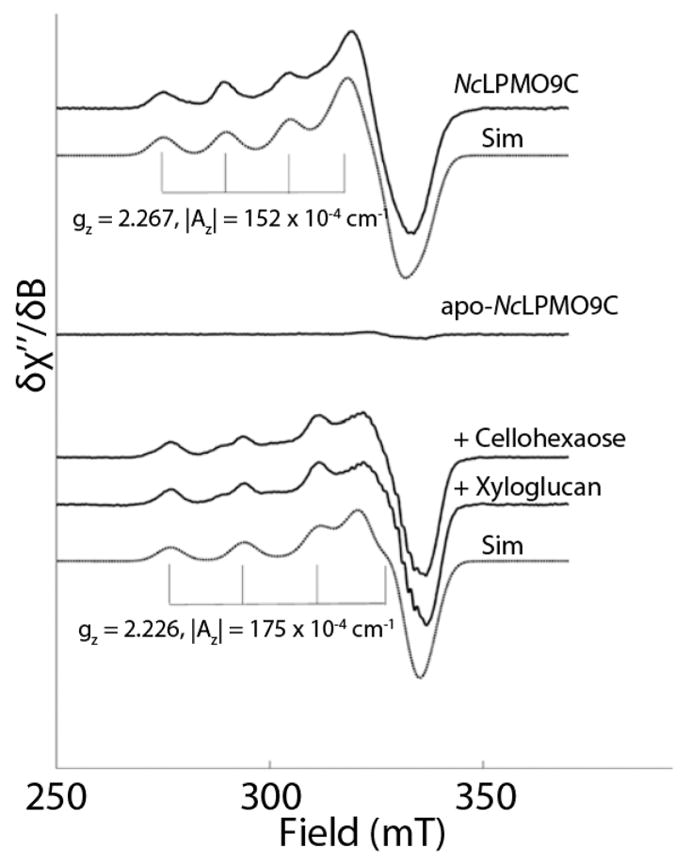
X-band EPR spectra of Cu(II)–NcLPMO9C recorded at 77 K in the absence (top) or presence (bottom) of the soluble substrates cellohexaose (20 mg/mL) or xyloglucan (15 mg/mL). Experimental spectra are shown as black solid lines. Simulations of the experimental spectra are shown as dashed lines below the corresponding experimental spectra. This figure demonstrates the effects of polysaccharide substrate binding on the LPMO copper active site as monitored by EPR spectroscopy. Reprinted in part with permission from ref 182. Copyright 2015 The Journal of Biological Chemistry.
As noted above, crystal structures with soluble oligosaccharides that bound near the active site were reported in the LPMO LsAA9A (Figure 26).180 These structures show specific binding of oligosaccharides with three and six glucose units, respectively, highlighting the importance of solvent-exposed aromatic groups (His1, Tyr203) and H-bonding residues (Ser77, Glu148, Asn28, His66, Asn67) that position the substrate in close proximity to the Cu cofactor. Several of these H-bonding interactions are shown in Figure 26A.
Figure 26.

(A) Crystal structure of Cu(I)–Ls(AA9)A LPMO with G3 substrate bound on the surface of the enzyme. Protein–substrate contacts are shown as black dashed lines, and key residues involved in substrate binding are labeled. (B) Electron density map of the protein–substrate crystal structure of Cu(I)–Ls(AA9)A LPMO with G3 substrate under low X-ray dose conditions. Note the chloride ion that is nestled in the active site. Reprinted in part with permission from ref 180. Copyright 2016 Nature Publishing Group.
Comparison of the structures of LsAA9A and TaAA9A reveals a lack of the residues responsible for hydrogen bonding to the substrate (specifically Asn28, His66, and Asn67), though these residues were found to be conserved in NcLPMO9C.180 Additionally, earlier studies showed that the spacing of these residues was different for each AA9 studied.192 Furthermore, the lack of activity observed in NcLPMO9A, for example, toward the shortest oligosaccharides (DP < 4) has been attributed to a minimum substrate length needed to span the enzyme-binding sites.119 These differences likely contribute to the observed selectivity for certain types of polysaccharides across the LPMO families.
During turnover, the β-1,4 glycoside substrate C–H bond that is cleaved is thought to interact with the equatorial O2-derived ligand of the copper, as the axial binding site would be blocked by the polysaccharide substrate. Interestingly, the substrate-bound crystal structures of LsAA9A revealed that upon substrate binding the equatorial water-derived ligand is replaced by a chloride ligand even under low chloride conditions with a Cu(II)–Cl distance of 2.3 Å (Figure 26B). This chloride ligand was resolved at distances of 3.7 and 3.9 Å from the C4 and C1 positions, respectively, of the substrate. Figure 27 shows a hypothetical model based on the LsAA9 active site where the Cl− ligand has been replaced with a superoxide ligand. The O–O and Cu–O bond distances were taken from optimized structures calculated in ref 251. Minimum distances between the distal oxygen and the C1 or C4 (obtained by rotating the Nπ(His1)–Cu–O–O dihedral angle) are 2.8 and 2.5 Å. It has been shown that this enzyme oxidizes at the C4 position,180 and although it is slightly closer to this carbon atom, a bound oxygen species would presumably be capable of abstracting an H atom from either position during turnover (vide infra). It is still unknown how the regioselectivity of substrate oxygenation results from the orientation of the substrate arising from specific binding residues on the enzyme surface.
Figure 27.
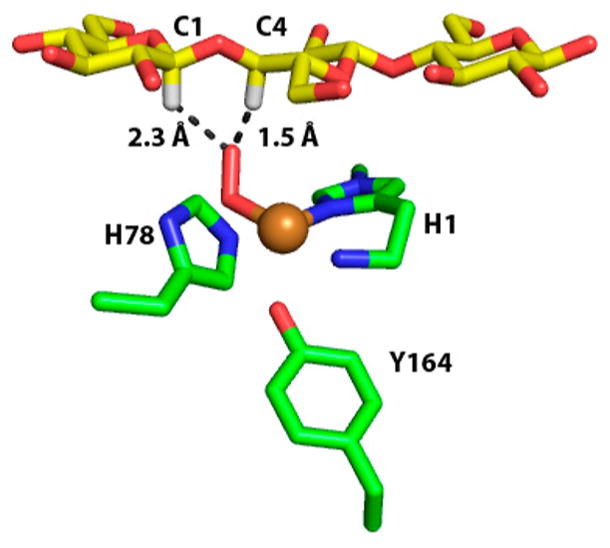
Hypothetical model of LsAA9 active site with the chloride ligand in Figure 26B replaced by O2. Orientation of the oxygen ligand is chosen to minimize C4–H distance. Initial coordinates from PDB ID 5ACF.
The presence of Cl− in the substrate-bound crystal structure of LsAA9A along with the increased substrate affinity in the presence of CN− for NcLPMO9C observed in ITC suggest a cooperative effect of anion and substrate binding, although the nature of the effect has not been explored.249 Interestingly, cyanide has been shown to inhibit LPMO turnover, though no such effect has been shown for chloride.3 The increased affinity for an anionic ligand (Cl−) to Cu(II) in the presence of substrate is consistent with favorable binding of a reduced oxygen-derived species during turnover. Currently, it is unclear whether anion binding enhances substrate binding or vice versa.
5.3. Oxygen Reaction Mechanism
The enzyme–substrate interactions, product profiles, and electron-transfer kinetics of the oxidative cleavage of polysaccharides by LPMOs have been described in many reports, yet the mechanism of substrate oxidation remains unresolved. Both dioxygen and hydrogen peroxide have been proposed, utilizing different mechanisms, to be the oxygen source for substrate hydroxylation.3,93,128,167,192,251,260,264 Initial reports favored dioxygen as the direct oxidant of polysaccharide substrates,3 though recent studies have implicated hydrogen peroxide as the cosubstrate in polysaccharide oxidation, which itself would be formed from molecular dioxygen under suitable reaction conditions (vide infra).167 The evidence for both O2 and H2O2 and their possible mechanisms for polysaccharide hydroxylation are outlined below.
In past reports of LPMO activity, researchers demonstrated the regioselective oxidation of crystalline polysaccharide substrates to yield soluble oligosaccharide products. Catalysis was not observed anaerobically nor in the absence of reductant, and isotopic labeling experiments revealed that molecular dioxygen was the source of the oxygen atom incorporated into the solubilized products3 (however, see the H2O2 discussion below). The reaction was inhibited by cyanide but not by catalase (however, see below) or superoxide dismutase, suggesting that O2 binding could be a critical step in turnover and that labile superoxide or peroxide species were not generated during catalysis.3 Lastly, peroxide formation, proposed to be a futile side reaction of Cu(I)–LPMOs with O2, was observed in the absence of suitable polysaccharide substrates.119 These observations led to a conclusion that molecular oxygen is used directly by LPMOs to oxidize polysaccharide substrates according to the monooxygenase reaction given in eq 1
| (1) |
Researchers have thus investigated Cu–oxygen adducts in LPMOs. Reports have been published on AA9s from N. crassa wherein dioxygen species were bound at or near the Cu(II) active site.192,264 In one study, two LPMOs, NcLPMO9D (PMO-2) and NcLPMO9M (PMO-3), were crystallized, and each was found to have electron density near the axial positions of the Cu active site that were best modeled as end-on dioxygen species.192 The O–O distance in the NcLPMO9D structure was 1.16 Å, which is even shorter than that of dioxygen. The Cu–O distances in this structure were 2.92–2.96 Å, which are not Cu–O2 bonds (Figure 28A).192 A second LPMO structure (NcLPMO9M) shows an O–O distance of 1.49 Å and a Cu–O distance of 3.44 Å (Figure 28B). This is most consistent with a peroxide moiety, yet the Cu–O distance is again too long to be a bound species. Another study reported the crystal structure of NcLPMO9D but with two different dioxygen molecules near the active site.264 In this case, one oxygen species was best modeled as a peroxide (O–O distance, 1.44 Å) bound in the equatorial position of the Cu(II) with a Cu–O distance of 1.9 Å (Figure 28C).264 The other enzyme monomer was modeled as having molecular oxygen near the Cu site but not directly bound to the metal center. This was described as “pre-bound” molecular oxygen (Figure 28D), though currently there is no experimental evidence to support or refute the existence of an O2 prebinding site. A third study used X-ray and neutron diffraction to solve high-resolution crystal structures of J. dentitrificans LPMO10A. The crystallography showed two molecules in the asymmetric unit, both with O2 bound to the Cu active site. Molecule A of the crystal structure (Figure 28E) showed the dioxygen species approximately side on with Cu–O distances of 1.8 and 2.1 Å, whereas molecule B (Figure 28F) was bound end on with Cu–O distances of 1.8 and 2.7 Å. Both molecules were solved with O–O distances of 1.5 Å, leading to an assignment of peroxide for this oxygen moieties. Incorporation of deuterium for hydrogen in exchangeable residues allowed for the determination of the protonation state of the N-terminal amine in the neutron diffraction data. Molecule A was found to have an NH2 ligand, but molecule B was in a mixed ND2/ND− state. Deprotonation of the Hisbrace amine has been suggested to stabilize higher Cu oxidation states (i.e., Cu(III) species) and may play an important role in the O2 reaction mechanism (vide infra).
Figure 28.

Renderings of Cu/O2 complexes reported to date with bond lengths given in Angstroms. (Left) Cu/O2 species reported by Li et al. in chain A of NcLPMO9D (PDB ID 4EIR) (A) and in chain A of NcLPMO9M (PDB ID 4EIS) (B). (C and D) Copper active site for NcLPMO9D treated with ascorbate and exposed to atmospheric O2 (PDB ID 5TKH). O–O electron density is oriented in an end-on geometry. In C the Cu–O distance is 1.9 Å. (E and F) Copper active site of JdLPMO10A.
While the assignment of peroxide bound to Cu(II) in these reports is interesting, it is important to remember that the catalytically relevant dioxygen intermediate (i.e., demonstrated to react with the C–H of substrate) has not yet been trapped nor identified and that the nature of the Cu–O2-bound structure has not been established. Several of these structures demonstrate axial Cu–O2 binding with Cu–O distances that are too long to be compatible with bonding. This binding motif is also at odds with both QM calculations showing equatorial binding to be preferred and with observed substrate-binding patterns that would block access to the axial position of the metal center. Further studies are necessary to confirm the identity of the dioxygen ligand and the nature of binding to the active site.251,260
Mechanisms that rely on O2 activation to provide the oxygen for substrate hydroxylation all share a common branch in which Cu(I) reacts with oxygen to form a Cu(II)–superoxide. From that point the mechanisms differ on the timing of electron and proton transfers and whether a Cu–superoxide or a Cu–oxyl is responsible for the H atom abstraction (HAA) from substrate.93,128,251,260 Figure 29 summarizes possible O2 reaction mechanisms as outlined below. Note that an important thermodynamic consideration for evaluating different molecular mechanisms is the ability of a given oxygen intermediate to abstract a hydrogen atom from the polysaccharide substrate. As mentioned in earlier sections of this review, LPMOs display selectivities for cleaving either the C1–H or the C4–H bond of their substrates. In a recent report, Hedegard and Ryde computationally calculated the C1–H and C4–H bond dissociation energies as 101.1 and 103.8 kcal/mol, respectively. 265
Figure 29.

Proposed LPMO mechanisms in which O2 is the direct oxidant of the polysaccharide substrate. Upper branch shows one pathway in which the Cu–superoxide is responsible for HAA. Lower branch utilizes a Cu–oxyl to perform HAA.
Kjaergaard et al. reported stopped-flow absorption and freeze–quench EPR spectroscopic data for the reaction of Cu(I)–AA9 with O2 in TaLPMO9A in which the Cu(II) signals of the resting enzyme were regained rapidly with an overall rate constant > 0.15 s−1.251 However, they did not observe a bound Cu(II)–superoxo intermediate. As the reduction potentials (E°) of the Cu site and for O2 reduction to O2− are known, the rate for an outer-sphere one-electron reduction of O2 could be calculated and is orders of magnitude slower than the O2 reaction observed for TaLPMO9A. The reported ET rate for the copper reflects an inner-sphere process involving a Cu(I) + O2 bound species. These results were coupled to DFT calculations supporting equatorial binding of an end-on Cu(II)–superoxide species, consistent with calculations by Hedegard and Ryde that have also shown equatorial superoxide binding to be more stable than axial binding.265 This calculated superoxide species could be easily displaced by an axial H2O. Figure 30 shows the calculated structures along the reaction coordinate of O2 binding and superoxide displacement. During turnover, however, the equatorial superoxide is proposed to remain bound since the polysaccharide substrate would presumably block the axial position and therefore preclude both axial oxygen binding and displacement of superoxide by water.
Figure 30.

DFT-optimized structures along the reaction coordinate of superoxide displacement by water. Reprinted with permission from ref 251. Copyright 2014 National Academy of Sciences of the United States of America.
It has generally been assumed that the reactive Cu/O2 intermediates formed in this reaction are fleeting, and researchers have turned to superoxide mimics such as cyanide3 and azide, both of which have been shown to bind to the Cu(II) site in LPMOs. Binding of these small molecules has been confirmed by EPR, where a decrease in g|| and increase in A|| have been reported.107 Of the two only cyanide has been shown to inhibit LPMO activity, yet as noted above, the addition of cyanide enhanced polysaccharide substrate binding. These findings suggest that an oxygen-bound intermediate may yet be trapped during turnover, but its stability may depend on the structural changes induced by substrate binding.
Several studies of model complexes have shown that binding of O2 to Cu(I) generates Cu(II)–superoxide intermediates; however, there are only two cases with crystallographic structures for the Cu(II)–superoxide species.266–280 To better understand the formation of this species, computational analysis of the thermodynamic properties associated with O2 binding in the AA9-LPMO TaGH61 were compared to those reported by Lanci et al. and Kunishita et al. for Cu(II)–superoxide model complexes.281,282 In calculations on both model complexes and on the enzyme active-site model, O2 binding to reduced copper sites was determined to be essentially thermoneutral, and the thermodynamic properties were in good agreement with experimental and literature values. Furthermore, it was determined that in TaLPMO9A Cu(I) undergoes rapid reoxidation via an inner-sphere mechanism to bind O2 as superoxide in the equatorial position to yield the Cu(II)–superoxide intermediate.251 In this case, the thermodynamically difficult one-electron reduction of O2 is driven by the strength of the Cu–O2 bond. In a separate study, Cowley et al. calculated O2 binding for noncoupled binuclear copper enzymes and found that O2 binding via water displacement was more favorable than an associative binding mechanism which has an unfavorable entropic contribution to the Gibbs free energy.283
When superoxide is bound to copper it can adopt one of two geometries: end on (η1) or side on (η2). The side-on superoxide complex supported by the tris(pyrazoyl)borate ligand has a singlet (S = 0) ground state attributable to the highly covalent bonding interaction between the copper and the side-on superoxo moiety, leading to the high energy of the lowest unoccupied molecular orbital (LUMO) being the antibonding combination of the Cudx2−y2 and superoxide π*σ orbitals that lie in the CuO2 plane (Figure 31, red).284 This large bonding/antibonding interaction splits the two highest energy valence orbitals in Figure 31 (right), resulting in a spinpaired ground state. The overwhelming majority of copper superoxo complexes published to date fall into the second category, where the superoxide binds in an end-on geometry (Figure 31, blue). End-on superoxide Cu(II) complexes have triplet (S = 1) ground states that result from two factors. First, the two LUMOs, Cudz2 and superoxo-based π*v, are orthogonal and therefore cannot antiferromagnetically couple.270 Second, in contrast to side-on superoxide binding, end-on coordination utilizes a single, weaker Cu–O bond that results in a lower energy antibonding Cudz2 superoxo-based π*σ molecular orbital that as a result of its low energy is populated. Thus, the π*v and Cudz2 orbitals in Figure 31 (left) are split by an amount less than the spin-pairing energy. These features are vital to our understanding of the reactivity of the relevant frontier molecular orbitals (FMOs) and clearly show that the end-on geometry poises the π*v FMO which is low in energy, unoccupied, and has a large O character on the distal oxygen for reactivity on the triplet surface.
Figure 31.
