

Summary
Lafora disease (LD) clinically appears in previously healthy teenagers as progressively worsening seizures, myoclonus, dementia, and ultimately a vegetative state leading to death within a decade of its onset. Here we present a typical case of LD in which the patient survived until the age of 40. Although the patient's brain was severely affected, other organs remained functional until her death. The field of LD research is approaching potentially curative therapies (eg, with antisense oligonucleotides or gene replacement) targeting only the central nervous system (CNS). Our case provides anecdotal evidence suggesting that a patient with typical LD can retain full bodily health aside from the effects of neurological damage.
Keywords: Adult polyglucosan disease, Epilepsy, Gene therapy, Lafora disease
Introduction
Lafora disease (LD) is a severe progressive myoclonic epilepsy caused by recessively inherited mutations in the EPM2A or EPM2B genes encoding the laforin glycogen phosphatase and malin E3 ubiquitin ligase, respectively. Both enzymes are essential to glycogen metabolism, with absence of either leading to glycogen with overlong strands, which precipitates, aggregates, and accumulates into neurotoxic Lafora bodies (LBs). LBs do form in organs outside the brain such as heart, liver, and skeletal muscle. First symptoms (myoclonus, seizures, or cognitive decline) occur by the mid‐teenage years, and death by age 25, commonly in aspiration pneumonia due to poor airway control and status epilepticus. During the decade of neurological disease, the other organs remain clinically unaffected. Most patients have full loss‐of‐function mutations and follow the above uniform course (typical LD). A handful have unique mild mutations associated with adult onset and long disease course (atypical LD).1 Starting in 2011, research in LD mouse models established that a mere reduction of brain glycogen synthesis by ~50% is therapeutic in LD and prevents LB formation and neurological disease.1, 2 Among other approaches, an antisense oligonucleotide (ASO) against brain glycogen synthase has proven highly efficacious in mice, and is actively being developed for human clinical trial.3 Because this and certain other potential therapies would be administered only to the brain (ASOs, eg, do not cross the blood–brain barrier and are delivered by lumbar puncture), the question arises whether the longevity imparted by rescue of the brain will associate with disease manifestation in other organs. We report a case of typical LD who, through heroic maternal efforts, was sustained until age 40, and who developed no extraneurological disease.
The female patient from Sweden (adopted from Ecuador at 10‐months‐old) experienced unexplained headaches one year prior to her first generalized seizure at age 13 years. Although she had had mild intention myoclonus when she was 1‐year‐old, her electroencephalography (EEG) results were normal at the time, and the following 12 years were uneventful. After the initial seizure, she had difficulty keeping up with peers both socially and academically. Another EEG was performed at age 13, which appeared highly pathological with epileptic activity. A few months later she had a generalized tonic–clonic seizure (GTCS) and was placed on 100 mg phenobarbital once a day. At age 15, a low dose of valproic acid was added to her regimen, as phenobarbital was reduced to 25 mg per day. Valproic acid was eventually raised to 1500 mg daily. At age 17 she started showing symptoms of ataxia, dysarthria, and sialorrhea. Valproic acid was quickly discontinued when it was suspected to be the cause of some of these disturbances, but when she started having progressively and aggressively worsening myoclonus, it was restarted. Her gait difficulties prevented her from being able to walk without support and signs of dementia were beginning to appear. She was given 30 mg of valium and 50 mg phenobarbital per day while valproic acid was once again reduced. Her GTCS became more frequent. She was given primidone, carbamazepine, and clonazepam, which stopped her myoclonic jerks. At age 20, her mobility was lost completely and her GTCS were occurring 4‐6 times per month. She was only able to speak 3‐4 consecutive words and was mostly incomprehensible.
A blood test revealed mildly lowered serum biotinidase, but this was determined to be due to the storage of lipid material in cells. Her liver function tests were normal, as were her lactate levels. Biotin was slightly low. Examination of the blood marrow revealed reduced myelocytes, but otherwise, nothing remarkable. An ophthalmologic examination revealed no cherry‐red spots. Excretion of mucopolysaccharides was normal as was excretion of lipids in urine.
An EEG study obtained at age 19 while the patient was awake and on treatment was highly abnormal with continuous low frequency bilateral epileptiform activity with no discernable background rhythm. Several episodes of bilateral synchronous spikes and slow wave activity were recorded along with side‐changing focal activity and occasional polyspikes. The results of the EEG study are typical of LD progression.4 The patient's last EEG at age 30 reported in part (translated): “The patient lies with closed eyes, with some eye twitching. Patient does not respond when her eyes are touched. General low frequency activity with amplitude up to 150 μV in amplitude without asymmetry…synchronous spikes or polyspikes and even moments of spike and wave activity are seen. No electrographic seizure activity is noted….“ A page of this EEG is shown in Figure 1.
