

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a major cause of liver damage and has a strong genetic component. The rs4841132 G>A variant, modulating the expression of protein phosphatase 1 regulatory subunit 3B (PPP1R3B), which is involved in glycogen synthesis, has been reported to reduce the risk of NAFLD but at the same time may favor liver disease by facilitating glycogen accumulation. The aim of this study was to assess the impact of rs4841132 on development of histologic steatosis and fibrosis in 1,388 European individuals in a liver biopsy cohort, on NAFLD hepatocellular carcinoma in a cross‐sectional Italian cohort (n = 132 cases), and on liver disease at the population level in the United Kingdom Biobank cohort. We investigated the underlying mechanism by examining the impact of the variant on gene expression profiles. In the liver biopsy cohort, the rs4841132 minor A allele was associated with protection against steatosis (odds ratio [OR], 0.63; 95% confidence interval [CI], 0.42‐0.95; P = 0.03) and clinically significant fibrosis (OR, 0.35; 95% CI, 0.14‐0.87; P = 0.02) and with reduced circulating cholesterol (P = 0.02). This translated into protection against hepatocellular carcinoma development (OR, 0.22; 95% CI, 0.07‐0.70; P = 0.01). At the population level, the rs4841132 variation was not associated with nonalcoholic or nonviral diseases of the liver but was associated with lower cholesterol (P = 1.7 × 10–8). In individuals with obesity, the A allele protecting against steatosis was associated with increased PPP1R3B messenger RNA expression and activation of lipid oxidation and with down‐regulation of pathways related to lipid metabolism, inflammation, and cell cycle. Conclusion: The rs4841132 A allele is associated with protection against hepatic steatosis and fibrosis in individuals at high risk of NAFLD but not in the general population and against dyslipidemia. The mechanism may be related to modulation of PPP1R3B expression and hepatic lipid metabolism. (Hepatology Communications 2018;2:666‐675)
Abbreviations
- CI
confidence interval
- HCC
hepatocellular carcinoma
- kb
kilobase
- LBC
liver biopsy cohort
- NAFLD
Nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OR
odds ratio
- PPP1R3B
protein phosphatase 1 regulatory subunit 3B
Nonalcoholic fatty liver disease (NAFLD) is becoming a leading cause of liver damage worldwide.1 NAFLD pathogenesis is strongly intertwined with excessive adiposity, insulin resistance, and dyslipidemia.2 Dietary factors, such as alcohol, fructose, and physical activity, are other major risk factors for this condition.3 NAFLD has a strong genetic component, and variants in proteins regulating hepatocellular lipid handling, including patatin‐like phospholipase domain‐containing 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), membrane bound O‐acyltransferase domain‐containing 7 (MBOAT7), and glucokinase regulator (GCKR), predispose to the development and progression to nonalcoholic steatohepatitis (NASH), which is the inflammatory form of NAFLD, and to hepatic fibrosis,4, 5 which is the major prognostic determinant in patients with NAFLD.6 By exploiting naturally occurring variation at these loci, we recently highlighted by a Mendelian randomization approach that hepatic fat accumulation is a key driver of liver disease progression and fibrosis development in at‐risk individuals.7 However, whether this is generalizable to other risk factors for NAFLD development and progression and for protective variants remains to be demonstrated.
Protein phosphatase 1 regulatory subunit 3B (PPP1R3B) has been identified as another locus associated with hepatic fat content, as estimated by computed tomography in genome‐wide association studies.8 PPP1R3B encodes for a protein involved in glycogen synthesis.9 Subsequent studies have been consistent with the hypothesis that the rs4240624 variant, which is located 175 kilobase (kb) upstream of the PPP1R3B coding region, associates with reduced fat as estimated by ultrasonography10, 11 and with increased hepatic glycogen content.12 However, when liver fat was directly measured by histology, the association with the PPP1R3B variation was not confirmed.8, 13 Furthermore, Stender et al.9 showed that the minor allele of rs4841132, which is in complete linkage disequilibrium with rs4240624, was associated with increased X‐ray attenuation but not with decreased hepatic fat content in population studies. On the contrary, the minor A allele was associated with increased aminotransferases and liver disease.9 Therefore, the overall impact of the PPP1R3B variation on hepatic fat and progressive liver damage remains controversial.
Within this context, we evaluated the impact of the rs4841132 G>A variant, located in LOC157273, 175 kb upstream of the PPP1R3B gene, on a histologic steatosis, metabolic profile and on liver damage in a large cohort of European individuals.14 In addition, we evaluated the impact of the variant on liver diagnoses in the publicly available United Kingdom Biobank cohort. Finally, we explored the mechanism underlying the epidemiological association by examining the impact of the variant on gene expression profiles.
