

Abstract
Quantification of gene expression is a crucial research tool in the life sciences, which makes it important to identify any factors that could compromise its accuracy. One of these factors are non‐polyadenylated (poly(A)−) transcripts, including circular RNAs (circRNAs) that can skew quantification of gene expression as they resemble messenger RNAs (mRNAs). Here, we highlight the impact circRNAs and other poly(A)− transcripts have on gene expression profiling and the biological conclusions drawn from such experiments. We also propose easily adoptable strategies to increase the accuracy of gene expression quantification.
Subject Categories: RNA Biology, Transcription, Methods & Resources
Introduction
Changes in mRNA expression in response to experimental perturbations are widely used as an indicator of biological effect. While a number of approaches exist to quantify mRNA abundance—including qRT–PCR, microarray and next‐generation sequencing (NGS)—all techniques involve mapping data to a reference genome (Rosenfeld et al, 2012). Reference genomes have traditionally been populated with all known mRNAs, but, more recently, the discovery of antisense RNAs and other non‐coding RNAs (ncRNAs) has led to an expansion of genome annotations (Pruitt et al, 2014). The increased awareness of genome‐wide ncRNA expression has also prompted the development of new methods to discriminate between RNA species. Antisense RNAs can now be distinguished from overlapping coding RNAs owing to the advent of strand‐specific/directional strategies. However, these techniques do not discriminate against non‐coding transcripts resembling mRNAs, including circRNAs, truncated transcripts and bimorphic transcripts (see Box 1), as we will discuss below.
Box 1.Confounding transcript types.
Circular RNAs (circRNAs): These arise through alternative back‐splicing of pre‐mRNA, whereby non‐canonical and non‐collinear exons (and/or introns) are co‐transcriptionally spliced to form covalently closed, non‐polyadenylated circular RNA transcripts. They are identified through their unique back‐splice junction sequence, but are otherwise indistinguishable from the mRNA sequence, yet are practically absent from mRNA‐seq.
Truncated transcripts: These early termination transcripts, present in approximately 20% of genes, arise through alternative cleavage and polyadenylation within the coding region and can significantly impact the transcriptome and functionally impact the proteome (Tian et al, 2007; Li et al, 2015). Like circRNAs, they are indistinguishable from the mRNA sequence, but are exclusively present in total RNA‐seq libraries.
Bimorphic RNA: These transcripts are defined as those that do not clearly fall into either poly(A)+ or ploy(A) − groups as they exist with and without poly(A) tails. The poly(A)+ form will be detected in mRNA‐seq but not so the poly(A)− form however both will be present in total RNA‐seq libraries (Yang et al, 2011).
Histone variants: Transcripts encoding the ~ 75 replication‐dependent histone genes invariantly terminate with a highly conserved 16 nt long stem‐loop structure, which binds stem‐loop binding protein (SLBP), the U7 snRNP, Sm proteins and zinc finger protein 100 (ZFP of 100 kDa) (Marzluff et al, 2008). This complex leads to the cleavage of the 3′ end of the histone transcript producing the only known cellular metazoan mRNAs to lack polyadenylate tails. The resultant abundant transcripts will align to the relevant exons in total RNA‐seq, but will be absent from mRNA‐seq datasets.
Long non‐coding RNAs (lncRNAs): These non‐coding RNAs commonly arise from intergenic regions, or in antisense orientation with respect to the sense transcript. They have been classified into four major subcategories by the ENCODE project: (i) antisense, (ii) large intergenic non‐coding RNAs (lincRNAs), (iii) sense intronic and (iv) processed transcripts. They commonly have their own promoter/enhancer can be either polyadenylated or non‐polyadenylated, and are emerging as functional transcripts in cellular homeostasis and disease contexts. They are identified through mapping to a unique DNA strand and region (stranded NGS).