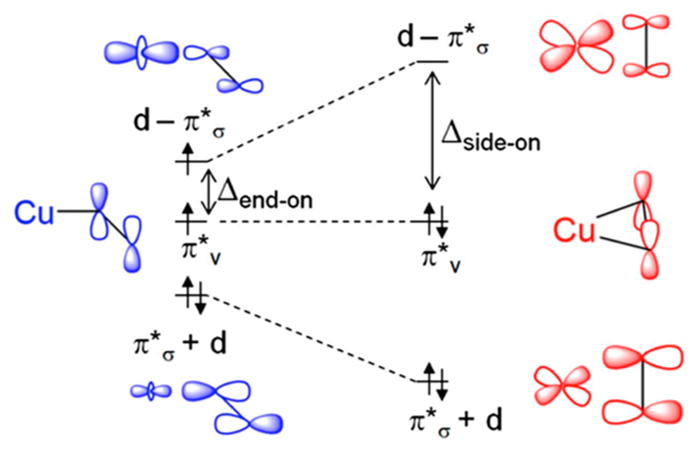
Molecular orbital diagram of the two possible orientations of superoxide binding to copper. End-on geometry is shown in blue on the left, and side-on geometry is shown in red on the right. Reprinted with permission from ref 270. Copyright 2013 American Chemical Society.
Detailed computational analyses by Cowley et al. revealed that end-on superoxo complexes have lower barriers for H atom abstraction as compared to their side-on counterparts, due to the lower energy and inherently high oxygen character in the π*v orbital.283 Therefore, the low-lying superoxo π*v orbital is the FMO that dictates reactivity in the end-on triplet Cu–O2·−. To this end, a large number of superoxo model complexes have demonstrated HAA or HAT (H atom transfer) reactivity on exogenous substrates or phenols to yield hydroxylated or hydroperoxylated products including two model complexes that mimic the reactivity of LPMOs toward inert C–H bonds.269,271–273,275–278,282,285,286 Itoh and Karlin each reported model complexes that are able to activate and hydroxylate C–H bonds in exogenous substrates, similar to LPMO action on cellulosic substrates.282,286 In a parallel study of the noncoupled binuclear copper enzyme, peptidylglycine α-hydroxylating monooxygenase (PHM), Cowley et al. calculated a barrier for HAA of +14.4 kcal/mol (~14 kcal/mol experimentally) and that the superoxide π*v orbital that lies perpendicular to the CuO2 plane is favored to participate in HAA by 10 kcal/mol over the superoxide π*σ orbital.283 Calculations using values reported for the model complex supported by the N-[2-(2-pyridyl)ethyl]-1,5-diazacyclooctane tridentate ligands show a barrier of +28.2 kcal/mol (+20.4 kcal/mol experimentally) for HAA.282 This result supports a mechanism whereby HAA is facilitated through the π*v orbital of the superoxide moiety.287 Substrate oxidation mechanisms are still under investigation by synthetic inorganic chemists and will inform future studies on the mechanisms of LPMOs.
Several mechanisms can be considered involving a Cu–O2− performing HAA to generate Cu(II)–O2H and then varying in the timing of the subsequent electron-transfer/proton transfer (ET/PT) and radical recombination steps. Phillips et al. proposed for LPMOs that a Cu(II)–hydroperoxide could undergo an intermolecular ET to cleave the O–OH bond, yielding water and a Cu–oxyl intermediate.93 This mechanism is analogous to that proposed by Klinman in the noncoupled binuclear Cu enzymes, PHM and DβM.288,289 The Cu–oxyl would then recombine with the substrate radical to form the bound oxygenated product (yellow pathway in Figure 29). An alternative mechanism for LPMOs was also initially considered for the noncoupled binuclear enzymes in which the substrate radical could instead couple with the distal O of the Cu(II)–hydroperoxide resulting in formation of a hydroxylated unbound product and a Cu(II)–oxyl species that was subsequently reduced to form water (blue pathway in Figure 29).288
The recent work of Cowley et al., also on the noncoupled binuclear Cu enzymes, suggested a variation of the latter pathway (red in Figure 29) that may be accessible to LPMOs.283 They proposed that after formation of a Cu(II)–hydroperoxide and substrate radical O–O bond cleavage occurs when the radical reacts with the proximal, nonprotonated oxygen atom rather than the distal oxygen of the hydroperoxide to form a Cu(II)–hydroxide and a bound oxygenated substrate radical. The calculated transition state for this radical rebound step invoked a side-on peroxide, similar to that recently observed in a crystal structure of a dioxygen species bound to the Cu(II) of JdLPMO10A290 (vide supra). A proton-coupled electron-transfer step would then quickly generate Cu(II)–H2O, and further protonation would yield the hydroxylated substrate product.
On the basis of DFT calculations of the LPMO mechanism, Kim et al. proposed an alternative mechanism in which a Cu(II)–oxyl would be the species responsible for HAA (green path in Figure 29).260 In their mechanism two electrons are transferred to the Cu(II)–superoxide prior to HAA to form a Cu(II)–oxyl (triplet ground state) with a substantially lower barrier for HAA than that calculated for a Cu(II)–superoxo species. This results in a substrate radical that then rebounds to form the hydroxylated product and regenerates the Cu(I)–LPMO. Although the reported barrier was lower for the Cu(II)–oxyl pathway in this study, the energies of the two protons and two electrons required to form the Cu(II)–oxyl from the Cu(II)–superoxo were not accounted for in the calculations. Additionally, the model included axial O2 binding which is at odds with crystallography and all other computational results. Support for HAA after O–O cleavage comes from Tolman and co-workers, who reported the one-electron oxidation of a tetragonal Cu(II) complex to form a Cu(III)–OH complex.267,291–294 While the Cu(III)–OH is formally a protonated form of Cu(II)–O·−, the former has a singlet ground state rather than the energetically favored triplet ground state of the Cu(II)–oxyl. However, it is interesting to note that a reactive, triplet Cu(II)–oxyl complex would likely be protonated in aqueous medium due to the high pKa (11.7 ± 2.2) of the Cu(III)–OH.293 H atom abstraction by a Cu(III)–OH would produce a Cu(II)–OH2 species that is not capable of rebound to form a hydroxylated product.
A report recently published by Bissaro and co-workers, originally available online in late 2016, argues that hydrogen peroxide, rather than molecular dioxygen, is the direct oxidant employed by LPMOs.101,167 They use several lines of evidence in support of their claim. First, they correlate increased LPMO reactivity with conditions in which low to moderate amounts of H2O2 are generated in situ or added to the reaction mixture. Note that excess hydrogen peroxide was found to be detrimental to activity and resulted in oxidative damage to the enzyme, with oxidation mostly restricted to residues near the active site. Second, they demonstrated that product formation using periodic additions of peroxide under anaerobic conditions matched product formation in the presence of dioxygen. Third, isotope-labeling experiments showed that the oxygen atoms from H2O2 were incorporated into the product even in the presence of dioxygen. Finally, they demonstrated that under turnover conditions using only O2 as the oxidant the addition of horseradish peroxidase inhibits LPMO activity. The lack of inhibition previously reported3 (and again demonstrated by Bissaro et al.) by catalase was attributed to the substantially lower Km of peroxidase, allowing it to compete more effectively with the LPMO for available peroxide. The catalytic mechanism would then follow the reaction in eq 2 rather than in eq 1. This study focused mainly on ScLPMO10C; however, increased activity in the presence of peroxide was also reported for three other LPMOs: PcLPMO9D, ScLPMO10B, and SmLPMO10A (CBP21).167 The results of their studies indicate that while utilization of H2O2 is common to many LPMOs, the activity enhancement was enzyme dependent, suggesting different peroxide reaction rates.
| (2) |
If the results of this study are germane to the LPMO family then the mechanistic picture outlined above would change. Low levels of H2O2 could be generated via several known mechanisms. The most obvious source of H2O2 generation would be by nonsubstrate-bound Cu(I)–LPMOs reacting with O2 to form superoxide which could then disproportionate to O2 and H2O2.119 As noted above, this reaction is already known to occur in the absence of substrate.4,295 In cases where excess reductant would be present under turnover conditions in the presence of O2, peroxide will be generated and also the initial reduction of Cu(II)–LPMO will take place to enable the reaction shown in eq 2. Note that an external reductant is required to activate the Cu(II) site (i.e., reduction to Cu(I)) but is not required for turnover with peroxide, as this cosubstrate contributes the 2 electron equivalents required for the reaction.
On the basis of their results, Bissaro et al. proposed several mechanisms for reactivity with peroxide (Figure 32) but favored one. In this proposal, a priming reduction step generates a Cu(I)–LPMO, which then binds substrate and reacts with H2O2 to homolytically cleave peroxide with protonation of the distal oxygen (Figure 32, top) to give off H2O and form a Cu(II)–oxyl (triplet). This would perform HAA on the substrate to give Cu(II)–OH and a substrate radical. Radical rebound of the hydroxyl with the substrate radical would produce the hydroxylated product and Cu(I). Note that if the Cu–oxyl triplet is further protonated, as mentioned above after HAA, it is likely not capable of rebound hydroxylation. Alternatively, binding of hydrogen peroxide to Cu(I) could be followed by protonation of the proximal oxygen (Figure 32, bottom), generating a hydroxyl radical and Cu(II)–hydroxide. On the basis of the observed selectivity, the hydroxyl radical would therefore have to be well positioned to rapidly abstract the correct H atom from the substrate to yield H2O. The Cu(II)–hydroxide formed with proximal oxygen protonation would then rebound to the carbon radical to hydroxylate the substrate and regenerate the Cu(I) state of LPMO. Importantly, in either pathway the reductant is an activator and is not required in the stoichiometry of the reaction.
Figure 32.

Proposed mechanisms for LPMO oxidation using H2O2 as the direct oxidant of polysaccharide substrate.
There are model systems where researchers report reaction of Cu(I) with H2O2. Model complex chemistry was reported using 6-[bis(phenylmethyl)amino]-N,N-bis(2-pyridinylmethyl)-2-pyridinemethanamine derivatives as ligands to Cu(I).273 Reaction of the Cu(I) complex with H2O2 formed a Cu(I)–OOH complex that can be protonated at either the proximal nonprotonated or the distal protonated O of the hydroperoxy moiety (Figure 33A). DFT calculations showed that distal protonation of the Cu(I)–OOH to yield a Cu(II)–oxyl species is thermodynamically more favorable than protonation at the proximal oxygen. However, there is a kinetic barrier (Figure 33B) associated with the intersystem crossing and spin state change for the conversion from the singlet Cu(I)–OOH to the triplet Cu(II)–oxyl. Although certain parallels can be drawn between these model complexes and the reactivity predicted for LPMOs, the calculated thermodynamic values and kinetic barriers would be dependent on the copper ligation and would be different for the LPMO active-site structure and ligation. Furthermore, hydroxyl radical generation would likely be less selective than the site-specific C1- or C4-hydroxylation observed for LPMOs; therefore, a mechanism invoking a hydroxyl radical has issues.
Figure 33.

(A) Reaction pathways proposed for Cu(I)–OOH showing both proximal and distal protonation possibilities. (B) 2-D potential energy surface of the S = 0 singlet Cu(I)–OOH and triplet S = 1 Cu(II)–O·− states. Reprinted with permission from ref 273. Copyright 2015 American Chemical Society.
Recently, Garcia-Bosch et al. reported reactivity data for Cu(I) complexes supported by tris(2-pyridylmethyl)amine or (2R,2′R)-1,1′-bis(2-pyridylmethyl)-2,2′-bipyrrolidine ligands which, when reacted with H2O2 and cyclohexane, gave the hydroperoxy product via a radical species in a Fenton-like reaction.269 In parallel, Concia et al. showed that tridentate complexes with both N-[(1-methyl-1H-imidazol-2-yl)-methylene]-2-pyridineethanamine (LAM) and N-[(1-methyl-1H-imidazol-2-yl)methylene]-2-pyridineethanimine (LIM) produced stable Cu(II)–hydroperoxide complexes that react with a model substrate (p-nitrophenyl-β-D-glycopyranoside) of cellulose at room temperature.296 The role(s) of this intermediate, however, were not uncoupled from alternative scenarios involving copper-generated radicals. Regardless, these data provide insight into a potential mechanism for LPMO function where H2O2 is utilized as a cosubstrate.
The mechanisms at work in LPMOs to selectively oxidize substrates without the release of reactive oxygen species, which would not be selective in C1/C4 hydroxylation, are presently the subject of intense study. Recent data suggest a role for hydrogen peroxide as cosubstrate, but the dependence of LPMO turnover on H2O2 has not been established. Furthermore, it may yet be determined that there is a pathway for LPMOs to use molecular oxygen directly without generation of potentially harmful oxidants such as H2O2. The possible mechanisms outlined above all rely on well-timed delivery of electrons and protons to specific Cu–oxygen species which could be influenced by the presence of a bound substrate limiting axial access.
5.4. Electron Transfer
A component in understanding the reaction mechanism(s) of LPMOs depends on identifying the source(s) of electrons required for catalysis. Many questions remain unanswered, but progress continues on understanding the thermodynamic and kinetic parameters of suitable electron donors and possible interactions with the LPMO protein during turnover.
The standard reduction potentials reported for AA9s at pH 6 are ~150–330 mV vs the standard hydrogen electrode (SHE),92,166,182 and potentials of AA10s have been reported in the range ~220–370 mV vs SHE.124,196,198 To date, no reduction potentials have been reported for AA11 or AA13 enzymes, though they are likely to be within the same range. In order to facilitate LPMO catalysis, an electron donor must have a potential that is close to or below that of the LPMO copper active site (≤200–300 mV). The inability of high potential electron donors to yield oxidized products has been demonstrated experimentally using a range of small molecule electron donors.4,5
Cellobiose dehydrogenase (CDH)93,99,297 is an extracellular flavocytochrome enzyme that is capable of storing two electrons and donating them to the LPMO active site (see section 4.5). The CDH consists of a flavin adenine dinucleotide (FAD)-binding domain, a cytochrome b domain, and some CDHs have a CBM domain. The FAD-binding domain is rapidly reduced by cellobiose (the disaccharide of two glucose units bound by β(1–4) glycosidic bonds found in cellulosic biomass). The FAD domain shuttles the electron to the cytochrome domain, which then reduces the LPMO copper center.145,298 In the absence of LPMOs, the cellobiose reduction rate for a CDH with a CBM was 42.7 s−1 and without a CBM was 1.68 s−1. Deletion of the CDH-encoding gene in LPMO-expressing fungi results in reduced cellulose activity that is partially restored upon reintroduction of a CDH to the deletion variants.93 Cyclic voltammetry4 and rapid kinetic measurements show that the cytochrome domain of the CDH is responsible for ET to the LPMO. Furthermore, reduction potentials (vs SHE) at pH 6 for the FAD domain are ~0 mV, and those of the cytochrome heme b cofactors are reported in the range from 99 to 163 mV, which are both well below those reported for Cu–LPMOs and appropriate for ET from the flavin to the cytochrome b.299
In the absence of substrate, the reduction rates of LPMO active sites by cytochrome domains of CDHs are comparable to CDH reduction by cellobiose, with rate constants reported in the range of 0.9–20.6 s−1 at pH 6, with others as high as 67.2 s−1 at pH 5.4 These ET rates show that CDHs are kinetically competent electron donors for LPMO catalysis given that turnover numbers for LPMOs are on the order of ~0.1 s−1. It is worth noting here that the reaction of the CDH with O2 was reported to be 105-fold slower than the rates reported for ET from the cytochrome domain to Cu–LPMO. The fact that the CDH is more efficient at reducing LPMOs argues against earlier proposals that CDHs contribute to cellulose degradation by promoting H2O2 formation upon reaction with O2, leading to uncontrolled Fenton chemistry.295 This, however, would not preclude the possibility that CDHs produce low levels of H2O2 upon reaction with O2 that would then function as the cosubstrate for LPMOs (vide supra).
The interactions between CDH and LPMOs have been investigated by NMR studies249 as well as site-directed mutagenesis and docking simulations (see section 4.5) leading to reports that the CDH interacts directly with the copper site of the LPMO via a patch of surface residues that include His-1, Ala-80, His-83, and Tyr-204. These are residues on the substrate-binding surface of LPMOs that have also been shown to interact directly with cellulosic substrates (Figure 34; NcLPMO9C numbering). While the evidence reported to date indicates that the CDH alone is capable of supplying the electron equivalents required for catalysis via access to the active-site surface, this interaction is likely not possible during the hydroxylation reaction, as the active-site surface will be blocked by substrate binding. In an effort to circumvent this complication of the O2 activation mechanism that requires two electrons, some have proposed7 that a nearby tyrosine or tryptophan residue is oxidized during turnover, perhaps by a copper–hydroperoxo species, and provides the second electron for the reaction. To complete the cycle, the CDH would donate two electrons: one to reduce Cu(II) to Cu(I) and another to quench the tyrosine or tryptophan radical.
Figure 34.
Crystal structure of NcLPMO9C showing surface residues common to both CDH and substrate binding. Side chains of residues His-1, His-64, Ala-80, His-83, and His-155 are shown as sticks (PDB ID 4D7U).
An alternative model exploits the aromatic residues in the vicinity of the copper site that form a through-protein ET pathway connecting the active site of the LPMO to a potential CDH-binding site on a solvent-exposed surface of the LPMO.107,192 Figure 35 shows an AA9 LPMO crystal structure (PDB ID 2YET) with the copper active site separated by 16.8 Å from a proposed conserved CDH-binding patch (magenta; Pro-221, Gly-222, Pro-223). Several conserved residues (cyan; Trp-129, Tyr-218, His-164) have been proposed as a potential ET pathway linking the conserved histidine of the active-site H-bonding network to the CDH-binding site on the opposite side of the protein.
Figure 35.