Figure 1.
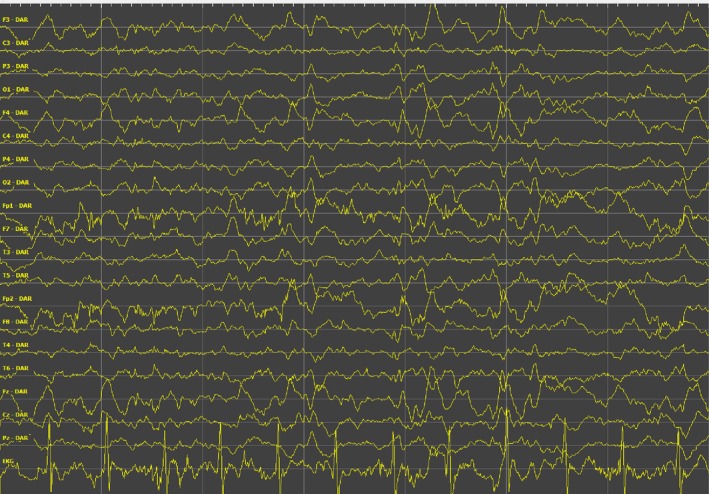
Sample of EEG at age 30 years. Patient in a state of unresponsive wakefulness. Time between vertical lines is 1 second. Voltage between horizontal lines is 50 μV.
Skin biopsy revealed LBs and genetic testing a homozygous truncating mutation in exon 1 of EPM2A, chr6:146056472G>A (GRCh37/hg19); c.163C>T; p.Q55*, expected to result in complete loss of function and typical LD.5 In effect, by age 21 the patient had intractable epilepsy and was in a state of perpetual unresponsive wakefulness. She was fed through a gastric tube and attended by her mother for her every need, especially constant suctioning and skin management against bed sores. Remarkably, at age 38 overt seizures stopped when 4 mg perampanel per night was added to her antiepileptic regimen (which consisted of 165 mg phenobarbital, 7.5 mg clonazepam, 2000 mg levetiracetam, 30 ml N‐acetylcysteine, and 100 mg zonisamide daily), but she remained in a vegetative state.6 However, mucous secretions increased, and at age 40 she was admitted to a hospital with difficulty breathing, where she passed away with a Pseudomonas pneumonia.
Through her disease course, even at last hospitalization, cardiac, hepatic, renal, and other organ functions remained normal. X‐ray prior to her death revealed severe osteoporosis, likely a result of long‐term bed rest and phenobarbital. Heart rhythm and rate were consistently normal. Renal function tests were normal except for low creatinine, likely due to decreased muscle mass. Liver function tests showed slightly elevated alkaline phosphatase. Thyroid function was normal. Bowel movements were slow but normalized with standard medications. Autopsy was not performed, and thus we were not able to determine the presence of storage material in extraneural organs. However, the literature on classical LD is clear on the invariant presence of LBs in peripheral organs in these patients,5 and our patient's positive skin biopsy suggests that she does not differ from this norm.
There are a number of reported cases of late‐onset, slow‐progressing LD with long survival (Table 1). These cases are caused by mild mutations, at least on one allele, that do not result in complete loss of protein product function. Table 1 compares these patients with ours to demonstrate that our patient is not one of those atypical cases.7, 8, 9, 10, 11, 12
Table 1.
