

ABSTRACT
Background
Chronic kidney disease (CKD) patients have deficient levels of glutathione peroxidase-3 (GPx3). We hypothesized that GPx3 deficiency may lead to cardiovascular disease in the presence of chronic kidney disease due to an accumulation of reactive oxygen species and decreased microvascular perfusion of the myocardium.
Methods. To isolate the exclusive effect of GPx3 deficiency in kidney disease–induced cardiac disease, we studied the GPx3 knockout mouse strain (GPx3−/−) in the setting of surgery-induced CKD.
Results. Ribonucleic acid (RNA) microarray screening of non-stimulated GPx3−/− heart tissue show increased expression of genes associated with cardiomyopathy including myh7, plac9, serpine1 and cd74 compared with wild-type (WT) controls. GPx3−/− mice underwent surgically induced renal mass reduction to generate a model of CKD. GPx3−/− + CKD mice underwent echocardiography 4 weeks after injury. Fractional shortening (FS) was decreased to 32.9 ± 5.8% in GPx3−/− + CKD compared to 62.0% ± 10.3 in WT + CKD (P < 0.001). Platelet aggregates were increased in the myocardium of GPx3−/− + CKD. Asymmetric dimethylarginine (ADMA) levels were increased in both GPx3−/− + CKD and WT+ CKD. ADMA stimulated spontaneous platelet aggregation more quickly in washed platelets from GPx3−/−. In vitro platelet aggregation was enhanced in samples from GPx3−/− + CKD. Platelet aggregation in GPx3−/− + CKD samples was mitigated after in vivo administration of ebselen, a glutathione peroxidase mimetic. FS improved in GPx3−/− + CKD mice after ebselen treatment.
Conclusion
These results suggest GPx3 deficiency is a substantive contributing factor to the development of kidney disease–induced cardiac disease.
Keywords: cardiorenal syndrome, chronic kidney disease, oxidative stress, platelet, thrombosis
INTRODUCTION
End-stage renal disease (ESRD) patients have an 8.8–10-fold higher incidence of cardiovascular disease compared with the general populations in the USA and Europe [1, 2]. Similar in magnitude to diabetes mellitus, chronic kidney disease (CKD) is recognized as an independent risk factor for cardiovascular disease [3]. The etiology of cardiovascular disease in ESRD patients is unclear, potentially resulting from the activation of several cell-signaling cascades [4–10].
Our previous work focused on the mechanism of the development of left ventricular hypertrophy (LVH) in CKD involving extracellular signal-regulated kinase (ERK) phosphorylation [6, 11]. In this current report we study the potential mechanism by which glutathione peroxidase-3 (GPx3) deficiency causes acute cardiac events in CKD patients. GPx3, previously known as extracellular glutathione peroxidase (Gpx), is a selenoprotein antioxidant enzyme that is synthesized in the basolateral compartment of the kidney [12]. It is transported to the systemic circulation with functions in the plasma, the vessel wall and circulating platelets [13, 14]. GPx3 is the only known GPx subtype with primary activity in the extracellular compartment. Patients with CKD are deficient in this enzyme [15–17]; however, the clinical consequences of this deficiency in CKD are unclear.
We observed that mice deficient in GPx3 due to deletion of the gpx3 gene were more susceptible to myocardial microthrombi, impaired ventricular function and fibrosis in the setting of surgically induced CKD. Extreme GPx3 deficiency was also associated with myocardial platelet activation in the setting of asymmetric dimethylarginine (ADMA) excess. Ebselen, a GPx mimetic, attenuated the pathophysiological effects of GPx3 deficiency, emphasizing the significance of heightened platelet activity in kidney disease-induced cardiac disease. These findings suggest that GPx replacement therapy (or its pharmacological equivalent) has the potential to offset the deleterious effect of GPx3 deficiency in CKD.
MATERIALS AND METHODS
(See supplementary materials for detailed methods.) on ndt online.
Statisical analysis
SPSS (version 22; IBM, Armonk, NY, USA) was used for all statistical analysis.
Genetic mouse model
Genotyping of GPx3−/− mice was performed as previously reported [13].
Gene expression analysis
Gene expression profiles were constructed using the GeneChip Mouse Genome 430 A 2.0 (Affymetrix, Southern Oaks, CA, USA) high-density olignouncleotide microarray technique. Human ventricular myocardium was obtained from the National Disease Research Interchange and was performed under the supervision of the Partners Healthcare Human Research Committee.
Renal injury model
All research involving the use of mice was performed in strict accordance with protocols approved by the Animal Studies Committee of Harvard Medical School. Surgically induced CKD was performed in two stages as previously described [18].
GPx activity assay
GPx3 activity was measured indirectly using blood plasma samples. Preparation of samples was performed in accordance with the manufacturer’s instructions (Cayman Chemical, Ann Arbor, MI, USA).
Murine echocardiography
Transthoracic echocardiography was performed in mice using a Vevo 2100 high-resolution in vivo micro-imaging system (VisualSonics, Toronto, ON, Canada) and a 30-MHz real-time microvisualization scan head as previously described [11].
Murine hemodynamic monitoring
Cardiac catheterization was performed on anesthetized mice as previously described [11, 19].
Cardiac CD41 quantification
Cardiac tissue was prepared in Optimal Cutting Temperature media. Anti-mouse CD41 antibody (Abcam, Cambridge, MA, USA) was applied (1:200). Using cy3 secondary (1:300), CD41+ aggregates were quantified per total cells per high power field (hpf). Aggregates were then averaged among 10 hpfs per animal.
Collagen quantification
Tissue was prepared as per the manufacturer’s recommendations (Chrondrex, Redmond, WA, USA) and Sirius Red staining was performed.
Serum creatinine measurement
Serum creatinine measurements were determined by liquid chromatography–tandem mass spectrometry at the University of Alabama in Birmingham core facility for Acute Kidney Injury Research as previously described [20].
Tissue morphometry
Heart tissue was procured after animals were euthanized. Tissue was dissected, rinsed in cold normal saline, patted dry, then weighed to obtain left ventricular mass.
ADMA serum measurement
ADMA levels were obtained using a mouse/rat ADMA direct ELISA kit (PromoCell, Heidelberg, Germany) according to the manufacturer’s instructions.
L-arginine plasma measurement
L-argine levels were obtained using a competitive arginine ELISA assay kit (Eagle Biosceinces, Nashua, NH, USA). Mouse ethylenediaminetetraacetic acid plasma was prepared according to the manufacturer’s instructions.