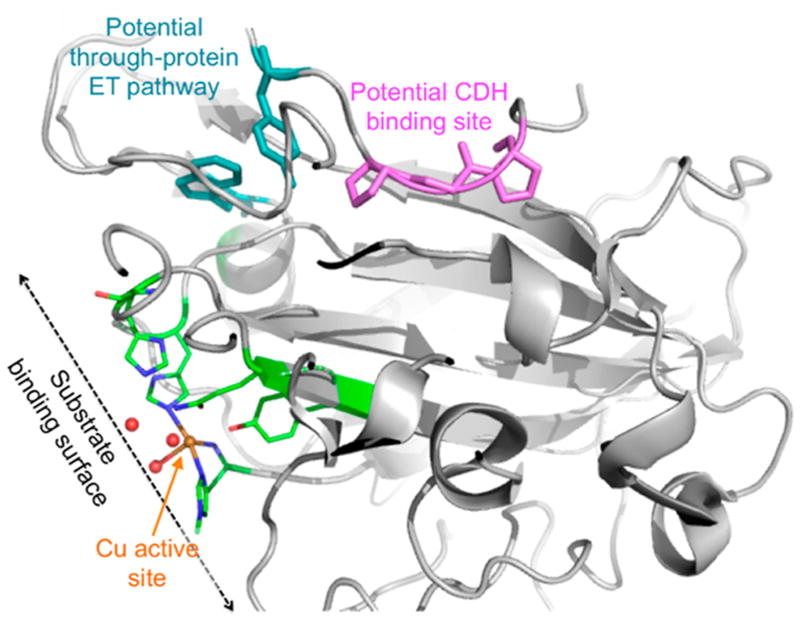
Cartoon representation of the T. aurentiacus LPMO9A (PDB ID 2YET) with the copper active site colored by atom type and shown in stick representation (green = C, red = O, blue = N, orange = Cu). Conserved patch of aromatic residues connecting the two sides of the structure are shown in stick representation (cyan). Proposed CDH-binding surface consisting of Pro-221 Gly-222 Pro-223 is shown in stick representation (magenta).
Since the potentials in the absence of substrate are known, the potential difference for ET between heme b (E° = 130 mV) and a typical AA9-LPMO (E° = 275 mV) is ~145 mV. If we assume the reorganization energy of Cu(II)/(I)–LPMO is comparable to that reported by Cowley et al. for the CuH site in the noncoupled binuclear copper enzymes PHM and DβM (1.33 eV) and that the reorganization energy for the heme b site in CDH is similar to the reorganization energy reported for heme c (1.2 eV), the total reorganization energy for the heme b CDH/Cu–LPMO ET can be estimated as 1.25 eV.300 Given the moderate thermodynamic driving force and significant reorganization energy, the fairly fast observed rate of 20.6 s−1 for the reduction of LPMO by the CDH cytochrome domain (at low protein concentrations) seems unlikely to be the result of a long-range ET (~17 Å) tunneling mechanism.
Although studies have focused on the involvement of the CDH in electron transfer, not all fungi-expressing LPMOs have genes to express CDH domains and bacteria-expressing LPMOs lack the CDH encoding gene. In some cases, researchers use small molecule reductants such as ascorbic acid.93 Other sources of reductants used include substituted phenols from lignin degradation or plant extractives, glucosemethanol-choline oxidoreductases that use plant-derived or fungal diphenols as redox mediators, and more recently lightactivated photosynthetic pigments.6,147,301 Of these reductants, the highest degree of activity and product formation was reported for those with standard reduction potentials close to or below those of the LPMOs (in the range from 75 to 260 mV vs SHE).4
An area of investigation is to what extent LPMO activity is a function of the potential of the electron donor used in catalysis. A study by Kracher et al. looked at a series of reductants and found that those with lower potentials could be correlated with higher LPMO turnover rates.4 A separate study utilizing three LPMOs from M. thermophila C1 reported oligosaccharide product yields using 34 different reductants, including mono-phenols, benzenediols and -triols, and sulfur-containing compounds.5 The fraction of those reductants that were capable of generating oxidized products differed among the three LPMOs as monitored by the amount of soluble oligosaccharides in the reaction mixture. These reactivity differences were correlated to differences in the surface charge distribution of the three enzymes. The homologue that showed significant activity with the largest number of reductants had the most positive charge on the active-site surface, and the homologue that reacted with the fewest number of reductants had the least positive charge near the active site. This correlation may be the result of more favorable interactions of negatively charged electron donors with the active-site surface. Alternatively, structural models of these three enzymes show that the active-site Cu of the most active enzyme is the most solvent exposed, and the least active enzyme is the least solvent exposed.
Taken together, these studies show a dependence of LPMO reactivity on the reduction potential differences between LPMOs and electron donors. While these studies are compelling, it is currently unclear whether in the presence of substrate these small molecule reductants would have direct access to the active site and be influenced by the surface charge of the protein near the active site or the accessibility of the Cu site or if they utilize a through-protein ET pathway as invoked for CDH turnover. Further studies are warranted to identify the nature and timing of the interaction between LPMOs and these electron donors.
5.5. Comparison to Other O2-Activating Enzymes: An Active Site Perspective
Comparison of the LPMO active site and reactivity to those of galactose oxidase (GalOx) and noncoupled binuclear copper enzymes, PHM, TβM, and DβM, highlights several key parallels and differences that are useful to note. First, the active site involved in cofactor biogenesis in GalOx utilizes a single reduced copper that is thought to react with O2 to yield a Cu(II)–superoxy intermediate.302,303 The copper–superoxo triplet then abstracts an H atom from a nearby cysteine residue to yield a radical that cross-links with a neighboring tyrosine residue. From here the resulting reduced Cu center can react with O2 to give a second H2O2 and generate the 2-electron-oxidized coupled Cu(II)–O(Tyr radical)--S(Cys) active site. Despite some similarities to proposed mechanisms for LPMOs, GalOx cofactor biogenesis is stoichiometric in generating the Cu(I)-bound cystinated tyrosinate and subsequent steps in the actual reaction of GalOx with galactose diverge from those of LPMOs. The copper-dependent enzymes PHM, TβM, and DβM, on the other hand, have two nonmagnetically coupled copper sites, CuH and CuM, separated by a solvent cleft spanning 11 Å.255,304 The O2 reaction in these enzymes takes place at the CuM site to likely form a superoxo level intermediate that can hydroxylate substrate. Despite the O2 activation and reaction step in the noncoupled binuclear enzymes being similar to those proposed for LPMOs in the O2 activation mechanism (Figure 29), the origin of the second electron required for catalysis is clearly defined as it comes from the nearby CuH site. In the case of LPMOs there is no second redox site (i.e., a Cys-Tyr or CuH) to donate electrons, and it is thus not clear where the second electron would come from or how it would be stored. Intriguingly, proponents of mechanisms invoking H2O2 as a cosubstrate to Cu(I)–LPMO point out that involvement of H2O2 would alleviate the need to store a second electron and would provide a clear picture of the electron source. H2O2 involvement will likely be the focus of future research into LPMO reactivity.
6. CONCLUDING COMMENTS
Natural polysaccharides provide carbon sources for organisms to support growth and can provide a source for renewable fuels and chemicals. The breadth of LPMO literature reflects the diverse structure and organization of natural polysaccharides with some organisms having evolved enzyme systems with multiple enzymatic activities for polysaccharide degradation. Early studies related to the conversion of polysaccharides to fermentable sugars focused on glycoside hydrolases. However, recent years have seen a surge in the number of LPMOs identified that are able to break the strong C–H bonds in the highly recalcitrant polysaccharide chains via an oxidative mechanism.
LPMOs form a large and diverse class of enzymes that encompasses a wide array of substrate specificities. Although most are small proteins with a single redox-active catalytic domain, some LPMOs also have small carbohydrate-binding domains. The genes encoding for these enzymes have been found in bacteria, fungi, plants, animals, and even viruses, with some organisms having a few dozen genes encoding LPMOs. These enzymes oxidize the C1 or C4 of the sugar units of polysaccharides, weakening the glycosidic bond. Although the LPMO nomenclature can be rather cumbersome, current convention sorts LPMOs into Auxiliary Activity Families AA9, AA10, AA11, and AA13 in the CAZY database. These LPMO families differ in terms of substrate and reaction specificity. A new family, AA14, was identified recently for which substrate specificities have been predicted but are not yet confirmed.
LPMOs have found applications in the degradation of lignocellulosic biomass and its conversion to fermentable sugars, often as part of a complex enzymatic cocktail. Despite protein-engineering efforts to understand the mechanism and improve the performance of LPMOs for lignocellulosic biomass conversion, the reaction mechanism is still unclear. What is known is that LPMO degradation of lignocellulosic biomass requires reducing equivalents as well as oxygen, which may place limitations or restrictions on the use of these enzymes in an industrial process. Future efforts in this area will benefit from tailoring both processes and enzymes for maximum efficiency of the lignocellulosic biomass conversion process.
Structure elucidations of LPMOs played a role in understanding this class of enzymes. Structural studies of LPMOs have, among other things, led to the elucidation of substrate specificities in this enzyme class and allowed for detailed snapshots of enzyme–substrate interactions. Several of the existing structures provide details of metal coordination that assists in interpretation of the spectroscopic measurements helping to understand the mechanism of oxygen reactivity in this class of enzyme.
A look at these small, simple, and powerful redox active proteins capable of the high-energy process of H atom abstraction at the molecular level provides key insight into polysaccharide degradation processes. The open, single copper reactive center of LPMOs provides opportunity for the study of oxygen activation mechanisms of copper without the complexity of interacting protein domains and additional metal centers. Spectroscopic measurements are key to developing active-site mechanistic insight on a molecular level. The variety of enzymes and small molecules that can donate electrons to LPMOs for oxidation of substrates allow flexibility in studying the reduction of the copper center. Unfortunately, small oligosaccharide binding is not common with LPMOs, limiting opportunities for spectroscopic definition of enzyme–substrate complexes.
Our groups have found working with these enzymes to be intellectually rewarding from both fundamental and applied sciences stances, and we look forward to the continued progress in elucidating the catalytic mechanism of LPMOs as well as their application in an industrial capacity.
Acknowledgments
This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under award number R01DK031450 (to E.I.S.) and a Ruth L. Kirschstein National Research Service Award from the National Institute of General Medical Sciences of the National Institutes of Health under award number F32GM116240 (to K.K.M.). This work was partly supported by the Swedish Energy Agency, through grant 40144-1 to M.S., and by the Faculty for Natural Resources and Agriculture, Swedish University of Agricultural Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. T.K., M.K., and B.K. are employees of DuPont Industrial Biosciences, a producer of enzymes for industrial use.
Biographies
Katlyn K. Meier is a Postdoctoral Research Fellow (with E.I. Solomon) in the Department of Chemistry at Stanford University and recipient of the NIH F32-Ruth L. Kirschstein National Research Service Award. She obtained her Ph.D. degree from Carnegie Mellon University (with E. Münck) in 2015 in Physical Bioinorganic Chemistry for research utilizing Mössbauer, EPR, and DFT methods to trap and characterize reactive intermediates in the reaction cycles of nonheme iron model complexes and enzyme systems. Her current research at Stanford University focuses on using a combination of spectroscopic techniques and computational methods to detect, trap, and characterize copper–oxygen intermediates in the catalytic cycles of enzyme systems with special emphasis on understanding how mononuclear copper sites are able to activate and react with O2.
Stephen M. Jones obtained his B.A. degree in Global Studies from UC Santa Barbara before obtaining his B.S. degree in Chemistry from UC Berkeley where he worked with Professor Heino Nitsche studying the interaction of radionuclides with ordered mesoporous carbon materials. He is currently a doctoral candidate advised by Edward I. Solomon at Stanford University. His research focuses on using an array of spectroscopic methods to elucidate the mechanisms of metalloenzymes that perform oxidase and oxygenase chemistry.
Thijs Kaper is a Senior Scientist at DuPont Industrial Biosciences, Palo Alto, CA. He obtained his Ph.D. degree from Wageningen University in 2001 in Life Sciences for research into β-glycosidases from hyperthermophilic Archaea. In 2007, he joined Genencor, A Danisco Division, in Palo Alto, CA, which was acquired by DuPont in 2011. His research activities have focused on enzyme engineering and the development of enzyme cocktails for degradation of lignocellulosic biomass.
Henrik Hansson is a Researcher at the Department of Molecular Sciences, Swedish University of Agricultural Sciences, Uppsala, Sweden. He obtained his Ph.D. degree from the Royal Institute of Technology, Stockholm, in 2002 for research in protein–protein interaction using NMR and other biophysical techniques. A postdoctoral period followed at Uppsala University, Uppsala, Sweden, working with first Dr. Gerard J. Kleywegt and then Dr. Mats Sandgren in the laboratory of Professor T. Alwyn Jones. In 2006, he went to the Swedish University of Agricultural Sciences and the Department of Molecular Biology, now the Department of Molecular Sciences, to use crystallography and structural biology to study fungal enzymes involved in the cellulose degradation.
Martijn Koetsier is a Scientist at DuPont Industrial Biosciences, Wageningen, The Netherlands. He obtained his Master’s degree in Chemistry from Groningen University in 2002 and was a group member there until he joined Dyadic Netherlands in 2010. At Dyadic his research activities have focused on the development of oxidative enzymes from Myceliophthora thermophile C1 as well as on enzyme cocktail development for the degradation of lignocellulosic biomass. He joined DuPont at the start of 2016 when DuPont acquired enzyme and technology assets from Dyadic International, Inc.
Saeid Karkehabadi earned his B.S. degree at Virginia Tech (Virginia Polytechnic Institute and State University) and his Master of Science degree at Uppsala University, Uppsala, Sweden. He received his Ph.D. deree from the Swedish University of Agricultural Sciences (SLU), Uppsala, in 2004 working with Professor Inger Anedrsson. Using protein X-ray crystallography, he carried out detailed studies into the structure–function relationship of ribulose-1,5-bisphosphate carboxylase/oxygenase (RUBISCO). He works currently at the Swedish University of Agricultural Sciences (SLU) Department of Molecular Sciences as a researcher and protein crystallographer.
Edward I. Solomon grew up in North Miami Beach, FL, received his Ph.D. degree at Princeton (with D. S. McClure), and was a postdoctoral fellow at The Ørsted Institute (with C. J. Ballhausen) and then at Caltech (with H. B. Gray). He was a professor at the Massachusetts Institute of Technology until 1982, when he joined the faculty at Stanford University, where he is now the Monroe E. Spaght Professor of Humanities and Sciences and Professor of Photon Science at the SLAC National Accelerator Lab. He has been an invited professor in Argentina, Australia, Brazil, China, France, India, and Japan. His research is in the fields of physical inorganic chemistry and bioinorganic chemistry with emphasis on the application of a wide range of spectroscopic methods combined with QM calculations to elucidate the electronic structure of transition-metal sites and its contribution to physical properties and reactivity. He has received a wide range of medals and awards and is a member of the National Academy of Sciences and the American Academy of Arts and Sciences and a fellow in the American Association for the Advancement of Science and the American Chemical Society
Mats Sandgren is an Associate Professor at the Department of Molecular Sciences, Swedish University of Agricultural Sciences, Uppsala, Sweden. He received his Ph.D. degree in Molecular Biology from Uppsala University in 2003 in the group of Alwyn Jones for research on the structure–function relationships in cellulases. After defending his Ph.D. degree he started to build his independent research group at Uppsala University. His research interests have since then been focused on understanding the structure–function relationships in carbohydrate-active enzymes, catalysis, and process development for carbohydrate and lignocellulose valorization. He and his research group moved to the Swedish University of Agricultural Sciences in 2007.
Bradley Kelemen is a Senior Scientist at DuPont in the Industrial Biosciences division at the Genencor Technology Center in Palo Alto, CA. He obtained his Ph.D. degree from the University of Wisconsin—Madison in 1999 studying the enzymology, structure, and function of ribonuclease A. He is an inventor on numerous patent applications and issued patents. He leads a team of biochemists who developed and launched enzyme products for lignocellulosic biomass hydrolysis.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Vermaas JV, Crowley MF, Beckham GT, Payne CM. Effects of Lytic Polysaccharide Monooxygenase Oxidation on Cellulose Structure and Binding of Oxidized Cellulose Oligomers to Cellulases. J Phys Chem B. 2015;119:6129–6143. doi: 10.1021/acs.jpcb.5b00778. [DOI] [PubMed] [Google Scholar]
- 2.Davies G, Henrissat B. Structures and Mechanisms of Glycosyl Hydrolases. Structure. 1995;3:853–859. doi: 10.1016/S0969-2126(01)00220-9. [DOI] [PubMed] [Google Scholar]
- 3.Vaaje-Kolstad G, Westereng B, Horn SJ, Liu Z, Zhai H, Sørlie M, Eijsink VGH. An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science. 2010;330:219–222. doi: 10.1126/science.1192231. [DOI] [PubMed] [Google Scholar]
- 4.Kracher D, Scheiblbrandner S, Felice AK, Breslmayr E, Preims M, Ludwicka K, Haltrich D, Eijsink VG, Ludwig R. Extracellular Electron Transfer Systems Fuel Cellulose Oxidative Degradation. Science. 2016;352:1098–1101. doi: 10.1126/science.aaf3165. [DOI] [PubMed] [Google Scholar]
- 5.Frommhagen M, Koetsier MJ, Westphal AH, Visser J, Hinz SW, Vincken JP, van Berkel WJ, Kabel MA, Gruppen H. Lytic Polysaccharide Monooxygenases from Myceliophthora Thermophila C1 Differ in Substrate Preference and Reducing Agent Specificity. Biotechnol Biofuels. 2016;9:186–203. doi: 10.1186/s13068-016-0594-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westereng B, Cannella D, Wittrup Agger J, Jørgensen H, Larsen Andersen M, Eijsink VG, Felby C. Enzymatic Cellulose Oxidation Is Linked to Lignin by Long-Range Electron Transfer. Sci Rep. 2016;5:18561–18570. doi: 10.1038/srep18561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beeson WT, Vu VV, Span EA, Phillips CM, Marletta MA. Cellulose Degradation by Polysaccharide Monooxygenases. Annu Rev Biochem. 2015;84:923–946. doi: 10.1146/annurev-biochem-060614-034439. [DOI] [PubMed] [Google Scholar]
- 8.Vu VV, Marletta MA. Starch-Degrading Polysaccharide Monooxygenases. Cell Mol Life Sci. 2016;73:2809–2819. doi: 10.1007/s00018-016-2251-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frandsen KE, Lo Leggio L. Lytic Polysaccharide Monooxygenases: A Crystallographer’s View on a New Class of Biomass-Degrading Enzymes. IUCrJ. 2016;3:448–467. doi: 10.1107/S2052252516014147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaaje-Kolstad G, Forsberg Z, Loose JS, Bissaro B, Eijsink VG. Structural Diversity of Lytic Polysaccharide Monooxygenases. Curr Opin Struct Biol. 2017;44:67–76. doi: 10.1016/j.sbi.2016.12.012. [DOI] [PubMed] [Google Scholar]
- 11.Johansen KS. Discovery and Industrial Applications of Lytic Polysaccharide Mono-Oxygenases. Biochem Soc Trans. 2016;44:143–149. doi: 10.1042/BST20150204. [DOI] [PubMed] [Google Scholar]
- 12.Johansen KS. Lytic Polysaccharide Monooxygenases: The Microbial Power Tool for Lignocellulose Degradation. Trends Plant Sci. 2016;21:926–936. doi: 10.1016/j.tplants.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 13. [accessed Jan 29, 2017];World Agricultural Supply and Demand Estimates. http://usda.mannlib.cornell.edu/MannUsda/viewDocumentInfo.do?documentID=1194.