Patients and Methods
THE LIVER BIOPSY COHORT
The liver biopsy cohort (LBC) has been described.5, 15 A total of 1,388 adult individuals of European descent were consecutively enrolled from the Metabolic Liver Diseases outpatient service and bariatric surgery center, Fondazione IRCCS Ca' Granda Ospedale Policlinico Milano, Milan, Italy, and from the Northern Savo Hospital District, Kuopio, Finland. Inclusion criteria were availability of a liver biopsy for suspected NASH or severe obesity, DNA samples, and clinical data. Individuals with excessive alcohol intake (men, >30 g/day; women, >20 g/day), viral and autoimmune hepatitis, or other causes of liver disease were excluded. The study conformed to the Declaration of Helsinki and was approved by the Institutional Review Board of the Fondazione Ca' Granda IRCCS of Milan and relevant Institutions. All participants gave written informed consent. The clinical features of individuals evaluated in the study are presented in Table 1.
Table 1.
Demographic, Anthropometric, and Clinical Features of the Liver Biopsy Cohort Stratified for Enrollment Criteria (Liver Clinic, Bariatric Surgery) and the NAFLD‐HCC Cohort
|
Liver Biopsy Cohort (N = 1,388) |
|||||
|---|---|---|---|---|---|
| Liver Clinic | Bariatric Surgery | NAFLD‐HCC | |||
| (n = 509) | (n = 879) | (n = 132) | P Value* | P Value† | |
| Sex, M | 409 (80) | 218 (25) | 101 (77) | <0.0001 | <0.0001 |
| Age, years | 49.9 ± 12.4 | 44.9 ± 10.3 | 67.5 ± 1.05 | <0.0001 | <0.0001 |
| BMI, kg/m2 | 28.4 ± 4.4 | 42.3 ± 6.7 | 29.3 ± 0.8 | <0.0001 | <0.0001 |
| T2D, yes | 111 (22) | 227 (26) | 76 (58) | 0.06 | <0.0001 |
| Total cholesterol, mg/dL | 201.1 ± 42.5 | 185.6 ± 46.4 | 170 ± 7.7 | <0.0001 | <0.0001 |
| LDL cholesterol, mg/dL | 124 ± 38.6 | 108.2 ± 38.6 | 100.5 ± 7.7 | <0.0001 | 0.0008 |
| HDL cholesterol, mg/dL | 46.4 ± 15.4 | 50.3 ± 19.3 | 50.2 ± 3.9 | 0.41 | 0.61 |
| Triglycerides, mg/dL | 148.7 ± 78.7 | 139.9 ± 69.9 | 139.9 ± 017.5 | 0.03 | 0.79 |
| ALT, IU/L‡ | 47 (29‐74) | 30 (23‐41) | 45 (28‐69) | <0.0001 | 0.11 |
| AST, IU/L‡ | 20 (16‐27) | 27 (18‐42) | 44 (30‐61) | <0.0001 | <0.0001 |
| PNPLA3, I148M | <0.0001 | <0.0001 | |||
| I/I | 180 (35.4) | 464 (52.8) | 31 (23.5) | ||
| I/M | 227 (44.6) | 317 (36) | 54 (419 | ||
| M/M | 101 (19.8) | 70 (8) | 49 (37) | ||
| TM6SF2, E167K | 0.003 | 0.59 | |||
| E/E | 431 (84.7) | 757 (86.1) | 113 (85.6) | ||
| E/K | 70 (13.8) | 87 (9.9) | 19 (14.4) | ||
| K/K | 8 (1.6) | 2 (0.2) | 2 (1.5) | ||
| MBOAT7, rs641738 C>T | 0.98 | 0.04 | |||
| C/C | 179 (35) | 295 (34) | 28 (21.2) | ||
| C/T | 225 (44) | 367 (42) | 69 (52.2) | ||
| T/T | 105 (21) | 172 (20) | 35 (26.5) | ||
| GCKR, P446L | 0.0001 | 0.64 | |||
| C/C | 112 (22) | 215 (24) | 32 (24.2) | ||
| C/T | 235 (46) | 368 (42) | 65 (49.2) | ||
| T/T | 162 (32) | 171 (19) | 33 (25) | ||
| PPP1R3B, rs4841132 G>A | 0.01 | 0.04 | |||
| G/G | 433 (85) | 698 (79) | 124 (93.9) | ||
| G/A | 72 (14) | 173 (20) | 8 (6.1) | ||
| A/A | 4 (0.8) | 8 (0.9) | 0 | ||
Values are reported as mean ± SD and number (%), except where noted. *Liver clinic versus bariatric surgery; †Milan liver biopsy cohort versus Italian NAFLD‐HCC cohort; ‡median (interquartile range).
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; GCKR, glucokinase regulator; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; MBOAT7, membrane bound O‐acyltransferase domain‐containing 7; PNPLA3, patatin‐like phospholipase domain‐containing 3; T2D, type 2 diabetes; TM6SF2, transmembrane 6 superfamily member 2.
HISTOLOGIC AND LIVER DAMAGE EVALUATION
Steatosis was graded into the following four categories based on the percentage of affected hepatocytes: 0, 0%‐4%; 1, 5%‐32%; 2, 33%‐65%; and 3, 66%‐100%. Disease activity was assessed according to the NAFLD activity score, with systematic evaluation of hepatocellular ballooning and necroinflammation; fibrosis was also staged according to the recommendations of the NAFLD Clinical Research Network.16 The scoring of liver biopsies was performed by independent pathologists unaware of patient status and genotype.15, 17 NASH was diagnosed in the presence of steatosis, lobular necroinflammation, and hepatocellular ballooning.