Platelet aggregation
After animals were sedated with vaporized isoflurane (1.5% distributed by 1.5 L/min oxygen), blood was drawn by mandibular vein access. Whole blood was anticoagulated using buffer A (65 mM trisodium citrate, 70 mM citric acid, 100 mM dextrose, pH 6.4). Following platelet isolation, platelet aggregation was monitored in a 70-μL volume at 37°C by optical density (OD) after 4 min of agitation.
Ebselen treatment
On postoperative day (POD) 0, animals were treated with ebselen (30 mg/kg) (Sigma Aldrich, St. Louis, MO, USA) by gavage. Doses were given every 3 days, i.e. PODs 0, 3, 6, 9, 12, 15, 18, 21, 24 and 27.
RESULTS
RNA was extracted from GPx3−/− cardiac tissue and compared with wild-type (WT) controls by microarray analysis. Of 14 000 differentially expressed genes, myh7 was the most highly upregulated gene in GPx3−/− heart (2.39-fold, P = 0.0029) (Figure 1A, Supplementary Table S1) and has been associated with pathologic hypertrophic cardiomyopathy [21]. Additionally plac9, associated with familial dilated cardiomyopathy (CMD1C) [22]; serpine1, a prothrombotic factor [23]; and cd74, an adenosine monophosphate–activated protein kinase activator in cardiac ischemia [24] were also upregulated in GPx3−/− heart (Supplementary Figure S1, Supplementary Table S1). We next compared MYH7 expression in the myocardium of 17 patients receiving hemodialysis to that in the myocardium of 13 patients with no kidney disease. MYH7 was increased 11.3-fold in renal disease patients compared with healthy controls (Figure 1B). From these initial results, we inferred that GPx3−/− animals were susceptible to cardiac dysfunction that may have clinical relevance. GPx3−/− with normal renal function showed no evidence of cardiac dysfunction (data not shown). We utilized a surgical kidney mass reduction model to stimulate CKD in GPx3−/− and study cardiac function in a uremic environment with severe GPx3 deficiency.
FIGURE 1.

Gene expression in nonstimulated GPx3−/−. (A) Volcano plot of relative RNA from cardiac tissue identified with an RNA microarray screening platform comparing nonstimulated wild-type (n = 3) and GPx3−/− animals (n = 3). (B) Gene expression by quantitative polymerase chain reaction using comparative cycle time (2−ΔΔCT) normalized to actin for myh7 in mice (n = 4 per group) [nonstimulated WT and GPx3−/− animals (1.00 ± 0.34 versus 2.86 ± 0.93), respectively; *P = 0.01] and humans [healthy control (n = 13), ESRD (n = 17), 0.63 ± 0.76 versus 7.14 ± 4.11, respectively].
Renal-induced cardiac disease in GPx3-deficient animals
In WT C57/B6 animals, GPx3 localized to the intima of the renal microvasculature and colocalized with endothelial cells (CD31) (Figure 2). GPx3 deficiency was assessed in a C57/B6 mouse line that underwent homologous deletion of the GPx3 allele (GPx3−/−) [13]. GPx3−/− and WT mice were subject to a surgical form of severe CKD (see ‘Materials and Methods’ section) [18, 25]. Two weeks after injury, GPx3 activity levels in plasma were reduced by 34.2% in WT littermate controls undergoing surgically induced CKD compared with sham-operated WT mice (Figure 3). Levels in GPx3−/− mice were 96.3% lower than those measured in sham-operated WT mice (58.4 ± 14.7 nmol/min/mL), overlapped with the assay’s negative control values (50.6 ± 2.6) and were not decreased further by CKD surgery. More severe CKD surgeries were performed to further decrease GPx3 levels in WT animals; however, this resulted in animal death within 5 days (data not shown). Steady-state renal function was observed 4 weeks after surgery with equivalent function 8 weeks after surgery. Similar elevations of serum creatinine were maintained in both GPx3−/− and WT mice over this time frame (Figure 4A) using a model we previously reported [18]. ADMA, an endogenous inhibitor of nitric oxide (NO) synthase known to accumulate in CKD [26], was also elevated after 4 and 8 weeks of surgically induced CKD compared to L-arginine plasma levels, which was not significantly altered (Figure 4B, C).
FIGURE 2.

Localized GPx3 expression in the kidney. GPx3 is concentrated in the interstitium of the kidney of WT mice and colocalizes with cells that display CD31: (A) low power (scale bar = 100 μm), (B) high power (scale bar = 10 μm).
FIGURE 3.

Plasma GPx3 activity in GPx3−/− and littermate controls 2 weeks after surgically induced CKD. WT + sham (266.1 ± 44.2) versus WT + CKD (175.2 ± 29.0) (*P < 0.001), WT + CKD versus GPx3−/− + CKD (56.2 ± 2.6) (**P < 0.001) and WT + sham versus GPx3−/− + CKD (†P < 0.001).
FIGURE 4.

Serum creatinine and ADMA levels 4 and 8 weeks after surgically induced CKD in GPx3−/− from nonserial blood draws. (A) Serum creatinine (mg/dL) at 4 weeks WT + sham (0.11 ± 0.02) (n = 6) versus WT + CKD (0.21 ± 0.02) (n = 6) (*P < 0.001) and 8 weeks 0.11 ± 0.02 (n = 6) versus 0.20 ± 0.02 (n = 6) (**P < 0.001), respectively; GPx3−/− + sham (0.13 ± 0.02) (n = 6) versus GPx3−/− + CKD (0.22 ± 0.02) (n = 6) (†P < 0.001) and 8 weeks [0.12 ± 0.02 (n = 6) versus 0.21 ± 0.02 (n = 6)] (‡P < 0.001), respectively. (B) Serum ADMA (μM) at 4 weeks WT + sham (0.47 ± 0.47) (n = 4) versus WT + CKD (5.00 ± 0.80) (n = 4) (*P < 0.001) and GPx3−/− + sham (0.39 ± 0.39) (n = 6) versus GPx3−/− + CKD (7.08 ± 0.66) (n = 7) (**P = 0.019) and 8 weeks WT + sham [0.39 ± 0.38 (n = 6)] versus WT + CKD [5.40 ± 4.07 (n = 7)] (†P = 0.030) and GPx3−/− + sham [0.59 ± 0.57 (n = 6)] versus GPx3−/− + CKD [5.34 ± 4.04 (n = 7)] (‡P = 0.024), respectively. (C) Plasma L-arginine (μM) at 4 weeks WT + sham (44 ± 16) (n = 4) versus WT + CKD (41 ± 30) (n = 5) (P = NS) and GPx3−/− + sham (41 ± 19) (n = 4) versus GPx3−/− + CKD (51 ± 30) (n = 4)] (P = NS) and 8 weeks WT + sham [38 ± 13 (n = 4)] versus WT + CKD [23 ± 10 (n = 5)] (P = NS) and GPx3−/− + sham [35 ± 17 (n = 4)] versus GPx3−/− + CKD [21 ± 8 (n = 4)] (P = NS), respectively.