- 14.Pfister B, Zeeman SC. Formation of Starch in Plant Cells. Cell Mol Life Sci. 2016;73:2781–2807. doi: 10.1007/s00018-016-2250-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolfenden R, Lu X, Young G. Spontaneous Hydrolysis of Glycosides. J Am Chem Soc. 1998;120:6814–6815. [Google Scholar]
- 16.Richmond TA, Somerville CR. The Cellulose Synthase Superfamily. Plant Physiol. 2000;124:495–498. doi: 10.1104/pp.124.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill JL, Jr, Hammudi MB, Tien M. The Arabidopsis Cellulose Synthase Complex: A Proposed Hexamer of Cesa Trimers in an Equimolar Stoichiometry. Plant Cell. 2014;26:4834–4842. doi: 10.1105/tpc.114.131193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hallac BB, Ragauskas AJ. Analyzing Cellulose Degree of Polymerization and Its Relevancy to Cellulosic Ethanol. Biofuels, Bioprod Biorefin. 2011;5:215–225. [Google Scholar]
- 19.Deguchi S, Tsujii K, Horikoshi K. Cooking Cellulose in Hot and Compressed Water. Chem Commun. 2006;0:3293–3295. doi: 10.1039/b605812d. [DOI] [PubMed] [Google Scholar]
- 20.Gomes TC, Skaf MS. Cellulose-Builder: A Toolkit for Building Crystalline Structures of Cellulose. J Comput Chem. 2012;33:1338–1346. doi: 10.1002/jcc.22959. [DOI] [PubMed] [Google Scholar]
- 21.The Pymol Molecular Graphics System, Version 1.8. Schrödinger, LLC; [Google Scholar]
- 22.Atalla RH, Vanderhart DL. Native Cellulose: A Composite of Two Distinct Crystalline Forms. Science. 1984;223:283–285. doi: 10.1126/science.223.4633.283. [DOI] [PubMed] [Google Scholar]
- 23.Stipanovic A, Sarko A. Packing Analysis of Carbohydrates and Polysaccharides. 6. Molecular and Crystal Structure of Regenerated Cellulose Ii. Macromolecules. 1976;9:851–857. [Google Scholar]
- 24.Kolpak FJ, Blackwell J. Determination of the Structure of Cellulose Ii. Macromolecules. 1976;9:273–278. doi: 10.1021/ma60050a019. [DOI] [PubMed] [Google Scholar]
- 25.Sarko A, Southwick J, Hayashi J. Packing Analysis of Carbohydrates and Polysaccharides. 7. Crystal Structure of Cellulose 111(1) and Its Relationship to Other Cellulose Polymorphs. Macromolecules. 1976;9:857–863. [Google Scholar]
- 26.Wada M, Chanzy H, Nishiyama Y, Langan P. Cellulose Iii(I) Crystal Structure and Hydrogen Bonding by Synchrotron X-Ray and Neutron Fiber Diffraction. Macromolecules. 2004;37:8548–8555. [Google Scholar]
- 27.Hutino K, Sakurada I. Uber Die Existenz Einer Vierten Modifikation Der Cellulose. Naturwissenschaften. 1940;28:577–578. [Google Scholar]
- 28.Wada M, Heux L, Sugiyama J. Polymorphism of Cellulose I Family: Reinvestigation of Cellulose Ivi. Biomacromolecules. 2004;5:1385–1391. doi: 10.1021/bm0345357. [DOI] [PubMed] [Google Scholar]
- 29.Wood TM. Preparation of Crystalline, Amorphous, and Dyed Cellulase Substrates. Methods Enzymol. 1988;160:19–25. [Google Scholar]
- 30.Ebringerova A. Structural Diversity and Application Potential of Hemicelluloses. Macromol Symp. 2006;232:1–12. [Google Scholar]
- 31.Ebringerova A, Heinze T. Xylan and Xylan Derivatives – Biopolymers with Valuable Properties, 1: Naturally Occurring Xylans Structures, Isolation Procedures and Properties. Macromol Rapid Commun. 2000;21:542–556. [Google Scholar]
- 32.de Vries RP, Visser J. Aspergillus Enzymes Involved in Degradation of Plant Cell Wall Polysaccharides. Microbiol Mol Biol Rev. 2001;65:497–522. doi: 10.1128/MMBR.65.4.497-522.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burton RA, Fincher GB. Current Challenges in Cell Wall Biology in the Cereals and Grasses. Front Plant Sci. 2012;3:130–136. doi: 10.3389/fpls.2012.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheller HV, Ulvskov P. Hemicelluloses. Annu Rev Plant Biol. 2010;61:263–289. doi: 10.1146/annurev-arplant-042809-112315. [DOI] [PubMed] [Google Scholar]
- 35.Burton RA, Fincher GB. Evolution and Development of Cell Walls in Cereal Grains. Front Plant Sci. 2014;5:456–471. doi: 10.3389/fpls.2014.00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burton RA, Gidley MJ, Fincher GB. Heterogeneity in the Chemistry, Structure and Function of Plant Cell Walls. Nat Chem Biol. 2010;6:724–732. doi: 10.1038/nchembio.439. [DOI] [PubMed] [Google Scholar]
- 37.Willats WG, McCartney L, Mackie W, Knox JP. Pectin: Cell Biology and Prospects for Functional Analysis. Plant Mol Biol. 2001;47:9–27. [PubMed] [Google Scholar]
- 38.Voragen AGJ, Coenen GJ, Verhoef RP, Schols HA. Pectin, a Versatile Polysaccharide Present in Plant Cell Walls. Struct Chem. 2009;20:263–275. [Google Scholar]
- 39.Lewis NG. A 20(Th) Century Roller Coaster Ride: A Short Account of Lignification. Curr Opin Plant Biol. 1999;2:153–162. doi: 10.1016/S1369-5266(99)80030-2. [DOI] [PubMed] [Google Scholar]
- 40.Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, Marita JM, Hatfield RD, Ralph SA, Christensen JH, et al. Lignins: Natural Polymers from Oxidative Coupling of 4-Hydroxyphenylpropanoids. Phytochem Rev. 2004;3:29–60. [Google Scholar]
- 41.Henrikson G. Lignin. In: Ek M, Gellerstedt G, Henrikson G, editors. Wood Chemistry and Wood Biotechnology. Walter de Gruyter GmbH & Co; Berlin: 2009. pp. 121–146. [Google Scholar]
- 42.Tolbert A, Akinosho H, Khunsupat R, Naskar A, Ragauskas AJ. Characterization and Analysis of the Molecular Weight of Lignin for Biorefining Studies. Biofuels, Bioprod Biorefin. 2014;8:836–856. [Google Scholar]
- 43.Dong X, Dong M, Lu Y, Turley A, Jin T, Wu C. Antimicrobial and Antioxidant Activities of Lignin from Residue of Corn Stover to Ethanol Production. Ind Crops Prod. 2011;34:1629–1634. [Google Scholar]
- 44.Ponomarenko J, Lauberts M, Dizhbite T, Lauberte L, Jurkjane V, Telysheva G. Antioxidant Activity of Various Lignins and Lignin-Related Phenylpropanoid Units with High and Low Molecular Weight. Holzforschung. 2015;69:795–805. [Google Scholar]
- 45.Crouvisier-Urion K, Bodart PR, Winckler P, Raya J, Gougeon RD, Cayot P, Domenek S, Debeaufort F, Karbowiak T. Biobased Composite Films from Chitosan and Lignin: Antioxidant Activity Related to Structure and Moisture. ACS Sustainable Chem Eng. 2016;4:6371–6381. [Google Scholar]
- 46.Loqué D, Scheller HV, Pauly M. Engineering of Plant Cell Walls for Enhanced Biofuel Production. Curr Opin Plant Biol. 2015;25:151–161. doi: 10.1016/j.pbi.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 47.Carpita NC. Update on Mechanisms of Plant Cell Wall Biosynthesis: How Plants Make Cellulose and Other (1->4)-Beta-D-Glycans. Plant Physiol. 2011;155:171–184. doi: 10.1104/pp.110.163360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keegstra K. Plant Cell Walls. Plant Physiol. 2010;154:483–486. doi: 10.1104/pp.110.161240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pattathil S, Avci U, Zhang T, Cardenas CL, Hahn MG. Immunological Approaches to Biomass Characterization and Utilization. Front Bioeng Biotechnol. 2015;3:173–187. doi: 10.3389/fbioe.2015.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fangel JU, Ulvskov P, Knox JP, Mikkelsen MD, Harholt J, Popper ZA, Willats WG. Cell Wall Evolution and Diversity. Front Plant Sci. 2012;3:152–160. doi: 10.3389/fpls.2012.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vogel J. Unique Aspects of the Grass Cell Wall. Curr Opin Plant Biol. 2008;11:301–307. doi: 10.1016/j.pbi.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 52.Tan Z, Liu S, Bliss N, Tieszen LL. Current and Potential Sustainable Corn Stover Feedstock for Biofuel Production in the United States. Biomass Bioenergy. 2012;47:372–386. [Google Scholar]
- 53.Karlen DL, Birell SJ, Hess JR. A Five-Year Assessment of Corn Stover Harvest in Central Iowa, USA. Soil Tillage Res. 2011;115–116:47–55. [Google Scholar]
- 54.Dias MOS, Cunha MP, Jesus CDF, Rocha GJM, Pradella JGC, Rossell CEV, Maciel Filho R, Bonomi A. Second Generation Ethanol in Brazil: Can It Compete with Electricity Production? Bioresour Technol. 2011;102:8964–8971. doi: 10.1016/j.biortech.2011.06.098. [DOI] [PubMed] [Google Scholar]
- 55.Dantas GA, Legey LFL, Mazzone A. Energy from Sugarcane Bagasse in Brazil: An Assessment of the Productivity and Cost of Different Technological Routes. Renewable Sustainable Energy Rev. 2013;21:356–364. [Google Scholar]
- 56.Alvira P, Tomas-Pejo E, Ballesteros M, Negro MJ. Pretreatment Technologies for an Efficient Bioethanol Production Process Based on Enzymatic Hydrolysis: A Review. Bioresour Technol. 2010;101:4851–4861. doi: 10.1016/j.biortech.2009.11.093. [DOI] [PubMed] [Google Scholar]
- 57.Galbe M, Zacchi G. Pretreatment of Lignocellulosic Materials for Efficient Bioethanol Production. Adv Biochem Eng /Biotechnol. 2007;108:41–65. doi: 10.1007/10_2007_070. [DOI] [PubMed] [Google Scholar]
- 58.Chandra RP, Bura R, Mabee WE, Berlin A, Pan X, Saddler JN. Substrate Pretreatment: The Key to Effective Enzymatic Hydrolysis of Lignocellulosics? Adv Biochem Eng /Biotechnol. 2007;108:67–93. doi: 10.1007/10_2007_064. [DOI] [PubMed] [Google Scholar]
- 59.Wyman CE, Dale BE, Elander RT, Holtzapple M, Ladisch MR, Lee YY. Coordinated Development of Leading Biomass Pretreatment Technologies. Bioresour Technol. 2005;96:1959–1966. doi: 10.1016/j.biortech.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 60.da Costa Sousa L, Jin M, Chundawat SPS, Bokade V, Tang X, Azarpira A, Lu F, Avci U, Humpula J, Uppugundla N, et al. Next-Generation Ammonia Pretreatment Enhances Cellulosic Biofuel Production. Energy Environ Sci. 2016;9:1215–1223. [Google Scholar]
- 61.Selig MJ, Viamajala S, Decker SR, Tucker MP, Himmel ME, Vinzant TB. Deposition of Lignin Droplets Produced During Dilute Acid Pretreatment of Maize Stems Retards Enzymatic Hydrolysis of Cellulose. Biotechnol Prog. 2007;23:1333–1339. doi: 10.1021/bp0702018. [DOI] [PubMed] [Google Scholar]
- 62.Merzendorfer H. Insect Chitin Synthases: A Review. J Comp Physiol, B. 2006;176:1–15. doi: 10.1007/s00360-005-0005-3. [DOI] [PubMed] [Google Scholar]
- 63.Sikorski P, Hori R, Wada M. Revisit of Alpha-Chitin Crystal Structure Using High Resolution X-Ray Diffraction Data. Biomacromolecules. 2009;10:1100–1105. doi: 10.1021/bm801251e. [DOI] [PubMed] [Google Scholar]
- 64.Kumar MN, Muzzarelli RA, Muzzarelli C, Sashiwa H, Domb AJ. Chitosan Chemistry and Pharmaceutical Perspectives. Chem Rev. 2004;104:6017–6084. doi: 10.1021/cr030441b. [DOI] [PubMed] [Google Scholar]
- 65. [accessed Jan 29, 2017];Global Aquaculture Production. http://www.fao.org/fishery/statistics/global-aquaculture-production/en.