NAFLD‐HEPATOCELLULAR CARCINOMA COHORT
Part of the NAFLD‐hepatocellular carcinoma (NAFLD‐HCC) cohort of the exome sequencing for the identification of inherited variants associated with hepatocellular carcinoma in NAFLD (EPIDEMIC‐NAFLD) study has been described.18, 19 Briefly, this is a case‐control study in which we genotyped Italian patients with NAFLD‐HCC and compared the frequency of risk variants with healthy individuals and with controls with advanced fibrosis due to NAFLD. The NAFLD‐HCC patients of Italian descent were enrolled between January 2008 and January 2015 at the Milan, Udine, Turin, Rome, and Palermo hospitals. Diagnosis of HCC was based on the European Association for the Study of the Liver –European Organization for Research and Treatment of Cancer Clinical Practice Guidelines.20 In the absence of liver biopsy, diagnosis of NAFLD required detection of ultrasonographic steatosis plus at least one criterion of the metabolic syndrome. Clinical features of these individuals are presented in Table 1.
GENOTYPING
The LBC cohort was genotyped for the rs738409 C>G (PNPLA3 I148M), rs58542926 C>T (TM6SF2 E167K), rs1260326 C>T (GCKR P446L), rs641738 C>T MBOAT7, and rs4841132 G>A (LOC157273‐PPP1R3B) variants as described.5, 15 Genotyping of the LBC was performed in duplicate using TaqMan 5′‐nuclease assays (Life Technologies, Carlsbad, CA). Genotype frequencies of the four variants were in agreement with Hardy‐Weinberg proportions (P > 0.1).
UNITED KINGDOM BIOBANK COHORT
The associations of the rs4841132 variant with phenotype related to metabolic disorders and liver disease were evaluated in the United Kingdom Biobank cohort, a prospective population study of half a million subjects not selected for liver diseases. These subjects were identified at 22 centers across the United Kingdom during 2006 to 2010, and almost all were aged 40‐69 years old. Freely available basic association data were downloaded from Neale Lab (http://geneatlas.roslin.ed.ac.uk).
TRANSCRIPTOMIC ANALYSIS
Gene expression was measured in a subset of 125 patients with obesity belonging to the Milan cohort, after percutaneous liver biopsy performed during bariatric surgery. Their clinical features are presented in http://onlinelibrary.wiley.com/doi/10.1002/hep4.1192/full. Total RNA was isolated using the miRNeasy mini‐kit (Qiagen, Hulsterweg, Germany), according to the manufacturer's instructions. RNA quality was assessed using the Agilent 2100 Bioanalyzer, and samples with RNA integrity numbers greater than or equal to 7 were used for library preparation. RNA sequencing was performed in paired‐end mode with a read length of 150 nucleotides using the Illumina HiSeq 4000 (Novogene, Hong Kong, China). Raw reads were mapped against the Human Genome,21 using a custom pipeline based on the standard primary analysis procedure. The pipeline performed the primary analysis step, including FASTQ quality check (FastQC software; Babraham Bioinformatics, Cambridge, United Kingdom), low‐quality reads trimming with Trimmomatic,22 and mapping on the GRCh37 reference genome using STAR mapper.23 RNA sequencing quality control was performed, and samples with <10 million reads uniquely mapped or with <60% uniquely mapped/mapped reads were excluded from the analysis. Gene reads count (Ensemble human transcript reference assembly, version 75) was performed using RSEM software.24 To quantify gene expression, RSEM per gene counts were normalized using the DESeq2 package.25
STATISTICAL ANALYSIS
For descriptive statistics, continuous variables were shown as mean and SD or median and interquartile range for highly skewed biological variables. Variables with skewed distributions were log‐transformed before analyses. Categorical variables were presented as number and proportion. All genetic analyses were performed under additive models.
Analyses were performed by fitting data into generalized linear regression models. In particular, general linear models were fit to examine continuous traits. Logistic regression models were fit to examine binary traits (NASH, severe fibrosis stage F3‐F4), and ordinal regression models were fit for ordinal traits (components of the NAFLD activity score: severity of steatosis, necroinflammation and hepatocellular ballooning, stage of fibrosis). When specified, confounding factors (including recruitment center) were included in a model.
Statistical analyses were carried out using JMP 13.0 (SAS Institute, Cary, NC) and R statistical analysis software, version 3.3.2 (http://www.R-project.org/). P < 0.05 (two‐tailed) was considered statistically significant.
Results
PPP1R3B GENE VARIATION IS ASSOCIATED WITH PROTECTION AGAINST HISTOLOGIC STEATOSIS
The frequency distribution of the rs4841132 G>A variant was in Hardy‐Weinberg equilibrium (P = 0.75; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1192/full). The independent predictors of liver damage in the liver biopsy cohort are shown in Table 2. Logistic regression analysis adjusted for sex, age, body mass index, type 2 diabetes mellitus, indication for liver biopsy, and known genetic risk variants for NAFLD showed the rs4841132 minor A allele was associated with protection against steatosis (OR, 0.63; 95% confidence interval [CI], 0.42‐0.95; P = 0.03) and against clinically significant fibrosis (OR, 0.35; 95% CI, 0.14‐0.87; P = 0.02). With ordinal regression analysis adjusted as above, the minor A allele was associated with reduced steatosis grade (estimated beta, –0.36; 95% CI, –0.64 to –0.08; P = 0.01; Fig. 1).