Echocardiographic and hemodynamic measures of heart failure
Animals underwent echocardiography 4 weeks after surgery. Left ventricular mass increased in both WT and GPx3−/− mice undergoing CKD surgery; however, left ventricular fractional shortening (FS) was decreased to 32.9 ± 5.8% in GPx3−/− + CKD compared to 62.0 ± 10.3% (normal) in WT + CKD mice (P < 0.05) (Figure 5, Table 1). Left ventricular cardiac catheterization was also performed 4 weeks after surgery. Although there was no significant difference in left ventricular end-diastolic pressure (LVEDP), the maximum left ventricular pressure (LVPmax) during systole and mean systemic arterial pressure were significantly decreased in GPx3−/− + CKD compared with WT + CKD mice (Figure 6). Gram weight of animals at the time of cardiac catheterization was equivalent to that of animals prior to injury, inferring that volume overload did not accompany decreased LVPmax. Eight weeks after surgery, a significantly different proportion of GPx3−/− + CKD mice died (50.0%) compared with WT + CKD mice (10.0%; Figure 7) (P = 0.047).
FIGURE 5.

Echocardiography after CKD. Representative left ventricular wall motion per unit time (milliseconds) in WT + CKD versus GPx3−/− + CKD mice 4 weeks after surgically induced CKD.
Table 1.
Morphologic and echocardiographic analysis of GPx3−/− mice 4 weeks after surgically induced CKD
| Morphologic and echocardiographic analysis of GPx3−/− mice 4 weeks after CKD surgery | ||||
|---|---|---|---|---|
| WT +sham | GPx3−/− + sham | WT + CKD | GPx3−/− + CKD | |
| (n = 5) | (n = 5) | (n = 6) | (n = 6) | |
| BW (g) | 24.7 ± 2.8 | 24.8 ± 0.5 | 25.3 ± 0.9 | 25.8 ± 0.9 |
| HR (bpm) | 593 ± 35 | 566 ± 26 | 580 ± 14* | 542 ± 24** |
| LVM (mg) | 86.1 ± 0.4 | 88.2 ± 8.7 | 99.2 ± 3.4* | 96.4 ± 15.1** |
| FS (%) | 68.8 ± 2.4 | 73.1 ± 12.3 | 62 ± 10.3 | 32.9 ± 5.8*** |
| LVM/BW | 3.4 ± 0.5 | 3.6 ± 0.3 | 4 ± 0.2* | 3.7 ± 0.4 |
BW, body weight; bpm, beats per minute; HR, heart rate; FS, fractional shortening; LVM, left ventricular mass by dry weight.
P < 0.05, WT + sham versus WT + CKD, solid line.
P < 0.05, GPx3−/− + sham versus GPx3−/− + CKD, dotted line.
P < 0.05, WT + CKD versus GPx3−/− + CKD, red line.
FIGURE 6.

Aortic and left ventricular cardiac catheterization in GPx3−/− 4 weeks after surgically induced CKD. Continuous blood pressure measurements were taken in the aortic root and left ventricle in (A) WT + CKD and (B) GPx3−/− + CKD mice. (C) Mean arterial pressure (MAP) in WT + CKD (64.85 ± 0.48) (n = 6) versus GPx3−/− + CKD (55.78 ± 1.89) (n = 6) (P = 0.001) mice, LVPmax in WT and CKD (90.86 ± 1.04) (n = 6) versus GPx3−/− + CKD (75.72 ± 1.50) (n = 6) (P < 0.001) mice and LVEDP in WT + CKD (10.34 ± 0.58) (n = 6) versus GPx3−/− + CKD (10.23 ± 0.55) (n = 6) (P = NS) mice.
FIGURE 7.

Survival analysis after surgically induced CKD in GPx3−/− mice. Groups were compared by log-rank (Mantel–Cox) test. Six of 12 GPx3−/− animals (50.0%) survived 8 weeks after injury compared to 9 of 10 littermate control animals (90.0%) undergoing injury (P = 0.047).
Platelet dysfunction in the setting of CKD and GPx3 deficiency
To understand better the cause of early mortality in GPx3−/− + CKD mice, cardiac tissue was evaluated histologically. Using a murine-specific monoclonal CD41 antibody (to integrin alpha-IIb), intracardiac platelet microthrombi were detected 4 weeks after CKD in GPx3−/− mice at levels significantly greater than controls (P < 0.001) and WT + CKD mice (P < 0.001) (Figure 8A). Collagen type I–V staining was increased after 8 weeks in GPx3−/− + CKD mice compared with GPx3−/− + sham mice (Figure 8B). WT + CKD mice also developed collagen deposition, but it was less extensive than that detected in GPx3−/− + CKD mice.
FIGURE 8.

CD41 and collagen content in GPx3−/− myocardium after surgically induced CKD. (A) four weeks after CKD, CD41+ thrombi per total cell number per hpf in GPx3−/− + sham (0.03 ± 0.01) (n = 9) versus GPx3−/− + CKD (0.17 ± 0.05) (n = 9) mice (*P < 0.001) and WT + CKD (0.03 ± 0.01) (n = 9) versus GPx3−/− + CKD mice (**P < 0.001). (B) Collagen content per total pixel area after 8 weeks: WT + sham (0.19 ± 0.04) (n = 10) mice versus WT + CKD (0.38 ± 0.07) (n = 10) mice (*P < 0.001), GPx3−/− + sham (0.21 ± 0.05) (n = 10) versus GPx3−/− + CKD (0.51 ± 0.08) (n = 10) mice (**P < 0.001) and WT + CKD versus GPx3−/− + CKD mice (†P = 0.002).