- 66.Sharp RG. A Review of the Applications of Chitin and Its Derivatives in Agriculture to Modify Plant-Microbial Interactions and Improve Crop Yields. Agronomy. 2013;3:757–793. [Google Scholar]
- 67.Payne CM, Knott BC, Mayes HB, Hansson H, Himmel ME, Sandgren M, Sta hlberg J, Beckham GT. Fungal Cellulases. Chem Rev. 2015;115:1308–1448. doi: 10.1021/cr500351c. [DOI] [PubMed] [Google Scholar]
- 68.van der Maarel MJ, van der Veen B, Uitdehaag JC, Leemhuis H, Dijkhuizen L. Properties and Applications of Starch-Converting Enzymes of the Alpha-Amylase Family. J Biotechnol. 2002;94:137–155. doi: 10.1016/s0168-1656(01)00407-2. [DOI] [PubMed] [Google Scholar]
- 69.Dahiya N, Tewari R, Hoondal GS. Biotechnological Aspects of Chitinolytic Enzymes: A Review. Appl Microbiol Biotechnol. 2006;71:773–782. doi: 10.1007/s00253-005-0183-7. [DOI] [PubMed] [Google Scholar]
- 70.Henrissat B. Cellulases and Their Interaction with Cellulose. Cellulose. 1994;1:169–196. [Google Scholar]
- 71.Voshol GP, Vijgenboom E, Punt PJ. The Discovery of Novel Lpmo Families with a New Hidden Markov Model. BMC Res Notes. 2017;10:105–113. doi: 10.1186/s13104-017-2429-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berka RM, Grigoriev IV, Otillar R, Salamov A, Grimwood J, Reid I, Ishmael N, John T, Darmond C, Moisan MC, et al. Comparative Genomic Analysis of the Thermophilic Biomass-Degrading Fungi Myceliophthora Thermophila and Thielavia Terrestris. Nat Biotechnol. 2011;29:922–927. doi: 10.1038/nbt.1976. [DOI] [PubMed] [Google Scholar]
- 73.Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the Enzymatic Repertoire of the Cazy Database to Integrate Auxiliary Redox Enzymes. Biotechnol Biofuels. 2013;6:41–55. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Espagne E, Lespinet O, Malagnac F, Da Silva C, Jaillon O, Porcel BM, Couloux A, Aury JM, Segurens B, Poulain J, et al. The Genome Sequence of the Model Ascomycete Fungus Podospora Anserina. Genome Biol. 2008;9:R77–R99. doi: 10.1186/gb-2008-9-5-r77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Riley R, Salamov AA, Brown DW, Nagy LG, Floudas D, Held BW, Levasseur A, Lombard V, Morin E, Otillar R, et al. Extensive Sampling of Basidiomycete Genomes Demonstrates Inadequacy of the White-Rot/Brown-Rot Paradigm for Wood Decay Fungi. Proc Natl Acad Sci U S A. 2014;111:9923–9928. doi: 10.1073/pnas.1400592111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Book AJ, Yennamalli RM, Takasuka TE, Currie CR, Phillips GN, Jr, Fox BG. Evolution of Substrate Specificity in Bacterial Aa10 Lytic Polysaccharide Monooxygenases. Biotechnol Biofuels. 2014;7:109–123. doi: 10.1186/1754-6834-7-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bai Y, Eijsink VG, Kielak AM, van Veen JA, de Boer W. Genomic Comparison of Chitinolytic Enzyme Systems from Terrestrial and Aquatic Bacteria. Environ Microbiol. 2016;18:38–49. doi: 10.1111/1462-2920.12545. [DOI] [PubMed] [Google Scholar]
- 78.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. The Pfam Protein Families Database: Towards a More Sustainable Future. Nucleic Acids Res. 2016;44:D279–285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vaaje-Kolstad G, Horn SJ, van Aalten DM, Synstad B, Eijsink VG. The Non-Catalytic Chitin-Binding Protein Cbp21 from Serratia Marcescens Is Essential for Chitin Degradation. J Biol Chem. 2005;280:28492–28497. doi: 10.1074/jbc.M504468200. [DOI] [PubMed] [Google Scholar]
- 80.Chiu E, Hijnen M, Bunker RD, Boudes M, Rajendran C, Aizel K, Olieric V, Schulze-Briese C, Mitsuhashi W, Young V, et al. Structural Basis for the Enhancement of Virulence by Viral Spindles and Their in Vivo Crystallization. Proc Natl Acad Sci U S A. 2015;112:3973–3978. doi: 10.1073/pnas.1418798112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nekiunaite L, Arntzen MO, Svensson B, Vaaje-Kolstad G, Abou Hachem M. Lytic Polysaccharide Monooxygenases and Other Oxidative Enzymes Are Abundantly Secreted by Aspergillus Nidulans Grown on Different Starches. Biotechnol Biofuels. 2016;9:187–203. doi: 10.1186/s13068-016-0604-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Raguz S, Yaguea E, Wood DA, Thurston CF. Isolation and Characterization of a Cellulose-Growth-Specific Gene from Agaricus Bisporus. Gene. 1992;119:183–190. doi: 10.1016/0378-1119(92)90270-y. [DOI] [PubMed] [Google Scholar]
- 83.Armesilla AL, Thurston CF, Yague E. Cel1: A Novel Cellulose Binding Protein Secreted by Agaricus Bisporus During Growth on Crystalline Cellulose. FEMS Microbiol Lett. 1994;116:293–299. doi: 10.1111/j.1574-6968.1994.tb06718.x. [DOI] [PubMed] [Google Scholar]
- 84.Foreman PK, Brown D, Dankmeyer L, Dean R, Diener S, Dunn-Coleman NS, Goedegebuur F, Houfek TD, England GJ, Kelley AS, et al. Transcriptional Regulation of Biomass-Degrading Enzymes in the Filamentous Fungus Trichoderma Reesei. J Biol Chem. 2003;278:31988–31997. doi: 10.1074/jbc.M304750200. [DOI] [PubMed] [Google Scholar]
- 85.Chen S, Wilson DB. Proteomic and Transcriptomic Analysis of Extracellular Proteins and Mrna Levels in Thermobifida Fusca Grown on Cellobiose and Glucose. J Bacteriol. 2007;189:6260–6265. doi: 10.1128/JB.00584-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jagadeeswaran G, Gainey L, Prade R, Mort AJ. A Family of Aa9 Lytic Polysaccharide Monooxygenases in Aspergillus Nidulans Is Differentially Regulated by Multiple Substrates and at Least One Is Active on Cellulose and Xyloglucan. Appl Microbiol Biotechnol. 2016;100:4535–4547. doi: 10.1007/s00253-016-7505-9. [DOI] [PubMed] [Google Scholar]
- 87.Schnellmann J, Zeltins A, Blaak H, Schrempf H. The Novel Lectin-Like Protein Chb1 Is Encoded by a Chitin-Inducible Streptomyces Olivaceoviridis Gene and Binds Specifically to Crystalline A-Chitin of Fungi and Other Organisms. Mol Microbiol. 1994;13:807–819. doi: 10.1111/j.1365-2958.1994.tb00473.x. [DOI] [PubMed] [Google Scholar]
- 88.Schrempf H. Recognition and Degradation of Chitin by Streptomycetes. Antonie van Leeuwenhoek. 2001;79:285–289. doi: 10.1023/a:1012058205158. [DOI] [PubMed] [Google Scholar]
- 89.Watanabe T, Kimura K, Sumiya T, Nikaidou N, Suzuki K, Suzuki M, Taiyoji M, Ferrer S, Regue M. Genetic Analysis of the Chitinase System of Serratia Marcescens 2170. J Bacteriol. 1997;179:7111–7117. doi: 10.1128/jb.179.22.7111-7117.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reese ET, Siu RG, Levinson HS. The Biological Degradation of Soluble Cellulose Derivatives and Its Relationship to the Mechanism of Cellulose Hydrolysis. J Bacteriol. 1950;59:485–497. doi: 10.1128/jb.59.4.485-497.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reese ET. Enzymatic Degradation of Cellulose. Appl Microbiol. 1956;4:39–45. doi: 10.1128/am.4.1.39-45.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Quinlan RJ, Sweeney MD, Lo Leggio L, Otten H, Poulsen JCN, Johansen KS, Krogh KB, Jørgensen CI, Tovborg M, Anthonsen A, et al. Insights into the Oxidative Degradation of Cellulose by a Copper Metalloenzyme That Exploits Biomass Components. Proc Natl Acad Sci U S A. 2011;108:15079–15084. doi: 10.1073/pnas.1105776108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Phillips CM, Beeson WT, IV, Cate JH, Marletta MA. Cellobiose Dehydrogenase and a Copper-Dependent Polysaccharide Monooxygenase Potentiate Cellulose Degradation by Neurospora Crassa. ACS Chem Biol. 2011;6:1399–1406. doi: 10.1021/cb200351y. [DOI] [PubMed] [Google Scholar]
- 94.Jermyn MA. Fungal Cellulases I: General Propeerties of Unpurified Enzyme Preparations from Aspergillus Oryzae. Aust J Biol Sci. 1952;5:409–432. [Google Scholar]
- 95.Harris PV, Welner D, McFarland KC, Re E, Navarro Poulsen JC, Brown K, Salbo R, Ding H, Vlasenko E, Merino S, et al. Stimulation of Lignocellulosic Biomass Hydrolysis by Proteins of Glycoside Hydrolase Family 61: Structure and Function of a Large, Enigmatic Family. Biochemistry. 2010;49:3305–3316. doi: 10.1021/bi100009p. [DOI] [PubMed] [Google Scholar]
- 96.Sampathnarayanan A, Shanmugasundaram ERB. Studies on Cellulase of the Cotton Wilt Pathogen Fusarium Vasinfectum Atk. Mycopathologia. 1970;41:223–232. doi: 10.1007/BF02051100. [DOI] [PubMed] [Google Scholar]
- 97.Müller G, Várnai A, Johansen KS, Eijsink VGH, Horn SJ. Harnessing the Potential of Lpmo-Containing Cellulase Cocktails Poses New Demands on Processing Conditions. Biotechnol Biofuels. 2015;8:187–196. doi: 10.1186/s13068-015-0376-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Eriksson KE, Pettersson B, Westermark U. Oxidation: An Important Enzyme Reaction in Fungal Degradation of Cellulose. FEBS Lett. 1974;49:282–285. doi: 10.1016/0014-5793(74)80531-4. [DOI] [PubMed] [Google Scholar]
- 99.Kittl R, Kracher D, Burgstaller D, Haltrich D, Ludwig R. Production of Four Neurospora Crassa Lytic Polysaccharide Monooxygenases in Pichia Pastoris Monitored by a Fluorimetric Assay. Biotechnol Biofuels. 2012;5:79–92. doi: 10.1186/1754-6834-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bennati-Granier C, Garajova S, Champion C, Grisel S, Haon M, Zhou S, Fanuel M, Ropartz D, Rogniaux H, Gimbert I, et al. Substrate Specificity and Regioselectivity of Fungal Aa9 Lytic Polysaccharide Monooxygenases Secreted by Podospora Anserina. Biotechnol Biofuels. 2015;8:90–104. doi: 10.1186/s13068-015-0274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bissaro B, Rohr AK, Muller G, Chylenski P, Skaugen M, Forsberg Z, Horn SJ, Vaaje-Kolstad G, Eijsink VGH. Oxidative Cleavage of Polysaccharides by Monocopper Enzymes Depends on H2o2. Nat Chem Biol. 2017;13:1123–1128. doi: 10.1038/nchembio.2470. [DOI] [PubMed] [Google Scholar]
- 102.Arantes V, Jellison J, Goodell B. Peculiarities of Brown-Rot Fungi and Biochemical Fenton Reaction with Regard to Their Potential as a Model for Bioprocessing Biomass. Appl Microbiol Biotechnol. 2012;94:323–338. doi: 10.1007/s00253-012-3954-y. [DOI] [PubMed] [Google Scholar]
- 103.Henrissat B. A Classification of Glycosyl Hydrolases Based on Amino Acid Sequence Similarities. Biochem J. 1991;280:309–316. doi: 10.1042/bj2800309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The Carbohydrate-Active Enzymes Database (Cazy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lo Leggio L, Simmons TJ, Poulsen JC, Frandsen KE, Hemsworth GR, Stringer MA, von Freiesleben P, Tovborg M, Johansen KS, De Maria L, et al. Structure and Boosting Activity of a Starch-Degrading Lytic Polysaccharide Monooxygenase. Nat Commun. 2015;6:5961–5970. doi: 10.1038/ncomms6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Karkehabadi S, Hansson H, Kim S, Piens K, Mitchinson C, Sandgren M. The First Structure of a Glycoside Hydrolase Family 61 Member, Cel61b from Hypocrea Jecorina, at 1.6 a Resolution. J Mol Biol. 2008;383:144–154. doi: 10.1016/j.jmb.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 107.Hemsworth GR, Henrissat B, Davies GJ, Walton PH. Discovery and Characterization of a New Family of Lytic Polysaccharide Monooxygenases. Nat Chem Biol. 2014;10:122–126. doi: 10.1038/nchembio.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. [accessed Nov 22, 2011];Pfam Database Cbm33/Lpmo_10 (Pf03067) http://pfam.xfam.org/family/PF03067.
- 109.Moses V, Hatherley R, Tastan Bishop O. Bioinformatic Characterization of Type-Specific Sequence and Structural Features in Auxiliary Activity Family 9 Proteins. Biotechnol Biofuels. 2016;9:239–256. doi: 10.1186/s13068-016-0655-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vu VV, Beeson WT, Phillips CM, Cate JH, Marletta MA. Determinants of Regioselective Hydroxylation in the Fungal Polysaccharide Monooxygenases. J Am Chem Soc. 2014;136:562–565. doi: 10.1021/ja409384b. [DOI] [PubMed] [Google Scholar]
- 111.Busk PK, Lange L. Classification of Fungal and Bacterial Lytic Polysaccharide Monooxygenases. BMC Genomics. 2015;16:368–381. doi: 10.1186/s12864-015-1601-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koseki T, Mese Y, Fushinobu S, Masaki K, Fujii T, Ito K, Shiono Y, Murayama T, Iefuji H. Biochemical Characterization of a Glycoside Hydrolase Family 61 Endoglucanase from Aspergillus Kawachii. Appl Microbiol Biotechnol. 2008;77:1279–1285. doi: 10.1007/s00253-007-1274-4. [DOI] [PubMed] [Google Scholar]
- 113.Karlsson J, Saloheimo M, Siika-Aho M, Tenkanen M, Penttila M, Tjerneld F. Homologous Expression and Characterization of Cel61a (Eg Iv) of Trichoderma Reesei. Eur J Biochem. 2001;268:6498–6507. doi: 10.1046/j.0014-2956.2001.02605.x. [DOI] [PubMed] [Google Scholar]
- 114.Bauer S, Vasu P, Persson S, Mort AJ, Somerville CR. Development and Application of a Suite of Polysaccharide-Degrading Enzymes for Analyzing Plant Cell Walls. Proc Natl Acad Sci U S A. 2006;103:11417–11422. doi: 10.1073/pnas.0604632103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Saloheimo M, Nakari-Setala T, Tenkanen M, Penttila M. Cdna Cloning of a Trichoderma Reesei Cellulase and Demonstration of Endoglucanase Activity by Expression in Yeast. Eur J Biochem. 1997;249:584–591. doi: 10.1111/j.1432-1033.1997.00584.x. [DOI] [PubMed] [Google Scholar]
- 116.Agger JW, Isaksen T, Várnai A, Vidal-Melgosa S, Willats WG, Ludwig R, Horn SJ, Eijsink VG, Westereng B. Discovery of Lpmo Activity on Hemicelluloses Shows the Importance of Oxidative Processes in Plant Cell Wall Degradation. Proc Natl Acad Sci U S A. 2014;111:6287–6292. doi: 10.1073/pnas.1323629111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kojima Y, Varnai A, Ishida T, Sunagawa N, Petrovic DM, Igarashi K, Jellison J, Goodell B, Alfredsen G, Westereng B, et al. A Lytic Polysaccharide Monooxygenase with Broad Xyloglucan Specificity from the Brown-Rot Fungus Gloeophyllum Trabeum and Its Action on Cellulose-Xyloglucan Complexes. Appl Environ Microbiol. 2016;82:6557–6572. doi: 10.1128/AEM.01768-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Frommhagen M, Sforza S, Westphal AH, Visser J, Hinz SW, Koetsier MJ, van Berkel WJ, Gruppen H, Kabel MA. Discovery of the Combined Oxidative Cleavage of Plant Xylan and Cellulose by a New Fungal Polysaccharide Monooxygenase. Biotechnol Biofuels. 2015;8:101–113. doi: 10.1186/s13068-015-0284-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Isaksen T, Westereng B, Aachmann FL, Agger JW, Kracher D, Kittl R, Ludwig R, Haltrich D, Eijsink VG, Horn SJ. A C4-Oxidizing Lytic Polysaccharide Monooxygenase Cleaving Both Cellulose and Cello-Oligosaccharides. J Biol Chem. 2014;289:2632–2642. doi: 10.1074/jbc.M113.530196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vaaje-Kolstad G, Bohle LA, Gaseidnes S, Dalhus B, Bjoras M, Mathiesen G, Eijsink VG. Characterization of the Chitinolytic Machinery of Enterococcus Faecalis V583 and High-Resolution Structure of Its Oxidative Cbm33 Enzyme. J Mol Biol. 2012;416:239–254. doi: 10.1016/j.jmb.2011.12.033. [DOI] [PubMed] [Google Scholar]
- 121.Forsberg Z, Nelson CE, Dalhus B, Mekasha S, Loose JS, Crouch LI, Rohr AK, Gardner JG, Eijsink VG, Vaaje-Kolstad G. Structural and Functional Analysis of a Lytic Polysaccharide Monooxygenase Important for Efficient Utilization of Chitin in Cellvibrio Japonicus. J Biol Chem. 2016;291:7300–7312. doi: 10.1074/jbc.M115.700161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kolbe S, Fischer S, Becirevic A, Hinz P, Schrempf H. The Streptomyces Reticuli Alpha-Chitin-Binding Protein Chb2 and Its Gene. Microbiology. 1998;144:1291–1297. doi: 10.1099/00221287-144-5-1291. [DOI] [PubMed] [Google Scholar]
- 123.Mekasha S, Forsberg Z, Dalhus B, Bacik JP, Choudhary S, Schmidt-Dannert C, Vaaje-Kolstad G, Eijsink VG. Structural and Functional Characterization of a Small Chitin-Active Lytic Polysaccharide Monooxygenase Domain of a Multi-Modular Chitinase from Jonesia Denitrificans. FEBS Lett. 2016;590:34–42. doi: 10.1002/1873-3468.12025. [DOI] [PubMed] [Google Scholar]