Table 2.
Independent Predictors of Liver Damage (Presence of Steatosis, NASH, and Fibrosis Stage F2‐F4) in the Liver Biopsy Cohort
| Steatosis | NASH | Fibrosis F2‐F4 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | P Value | OR | 95% CI | P Value | OR | 95% CI | P Value | |
| Age, years | 1.02 | 0.99‐1.04 | 0.07 | 1.01 | 1.00‐1.02 | 0.09 | 1.05 | 1.03‐1.08 | <0.001 |
| Sex, M | 1.49 | 0.94‐2.39 | 0.09 | 1.61 | 1.19‐2.17 | 0.002 | 0.99 | 0.54‐1.84 | 0.99 |
| BMI, kg/m2 | 1.07 | 1.03‐1.10 | <0.001 | 1.07 | 1.04‐1.09 | <0.001 | 1.03 | 0.99‐1.08 | 0.17 |
| IFG/T2DM, yes | 2.18 | 1.29‐3.67 | 0.004 | 2.06 | 1.49‐2.86 | <0.001 | 3.43 | 1.93‐6.11 | <0.001 |
| PNPLA3, I148M alleles | 1.39 | 1.02‐1.90 | 0.04 | 1.51 | 1.25‐1.82 | <0.001 | 2.24 | 1.54‐3.26 | <0.001 |
| TM6SF2, E167K | 1.62 | 0.83‐3.13 | 0.16 | 1.88 | 1.32‐2.68 | <0.001 | 1.58 | 0.80‐3.09 | 0.19 |
| MBOAT7 rs641738 T | 1.06 | 0.82‐1.39 | 0.65 | 1.06 | 0.89‐1.27 | 0.48 | 0.91 | 0.61‐1.33 | 0.62 |
| GCKR, L446P | 1.06 | 0.80‐1.39 | 0.69 | 1.05 | 0.88‐1.25 | 0.62 | 0.77 | 0.53‐1.12 | 0.17 |
| PPP1R3B, rs4841132 A alleles | 0.63 | 0.42‐0.95 | 0.03 | 0.79 | 0.57‐1.09 | 0.16 | 0.35 | 0.14‐0.87 | 0.02 |
Logistic regression models were used to test the association of demographic, clinical, and genetic factors with liver damage. In addition to the predictors shown above, models were adjusted for indication for liver biopsy (severe obesity versus increased liver enzymes in NAFLD).
Abbreviations: BMI, body mass index; GCKR, glucokinase regulator; IFG, impaired fasting glucose; MBOAT7, membrane bound O‐acyltransferase domain‐containing 7; PNPLA3, patatin‐like phospholipase domain‐containing 3; TM6SF2, transmembrane 6 superfamily member 2; T2DM: type 2 diabetes mellitus.
Figure 1.
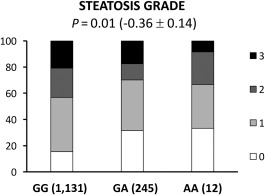
In the LBC, the PPP1R3B rs4841132 variant is associated with protection against steatosis development. The association between the PPP1R3B rs4841132 variant and the risk of NAFLD was tested by multivariate ordinal regression analysis adjusted for age, sex, body mass index, presence of impaired fasting glucose/type 2 diabetes mellitus, PNPLA3 I148M alleles, presence of the TM6SF2 E167K variant, MBOAT7/TMC4 T allele, GCKR P446L variant, and indication of liver biopsy (severe obesity versus nonalcoholic fatty liver with increased liver enzymes. Abbreviations: AA, homozygotes for the A allele (carriers); GA, heterozygotes; GG, homozygotes for the G allele.
Findings were generally consistent after stratification of patients for the enrollment criteria. The rs4841132 A allele was associated with protection against fibrosis (OR, 0.26; 95% CI, 0.08‐0.80; P = 0.02) in the liver clinic cohort (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1192/full). In the bariatric cohort, the A allele was associated with protection against steatosis (OR, 0.61; 95% CI, 0.40‐0.93; P = 0.02; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1192/full; estimated beta, –0.50; 95% CI, –0.86 to –0.15; P = 0.006; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1192/full) and lobular inflammation (estimated beta –0.41; 95% CI, –0.83 to –0.02; P = 0.04; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1192/full).
In the overall series of patients at risk of NASH, the A allele was associated with reduced total and high‐density lipoprotein cholesterol (P < 0.05; Table 3). After stratification for enrollment criteria, the impact of the variant on circulating lipids was larger in the bariatric cohort (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1192/full).
Table 3.