GPx3−/− animals have previously shown to be susceptible to thrombosis following platelet activation; this effect was shown to be a consequence of impaired metabolism of reactive oxygen species leading to enhanced oxidative inactivation of NO [13, 27, 28]. As the increased concentration of microthrombi in GPx3−/− mice was observed only after kidney injury and not in sham-treated mice, we concluded that this was due to the presence of an additional prothrombotic determinant in the uremic milieu that triggered platelet activation. As described above, levels of ADMA, an NO synthase inhibitor, were significantly elevated in WT + CKD and GPx3−/− + CKD mice compared with age-matched sham controls at 4 weeks and 8 weeks after surgery (Figure 4B). Washed platelet studies showed increased platelet aggregation, based on decreased OD, when platelets derived from GPx3−/− sham controls compared with WT sham-treated mice were exposed to 1 μM ADMA (0.62 ± 0.05 versus 0.70 ± 0.06 OD; P = 0.028) and 10 μM ADMA (0.47 ± 0.04 versus 0.57 ± 0.04 OD; P = 0.004), respectively (Figure 9A). ADMA concentrations used for the in vitro studies were consistent with levels measured in the animal models.
FIGURE 9.
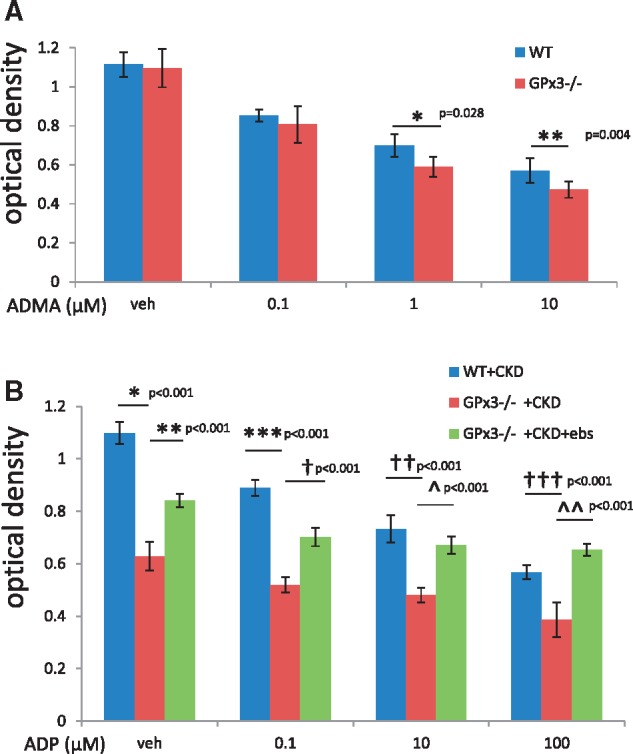
Platelet aggregation in GPx3−/− in the setting of chronic kidney injury. (A) OD of washed platelets derived from WT + sham versus GPx3−/− + sham following treatment with 1 μM ADMA (0.70 ± 0.06) (n = 6 animals) versus (0.62 ± 0.05) (n = 6 animals) (*P = 0.028) and 10 μM ADMA (0.57 ± 0.04) (n = 6 animals) versus (0.47 ± 0.04) (n = 6 animals) (**P = 0.004). (B) OD after treatment with vehicle, WT + CKD (1.10 ± 0.04) (n = 6 animals) versus GPx3−/−+ CKD (0.63 ± 0.05) mice (*P < 0.001), GPx3−/− + CKD + vehicle versus GPx3−/− + CKD + ebselen (0.84 ± 0.03) mice (**P < 0.001) after 0.1 μM ADP; WT + CKD (0.89 ± 0.03) (n = 7 animals) versus GPx3−/− + CKD (0.52 ± 0.03) mice (***P < 0.001), GPx3−/− + CKD + vehicle versus GPx3−/− + CKD + ebselen (0.70 ± 0.03) mice (†P < 0.001) after 10 μM ADP, WT + CKD (0.73 ± 0.05) (n = 7 animals) versus GPx3−/− + CKD + vehicle (0.48 ± 0.03) mice (††P < 0.001), GPx3−/− + CKD + vehicle versus GPx3−/− + CKD + ebselen (0.67 ± 0.03) mice (^P < 0.001) and after 100 μM ADP WT + CKD (0.57 ± 0.03) (n = 7 animals) versus GPx3−/− + CKD + vehicle (0.39 ± 0.07) (†††P < 0.001), GPx3−/− + CKD + vehicle versus GPx3−/− + CKD + ebselen (0.65 ± 0.02) (^^P < 0.001).
Treatment with the GPx mimetic ebselen
The rate of platelet aggregation with adenosine diphosphate (ADP) stimulation of P2Y12 receptor was increased for platelets isolated from GPx3−/− + CKD compared with WT + CKD mice (Figure 9B). Recognizing that ebselen is a GPx mimetic, GPx3−/− + CKD mice were next treated with ebselen for 4 weeks. Platelets isolated from GPx3−/− + CKD + ebselen mice showed decreased aggregation at each ADP dose compared with platelets isolated from vehicle-treated GPx3−/− + CKD mice. GPx3−/− + CKD + ebselen mice also showed an increase in left ventricular FS compared with GPx3−/− + CKD + vehicle mice 4 weeks after injury (55.0 ± 13.0% versus 30.6 ± 9.5%, respectively; P = 0.003) (Table 2). Histologic analysis of hearts (Figure 10) showed a decreased number of CD41 platelet aggregates per total cells per hpf in ebselen-treated GPx3−/− + CKD mice compared with vehicle-treated GPx3−/− + CKD mice after 4 weeks (0.171 ± 0.017 versus 0.058 ± 0.005; P < 0.001). Myh7 expression was not decreased in treated animals, however, Col3a1 and SOD3, markers of fibrosis and oxidative stress, respectively, were both downregulated with ebselen treatment. Eight weeks after therapy, absolute collagen content was decreased compared with controls; however, this change was not statistically significant.
Table 2.
Morphologic and echocardiographic analysis of GPx3-/- mice 4 weeks after surgically induced CKD and 4 weeks of ebselen treatment
| GPx3−/− +CKD + vehicle | GPx3−/− +CKD + ebselen | |
|---|---|---|
| (n = 7) | (n = 7) | |
| BW (g) | 24.8 ± 2.3 | 24.2 ± 1 |
| HR (bpm) | 549 ± 22 | 545.6 ± 19.3 |
| LVM (mg) | 98.2 ± 7.4 | 97.4 ± 5.2 |
| FS (%) | 30.1 ± 3.8 | 49.4 ± 9.8* |
| LVM/BW | 4.1 ± 0.2 | 3.9 ± 0.3 |
BW, body weight; bpm, beats per minute; HR, heart rate; LVM, left ventricular mass by dry weight (mg).
P < 0.05
FIGURE 10.