Distinct RNA isoforms from a single genetic locus
All RNAs transcribed from a single gene can be classified as polyadenylated (poly(A)+) or non‐polyadenylated (poly(A)−) transcripts (Fig 1A). While mRNAs comprise the bulk of the coding poly(A)+ transcript pool, some RNA transcripts are defined as bimorphic, in that they can exist in both poly(A)+ and poly(A)− populations, and include antisense RNAs and ncRNAs (Fig 1A; Yang et al, 2011). CircRNAs are a recently discovered class of RNAs derived from pre‐mRNAs through a process known as back‐splicing (Jeck et al, 2013). During back‐splicing, a donor splice site fuses with an upstream acceptor splice site to generate a covalently closed, exon‐rich circular RNA that lacks a poly(A) tail (Fig 1A and B; Ashwal‐Fluss et al, 2014). Although the circular form of an RNA possesses a unique back‐splice junction sequence, which can be used to discriminate it from the cognate linear mRNA, the rest of the circRNA is identical to the cognate linear mRNA. As a consequence, current gene expression analyses cannot automatically distinguish circRNAs from mRNAs.
Figure 1. RNA variants and RNA fractionation for mRNA‐seq versus total RNA‐seq.
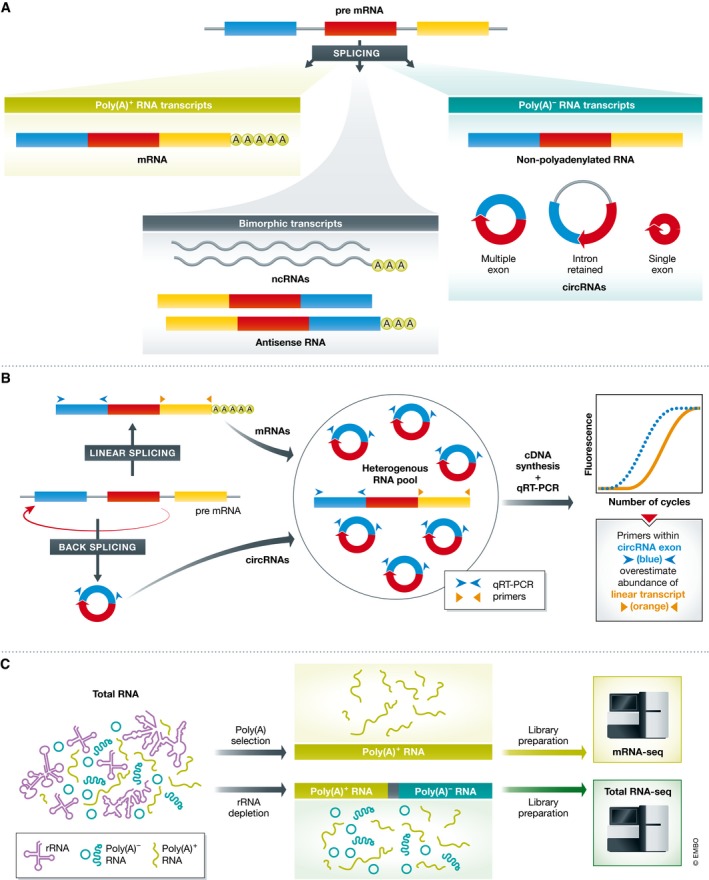
(A) Schematic of a three‐exon gene and the potential RNA molecules produced from it, stratified into poly(A)+ RNA, poly(A)− RNA and bimorphic RNA transcripts. Note circular RNAs can be single or multi‐exon, and can retain intervening introns. (B) Linear splicing of RNA into mRNA and back‐splicing to produce circRNAs. Schematic showing how circRNAs increase amplification of target transcripts by qRT–PCR. (C) Library preparation methods for mRNA‐seq and total RNA‐seq, highlighting RNA species which persist in the preparations.
Quantifying RNA transcripts: finding a needle in a large pile of needles
qRT–PCR
The standard approach for quantifying individual candidate gene expression is qRT–PCR. Irrespective of the detection chemistry used, the approach requires a pair of specific primers designed to exclusively amplify the target gene. The quantitative basis of qRT–PCR relies on primer specificity producing two amplicons from each target with every amplification cycle. As a result, qRT–PCR is sensitive to anything that alters copy numbers in the starting material as can occur when the primers amplify from a circRNA‐containing region of an mRNA (Fig 1B, blue trace). The circRNAs constitute a non‐mRNA template that will result in earlier amplification and therefore an overestimation of mRNA abundance within a particular sample (Fig 1B). Designing primers outside known circRNA regions (Fig 1B, orange trace) and/or priming reverse transcription with recursive deoxy‐thymidine primers (oligo(dT)), which cannot prime the majority of circRNAs, are strategies to limit/avoid overestimating mRNA abundance.