Demographic, Anthropometric, and Clinical Features of the Liver Biopsy Cohort (N = 1,388) Stratified by PPP1R3B rs4841132 Genotype
|
GG (n = 1,131) |
GA (n = 245) |
AA (n = 12) |
P Value* | |
|---|---|---|---|---|
| Sex, M | 522 (46) | 100 (41) | 5 (42) | 0.36 |
| Age, years | 47 ± 11 | 47 ± 11 | 54 ± 6 | 0.12 |
| BMI, kg/m2 | 37 ± 9 | 38 ± 8 | 38 ± 7 | 0.64 |
| IFG/T2D, yes | 276 (24) | 60 (24) | 2 (17) | 0.08 |
| HOMA‐IR | 6.2 ± 15 | 6.9 ± 13 | 4.1 ± 1.9 | 0.92 |
| Insulin, IU/mL | 22 ± 33 | 23 ± 27 | 17 ± 7.6 | 0.99 |
| Total cholesterol, mg/dL | 193.3 ± 42.5 | 181.7 ± 42.5 | 170.1 ± 50.3 | 0.02 |
| LDL cholesterol, mg/dL | 116 ± 38.6 | 112.1 ± 38.6 | 96.7 ± 34.8 | 0.18 |
| HDL cholesterol, mg/dL | 50.3 ± 15.5 | 46.4 ± 11.6 | 46.4 ± 11.6 | 0.03 |
| Triglycerides, mg/dL | 139.9 ± 78.7 | 131.2 ± 61.2 | 113.7 ± 35 | 0.07 |
| ALT, IU/L† | 32 (20‐54) | 36 (24‐55) | 32 (22‐43) | 0.78 |
| AST, IU/L† | 24 (18‐34) | 26 (20‐36) | 27 (21‐40) | 0.87 |
Values are reported as mean ± SD or number (%), except where indicated. Characteristics of participants were compared across rs236918 genotypes using linear regression model (for continuous characteristics) or logistic regression model (for categorical characteristics). *Models were adjusted for sex, age, BMI, PNPLA3 I148M alleles, TM6SF2 E167K alleles, MBOAT7 rs641738 T, GCKR P446L variant and statin use. †median (IQR).
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; HDL, high‐density lipoprotein; HOMA‐IR, homeostasis model assessment of insulin resistance; IFG, impaired fasting glucose; LDL, low‐density lipoprotein; T2D, type 2 diabetes.
PPP1R3B GENE VARIATION IS ASSOCIATED WITH REDUCED RISK OF NAFLD‐HCC
To examine the impact of the variant on specific clinically relevant outcomes, we assessed the association with NAFLD‐HCC in the Italian EPIDEMIC case controls (n = 132 cases, n = 995 controls) whose characteristics and prevalence of the PPP1R3B variant are shown in Table 1 and Fig. 2. The independent predictors of NAFLD‐HCC are reported in Table 4. We detected a reduced frequency of the minor A allele in patients with HCC than in those without HCC (P = 0.02; Fig. 2), and the minor A allele was independently associated with protection against HCC development (OR, 0.22; 95% CI, –2.6 to –0.4; P = 0.01). Notably, the protective effect of the variant seemed to depend on liver disease severity as adjustment for advanced fibrosis abolished the association of the A allele with NAFLD‐HCC (Table 4). These findings suggest that the rs4841132 variation does not protect against HCC independently of hepatic fat accumulation and fibrosis development.
Figure 2.
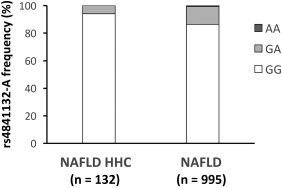
Frequency distribution of the rs4841132 A allele in 1,127 Italian patients with NAFLD stratified by the presence of HCC. Comparisons were performed by logistic regression setting HCC as the dependent variable, and the association with the PPP1R3B variant was analyzed assuming an additive model. Abbreviations: AA, homozygotes for the A allele; GA, heterozygotes; GG, homozygotes for the G allele.
Table 4.
Independent Predictors of NAFLD‐HCC in 1,127 Patients with NAFLD (cases = 132)
| Model 1 | Model 2 | |||||
|---|---|---|---|---|---|---|
| OR | 95% CI | P Value | OR | 95% CI | P Value | |
| Age, years | 1.18 | 1.13‐1.22 | <0.0001 | 1.18 | 1.09‐1.28 | <0.0001 |
| Sex, M | 1.21 | 0.65‐2.26 | 0.54 | 0.37 | 0.11‐1.30 | 0.12 |
| BMI, kg/m2 | 0.92 | 0.87‐0.96 | 0.002 | 0.89 | 0.78‐1.02 | 0.09 |
| IFG/T2DM, yes | 4.61 | 2.56‐8.28 | <0.0001 | 4.2 | 1.07‐16.5 | 0.04 |
| PNPLA3, I148M alleles | 1.68 | 1.14‐2.46 | 0.008 | 1.14 | 0.51‐2.53 | 0.75 |
| Advanced fibrosis, F3‐F4 | NA | NA | NA | 91.4 | 10.6‐790 | <0.0001 |
| PPPR13B rs4841132, A alleles | 0.22 | 0.07‐0.70 | 0.01 | 1.76 | 0.30‐10.3 | 0.53 |
Comparisons were performed by logistic regression analysis setting HCC as the dependent variable. Model 1 was adjusted for age, sex, BMI and T2DM, PNPLA3, TM6SF2, MBOAT7 rs643718, and PPP1R3B rs4841132 genetic variants. Model 2 was further adjusted for presence of advanced fibrosis.