Cardiac phenotype of GPx3−/− after surgically induced CKD in animals treated with ebselen. (A) CD41 content (platelet aggregates/total cells) in GPx3−/− + CKD + vehicle (n = 7) versus GPx3−/− + CKD + ebselen (n = 6), (0.171 ± 0.017 versus 0.058 ± 0.005; *P < 0.001), respectively. (B) mRNA expression 4 weeks after surgery in GPx3−/− + CKD + vehicle (n = 5) versus GPx3−/− + CKD + ebselen (n = 5) mice, myh7 [1.15 ± 0.58 (2−ΔΔCT) versus 1.86 ± 1.11] (P = NS), col3a1 (1.00 ± 0.25 versus 0.68 ± 0.16) (P = 0.042), SOD3 (1.07 ± 0.32 versus 0.69 ± 0.13) (P = 0.039), respectively. (C) Collagen content per total pixel area 8 weeks after surgery in GPx3−/− + CKD + vehicle (n = 7) versus GPx3−/− + CKD + ebselen (n = 7) (0.54 ± 0.09 versus 0.47 ± 0.05; P = NS), respectively.
DISCUSSION
GPx3 deficiency has previously been investigated in hemodialysis-dependent patients and those with CKD, but studies of its association with cardiovascular disease in these patients has thus far been limited [15, 29, 30]. CKD promotes significant oxidative stress [31, 32] that leads to endothelial dysfunction [33], reduced levels of NO [34], vascular disease and hence a parallel tendency toward thrombosis [35]. Furthermore, it is recognized that the frequency of thromboembolic events in patients with CKD is increased compared with age-matched controls, occurring 2.34 times more frequently than in non-CKD patients [36]. In this context, we report an association between GPx3 deficiency and cardiac microthrombosis in the setting of CKD. We demonstrate for the first time the significance of severe GPx3 deficiency in a preclinical GPx3−/− rodent model of CKD.
GPx proteins are selenoproteins capable of reducing hydrogen peroxide and organic peroxides [37]. This family of enzymes is typified by glutathione peroxidase 1 (GPx1) and GPx3. Widely distributed in the cytosolic compartment of most cells, decreased GPx1 activity has been linked to cardiovascular disease burden [38, 39]. It has been described as a cardiovascular risk factor in CKD; however, the mechanism of systemic deficiency in CKD remains unclear, as GPx1 synthesis is not exclusive to the kidney [40]. In contrast, GPx3 is a selenoprotein primarily synthesized by epithelial cells in the proximal renal tubule, then secreted and distributed to the plasma and circulating platelet and endothelial microenvironment [12, 41–44]. GPx3 is the primary selenoprotein found in the circulation, and its deficiency results in the accumulation of circulating reactive oxygen species, including hydrogen peroxide and organic hydroperoxides [13].
In this study we have overcome the challenge of recapitulating GPx3 levels <175 nmol/min/mL in preclinical CKD models. Until now, such GPx3 deficiency has only been achievable in models of bilateral nephrectomy. In WT mice, aggressive renal ablation techniques are needed to achieve this level of reduction, which at the same time, incurs significant mortality. This limitation was obviated in our studies by utilizing the GPx3−/− mouse strain, where GPx3 activity levels overlapped with the assay’s negative control (see Figure 3 and the ‘Materials and Methods’ section).
Prior to this report, thrombosis of the coronary microvasculature had not been described as a phenotype of the GPx3−/− mouse strain. GPx3−/− animals were first found to be susceptible to thrombotic stroke following cerebral ischemia/reperfusion injury [13]. In our report we showed that microthrombi in the myocardium were increased in GPx3−/− mice in the context of CKD, a phenotype not found in WT mice with CKD. This implicated the additive effect of a uremic factor exacerbating the prothrombotic state of GPx3 deficiency in GPx3−/− mice.
In the environment of GPx3 deficiency, we observed an accumulation of plasma ADMA after CKD similar to the levels found in WT mice with CKD. However, we also showed in washed platelet aggregation studies that GPx3−/− platelets isolated from mice not undergoing CKD were more susceptible to spontaneous aggregation in the presence of ADMA than WT platelets. Further, washed platelets from GPx3−/− + CKD were already susceptible to aggregation prior to ADP stimulation (Figure 9B). Thus platelet aggregation studies suggest that ADMA (an inhibitor of NO synthase) had a direct effect on platelet aggregation by inhibiting intraplatelet NO synthase and promoting platelet activation in the setting of severe GPx3 deficiency [45, 46]. These findings implicate GPx3 activity <175 nmol/min/mL in combination with ADMA levels ≥5 pg/mL as factors necessary to initiate microthrombosis in vivo.
ADMA is an inhibitor of NO synthase, and patients with declining glomerular and tubular function are known to have elevated plasma ADMA levels [47–49]. The proven clinical relevance of ADMA in patients with CKD influenced our decision to study this NO synthase inhibitor rather than other uremic toxins (such as phenyl acetic acid, indoxyl sulfate, p-cresyl sulfate and homocysteine) in platelet activation studies [50–55]. Reduced concentrations of NO perpetuate platelet activation through a phosphoinositide kinase 3 (PI3K)-dependent pathway. NO prevents the formation of a complex between intracellular PI3K and the src family protein lyn. In a NO-depleted state, the extracellular thrombin receptor can then transmit an intracellular signal for platelet activation through PI3K while at the same time inhibiting guanylyl cyclase–dependent platelet homeostasis [56, 57]. In the composite setting of elevated ADMA and severely depressed GPx3, we identified an increased concentration of microthrombi in the cardiac vasculature that could be explained by impaired NO synthase activity and an increased pool of oxidants, respectively.
In addition to intracardiac microthrombi, we found that GPx3 deficiency was associated with decreased myocardial FS by echocardiography in GPx3−/− + CKD mice and increased collagen deposition. Microthrombi, which were not present in sham-operated animals or WT animals, suggested a mechanism for systolic dysfunction. GPx3−/− mice undergoing the sham procedure manifest no excess microthrombi or systolic dysfunction. Similar to the lack of effect of GPx3 deficiency on cerebral blood flow in unstressed animals [13], we did not observe a baseline decrease in left ventricular systolic function in knockout animals or animals undergoing sham surgery.
GPx3−/− + CKD rapidly developed a decline in FS over 4 weeks along with cardiac fibrosis, hypotension and a high mortality rate, pathological changes consistent with severe systolic heart failure [58–60]. It has been recognized by several groups that CKD-induced cardiac disease can be characterized by cardiac fibrosis in the absence of hypertension [11, 61–63]. Our model recapitulates these findings while adding the additional characteristics of severe systolic dysfunction and microvascular coagulopathy often seen in clinical presentation.