- 124.Forsberg Z, Mackenzie AK, Sorlie M, Rohr AK, Helland R, Arvai AS, Vaaje-Kolstad G, Eijsink VG. Structural and Functional Characterization of a Conserved Pair of Bacterial Cellulose-Oxidizing Lytic Polysaccharide Monooxygenases. Proc Natl Acad Sci U S A. 2014;111:8446–8451. doi: 10.1073/pnas.1402771111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Forsberg Z, Røhr ÅK, Mekasha S, Andersson KK, Eijsink VG, Vaaje-Kolstad G. Comparative Study of Two Chitin-Active and Two Cellulose-Active Aa10-Type Lytic Polysaccharide Monooxygenases. Biochemistry. 2014;53:1647–1656. doi: 10.1021/bi5000433. [DOI] [PubMed] [Google Scholar]
- 126.Horn SJ, Vaaje-Kolstad G, Westereng B, Eijsink VGH. Novel Enzymes for the Degradation of Cellulose. Biotechnol Biofuels. 2012;5:45–57. doi: 10.1186/1754-6834-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vu VV, Beeson WT, Span EA, Farquhar ER, Marletta MA. A Family of Starch-Active Polysaccharide Monooxygenases. Proc Natl Acad Sci U S A. 2014;111:13822–13827. doi: 10.1073/pnas.1408090111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Beeson WT, Phillips CM, Cate JH, Marletta MA. Oxidative Cleavage of Cellulose by Fungal Copper-Dependent Polysaccharide Monooxygenases. J Am Chem Soc. 2012;134:890–892. doi: 10.1021/ja210657t. [DOI] [PubMed] [Google Scholar]
- 129.Phillips CM, Iavarone AT, Marletta MA. Quantitative Proteomic Approach for Cellulose Degradation by Neurospora Crassa. J Proteome Res. 2011;10:4177–4185. doi: 10.1021/pr200329b. [DOI] [PubMed] [Google Scholar]
- 130.Bey M, Zhou S, Poidevin L, Henrissat B, Coutinho PM, Berrin JG, Sigoillot JC. Cello-Oligosaccharide Oxidation Reveals Differences between Two Lytic Polysaccharide Monooxygenases (Family Gh61) from Podospora Anserina. Appl Environ Microbiol. 2013;79:488–496. doi: 10.1128/AEM.02942-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Takamine J. Process of Making Diastatic Enzyme. 525,823. US Patent. 1894 Sep 11;
- 132.Oshikawa Y. Process for the Production of Seasonings from Mushrooms with Cellulase Produced by Microorganisms and the Resulting Product. 3,150,983. US Patent. 1964 Sep 29;
- 133.Toyama N. Method of Removing Seed Coats of Soybeans with Cellulase Produced by Microorganisms. 3,160,569. US Patent. 1964 Dec 8;
- 134.Kirk O, Borchert TV, Fuglsang CC. Industrial Enzyme Applications. Curr Opin Biotechnol. 2002;13:345–351. doi: 10.1016/s0958-1669(02)00328-2. [DOI] [PubMed] [Google Scholar]
- 135.Bhat MK. Cellulases and Related Enzymes in Biotechnology. Biotechnol Adv. 2000;18:355–383. doi: 10.1016/s0734-9750(00)00041-0. [DOI] [PubMed] [Google Scholar]
- 136.Polizeli ML, Rizzatti AC, Monti R, Terenzi HF, Jorge JA, Amorim DS. Xylanases from Fungi: Properties and Industrial Applications. Appl Microbiol Biotechnol. 2005;67:577–591. doi: 10.1007/s00253-005-1904-7. [DOI] [PubMed] [Google Scholar]
- 137.Allen F, Andreotti R, Eveleigh DE, Nystrom J. Mary Elizabeth Hickox Mandels, 90, Bioenergy Leader. Biotechnol Biofuels. 2009;2:22–28. doi: 10.1186/1754-6834-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Seiboth B, Ivanova C, Seidl-Seiboth V. Trichoderma Reesei: A Fungal Enzyme Producer for Cellulosic Biofuels. In: Dos Santos Bernardes MA, editor. Biofuel Production-Recent Developments and Prospects. InTech; London: 2011. [Google Scholar]
- 139.Coutts AD, Smith RE. Factors Influencing the Production of Cellulases by Sporotrichum Thermophile. Appl Environ Microbiol. 1976;31:819–825. doi: 10.1128/aem.31.6.819-825.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hinz SWA, Pouvreau L, Joosten R, Bartels J, Jonathan MC, Wery J, Schols HA. Hemicellulase Production in Chrysosporium Lucknowense C1. J Cereal Sci. 2009;50:318–323. [Google Scholar]
- 141.Visser H, Joosten V, Punt PJ, Gusakov AV, Olson PT, Joosten R, Bartels J, Visser J, Sinitsyn AP, Emalfarb MA, et al. Development of a Mature Fungal Technology and Production Platform for Industrial Enzymes Based on a Myceliophthora Thermophila Isolate, Previously Known as Chrysosporium Lucknowense C1. Ind Biotechnol. 2011;7:214–223. [Google Scholar]
- 142.Miehe H. Die Selbsterhitzung Des Heus: Eine Biologische Studie. Verlag von Gustav Fischer; Jena, Germany: 1907. [Google Scholar]
- 143.Schuerg T, Gabriel R, Baecker N, Baker SE, Singer SW. Thermoascus Aurantiacus Is an Intriguing Host for the Industrial Production of Cellulases. Curr Biotechnol. 2017;6:89–97. [Google Scholar]
- 144.Tansey MR. Isolation of Thermophilic Fungi from Self-Heated, Industrial Wood Chip Piles. Mycologia. 1971;63:537–547. [Google Scholar]
- 145.Tan TC, Kracher D, Gandini R, Sygmund C, Kittl R, Haltrich D. Structural Basis for Cellobiose Dehydrogenase Action During Oxidative Cellulose Degradation. Nat Commun. 2015;6:7542–7553. doi: 10.1038/ncomms8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Takeda K, Matsumura H, Ishida T, Samejima M, Ohno H, Yoshida M, Igarashi K, Nakamura N. Characterization of a Novel Pqq-Dependent Quinohemoprotein Pyranose Dehydrogenase from Coprinopsis Cinerea Classified into Auxiliary Activities Family 12 in Carbohydrate-Active Enzymes. PLoS One. 2015;10:e0115722–e0115738. doi: 10.1371/journal.pone.0115722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cannella D, Mollers KB, Frigaard NU, Jensen PE, Bjerrum MJ, Johansen KS, Felby C. Light-Driven Oxidation of Polysaccharides by Photosynthetic Pigments and a Metalloenzyme. Nat Commun. 2016;7:11134–11142. doi: 10.1038/ncomms11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Frommhagen M, Mutte SK, Westphal AH, Koetsier MJ, Hinz SWA, Visser J, Vincken JP, Weijers D, van Berkel WJH, Gruppen H, et al. Boosting Lpmo-Driven Lignocellulose Degradation by Polyphenol Oxidase-Activated Lignin Building Blocks. Biotechnol Biofuels. 2017;10:121–137. doi: 10.1186/s13068-017-0810-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Forsberg Z, Vaaje-Kolstad G, Westereng B, Bunæs AC, Stenstrøm Y, MacKenzie A, Sørlie M, Horn SJ, Eijsink VG. Cleavage of Cellulose by a Cbm33 Protein. Protein Sci. 2011;20:1479–1483. doi: 10.1002/pro.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dimarogona M, Topakas E, Olsson L, Christakopoulos P. Lignin Boosts the Cellulase Performance of a Gh-61 Enzyme from Sporotrichum Thermophile. Bioresour Technol. 2012;110:480–487. doi: 10.1016/j.biortech.2012.01.116. [DOI] [PubMed] [Google Scholar]
- 151.Rodriguez-Zuniga UF, Cannella D, Giordano RC, Giordano RLC, Jorgensen H, Felby C. Lignocellulose Pretreatment Technologies Affect the Level of Enzymatic Cellulose Oxidation by Lpmo. Green Chem. 2015;17:2896–2903. [Google Scholar]
- 152.Hu J, Arantes V, Pribowo A, Gourlay K, Saddler JN. Substrate Factors That Influence the Synergistic Interaction of Aa9 and Cellulases During the Enzymatic Hydrolysis of Biomass. Energy Environ Sci. 2014;7:2308–2315. [Google Scholar]
- 153.Bao W, Renganathan V. Cellobiose Oxidase of Phanerochaete Chrysosporium Enhances Crystalline Cellulose Degradation by Cellulases. FEBS Lett. 1992;302:77–80. doi: 10.1016/0014-5793(92)80289-s. [DOI] [PubMed] [Google Scholar]
- 154.Henriksson G, Ander P, Pettersson B, Pettersson G. Cellobiose Dehydrogenase (Cellobiose Oxidase) from Phanerochaete Chrysosporium as a Wood-Degrading Enzyme. Studies on Cellulose, Xylan and Synthetic Lignin. Appl Microbiol Biotechnol. 1995;42:790–796. [Google Scholar]
- 155.Zamocky M, Ludwig R, Peterbauer C, Hallberg BM, Divne C, Nicholls P, Haltrich D. Cellobiose Dehydrogenase–a Flavocytochrome from Wood-Degrading, Phytopathogenic and Saprotropic Fungi. Curr Protein Pept Sci. 2006;7:255–280. doi: 10.2174/138920306777452367. [DOI] [PubMed] [Google Scholar]
- 156.Harreither W, Sygmund C, Augustin M, Narciso M, Rabinovich ML, Gorton L, Haltrich D, Ludwig R. Catalytic Properties and Classification of Cellobiose Dehydrogenases from Ascomycetes. Appl Environ Microbiol. 2011;77:1804–1815. doi: 10.1128/AEM.02052-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Langston JA, Shaghasi T, Abbate E, Xu F, Vlasenko E, Sweeney MD. Oxidoreductive Cellulose Depolymerization by the Enzymes Cellobiose Dehydrogenase and Glycoside Hydrolase 61. Appl Environ Microbiol. 2011;77:7007–7015. doi: 10.1128/AEM.05815-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Loose JSM, Forsberg Z, Kracher D, Scheiblbrandner S, Ludwig R, Eijsink VGH, Vaaje-Kolstad G. Activation of Bacterial Lytic Polysaccharide Monooxygenases with Cellobiose Dehydrogenase. Protein Sci. 2016;25:2175–2186. doi: 10.1002/pro.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Saibi W, Gargouri A. Cellobiose Dehydrogenase Influences the Production of S. Microspora Beta-Glucosidase. World J Microbiol Biotechnol. 2012;28:23–29. doi: 10.1007/s11274-011-0787-2. [DOI] [PubMed] [Google Scholar]
- 160.Holtzapple M, Cognata M, Shu Y, Hendrickson C. Inhibition of Trichoderma Reesei Cellulase by Sugars and Solvents. Biotechnol Bioeng. 1990;36:275–287. doi: 10.1002/bit.260360310. [DOI] [PubMed] [Google Scholar]
- 161.Beeson W, Iavarone AT, Hausmann CD, Cate JH, Marletta MA. Extracellular Aldonolactonase from Myceliophthora Thermophila. Appl Environ Microbiol. 2011;77:650–656. doi: 10.1128/AEM.01922-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Peng S, Cao Q, Qin Y, Li X, Liu G, Qu Y. An Aldonolactonase Alta from Penicillium Oxalicum Mitigates the Inhibition of Beta-Glucosidase During Lignocellulose Biodegradation. Appl Microbiol Biotechnol. 2017;101:3627–3636. doi: 10.1007/s00253-017-8134-7. [DOI] [PubMed] [Google Scholar]
- 163.Turbe-Doan A, Arfi Y, Record E, Estrada-Alvarado I, Levasseur A. Heterologous Production of Cellobiose Dehydrogenases from the Basidiomycete Coprinopsis Cinerea and the Ascomycete Podospora Anserina and Their Effect on Saccharification of Wheat Straw. Appl Microbiol Biotechnol. 2013;97:4873–4885. doi: 10.1007/s00253-012-4355-y. [DOI] [PubMed] [Google Scholar]
- 164.Matsumura H, Umezawa K, Takeda K, Sugimoto N, Ishida T, Samejima M, Ohno H, Yoshida M, Igarashi K, Nakamura N. Discovery of a Eukaryotic Pyrroloquinoline Quinone-Dependent Oxidoreductase Belonging to a New Auxiliary Activity Family in the Database of Carbohydrate-Active Enzymes. PLoS One. 2014;9:e104851–e104859. doi: 10.1371/journal.pone.0104851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yoshida M, Igarashi K, Wada M, Kaneko S, Suzuki N, Matsumura H, Nakamura N, Ohno H, Samejima M. Characterization of Carbohydrate-Binding Cytochrome B562 from the White-Rot Fungus Phanerochaete Chrysosporium. Appl Environ Microbiol. 2005;71:4548–4555. doi: 10.1128/AEM.71.8.4548-4555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Garajova S, Mathieu Y, Beccia MR, Bennati-Granier C, Biaso F, Fanuel M, Ropartz D, Guigliarelli B, Record E, Rogniaux H, et al. Single-Domain Flavoenzymes Trigger Lytic Polysaccharide Monooxygenases for Oxidative Degradation of Cellulose. Sci Rep. 2016;6:28276–28285. doi: 10.1038/srep28276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bissaro B, Røhr AsK, Skaugen M, Forsberg Z, Horn SJ, Vaaje-Kolstad G, Eijsink VGH. Fenton-type chemistry by a copper enzyme: molecular mechanism of polysaccharide oxidative cleavage. bioRxiv. 2016 doi: 10.1101/097022. [DOI] [Google Scholar]
- 168.Möllers KB, Mikkelsen H, Simonsen TI, Cannella D, Johansen KS, Bjerrum MJ, Felby C. On the Formation and Role of Reactive Oxygen Species in Light-Driven Lpmo Oxidation of Phosphoric Acid Swollen Cellulose. Carbohydr Res. 2017;448:182–186. doi: 10.1016/j.carres.2017.03.013. [DOI] [PubMed] [Google Scholar]
- 169.Loose JSM, Forsberg Z, Fraaije MW, Eijsink VGH, Vaaje-Kolstad G. A Rapid Quantitative Activity Assay Shows That the Vibrio Cholerae Colonization Factor Gbpa Is an Active Lytic Polysaccharide Monooxygenase. FEBS Lett. 2014;588:3435–3440. doi: 10.1016/j.febslet.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 170.Tang X, Liu G, Tian S, Zhang Y, Xing M. Expression of the Trichoderma Reesei Endoglucanse Iv in Pichia Pastoris. Weishengwuxue Tongbao. 2005;32:47–51. [Google Scholar]
- 171.Banerjee G, Car S, Scott-Craig JS, Borrusch MS, Walton JD. Rapid Optimization of Enzyme Mixtures for Deconstruction of Diverse Pretreatment/Biomass Feedstock Combinations. Biotechnol Biofuels. 2010;3:22–37. doi: 10.1186/1754-6834-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tanghe M, Danneels B, Camattari A, Glieder A, Vandenberghe I, Devreese B, Stals I, Desmet T. Recombinant Expression of Trichoderma Reesei Cel61a in Pichia Pastoris: Optimizing Yield and N-Terminal Processing. Mol Biotechnol. 2015;57:1010–1017. doi: 10.1007/s12033-015-9887-9. [DOI] [PubMed] [Google Scholar]
- 173.Marinai SL. Masters Thesis. Norwegian University of Life Sciences; 2013. Exploring Methods for Functional Studies of Cbm33-Type Lytic Polysaccharide Monooxygenases. [Google Scholar]
- 174.Yu M-J, Yoon S-H, Kim Y-W. Overproduction and Characterization of a Lytic Polysaccharide Monooxygenase in Bacillus Subtilis Using an Assay Based on Ascorbate Consumption. Enzyme Microb Technol. 2016;93–94:150–156. doi: 10.1016/j.enzmictec.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 175.Span EA, Suess DLM, Deller MC, Britt RD, Marletta MA. The Role of the Secondary Coordination Sphere in a Fungal Polysaccharide Monooxygenase. ACS Chem Biol. 2017;12:1095–1103. doi: 10.1021/acschembio.7b00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Westereng B, Agger JW, Horn SJ, Vaaje-Kolstad G, Aachmann FL, Stenstrøm YH, Eijsink VGH. Efficient Separation of Oxidized Cello-Oligosaccharides Generated by Cellulose Degrading Lytic Polysaccharide Monooxygenases. J Chromatogr. 2013;1271:144–152. doi: 10.1016/j.chroma.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 177.Hansson H, Karkehabadi S, Mikkelsen N, Douglas NR, Kim S, Lam A, Kaper T, Kelemen B, Meier KK, Jones SM, et al. High-Resolution Structure of a Lytic Polysaccharide Monooxygenase from Hypocrea Jecorina Reveals a Predicted Linker as an Integral Part of the Catalytic Domain. J Biol Chem. 2017 doi: 10.1074/jbc.M117.799767. 799767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Eibinger M, Ganner T, Bubner P, Rosker S, Kracher D, Haltrich D, Ludwig R, Plank H, Nidetzky B. Cellulose Surface Degradation by a Lytic Polysaccharide Monooxygenase and Its Effect on Cellulase Hydrolytic Efficiency. J Biol Chem. 2014;289:35929–35938. doi: 10.1074/jbc.M114.602227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Villares A, Moreau C, Bennati-Granier C, Garajova S, Foucat L, Falourd X, Saake B, Berrin JG, Cathala B. Lytic Polysaccharide Monooxygenases Disrupt the Cellulose Fibers Structure. Sci Rep. 2017;7:40262–40271. doi: 10.1038/srep40262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Frandsen KEH, Simmons TJ, Dupree P, Poulsen JCN, Hemsworth GR, Ciano L, Johnston EM, Tovborg M, Johansen KS, von Freiesleben P, et al. The Molecular Basis of Polysaccharide Cleavage by Lytic Polysaccharide Monooxygenases. Nat Chem Biol. 2016;12:298–303. doi: 10.1038/nchembio.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhang YH, Lynd LR. A Functionally Based Model for Hydrolysis of Cellulose by Fungal Cellulase. Biotechnol Bioeng. 2006;94:888–898. doi: 10.1002/bit.20906. [DOI] [PubMed] [Google Scholar]
- 182.Borisova AS, Isaksen T, Dimarogona M, Kognole AA, Mathiesen G, Varnai A, Rohr AK, Payne CM, Sorlie M, Sandgren M, et al. Structural and Functional Characterization of a Lytic Polysaccharide Monooxygenase with Broad Substrate Specificity. J Biol Chem. 2015;290:22955–22969. doi: 10.1074/jbc.M115.660183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Vaaje-Kolstad G, Houston DR, Riemen AHK, Eijsink VGH, van Aalten DMF. Crystal Structure and Binding Properties of the Serratia Marcescens Chitin-Binding Protein Cbp21. J Biol Chem. 2005;280:11313–11319. doi: 10.1074/jbc.M407175200. [DOI] [PubMed] [Google Scholar]
- 184.Kruer-Zerhusen N. PhD Thesis. Cornell University; 2016. Studies of Thermobifida Fusca Lytic Polysaccharide Monooxygenases Aa10a and Aa10b for Cleavage of Recalcitrant Cellulose. [Google Scholar]
- 185.Marletta MA, Doudna CJH, Beeson WT, Phillips CM. The Regents of the University of California. WO 2012/138772 A1. Enhanced Cellulose Degradation. 2012 Oct 11;