Abbreviations: BMI, body mass index; IFG, impaired fasting glucose; NA, not addressed; T2DM, type 2 diabetes mellitus.
PPP1R3B GENE VARIATION IS NOT ASSOCIATED WITH INCREASED LIVER DISEASE RISK IN THE UNITED KINGDOM BIOBANK COHORT
We next examined the impact of the rs4841132 minor A allele on liver disease and metabolic traits in the United Kingdom Biobank cohort, which is representative of the general population (N = 500,000; Table 5). Here, we did not find any significant association between the minor A allele and the risk of liver disease. These findings suggest that carriage of the A allele does not reduce the predisposition to develop liver disease at the population level, despite seeming to protect against the development of NAFLD in high‐risk individuals. However, consistent with results observed in the LBC, the A allele was associated with lower circulating cholesterol levels (P = 8.4 × 10–8).
Table 5.
Association of the PPP1R3B Variant With Liver‐Related Outcomes in the United Kingdom Biobank Cohort (N = 500,000)
| Phenotype | n Cases | Beta | P value |
|---|---|---|---|
| K70: Alcoholic liver disease | 808 | –0.00014799 | 0.39 |
| K74: Fibrosis and cirrhosis of liver | 805 | –0.0001773 | 0.30 |
| K75: Other inflammatory liver diseases | 662 | –0.00026552 | 0.09 |
| K76: Other diseases of liver | 3,351 | 0.00035659 | 0.31 |
| High cholesterol | 50,421 | –0.0064655 | 8.4 × 10–8 |
PPP1R3B rs4841132 IMPACTS ON PPP1R3B AND LOC157273 EXPRESSION
The rs4841132 variant is located in exon 2 of LOC157273, a liver‐specific long noncoding RNA, 175 kb upstream of the PPP1R3B gene, which encodes a protein that promotes hepatic glycogen synthesis. To determine whether the epidemiological association of rs4841132 with hepatic steatosis is mediated by modulation of transcription at this locus, we examined the hepatic expression of LOC157273, PPP1R3B, and tankyrase (TNKS) in a subset of patients with available liver samples (n = 125; Fig. 3A). The rs4841132 A allele was associated with higher PPP1R3B messenger RNA levels (P = 0.04) and with reduced LOC157273 expression (P = 0.008), whereas TNKS expression was not affected (P value not significant).
Figure 3.
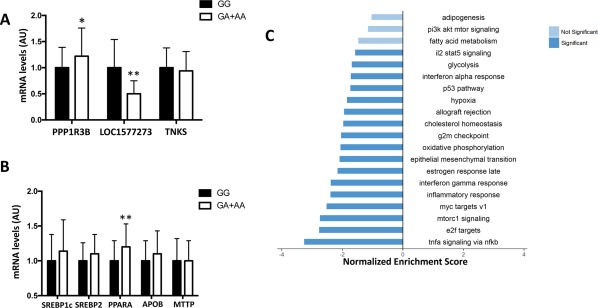
Gene expression in human liver samples collected from liver biopsies was evaluated by transcriptome analysis. (A) PPP1R3B, LOC157273, TNKS; (B) beta‐oxidation (PPARα), lipogenesis (SREBP1c), cholesterol synthesis (SREBP2), and lipoprotein secretion (MTTP, APOB). *P < 0.05, **P < 0.01 compared to noncarriers (n = 125). (C) Hallmark molecular functions enriched in patients who carried the rs4841132 A allele, by RNA sequencing analysis. Bar plots show the gene set enrichment analysis hallmark gene sets that were found down‐regulated (FDR <0.1). Light blue bars indicate gene sets slightly below the significance (FDR between 0.1 and 0.6). Bars were ordered by normalized enrichment score that indicated the strength of the enrichment. Data are presented as beta coefficient ± SE. Abbreviations: AA, homozygotes for the A allele; APOB apolipoprotein B; AU, arbitrary unit; FDR, false discovery rate; GA, heterozygotes; GG, homozygotes for the G allele; il2 stat5, interleukin 2 signal transducer and activator of transcription 5; mRNA, messenger RNA; mtorc1, mammalian target of rapamycin complex 1; MTTP, microsomal triglyceride transfer protein; nfkb, nuclear factor kappa β; pi3k akt mtor; phosphoinositide 3‐kinase–protein kinase B–mammalian target of rapamycin; PPARα, peroxisome proliferator‐activated receptor alpha; SREBP, sterol regulatory element binding protein; tnfa, tumor necrosis factor α; TNKS, tankyrase.
Next, we assessed whether the PPP1R3B variation was associated with altered expression of genes involved in hepatic lipid metabolism. Notably, we found that the minor A allele was associated with up‐regulation of peroxisome proliferator‐activated receptor alpha (P = 0.004), whereas expressions of sterol regulatory element binding protein 1 and 2, microsomal triglyceride transfer protein, and apolipoprotein B were not affected (Fig. 3B).