Ebselen proved to be an effective treatment for the model of CKD and severe GPx3 deficiency. Ritz et al. [61] inferred this mechanism several years ago and showed a reduction in medial thickness of cardiac arterioles following antioxidant therapy in a model of uninephrectomy in ApoE−/− mice. In contrast to these earlier studies, our investigations focused on the intravascular effect of ebselen on platelet activity. We showed that washed platelets that were collected from GPx3−/− + CKD + ebselen mice were less activated following ADP stimulation at multiple concentrations than vehicle-treated controls. In vivo, the cardiac pathology observed in GPx3−/− + CKD mice was partially reversed by ebselen with an improvement in FS. Cardiac tissue from these animals revealed a decrease in col3a1 and SOD3 gene expression, consistent with a system under decreased oxidative stress [64, 65]. Histologically, the same tissue showed fewer platelet aggregates in the ebselen-treated group.
These findings suggest that ebselen overcomes the effects of severe GPx3 deficiency, attenuating the initiation of platelet activation in the context of elevated plasma ADMA levels. In contrast, these findings also suggest that persistently low GPx3 levels promote NO consumption while ADMA inhibits NO synthase, promoting endothelial dysfunction [13], and thereby enhancing platelet susceptibility to agonist-induced activation. The combined effect of GPx3 gene disruption and ADMA excess in our preclinical model shows a remarkable display of cardiac pathology that cannot otherwise be generated in a single-mammalian model system and often takes several years to develop in the clinical setting of progressive renal failure.
The limitation of our study centers on the persistence of LVH following ebselen administration. Reduced thrombotic burden in the cardiac microvasculature by administration of a GPx mimetic was anticipated, given the previous reports that GPx3 deficiency promoted injury in the context of acute cerebral ischemia [13]; however, it is unclear how GPx3 deficiency may contribute to fibrosis and LVH. We previously found that LVH due to CKD can be treated with mammalian target of rapamycin (mTOR) blockade and inhibition of ERK phosphorylation. Overall, the mechanism for the development of normotensive LVH remains unknown. Our current working model thus depicts two pathologic processes that separately involve microthrombosis due to GPx3 deficiency and cardiac hypertrophy due to mTOR pathway activation. New models of kidney disease–induced cardiac disease may benefit from incorporating both processes in phenotypic analyses.
In summary, patients with CKD have deficient levels of circulating GPx3, which may contribute to cardiovascular disease risk due to the accumulation of reactive oxygen species that exacerbate inflammatory signaling and platelet activation in the setting of ADMA-impaired NO generation in the microvasculature of the myocardium (Figure 11). This prothrombotic state can be counteracted by oral GPx mimetic supplementation. Such a condition warrants future clinical studies that prospectively study the incidence of cardiac events in CKD patients with relative GPx3 deficiency.
FIGURE 11.
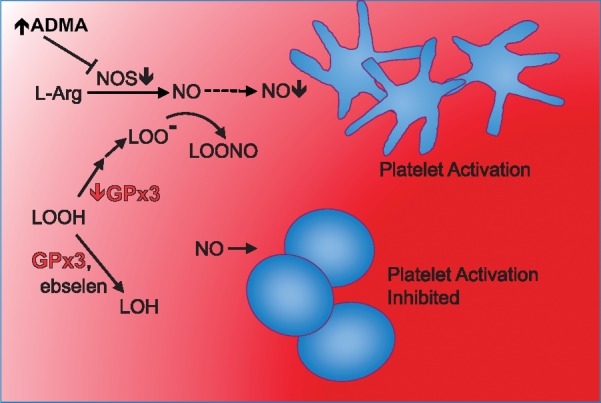
GPx3 converts hydrogen or lipid peroxides to their corresponding alcohols (i.e. water and lipid alcohols), inhibiting the cascade of platelet activation. ADMA inhibits NO synthase, which inhibits synthesis of NO (dotted line) from L-arginine. Platelet activation is greatest in an ADMA-rich, GPx3-deficient state consistent with the plasma environment of patients with severe CKD. In this environment, platelet inhibitory NO is decreased by reduced synthesis via ADMA inhibition of NO synthase and by increased oxidative consumption via reaction with peroxyl derivatives of hydrogen and lipid peroxides.
SUPPLEMENTARY DATA
Supplementary data are available at ndt online.
Supplementary Material
ACKNOWLEDGEMENTS
Material contained here was presented as an oral presentation at the 2013 American Society of Nephrology annual meeting as abstract 189. We thank Partners HealthCare for making the SPSS statistical software package available to physicians working for Partners HealthCare.
FUNDING
This work was supported by a National Institutes of Health grant (K08DK089002) (A.M.S.), a Brigham and Women’s Hospital Faculty Career Development Award (A.M.S.) and National Institutes of Health grants R37HL061795, P01HL048743, U01HL108630 and P50GM107618 (J.L.).