Patients with mild mutations in at least one disease gene allele and atypical LD course compared to the patient in the present study with gene‐inactivating mutation and typical course
| References | Sex | Mutation type/gene | Nucleotide (amino acid change) | Age of onset | Age at follow‐up | Clinical status at time of follow‐up |
|---|---|---|---|---|---|---|
| Present paper | F | Nonsense/EPM2A | c.163C>T (p.Gln55Ter) | 13 | 20 | Severe cognitive impairment; severe myoclonus; limited speech; unable to walk or feed |
| 7 | M | Missense/ EPM2B | c.436 G>A (D146N) | 19 | 32 | Mild myoclonus; normal cognition |
| 7 | M | Missense/ EPM2B | c.436 G>A (D146N) | 21 | 29 | Mild myoclonus; normal cognition |
| 7 | M | Missense/ EPM2B | c.436 G>A (D146N) | 17 | 28 | Mild myoclonus; normal cognition |
| 7 | M | Missense/ EPM2B | c.436 G>A (D146N) | 21 | 25 | Mild myoclonus; normal cognition |
| 8 | M | Missense/ EPMzx 2B | c.436 G>A (D146N) | 30 | 48 | Dementia; no disabling myoclonus |
| 9 | F | Missense/ EPM2B | c.436 G>A (D146N) | 19 | 22 | Cognitive impairment; no myoclonus |
| 10 | M | Missense/ EPM2B | c.436 G>A (D146N) | 15 | 29 | Mild cognitive impairment; no interference with daily living activities; mild myoclonus |
| 10 | M | Missense/ EPM2B | c.436 G>A (D146N) | 18 | 28 | Mild cognitive impairment; no interference with daily living activities |
| 10 | M | Missense/ EPM2B | c.436 G>A (D146N) | 13 | 32 | Mild cognitive impairment; no interference with daily living activities; absence seizures |
| 11 | F | Compound heterozygous/ EPM2A | c.721G> T (p.R241X) (nonsense) and c.835G> T (p.G279C) (missense) | 21 | 28 | Mild myoclonus; slow progressing ataxia; mild to moderate cognitive decline. |
| 11 | F | Compound heterozygous/ EPM2A | c.721G> T (p.R241X) (nonsense) and c.835G> T (p.G279C) (missense) | 25 | 30 | Mild cognitive impairment; no ataxia; some myoclonus and GTCS |
| 11 | F | Compound heterozygous/ EPM2A | c.721G> T (p.R241X) (nonsense) and c.835G> T (p.G279C) (missense) | 28 | 33 | No cognitive impairment; no ataxia; myoclonus only |
| 12 | F | Missense/ EPM2A | c.962T>G (p. F321C) | Early 20s | 53 | Slowly progressing bradykinesia; rigidity in limbs; mild cognitive decline |
With the imminence of therapies directed at the basic mechanisms of LD and to the affected organ, this lady's life is testament that organs outside the brain remain clinically preserved at least until age 40. Excessive secretions and airway control difficulty are common features of chronic neurodegeneration,13 that is, they are part of the neurological disease. This report supports accelerating the advancing of currently most developed, brain‐targeting, LD therapies until a systemic therapy can be devised.
Disclosure
Neither author has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Acknowledgments
This work was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (NIH) under award number P01 NS097197. BA Minassian holds the University of Texas Southwestern Jimmy Elizabeth Westcott Chair in Pediatric Neurology.
Biography
Danielle Goldsmith has been working on Lafora Disease research with Dr. Berge Minassian since 2012.

References
- 1. Ackerley CA, Carpenter S, Genton P, et al. Lafora disease. Epileptic Disord 2016;18:38–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Turnbull J, DePaoli‐Roach AA, Cortez MA, et al. PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PLoS Genet 2011;7:e1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grossman TR, Ahonen S, Turnbull J, et al. (Program #186). Presented at the 66th Annual Meeting of The American Society of Human Genetics, October 19, 2016, Vancouver, Canada.
- 4. Striano P, Zara F, Turnbull J, et al. Typical progression of myoclonic epilepsy of the Lafora type: a case report. Nat Clin Pract Neurool 2008;4:106–111. [DOI] [PubMed] [Google Scholar]
- 5. Minassian BA, Ianzano L, Meloche M, et al. Mutation spectrum and predicted function of laforin in Lafora's progressive myoclonus epilepsy. Neurol 2000;55:341–346. [DOI] [PubMed] [Google Scholar]
- 6. Goldsmith D, Minassian BA. Efficacy and tolerability of perampanel in 10 patients with Lafora disease. Epilepsy Behav 2016;62:132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baykan B, Striano P, Gianotti S, et al. Late‐onset and Slow‐progressing Lafora Disease in Four Siblings with EPM2B Mutation. Epilepsia 2005;46:1695–1697. [DOI] [PubMed] [Google Scholar]
- 8. Salar S, Yeni N, Gündüz A, et al. Four novel and two recurrent NHLRC1 (EPM2B) and EPM2A gene mutations leading to Lafora disease in six Turkish families. Epilepsy Res 2012;98:273–276. [DOI] [PubMed] [Google Scholar]
- 9. Lanoiselée HM, Genton P, Lesca G, et al. Are c. 436G> A mutations less severe forms of Lafora disease? A case report Epilepsy Behav Case Rep 2014;2:19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Franceschetti S, Gambardella A, Canafoglia L, et al. Clinical and genetic findings in 26 Italian patients with Lafora disease. Epilepsia 2006;47:640–643. [DOI] [PubMed] [Google Scholar]
- 11. Jara‐Prado A, Ochoa A, Alonso ME, et al. Late onset Lafora disease and novel EPM2A mutations: Breaking paradigms. Epilepsy Res 2014;108:1501–1510. [DOI] [PubMed] [Google Scholar]
- 12. Lynch DS, Wood NW, Houlden H. Late‐onset Lafora disease with prominent parkinsonism due to a rare mutation in EPM2A. Neurol Genet 2016;2:e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Daniels SK. Neurological disorders affecting oral, pharyngeal swallowing. GI Motility online, 2006;https://doi.org/10.1038/gimo34.