Gene set enrichment analysis on differentially expressed genes (n = 1,060; nominal P < 0.05) revealed down‐regulation of several pathways related to cell cycle and inflammation in patients who carried the A allele. Consistently, we found a lower expression of genes involved in inflammation and fibrogenesis, cholesterol homeostasis, oxidative phosphorylation, and glycolysis. We also found a nonsignificant down‐regulation of other metabolic pathways, including fatty acid metabolism, phosphoinositide 3‐kinase–protein kinase B–mammalian target of rapamycin, and lipogenesis (adipogenesis). Gene set enrichment analysis results are summarized in Fig. 3C and http://onlinelibrary.wiley.com/doi/10.1002/hep4.1192/full.
Finally, to further investigate the mechanism whereby the rs4841132 variation protects against hepatic fat accumulation, we performed a gene expression differential analysis of the whole transcriptome in patients carrying the A allele (n = 19) compared to noncarriers (n = 106). The analysis revealed that after adjustment for the false discovery rate the expression of PLEXIN A4 (PLXNA4) and protein kinase, membrane‐associated tyrosine/threonine 1 (PKMYT1) was lower in carriers of the A allele (P = 1.3 × 10–5 and P = 2.1 × 10–5, respectively; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1192/full).
Discussion
In this study, we examined whether PPP1R3B gene variation influences the susceptibility to liver damage in European individuals at high risk of NASH. Our results suggest that the minor rs4841132 A allele is associated with a reduced risk of histologic steatosis, which also resulted in protection against clinically significant fibrosis and HCC. Although the direction of the association is protective and the variant is rarer, the size effect was marked, making it very similar to that of the PNPLA3 I148M variant on these three major outcomes. Despite previous contrasting evidence,8, 9, 10, 11, 12, 13 these novel data lend strong support to the hypothesis that the PPP1R3B variation does protect against hepatic fat accumulation, at least in at‐risk individuals. Furthermore, they support the notion that steatosis is a major driver of liver disease progression to fibrosis7 and HCC18 in individuals with NAFLD.
However, in the United Kingdom Biobank cohort, a well‐powered population study, the A allele had a neutral impact on liver disease risk. It might be speculated the protective effect of the rs4841132 A allele on steatosis development can be observed only in individuals at high baseline risk for NASH and/or in the presence of unknown environmental triggers. Alternatively, in individuals at lower risk of NAFLD, predisposition to glycogen accumulation in carriers of the A allele may offset the protective effect on hepatic fat accumulation. Indeed, in two other population studies, namely the Copenhagen City Heart Study and in the Dallas Liver Study, carriage of the A allele was borderline associated with an increased risk of a composite outcome of liver‐related outcomes.9 Therefore, a limitation of this study is that we could not assess hepatic glycogen content.
Concerning the mechanism underpinning the epidemiological association, we confirmed previous findings indicating that rs4841132 variation determines down‐regulation of the expression of LOC157273 as well as and increased levels of PPP1R3B.9 However, we could not demonstrate whether this is mediated by an alteration of the chromatin structure of the whole locus or LOC157273 controls PPP1R3B transcription.
It is tempting to speculate that higher expression of PPP1R3B would favor energy storage under the form of glycogen in the liver.9 This may lead to shunting of glucose to glycogen synthesis from glycolysis and suppression of de novo lipogenesis in carriers of the protective variant. Interestingly, although we did not directly measure lipid fluxes, our novel gene expression data from transcriptomic analysis suggest that carriers of the variant had reduced glycolysis and lipid oxidation, which are associated with reduced activation of inflammatory and fibrogenic pathways. However, we cannot rule out that LOC157273 may directly affect the expression of genes involved in steatosis development and lipid handling. The association with down‐regulation of cholesterol metabolism genes is consistent with the reduced circulating levels of cholesterol in severely obese individuals carrying the A allele, which was also confirmed at the population level both in the United Kingdom Biobank and in previous studies.8 Moreover, differential analysis of gene expression revealed that two genes involved in tumor progression and inflammation26, 27 were significantly down‐regulated in patients who carried the A allele.
This study has other limitations. These include semiquantitative assessment of hepatic fat accumulation by histology, lack of prospective assessment of the impact of the variant on disease outcomes, and lack of availability of individual patient data in the United Kingdom Biobank cohort, precluding a formal evaluation of the interaction between PPP1R3B and adiposity in determining the risk of liver disease. Furthermore, results are only applicable to European individuals.
In conclusion, rs4841132 variation influencing PPP1R3B expression is associated with protection against hepatic steatosis and fibrosis in individuals at high risk of NAFLD and against dyslipidemia. The mechanism seems related to modulation of hepatic lipid metabolism.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1192/full.
Supporting Information 1
Potential conflict of interest: Nothing to report.
Supported by the Swedish Research Council (Vetenskapsrådet, 254439006, to S.R. and R.M.M.), the Swedish Heart Lung Foundation (244439007 to S.R. and R.M.M.), the Swedish Federal Government funding under the Agreement on Medical Training and Medical Research (76290 to A.L.F.), the Novonordisk Foundation Grant for Excellence in Endocrinology (244439012), the Swedish Diabetes Foundation (DIA 2014‐052 to S.R.), the Wilhelm and Martina Lundgren Science Fund (to R.M.M., P.P., B.M.M., and S.R.), the Nilsson‐Ehle funds from the Fysiografiska Sällsk‐apet in Lund, Ricerca Corrente Fondazione Ca' Granda IRCCS Policlinico of Milan (to L.V.), Associazione Malattie Metaboliche del Fegato ONLUS (to L.V.), and the Fondazione Policlinico‐INGM Molecular Medicine grant 2014‐2016, My First AIRC Grant (project code 16888 to L.V.).