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
- 1. Sarnak MJ, Levey AS.. Epidemiology, diagnosis, and management of cardiac disease in chronic renal disease. J Thromb Thrombolysis 2000; 10: 169–180 [DOI] [PubMed] [Google Scholar]
- 2. de Jager DJ, Grootendorst DC, Jager KJ. et al. Cardiovascular and noncardiovascular mortality among patients starting dialysis. JAMA 2009; 302: 1782–1789 [DOI] [PubMed] [Google Scholar]
- 3. Go AS, Chertow GM, Fan D. et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351: 1296–1305 [DOI] [PubMed] [Google Scholar]
- 4. Tsai YC, Lee CT, Huang TL. et al. Inflammatory marker but not adipokine predicts mortality among long-term hemodialysis patients. Mediators Inflamm 2007; 2007: 19891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stosovic M, Stanojevic M, Radovic M. et al. Hemodialysis modality, percentage of body fat, and patient survival. Int J Artif Organs 2009; 32: 20–30 [DOI] [PubMed] [Google Scholar]
- 6. de Mattos AM, Siedlecki A, Gaston RS. et al. Systolic dysfunction portends increased mortality among those waiting for renal transplant. J Am Soc Nephrol 2008; 19: 1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garibotto G, Sofia A, Procopio V. et al. Peripheral tissue release of interleukin-6 in patients with chronic kidney diseases: effects of end-stage renal disease and microinflammatory state. Kidney Int 2006; 70: 384–390 [DOI] [PubMed] [Google Scholar]
- 8. Psychari SN, Sinos L, Iatrou C. et al. Relations of inflammatory markers to lipid levels and autonomic tone in patients with moderate and severe chronic kidney disease and in patients under maintenance hemodialysis. Clin Nephrol 2005; 64: 419–427 [DOI] [PubMed] [Google Scholar]
- 9. Kimmel PL, Phillips TM, Simmens SJ. et al. Immunologic function and survival in hemodialysis patients. Kidney Int 1998; 54: 236–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oh HJ, Nam BY, Lee MJ. et al. Decreased circulating klotho levels in patients undergoing dialysis and relationship to oxidative stress and inflammation. Perit Dial Int 2015; 35: 43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Siedlecki AM, Jin X, Muslin AJ.. Uremic cardiac hypertrophy is reversed by rapamycin but not by lowering of blood pressure. Kidney Int 2009; 75: 800–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Avissar N, Ornt DB, Yagil Y. et al. Human kidney proximal tubules are the main source of plasma glutathione peroxidase. Am J Physiol 1994; 266: C367–C375 [DOI] [PubMed] [Google Scholar]
- 13. Jin RC, Mahoney CE, Coleman Anderson L. et al. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation 2011; 123: 1963–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Voetsch B, Jin RC, Bierl C. et al. Promoter polymorphisms in the plasma glutathione peroxidase (GPx-3) gene: a novel risk factor for arterial ischemic stroke among young adults and children. Stroke 2007; 38: 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roxborough HE, Mercer C, McMaster D. et al. Plasma glutathione peroxidase activity is reduced in haemodialysis patients. Nephron 1999; 81: 278–283 [DOI] [PubMed] [Google Scholar]
- 16. Wong FN, Tan JA, Keng TC. et al. Association between plasma soluble RAGE and renal function is unaffected by medication usage and enzymatic antioxidants in chronic kidney disease with type 2 diabetes. Clin Chim Acta 2016; 453: 56–61. [DOI] [PubMed] [Google Scholar]
- 17. Fassett RG, Robertson IK, Ball MJ. et al. Effects of atorvastatin on oxidative stress in chronic kidney disease. Nephrology (Carlton) 2015; 20: 697. [DOI] [PubMed] [Google Scholar]
- 18. Pang P, Abbott M, Chang SL. et al. Human vascular progenitor cells derived from renal arteries are endothelial-like and assist in the repair of injured renal capillary networks. Kidney Int 2017; 91: 129–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rogers JH, Tamirisa P, Kovacs A. et al. RGS4 causes increased mortality and reduced cardiac hypertrophy in response to pressure overload. J Clin Invest 1999; 104: 567–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pang P, Jin X, Proctor BM. et al. RGS4 inhibits angiotensin II signaling and macrophage localization during renal reperfusion injury independent of vasospasm. Kidney Int 2015; 87: 771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fananapazir L, Dalakas MC, Cyran F. et al. Missense mutations in the beta-myosin heavy-chain gene cause central core disease in hypertrophic cardiomyopathy. Proc Natl Acad Sci USA 1993; 90: 3993–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuhl A, Melberg A, Meinl E. et al. Myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7: corroboration and narrowing of the critical region on 10q22.3. Eur J Hum Genet 2008; 16: 367–373. [DOI] [PubMed] [Google Scholar]
- 23. Morange PE, Saut N, Alessi MC. et al. Association of plasminogen activator inhibitor (PAI)-1 (SERPINE1) SNPs with myocardial infarction, plasma PAI-1, and metabolic parameters: the HIFMECH study. Arterioscler Thromb Vasc Biol 2007; 27: 2250–2257 [DOI] [PubMed] [Google Scholar]
- 24. Miller EJ, Li J, Leng L. et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature 2008; 451: 578–582 [DOI] [PubMed] [Google Scholar]
- 25. Polichnowski AJ, Lan R, Geng H. et al. Severe renal mass reduction impairs recovery and promotes fibrosis after AKI. J Am Soc Nephrol 2014; 25: 1496–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kielstein JT, Boger RH, Bode-Boger SM. et al. Asymmetric dimethylarginine plasma concentrations differ in patients with end-stage renal disease: relationship to treatment method and atherosclerotic disease. J Am Soc Nephrol 1999; 10: 594–600 [DOI] [PubMed] [Google Scholar]
- 27. Freedman JE, Loscalzo J, Benoit SE. et al. Decreased platelet inhibition by nitric oxide in two brothers with a history of arterial thrombosis. J Clin Invest 1996; 97: 979–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Freedman JE, Loscalzo J, Barnard MR. et al. Nitric oxide released from activated platelets inhibits platelet recruitment. J Clin Invest 1997; 100: 350–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crawford A, Fassett RG, Coombes JS. et al. Glutathione peroxidase, superoxide dismutase and catalase genotypes and activities and the progression of chronic kidney disease. Nephrol DialTransplant 2011; 26: 2806–2813. [DOI] [PubMed] [Google Scholar]
- 30. Glorieux G, Mullen W, Duranton F. et al. New insights in molecular mechanisms involved in chronic kidney disease using high-resolution plasma proteome analysis. Nephrol Dial Transplant 2015; 30: 1842–1852 [DOI] [PubMed] [Google Scholar]
- 31. Passauer J, Pistrosch F, Bussemaker E. et al. Reduced agonist-induced endothelium-dependent vasodilation in uremia is attributable to an impairment of vascular nitric oxide. J Am Soc Nephrol 2005; 16: 959–965. [DOI] [PubMed] [Google Scholar]
- 32. Annuk M, Zilmer M, Lind L. et al. Oxidative stress and endothelial function in chronic renal failure. J Am Soc Nephrol 2001; 12: 2747–2752 [DOI] [PubMed] [Google Scholar]