- 186.Crouch LI, Labourel A, Walton PH, Davies GJ, Gilbert HJ. The Contribution of Non-Catalytic Carbohydrate Binding Modules to the Activity of Lytic Polysaccharide Monooxygenases. J Biol Chem. 2016;291:7439–7449. doi: 10.1074/jbc.M115.702365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Arfi Y, Shamshoum M, Rogachev I, Peleg Y, Bayer EA. Integration of Bacterial Lytic Polysaccharide Monooxygenases into Designer Cellulosomes Promotes Enhanced Cellulose Degradation. Proc Natl Acad Sci U S A. 2014;111:9109–9114. doi: 10.1073/pnas.1404148111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liang Y, Si T, Ang EL, Zhao H. Engineered Pentafunctional Minicellulosome for Simultaneous Saccharification and Ethanol Fermentation in Saccharomyces Cerevisiae. Appl Environ Microbiol. 2014;80:6677–6684. doi: 10.1128/AEM.02070-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wu M, Beckham GT, Larsson AM, Ishida T, Kim S, Payne CM, Himmel ME, Crowley MF, Horn SJ, Westereng B, et al. Crystal Structure and Computational Characterization of the Lytic Polysaccharide Monooxygenase Gh61d from the Basidiomycota Fungus Phanerochaete Chrysosporium. J Biol Chem. 2013;288:12828–12839. doi: 10.1074/jbc.M113.459396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Doan N, Gettins PG. Human Alpha2-Macroglobulin Is Composed of Multiple Domains, as Predicted by Homology with Complement Component C3. Biochem J. 2007;407:23–30. doi: 10.1042/BJ20070764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Murzin AG, Brenner SE, Hubbard T, Chothia C. Scop: A Structural Classification of Proteins Database for the Investigation of Sequences and Structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 192.Li X, Beeson WT, Phillips CM, Marletta MA, Cate JH. Structural Basis for Substrate Targeting and Catalysis by Fungal Polysaccharide Monooxygenases. Structure. 2012;20:1051–1061. doi: 10.1016/j.str.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Westereng B, Kracun SK, Dimarogona M, Mathiesen G, Willats WGT, Sandgren M, Aachmann FL, Eijsink VGH. 5FOH: Crystal Structure of the Catalytic Domain of NcLPMO9A. Protein Data Bank. 2016 doi: 10.2210/pdb5foh/pdb. [DOI] [Google Scholar]
- 194.Frandsen KEH, Poulsen JCN, Tandrup T, Lo Leggio L. Unliganded and Substrate Bound Structures of the Cellooligosaccharide Active Lytic Polysaccharide Monooxygenase Lsaa9a at Low Ph. Carbohydr Res. 2017;448:187–190. doi: 10.1016/j.carres.2017.03.010. [DOI] [PubMed] [Google Scholar]
- 195.Danneels B, Tanghe M, Joosten HJ, Gundinger T, Spadiut O, Stals I, Desmet T. A Quantitative Indicator Diagram for Lytic Polysaccharide Monooxygenases Reveals the Role of Aromatic Surface Residues in Hjlpmo9a Regioselectivity. PLoS One. 2017;12:e0178446–e0178461. doi: 10.1371/journal.pone.0178446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Aachmann FL, Sørlie M, Skjåk-Bræk G, Eijsink VG, Vaaje-Kolstad G. Nmr Structure of a Lytic Polysaccharide Monooxygenase Provides Insight into Copper Binding, Protein Dynamics, and Substrate Interactions. Proc Natl Acad Sci U S A. 2012;109:18779–18784. doi: 10.1073/pnas.1208822109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wong E, Vaaje-Kolstad G, Ghosh A, Hurtado-Guerrero R, Konarev PV, Ibrahim AF, Svergun DI, Eijsink VG, Chatterjee NS, van Aalten DM. The Vibrio Cholerae Colonization Factor Gbpa Possesses a Modular Structure That Governs Binding to Different Host Surfaces. PLoS Pathog. 2012;8:e1002373–e1002385. doi: 10.1371/journal.ppat.1002373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hemsworth GR, Taylor EJ, Kim RQ, Gregory RC, Lewis SJ, Turkenburg JP, Parkin A, Davies GJ, Walton PH. The Copper Active Site of Cbm33 Polysaccharide Oxygenases. J Am Chem Soc. 2013;135:6069–6077. doi: 10.1021/ja402106e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Gudmundsson M, Kim S, Wu M, Ishida T, Momeni MH, Vaaje-Kolstad G, Lundberg D, Royant A, Ståhlberg J, Eijsink VGH, et al. Structural and Electronic Snapshots During the Transition from a Cu(Ii) to Cu(I) Metal Center of a Lytic Polysaccharide Monooxygenase by X-Ray Photoreduction. J Biol Chem. 2014;289:18782–18792. doi: 10.1074/jbc.M114.563494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Chaplin AK, Wilson MT, Hough MA, Svistunenko DA, Hemsworth GR, Walton PH, Vijgenboom E, Worrall JA. Heterogeneity in the Histidine-Brace Copper Coordination Sphere in Auxiliary Activity Family 10 (Aa10) Lytic Polysaccharide Monooxygenases. J Biol Chem. 2016;291:12838–12850. doi: 10.1074/jbc.M116.722447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gregory RC, Hemsworth GR, Turkenburg JP, Hart SJ, Walton PH, Davies GJ. Activity, Stability and 3-D Structure of the Cu(Ii) Form of a Chitin-Active Lytic Polysaccharide Monooxygenase from Bacillus Amyloliquefaciens. Dalton Trans. 2016;45:16904–16912. doi: 10.1039/c6dt02793h. [DOI] [PubMed] [Google Scholar]
- 202.Takemoto Y, Mitsuhashi W, Murakami R, Konishi H, Miyamoto K. The N-Terminal Region of an Entomopoxvirus Fusolin Is Essential for the Enhancement of Peroral Infection, Whereas the C-Terminal Region Is Eliminated in Digestive Juice. J Virol. 2008;82:12406–12415. doi: 10.1128/JVI.01605-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Alahuhta PM, Lunin VV. 4GBO: Structure of Thermobifida Fusca E7. Protein Data Bank. 2012 doi: 10.2210/pdb4gbo/pdb. [DOI] [Google Scholar]
- 204.Zhao Y, Zhang H, Yin H. 5WSZ: Crystal Structure of a Lytic Polysaccharide Monooxygenase, Btlpmo10a, from Bacillus Thuringiensis. Protein Data Bank. 2016 doi: 10.2210/pdb5wsz/pdb. [DOI] [Google Scholar]
- 205.Fox D, III, Gardberg A, Armour B, Staker B, Stewart L. 3UAM: Crystal Structure of a Chitin Binding Domain from Burkholderia Pseudomallei. Protein Data Bank. 2011 doi: 10.2210/pdb3uam/pdb. [DOI] [Google Scholar]
- 206.Frandsen KE, Poulsen JC, Tovborg M, Johansen KS, Lo Leggio L. Learning from Oligosaccharide Soaks of Crystals of an Aa13 Lytic Polysaccharide Monooxygenase: Crystal Packing, Ligand Binding and Active-Site Disorder. Acta Crystallogr, Sect D: Struct Biol. 2017;73:64–76. doi: 10.1107/S2059798316019641. [DOI] [PubMed] [Google Scholar]
- 207.Bhowmick R, Ghosal A, Das B, Koley H, Saha DR, Ganguly S, Nandy RK, Bhadra RK, Chatterjee NS. Intestinal Adherence of Vibrio Cholerae Involves a Coordinated Interaction between Colonization Factor Gbpa and Mucin. Infect Immun. 2008;76:4968–4977. doi: 10.1128/IAI.01615-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active Enzymes Database (Cazy): An Expert Resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Happs RM, Guan X, Resch MG, Davis MF, Beckham GT, Tan Z, Crowley MF. O-Glycosylation Effects on Family 1 Carbohydrate-Binding Module Solution Structures. FEBS J. 2015;282:4341–4356. doi: 10.1111/febs.13500. [DOI] [PubMed] [Google Scholar]
- 210.Malecki PH, Vorgias CE, Petoukhov MV, Svergun DI, Rypniewski W. Crystal Structures of Substrate-Bound Chitinase from the Psychrophilic Bacterium Moritella Marina and Its Structure in Solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2014;70:676–684. doi: 10.1107/S1399004713032264. [DOI] [PubMed] [Google Scholar]
- 211.Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-Binding Modules: Fine-Tuning Polysaccharide Recognition. Biochem J. 2004;382:769–781. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Gilbert HJ, Knox JP, Boraston AB. Advances in Understanding the Molecular Basis of Plant Cell Wall Polysaccharide Recognition by Carbohydrate-Binding Modules. Curr Opin Struct Biol. 2013;23:669–677. doi: 10.1016/j.sbi.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 213.Guillen D, Sanchez S, Rodriguez-Sanoja R. Carbohydrate-Binding Domains: Multiplicity of Biological Roles. Appl Microbiol Biotechnol. 2010;85:1241–1249. doi: 10.1007/s00253-009-2331-y. [DOI] [PubMed] [Google Scholar]
- 214.Hashimoto H. Recent Structural Studies of Carbohydrate-Binding Modules. Cell Mol Life Sci. 2006;63:2954–2967. doi: 10.1007/s00018-006-6195-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Shoseyov O, Shani Z, Levy I. Carbohydrate Binding Modules: Biochemical Properties and Novel Applications. Microbiol Mol Biol Rev. 2006;70:283–295. doi: 10.1128/MMBR.00028-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gao PJ, Chen GJ, Wang TH, Zhang YS, Liu J. Non-Hydrolytic Disruption of Crystalline Structure of Cellulose by Cellulose Binding Domain and Linker Sequence of Cellobiohydrolase I from Penicillium Janthinellum. Shengwu Huaxue Yu Shengwu Wuli Xuebao (Shanghai) 2001;33:13–18. [PubMed] [Google Scholar]
- 217.Din N, Gilkes NR, Tekant B, Miller RC, Warren RAJ, Kilburn DG. Non-Hydrolytic Disruption of Cellulose Fibres by the Binding Domain of a Bacterial Cellulase. Bio/Technology. 1991;9:1096–1099. [Google Scholar]
- 218.Gourlay K, Arantes V, Saddler JN. Use of Substructure-Specific Carbohydrate Binding Modules to Track Changes in Cellulose Accessibility and Surface Morphology During the Amorphogenesis Step of Enzymatic Hydrolysis. Biotechnol Biofuels. 2012;5:51. doi: 10.1186/1754-6834-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Southall SM, Simpson PJ, Gilbert HJ, Williamson G, Williamson MP. The Starch-Binding Domain from Glucoamylase Disrupts the Structure of Starch. FEBS Lett. 1999;447:58–60. doi: 10.1016/s0014-5793(99)00263-x. [DOI] [PubMed] [Google Scholar]
- 220.Herve C, Rogowski A, Blake AW, Marcus SE, Gilbert HJ, Knox JP. Carbohydrate-Binding Modules Promote the Enzymatic Deconstruction of Intact Plant Cell Walls by Targeting and Proximity Effects. Proc Natl Acad Sci U S A. 2010;107:15293–15298. doi: 10.1073/pnas.1005732107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Nimlos MR, Beckham GT, Matthews JF, Bu L, Himmel ME, Crowley MF. Binding Preferences, Surface Attachment, Diffusivity, and Orientation of a Family 1 Carbohydrate-Binding Module on Cellulose. J Biol Chem. 2012;287:20603–20612. doi: 10.1074/jbc.M112.358184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Bolam DN, Ciruela A, McQueen-Mason S, Simpson P, Williamson MP, Rixon JE, Boraston A, Hazlewood GP, Gilbert HJ. Pseudomonas Cellulose-Binding Domains Mediate Their Effects by Increasing Enzyme Substrate Proximity. Biochem J. 1998;331:775–781. doi: 10.1042/bj3310775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.McLean BW, Bray MR, Boraston AB, Gilkes NR, Haynes CA, Kilburn DG. Analysis of Binding of the Family 2a Carbohydrate-Binding Module from Cellulomonas Fimi Xylanase 10a to Cellulose: Specificity and Identification of Functionally Important Amino Acid Residues. Protein Eng, Des Sel. 2000;13:801–809. doi: 10.1093/protein/13.11.801. [DOI] [PubMed] [Google Scholar]
- 224.Linder M, Teeri TT. The Cellulose-Binding Domain of the Major Cellobiohydrolase of Trichoderma Reesei Exhibits True Reversibility and a High Exchange Rate on Crystalline Cellulose. Proc Natl Acad Sci U S A. 1996;93:12251–12255. doi: 10.1073/pnas.93.22.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Linder M, Teeri TT. The Roles and Function of Cellulose-Binding Domains. J Biotechnol. 1997;57:15–28. doi: 10.1016/s0168-1656(97)00088-6. [DOI] [PubMed] [Google Scholar]
- 226.Lehtio J, Sugiyama J, Gustavsson M, Fransson L, Linder M, Teeri TT. The Binding Specificity and Affinity Determinants of Family 1 and Family 3 Cellulose Binding Modules. Proc Natl Acad Sci U S A. 2003;100:484–489. doi: 10.1073/pnas.212651999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Gilkes NR, Henrissat B, Kilburn DG, Miller RC, Jr, Warren RA. Domains in Microbial Beta-1, 4-Glycanases: Sequence Conservation, Function, and Enzyme Families. Microbiol Rev. 1991;55:303–315. doi: 10.1128/mr.55.2.303-315.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Meinke A, Gilkes NR, Kilburn DG, Miller RC, Jr, Warren RA. Bacterial Cellulose-Binding Domain-Like Sequences in Eucaryotic Polypeptides. Protein Sequences Data Anal. 1991;4:349–353. [PubMed] [Google Scholar]
- 229.van Tilbeurgh H, Tomme P, Claeyssens M, Bhikhabhai R, Pettersson G. Limited Proteolysis of the Cellobiohydrolase I from Trichoderma Reesei. FEBS Lett. 1986;204:223–227. [Google Scholar]
- 230.DePristo MA, Zilversmit MM, Hartl DL. On the Abundance, Amino Acid Composition, and Evolutionary Dynamics of Low-Complexity Regions in Proteins. Gene. 2006;378:19–30. doi: 10.1016/j.gene.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 231.Irwin D, Shin DH, Zhang S, Barr BK, Sakon J, Karplus PA, Wilson DB. Roles of the Catalytic Domain and Two Cellulose Binding Domains of Thermomonospora Fusca E4 in Cellulose Hydrolysis. J Bacteriol. 1998;180:1709–1714. doi: 10.1128/jb.180.7.1709-1714.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Sakon J, Irwin D, Wilson DB, Karplus PA. Structure and Mechanism of Endo/Exocellulase E4 from Thermomonospora Fusca. Nat Struct Biol. 1997;4:810–818. doi: 10.1038/nsb1097-810. [DOI] [PubMed] [Google Scholar]
- 233.van Aalten DM, Synstad B, Brurberg MB, Hough E, Riise BW, Eijsink VG, Wierenga RK. Structure of a Two-Domain Chitotriosidase from Serratia Marcescens at 1.9-a Resolution. Proc Natl Acad Sci U S A. 2000;97:5842–5847. doi: 10.1073/pnas.97.11.5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Abuja PM, Pilz I, Claeyssens M, Tomme P. Domain Structure of Cellobiohydrolase Ii as Studied by Small Angle X-Ray Scattering: Close Resemblance to Cellobiohydrolase I. Biochem Biophys Res Commun. 1988;156:180–185. doi: 10.1016/s0006-291x(88)80821-0. [DOI] [PubMed] [Google Scholar]
- 235.Abuja PM, Pilz I, Tomme P, Claeyssens M. Structural Changes in Cellobiohydrolase I Upon Binding of a Macromolecular Ligand as Evident by Saxs Investigations. Biochem Biophys Res Commun. 1989;165:615–623. doi: 10.1016/s0006-291x(89)80010-5. [DOI] [PubMed] [Google Scholar]
- 236.Sammond DW, Payne CM, Brunecky R, Himmel ME, Crowley MF, Beckham GT. Cellulase Linkers Are Optimized Based on Domain Type and Function: Insights from Sequence Analysis, Biophysical Measurements, and Molecular Simulation. PLoS One. 2012;7:e48615–e48629. doi: 10.1371/journal.pone.0048615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Spiro RG. Protein Glycosylation: Nature, Distribution, Enzymatic Formation, and Disease Implications of Glycopeptide Bonds. Glycobiology. 2002;12:43R–56R. doi: 10.1093/glycob/12.4.43r. [DOI] [PubMed] [Google Scholar]
- 238.Nothaft H, Szymanski CM. Protein Glycosylation in Bacteria: Sweeter Than Ever. Nat Rev Microbiol. 2010;8:765–778. doi: 10.1038/nrmicro2383. [DOI] [PubMed] [Google Scholar]
- 239.Goto M. Protein O-Glycosylation in Fungi: Diverse Structures and Multiple Functions. Biosci, Biotechnol Biochem. 2007;71:1415–1427. doi: 10.1271/bbb.70080. [DOI] [PubMed] [Google Scholar]
- 240.Hart GW, Copeland RJ. Glycomics Hits the Big Time. Cell. 2010;143:672–676. doi: 10.1016/j.cell.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Zhou F, Olman V, Xu Y. Large-Scale Analyses of Glycosylation in Cellulases. Genomics, Proteomics Bioinf. 2009;7:194–199. doi: 10.1016/S1672-0229(08)60049-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Varki A. Biological Roles of Oligosaccharides: All of the Theories Are Correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Jafari-Aghdam J, Khajeh K, Ranjbar B, Nemat-Gorgani M. Deglycosylation of Glucoamylase from Aspergillus Niger: Effects on Structure, Activity and Stability. Biochim Biophys Acta, Proteins Proteomics. 2005;1750:61–68. doi: 10.1016/j.bbapap.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 244.Sutthirak P, Dharmsthiti S, Lertsiri S. Effect of Glycation on Stability and Kinetic Parameters of Thermostable Glucoamylase from Aspergillus Niger. Process Biochem. 2005;40:2821–2826. [Google Scholar]