Contributor Information
Stefano Romeo, Email: stefano.romeo@wlab.gu.se.
Luca Valenti, Email: luca.valenti@unimi.it.
REFERENCES
- 1. Younossi Z, Henry L. Contribution of alcoholic and nonalcoholic fatty liver disease to the burden of liver‐related morbidity and mortality. Gastroenterology 2016;150:1778‐1785. [DOI] [PubMed] [Google Scholar]
- 2. Valenti L, Bugianesi E, Pajvani U, Targher G. Nonalcoholic fatty liver disease: cause or consequence of type 2 diabetes? Liver Int 2016;36:1563‐1579. [DOI] [PubMed] [Google Scholar]
- 3. Dongiovanni P, Valenti L. A nutrigenomic approach to non‐alcoholic fatty liver disease. Int J Mol Sci 2017;18pii:E1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dongiovanni P, Romeo S, Valenti L. Genetic factors in the pathogenesis of nonalcoholic fatty liver and steatohepatitis. Biomed Res Int 2015;2015:460190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, et al. The MBOAT7‐TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 2016;150:1219‐1230.e1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta‐analysis. Hepatology 2017;65:1557‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dongiovanni P, Stender S, Pietrelli A, Mancina RM, Cespiati A, Petta S, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med 2018;283:356‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Speliotes EK, Yerges‐Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al.; NASH CRN ; GIANT Consortium ; MAGIC Investigators ; GOLD Consortium . Genome‐wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stender S, Smagris E, Lauridsen BK, Kofoed KF, Nordestgaard BG, Tybjaerg‐Hansen A, et al. Relationship between genetic variation at PPP1R3B and liver glycogen and triglyceride levels. Hepatology 2017; doi: 10.1002/hep.29751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Palmer ND, Musani SK, Yerges‐Armstrong LM, Feitosa MF, Bielak LF, Hernaez R, et al. Characterization of European ancestry nonalcoholic fatty liver disease‐associated variants in individuals of African and Hispanic descent. Hepatology 2013;58:966‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hernaez R, McLean J, Lazo M, Brancati FL, Hirschhorn JN, Borecki IB, et al.; Genetics of Obesity‐Related Liver Disease (GOLD) Consortium . Association between variants in or near PNPLA3, GCKR, and PPP1R3B with ultrasound‐defined steatosis based on data from the third National Health and Nutrition Examination Survey. Clin Gastroenterol Hepatol 2013;11:1183‐1190.e1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dwyer A, Doppman JL, Adams AJ, Girton ME, Chernick SS, Cornblath M. Influence of glycogen on liver density: computed tomography from a metabolic perspective. J Comput Assist Tomogr 1983;7:70‐73. [DOI] [PubMed] [Google Scholar]
- 13. Gorden A, Yang R, Yerges‐Armstrong LM, Ryan KA, Speliotes E, Borecki IB, et al.; GOLD Consortium . Genetic variation at NCAN locus is associated with inflammation and fibrosis in non‐alcoholic fatty liver disease in morbid obesity. Hum Hered 2013;75:34‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg‐Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet 2017;49:842‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015;61:506‐514. [DOI] [PubMed] [Google Scholar]
- 16. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 17. Dongiovanni P, Petta S, Mannisto V, Mancina RM, Pipitone R, Karja V, et al. Statin use and non‐alcoholic steatohepatitis in at risk individuals. J Hepatol 2015;63:705‐712. [DOI] [PubMed] [Google Scholar]
- 18. Donati B, Dongiovanni P, Romeo S, Meroni M, McCain M, Miele L, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non‐cirrhotic individuals Sci Rep 2017;7:4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Donati B, Pietrelli A, Pingitore P, Dongiovanni P, Caddeo A, Walker L, et al. Telomerase reverse transcriptase germline mutations and hepatocellular carcinoma in patients with nonalcoholic fatty liver disease. Cancer Med 2017;6:1930‐1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. European Association for the Study of the Liver ; European Organisation for Research and Treatment of Cancer . EASL‐EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol 2012;56:908‐943. [DOI] [PubMed] [Google Scholar]
- 21. Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2015. Nucleic Acids Res 2015;43:D662‐D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114‐2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013;29:15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kigel B, Rabinowicz N, Varshavsky A, Kessler O, Neufeld G. Plexin‐A4 promotes tumor progression and tumor angiogenesis by enhancement of VEGF and bFGF signaling. Blood 2011;118:4285‐4296. [DOI] [PubMed] [Google Scholar]
- 27. Murata H, Yagi T, Iwagaki H, Ogino T, Sadamori H, Matsukawa H, et al. Mechanism of impaired regeneration of fatty liver in mouse partial hepatectomy model. J Gastroenterol Hepatol 2007;22:2173‐2180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1192/full.
Supporting Information 1