- 33. Linden E, Cai W, He JC. et al. Endothelial dysfunction in patients with chronic kidney disease results from advanced glycation end products (AGE)-mediated inhibition of endothelial nitric oxide synthase through RAGE activation. Clin J Am Soc Nephrol 2008; 3: 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bongartz LG, Braam B, Verhaar MC. et al. The nitric oxide donor molsidomine rescues cardiac function in rats with chronic kidney disease and cardiac dysfunction. Am J Physiol Heart Circ Physiol 2010; 299: H2037–H2045 [DOI] [PubMed] [Google Scholar]
- 35. Casserly LF, Dember LM.. Thrombosis in end-stage renal disease. Semin Dial 2003; 16: 245–256 [DOI] [PubMed] [Google Scholar]
- 36. Wattanakit K, Cushman M, Stehman-Breen C. et al. Chronic kidney disease increases risk for venous thromboembolism. J Am Soc Nephrol 2008; 19: 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Loscalzo J. Antioxidant enzyme deficiencies and vascular disease. Expert Rev Endocrinol Metab 2010; 5: 15–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paglia DE, Valentine WN.. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med 1967; 70: 158–169 [PubMed] [Google Scholar]
- 39. Blankenberg S, Rupprecht HJ, Bickel C. et al. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N Engl J Med 2003; 349: 1605–1613 [DOI] [PubMed] [Google Scholar]
- 40. Girelli D, Olivieri O, Stanzial AM. et al. Low platelet glutathione peroxidase activity and serum selenium concentration in patients with chronic renal failure: relations to dialysis treatments, diet and cardiovascular complications. Clin Sci 1993; 84: 611–617 [DOI] [PubMed] [Google Scholar]
- 41. Whitin JC, Tham DM, Bhamre S. et al. Plasma glutathione peroxidase and its relationship to renal proximal tubule function. Mol Gen Metab 1998; 65: 238–245 [DOI] [PubMed] [Google Scholar]
- 42. Olson GE, Whitin JC, Hill KE. et al. Extracellular glutathione peroxidase (Gpx3) binds specifically to basement membranes of mouse renal cortex tubule cells. Am J Physiol Renal Physiol 2010; 298: F1244–F1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Burk RF, Olson GE, Winfrey VP. et al. Glutathione peroxidase-3 produced by the kidney binds to a population of basement membranes in the gastrointestinal tract and in other tissues. Am J Physiol Gastrointest Liver Physiol 2011; 301: G32–G38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Malinouski M, Kehr S, Finney L. et al. High-resolution imaging of selenium in kidneys: a localized selenium pool associated with glutathione peroxidase 3. Antioxid Redox Signal 2012; 16: 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kanaya S, Ikeda H, Haramaki N. et al. Intraplatelet tetrahydrobiopterin plays an important role in regulating canine coronary arterial thrombosis by modulating intraplatelet nitric oxide and superoxide generation. Circulation 2001; 104: 2478–2484 [DOI] [PubMed] [Google Scholar]
- 46. Schoenfeld H, Muhm M, Doepfmer U. et al. Platelet activity in washed platelet concentrates. Anesth Analg 2004; 99: 17–20 [DOI] [PubMed] [Google Scholar]
- 47. Antoniades C, Shirodaria C, Leeson P. et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur Heart J 2009; 30: 1142–1150 [DOI] [PubMed] [Google Scholar]
- 48. Vallance P, Leone A, Calver A. et al. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992; 339: 572–575 [DOI] [PubMed] [Google Scholar]
- 49. Kielstein JT, Boger RH, Bode-Boger SM. et al. Marked increase of asymmetric dimethylarginine in patients with incipient primary chronic renal disease. J Am Soc Nephrol 2002; 13: 170–176 [DOI] [PubMed] [Google Scholar]
- 50. Luksha L, Stenvinkel P, Hammarqvist F. et al. Mechanisms of endothelial dysfunction in resistance arteries from patients with end-stage renal disease. PLoS One 2012; 7: e36056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin IC, Hsu CN, Lo MH. et al. Low urinary citrulline/arginine ratio associated with blood pressure abnormalities and arterial stiffness in childhood chronic kidney disease. J Am Soc Hypertens 2016; 10: 115–123. [DOI] [PubMed] [Google Scholar]
- 52. Hov GG, Sagen E, Hatlen G. et al. Arginine/asymmetric dimethylarginine ratio and cardiovascular risk factors in patients with predialytic chronic kidney disease. Clin Biochem 2011; 44: 642–646 [DOI] [PubMed] [Google Scholar]
- 53. Baylis C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat Clin Pract Nephrol 2006; 2: 209–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Itoh Y, Ezawa A, Kikuchi K. et al. Protein-bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ROS production. Anal Bioanal Chem 2012; 403: 1841–1850 [DOI] [PubMed] [Google Scholar]
- 55. Jamison RL, Hartigan P, Kaufman JS. et al. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: a randomized controlled trial. JAMA 2007; 298: 1163–1170 [DOI] [PubMed] [Google Scholar]
- 56. Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ Res 2001; 88: 756–762 [DOI] [PubMed] [Google Scholar]
- 57. Pigazzi A, Heydrick S, Folli F. et al. Nitric oxide inhibits thrombin receptor-activating peptide-induced phosphoinositide 3-kinase activity in human platelets. J Biol Chem 1999; 274: 14368–14375 [DOI] [PubMed] [Google Scholar]
- 58. De Pasquale CG, Dunne JS, Minson RB. et al. Hypotension is associated with diuretic resistance in severe chronic heart failure, independent of renal function. Eur J Heart Fail 2005; 7: 888–891 [DOI] [PubMed] [Google Scholar]
- 59. Patel PA, Heizer G, O'Connor CM. et al. Hypotension during hospitalization for acute heart failure is independently associated with 30-day mortality: findings from ASCEND-HF. Circ Heart Fail 2014; 7: 918–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Harjola VP, Lassus J, Sionis A. et al. Clinical picture and risk prediction of short-term mortality in cardiogenic shock. Eur J Heart Fail 2015; 17: 501–509 [DOI] [PubMed] [Google Scholar]
- 61. Piecha G, Koleganova N, Gross ML. et al. Oxidative stress after uninephrectomy alters heart morphology in the apolipoprotein E -/- mouse. J Hypertens 2008; 26: 2220–2229 [DOI] [PubMed] [Google Scholar]
- 62. Dahan M, Siohan P, Viron B. et al. Relationship between left ventricular hypertrophy, myocardial contractility, and load conditions in hemodialysis patients: an echocardiographic study. Am J Kidney Dis 1997; 30: 780–785. [DOI] [PubMed] [Google Scholar]
- 63. Hu MC, Shi M, Cho HJ. et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol 2015; 26: 1290–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lutucuta S, Tsybouleva N, Ishiyama M. et al. Induction and reversal of cardiac phenotype of human hypertrophic cardiomyopathy mutation cardiac troponin T-Q92 in switch on-switch off bigenic mice. J Am Coll Cardiol 2004; 44: 2221–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Estruch R, Sacanella E, Mota F. et al. Moderate consumption of red wine, but not gin, decreases erythrocyte superoxide dismutase activity: a randomised cross-over trial. Nutr Metab Cardiovasc Dis 2011; 21: 46–53 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.