- 245.Beckham GT, Dai Z, Matthews JF, Momany M, Payne CM, Adney WS, Baker SE, Himmel ME. Harnessing Glycosylation to Improve Cellulase Activity. Curr Opin Biotechnol. 2012;23:338–345. doi: 10.1016/j.copbio.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 246.Gusakov AV, Dotsenko AS, Rozhkova AM, Sinitsyn AP. N-Linked Glycans Are an Important Component of the Processive Machinery of Cellobiohydrolases. Biochimie. 2017;132:102–108. doi: 10.1016/j.biochi.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 247.Dotsenko AS, Gusakov AV, Volkov PV, Rozhkova AM, Sinitsyn AP. N-Linked Glycosylation of Recombinant Cellobiohydrolase I (Cel7a) from Penicillium Verruculosum and Its Effect on the Enzyme Activity. Biotechnol Bioeng. 2016;113:283–291. doi: 10.1002/bit.25812. [DOI] [PubMed] [Google Scholar]
- 248.Dotsenko AS, Gusakov AV, Rozhkova AM, Sinitsyna OA, Nemashkalov VA, Sinitsyn AP. Effect of N-Linked Glycosylation on the Activity and Other Properties of Recombinant Endoglucanase Iia (Cel5a) from Penicillium Verruculosum. Protein Eng, Des Sel. 2016;29:495–502. doi: 10.1093/protein/gzw030. [DOI] [PubMed] [Google Scholar]
- 249.Courtade G, Wimmer R, Rohr AK, Preims M, Felice AK, Dimarogona M, Vaaje-Kolstad G, Sorlie M, Sandgren M, Ludwig R, et al. Interactions of a Fungal Lytic Polysaccharide Monooxygenase with Beta-Glucan Substrates and Cellobiose Dehydrogenase. Proc Natl Acad Sci U S A. 2016;113:5922–5927. doi: 10.1073/pnas.1602566113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Davies GJ, Ducros V, Lewis RJ, Borchert TV, Schülein M. Oligosaccharide Specificity of a Family 7 Endoglucanase: Insertion of Potential Sugar-Binding Subsites. J Biotechnol. 1997;57:91–100. doi: 10.1016/s0168-1656(97)00092-8. [DOI] [PubMed] [Google Scholar]
- 251.Kjaergaard CH, Qayyum MF, Wong SD, Xu F, Hemsworth GR, Walton DJ, Young NA, Davies GJ, Walton PH, Johansen KS, et al. Spectroscopic and Computational Insight into the Activation of O2 by the Mononuclear Cu Center in Polysaccharide Monooxygenases. Proc Natl Acad Sci U S A. 2014;111:8797–8802. doi: 10.1073/pnas.1408115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Hemsworth GR, Davies GJ, Walton PH. Recent Insights into Copper-Containing Lytic Polysaccharide Mono-Oxygenases. Curr Opin Struct Biol. 2013;23:660–668. doi: 10.1016/j.sbi.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 253.Sirajuddin S, Rosenzweig AC. Enzymatic Oxidation of Methane. Biochemistry. 2015;54:2283–2294. doi: 10.1021/acs.biochem.5b00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Zhang L, Koay M, Maher MJ, Xiao Z, Wedd AG. Intermolecular Transfer of Copper Ions from the Copc Protein of Pseudomonas Syringae. Crystal Structures of Fully Loaded Cu(I)Cu-(Ii) Forms. J Am Chem Soc. 2006;128:5834–5850. doi: 10.1021/ja058528x. [DOI] [PubMed] [Google Scholar]
- 255.Chen P, Solomon EI. Oxygen Activation by the Non-coupled Binuclear Copper Site in Peptidylglycine A-Hydroxylating Monooxygenase. Reaction Mechanism and Role of the Noncoupled Nature of the Active Site. J Am Chem Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 256.Shepard EM, Dooley DM. Inhibition and Oxygen Activation in Copper Amine Oxidases. Acc Chem Res. 2015;48:1218–1226. doi: 10.1021/ar500460z. [DOI] [PubMed] [Google Scholar]
- 257.Li Y, Hodak M, Bernholc J. Enzymatic Mechanism of Copper-Containing Nitrite Reductase. Biochemistry. 2015;54:1233–1242. doi: 10.1021/bi5007767. [DOI] [PubMed] [Google Scholar]
- 258.Citek C, Lin BL, Phelps TE, Wasinger EC, Stack TD. Primary Amine Stabilization of a Dicopper(Iii) Bis(M-Oxo) Species: Modeling the Ligation in Pmmo. J Am Chem Soc. 2014;136:14405–14408. doi: 10.1021/ja508630d. [DOI] [PubMed] [Google Scholar]
- 259.Paiva AC, Juliano L, Boschcov P. Ionization of Methyl Derivatives of Imidazole, Histidine, Thyreotropin Releasing Factor, and Related Compounds. J Am Chem Soc. 1976;98:7645–7648. doi: 10.1021/ja00440a033. [DOI] [PubMed] [Google Scholar]
- 260.Kim S, Ståhlberg J, Sandgren M, Paton RS, Beckham GT. Quantum Mechanical Calculations Suggest That Lytic Polysaccharide Monooxygenases Use a Copper-Oxyl, Oxygen-Rebound Mechanism. Proc Natl Acad Sci U S A. 2014;111:149–154. doi: 10.1073/pnas.1316609111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Tickler AK, Smith DG, Ciccotosto GD, Tew DJ, Curtain CC, Carrington D, Masters CL, Bush AI, Cherny RA, Cappai R, et al. Methylation of the Imidazole Side Chains of the Alzheimer Disease Amyloid-Beta Peptide Results in Abolition of Superoxide Dismutase-Like Structures and Inhibition of Neurotoxicity. J Biol Chem. 2005;280:13355–13363. doi: 10.1074/jbc.M414178200. [DOI] [PubMed] [Google Scholar]
- 262.Whistler RL. Carbohydrates in Solution. American Chemical Society; 1973. Solubility of Polysaccharides and Their Behavior in Solution; pp. 242–255. [Google Scholar]
- 263.Westereng B, Ishida T, Vaaje-Kolstad G, Wu M, Eijsink VG, Igarashi K, Samejima M, Ståhlberg J, Horn SJ, Sandgren M. The Putative Endoglucanase Pcgh61d from Phanerochaete Chrysosporium Is a Metal-Dependent Oxidative Enzyme That Cleaves Cellulose. PLoS One. 2011;6:e27807–e27818. doi: 10.1371/journal.pone.0027807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.O’Dell WB, Agarwal PK, Meilleur F. Oxygen Activation at the Active Site of a Fungal Lytic Polysaccharide Monooxygenase. Angew Chem, Int Ed. 2017;56:767–770. doi: 10.1002/anie.201610502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Hedegard ED, Ryde U. Multiscale Modelling of Lytic Polysaccharide Monooxygenases. ACS Omega. 2017;2:536–545. doi: 10.1021/acsomega.6b00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Cramer CJ, Tolman WB. Mononuclear Cuo2 Complexes: Geometries, Spectroscopic Properties, Electronic Structures, and Reactivity. Acc Chem Res. 2007;40:601–608. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Donoghue PJ, Gupta AK, Boyce DW, Cramer CJ, Tolman WB. An Anionic, Tetragonal Copper(Ii) Superoxide Complex. J Am Chem Soc. 2010;132:15869–15871. doi: 10.1021/ja106244k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Fujisawa K, Tanaka M, Moro-oka Y, Kitajima N. A Monomeric Side-on Superoxocopper(Ii) Complex: Cu(O2)(Hb(3-Tbu-5-Iprpz)3) J Am Chem Soc. 1994;116:12079–12080. [Google Scholar]
- 269.Garcia-Bosch I, Siegler MA. Copper-Catalyzed Oxidation of Alkanes with H2o2 under a Fenton-Like Regime. Angew Chem, Int Ed. 2016;55:12873–12876. doi: 10.1002/anie.201607216. [DOI] [PubMed] [Google Scholar]
- 270.Ginsbach JW, Peterson RL, Cowley RE, Karlin KD, Solomon EI. Correlation of the Electronic and Geometric Structures in Mononuclear Copper(Ii) Superoxide Complexes. Inorg Chem. 2013;52:12872–12874. doi: 10.1021/ic402357u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Itoh S. Mononuclear Copper Active-Oxygen Complexes. Curr Opin Chem Biol. 2006;10:115–122. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 272.Itoh S. Developing Mononuclear Copper-Active-Oxygen Complexes Relevant to Reactive Intermediates of Biological Oxidation Reactions. Acc Chem Res. 2015;48:2066–2074. doi: 10.1021/acs.accounts.5b00140. [DOI] [PubMed] [Google Scholar]
- 273.Kim S, Ginsbach JW, Lee JY, Peterson RL, Liu JJ, Siegler MA, Sarjeant AA, Solomon EI, Karlin KD. Amine Oxidative N-Dealkylation Via Cupric Hydroperoxide Cu-Ooh Homolytic Cleavage Followed by Site-Specific Fenton Chemistry. J Am Chem Soc. 2015;137:2867–2874. doi: 10.1021/ja508371q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Komiyama K, Furutachi H, Nagatomo S, Hashimoto A, Hayashi H, Fujinami S, Suzuki M, Kitagawa T. Dioxygen Reactivity of Copper(I) Complexes with Tetradentate Tripodal Ligands Having Aliphatic Nitrogen Donors: Synthesis, Structures, and Properties of Peroxo and Superoxo Complexes. Bull Chem Soc Jpn. 2004;77:59–72. [Google Scholar]
- 275.Kunishita A, Kubo M, Sugimoto H, Ogura T, Sato K, Takui T, Itoh S. Mononuclear Copper(Ii)–Superoxo Complexes That Mimic the Structure and Reactivity of the Active Centers of Phm and Dβm. J Am Chem Soc. 2009;131:2788–2789. doi: 10.1021/ja809464e. [DOI] [PubMed] [Google Scholar]
- 276.Lee JY, Peterson RL, Ohkubo K, Garcia-Bosch I, Himes RA, Woertink J, Moore CD, Solomon EI, Fukuzumi S, Karlin KD. Mechanistic Insights into the Oxidation of Substituted Phenols Via Hydrogen Atom Abstraction by a Cupric-Superoxo Complex. J Am Chem Soc. 2014;136:9925–9937. doi: 10.1021/ja503105b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD. A 1:1 Copper-Dioxygen Adduct Is an End-on Bound Superoxo Copper(Ii) Complex Which Undergoes Oxygenation Reactions with Phenols. J Am Chem Soc. 2007;129:264–265. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]
- 278.Peterson RL, Ginsbach JW, Cowley RE, Qayyum MF, Himes RA, Siegler MA, Moore CD, Hedman B, Hodgson KO, Fukuzumi S, et al. Stepwise Protonation and Electron-Transfer Reduction of a Primary Copper-Dioxygen Adduct. J Am Chem Soc. 2013;135:16454–16467. doi: 10.1021/ja4065377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Woertink JS, Tian L, Maiti D, Lucas HR, Himes RA, Karlin KD, Neese F, Würtele C, Holthausen MC, Bill E, et al. Spectroscopic and Computational Studies of an End-on Bound Superoxo-Cu(Ii) Complex: Geometric and Electronic Factors That Determine the Ground State. Inorg Chem. 2010;49:9450–9459. doi: 10.1021/ic101138u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Würtele C, Gaoutchenova E, Harms K, Holthausen MC, Sundermeyer J, Schindler S. Crystallographic Characterization of a Synthetic 1:1 End-on Copper Dioxygen Adduct Complex. Angew Chem, Int Ed. 2006;45:3867–3869. doi: 10.1002/anie.200600351. [DOI] [PubMed] [Google Scholar]
- 281.Lanci MP, Smirnov VV, Cramer CJ, Gauchenova EV, Sundermeyer J, Roth JP. Isotopic Probing of Molecular Oxygen Activation at Copper(I) Sites. J Am Chem Soc. 2007;129:14697–14709. doi: 10.1021/ja074620c. [DOI] [PubMed] [Google Scholar]
- 282.Kunishita A, Ertem MZ, Okubo Y, Tano T, Sugimoto H, Ohkubo K, Fujieda N, Fukuzumi S, Cramer CJ, Itoh S. Active Site Models for the Cua Site of Peptidylglycine A-Hydroxylating Monooxygenase and Dopamine B-Monooxygenase. Inorg Chem. 2012;51:9465–9480. doi: 10.1021/ic301272h. [DOI] [PubMed] [Google Scholar]
- 283.Cowley RE, Tian L, Solomon EI. Mechanism of O2 Activation and Substrate Hydroxylation in Noncoupled Binuclear Copper Monooxygenases. Proc Natl Acad Sci U S A. 2016;113:12035–12040. doi: 10.1073/pnas.1614807113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Chen P, Root DE, Campochiaro C, Fujisawa K, Solomon EI. Spectroscopic and Electronic Structure Studies of the Diamagnetic Side-on Cuii-Superoxo Complex Cu(O2)[Hb(3-R-5-Iprpz)3]: Antiferromagnetic Coupling Versus Covalent Delocalization. J Am Chem Soc. 2003;125:466–474. doi: 10.1021/ja020969i. [DOI] [PubMed] [Google Scholar]
- 285.Maiti D, Lee DH, Gaoutchenova K, Würtele C, Holthausen MC, Narducci Sarjeant AA, Sundermeyer J, Schindler S, Karlin KD. Reactions of a Copper(Ii) Superoxo Complex Lead to C-H and O-H Substrate Oxygenation: Modeling Copper-Monooxygenase C-H Hydroxylation. Angew Chem. 2008;120:88–91. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]
- 286.Peterson RL, Himes RA, Kotani H, Suenobu T, Tian L, Siegler MA, Solomon EI, Fukuzumi S, Karlin KD. Cupric Superoxo-Mediated Intermolecular C-H Activation Chemistry. J Am Chem Soc. 2011;133:1702–1705. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Tano T, Okubo Y, Kunishita A, Kubo M, Sugimoto H, Fujieda N, Ogura T, Itoh S. Redox Properties of a Mononuclear Copper(Ii)-Superoxide Complex. Inorg Chem. 2013;52:10431–10437. doi: 10.1021/ic401261z. [DOI] [PubMed] [Google Scholar]
- 288.Chen P, Bell J, Eipper BA, Solomon EI. Oxygen Activation by the Noncoupled Binuclear Copper Site in Peptidylglycine Alpha-Hydroxylating Monooxygenase. Spectroscopic Definition of the Resting Sites and the Putative Cuii M-Ooh Intermediate. Biochemistry. 2004;43:5735–5747. doi: 10.1021/bi0362830. [DOI] [PubMed] [Google Scholar]
- 289.Klinman JP. The Copper-Enzyme Family of Dopamine Beta-Monooxygenase and Peptidylglycine Alpha-Hydroxylating Monooxygenase: Resolving the Chemical Pathway for Substrate Hydroxylation. J Biol Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 290.Bacik JP, Mekasha S, Forsberg Z, Kovalevsky AY, Vaaje-Kolstad G, Eijsink VGH, Nix JC, Coates L, Cuneo MJ, Unkefer CJ, et al. Neutron and Atomic Resolution X-Ray Structures of a Lytic Polysaccharide Monooxygenase Reveal Copper-Mediated Dioxygen Binding and Evidence for N-Terminal Deprotonation. Biochemistry. 2017;56:2529–2532. doi: 10.1021/acs.biochem.7b00019. [DOI] [PubMed] [Google Scholar]
- 291.DuBois JL, Mukherjee P, Stack TDP, Hedman B, Solomon EI, Hodgson KO. A Systematic K-Edge X-Ray Absorption Spectroscopic Study of Cu(Iii) Sites. J Am Chem Soc. 2000;122:5775–5787. [Google Scholar]
- 292.Sarangi R, Aboelella N, Fujisawa K, Tolman WB, Hedman B, Hodgson KO, Solomon EI. X-Ray Absorption Edge Spectroscopy and Computational Studies on Lcuo2 Species: Superoxide–Cuii Versus Peroxide–Cuiii Bonding. J Am Chem Soc. 2006;128:8286–8296. doi: 10.1021/ja0615223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Dhar D, Tolman WB. Hydrogen Atom Abstraction from Hydrocarbons by a Copper(Iii)-Hydroxide Complex. J Am Chem Soc. 2015;137:1322–1329. doi: 10.1021/ja512014z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Dhar D, Yee GM, Markle TF, Mayer JM, Tolman WB. Reactivity of the Copper(Iii)-Hydroxide Unit with Phenols. Chem Sci. 2017;8:1075–1085. doi: 10.1039/c6sc03039d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Wood PM. Pathways for Production of Fenton’s Reagent by Wood-Rotting Fungi. FEMS Microbiol Rev. 1994;13:313–320. [Google Scholar]
- 296.Concia AL, Beccia MR, Orio M, Ferre FT, Scarpellini M, Biaso F, Guigliarelli B, Réglier M, Simaan AJ. Copper Complexes as Bioinspired Models for Lytic Polysaccharide Monooxygenases. Inorg Chem. 2017;56:1023–1026. doi: 10.1021/acs.inorgchem.6b02165. [DOI] [PubMed] [Google Scholar]
- 297.Hallberg BM, Henriksson G, Pettersson G, Divne C. Crystal Structure of the Flavoprotein Domain of the Extracellular Flavocytochrome Cellobiose Dehydrogenase. J Mol Biol. 2002;315:421–434. doi: 10.1006/jmbi.2001.5246. [DOI] [PubMed] [Google Scholar]
- 298.Kracher D, Zahma K, Schulz C, Sygmund C, Gorton L, Ludwig R. Interdomain Electron Transfer in Cellobiose Dehydrogenase: Modulation by Ph and Divalent Cations. FEBS J. 2015;282:3136–3148. doi: 10.1111/febs.13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Sygmund C, Kracher D, Scheiblbrandner S, Zahma K, Felice AKG, Harreither W, Kittl R, Ludwig R. Characterization of the Two Neurospora Crassa Cellobiose Dehydrogenases and Their Connection to Oxidative Cellulose Degradation. Appl Environ Microbiol. 2012;78:6161–6171. doi: 10.1128/AEM.01503-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.DeBeer George S, Metz M, Szilagyi RK, Wang H, Cramer SP, Lu Y, Tolman WB, Hedman B, Hodgson KO, Solomon EI. A Quantitative Description of the Ground-State Wave Function of Cu Aby X-Ray Absorption Spectroscopy: Comparison to Plastocyanin and Relevance to Electron Transfer. J Am Chem Soc. 2001;123:5757–5767. doi: 10.1021/ja004109i. [DOI] [PubMed] [Google Scholar]
- 301.Westereng B, Arntzen MØ, Aachmann FL, Várnai A, Eijsink VGH, Agger JW. Simultaneous Analysis of C1 and C4 Oxidized Oligosaccharides, the Products of Lytic Polysaccharide Monooxygenases Acting on Cellulose. J Chromatogr. 2016;1445:46–54. doi: 10.1016/j.chroma.2016.03.064. [DOI] [PubMed] [Google Scholar]
- 302.Whittaker MM, Whittaker JW. Cu(I)-Dependent Biogenesis of the Galactose Oxidase Redox Cofactor. J Biol Chem. 2003;278:22090–22101. doi: 10.1074/jbc.M300112200. [DOI] [PubMed] [Google Scholar]
- 303.Cowley RE, Cirera J, Qayyum MF, Rokhsana D, Hedman B, Hodgson KO, Dooley DM, Solomon EI. Structure of the Reduced Copper Active Site in Preprocessed Galactose Oxidase: Ligand Tuning for One-Electron O2 Activation in Cofactor Biogenesis. J Am Chem Soc. 2016;138:13219–13229. doi: 10.1021/jacs.6b05792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Osborne RL, Klinman JP. Insights into the Proposed Copper-Oxygen Intermediates That Regulate the Mechanism of Reactions Catalyzed by Dopamine B-Monooxygenase, Peptidylglycine A-Hydroxylating Monooxygenase, and Tyramine B-Monooxygenase. In: Karlin KD, Itoh S, editors. Copper-Oxygen Chemistry. John Wiley & Sons, Inc; Hoboken, NJ: 2011. pp. 1–22. [Google Scholar]