If all things were equal and circular RNA expression was constant, confounding effects would exist but be minor or inconsequential. However, in addition to being abundant—on occasion up to 1,000 times the abundance of the cognate mRNA (Salzman et al, 2012; Jeck et al, 2013)—circRNAs are widespread (present in 17–30% of expressed genes), cell‐type specific (Salzman et al, 2013) and developmentally regulated, often independently of their cognate mRNAs (Conn et al, 2015). The consequence is that any claims made about changes in gene expression may be affected by changes in circRNA abundance as well as other “mRNA‐indistinguishable” RNA species including truncated and bimorphic transcripts. This phenomenon also extends to housekeeping genes: glyceraldehyde acid 3‐phosphate dehydrogenase (GAPDH) and elongation factor 1α (EF‐1α), which are frequently used to normalise between samples. Online databases such as circNet (Liu et al, 2016) show that both genes are replete with circRNAs that vary in expression between samples.
Next‐generation sequencing (NGS)
While NGS becomes more popular due to dropping costs, it is still necessary to enrich for certain transcript types prior to sequencing to focus on the most relevant populations. These enrichment approaches simplify bioinformatic analyses and enhance statistical power. (Muir et al, 2016). Separation of poly(A)+ and poly(A)− RNA populations is the most commonly employed strategy for enrichment, and transcriptome analysis for a given sample is normally performed on poly(A)+ RNA (hereafter called mRNA‐seq; Fig 1C). As the mRNA‐seq process precludes the detection of poly(A)− transcripts, such as circRNAs and highly abundant ribosomal RNA (rRNA) species, it provides increased sequencing depth for poly(A)+ transcripts and reduces “background noise”. However, mRNA‐seq comes at the cost of reduced coverage of transcripts from degraded samples (such as fixed human patient material) and an inability to quantify ncRNAs that lack poly(A) tails but may still act in gene regulation, cellular homeostasis and disease (Adiconis et al, 2013; Gallego Romero et al, 2014; Zhao et al, 2014). Consequently, strategies for sequencing poly(A)− RNA, including total RNA‐seq, are becoming more prevalent. Total RNA‐seq requires depletion of rRNA from the total RNA combined with random‐primed cDNA synthesis to capture both poly(A)+ and poly (A)− RNA populations (Fig 1C). A potential disadvantage of total RNA‐seq is that a greater sequencing depth is required to obtain a coverage of poly(A)+ transcripts comparable to mRNA‐seq from the same population, since many reads will be absorbed by poly(A)− transcripts.
To compare the efficiency and reliability of total RNA‐seq and mRNA‐seq for gene expression analysis, we reanalysed published datasets prepared from the same starting RNA (Kelly et al, 2015). This study treated human umbilical vein endothelial cells (HUVECs) with transforming growth factor beta (TGFβ) over 4 h (time 0, 60, 120, 240 min) in biological duplicates and generated both mRNA‐seq and total RNA‐seq libraries from the same starting RNA. If these two quantification methods were comparable, then comparing transcript abundances from the same starting material would manifest as a scatterplot with all transcripts tightly distributed along the parity line (a line drawn on the graph where at each point the x‐value is equal to the y‐value, Fig 2A). Deviation from the parity line for any one particular observation indicates a discrepancy in the abundance estimation between the two methods (Fig 2A). Certainly comparisons by scatter plot within either mRNA‐seq or total RNA‐seq libraries found the data clustered around the parity line (Fig 2B). However, comparing between mRNA‐seq and total RNA‐seq for the same RNA generated two obvious disparities (Fig 2B):
Figure 2. Comparative expression profiling between total RNA‐seq and mRNA‐seq on the same RNA input.
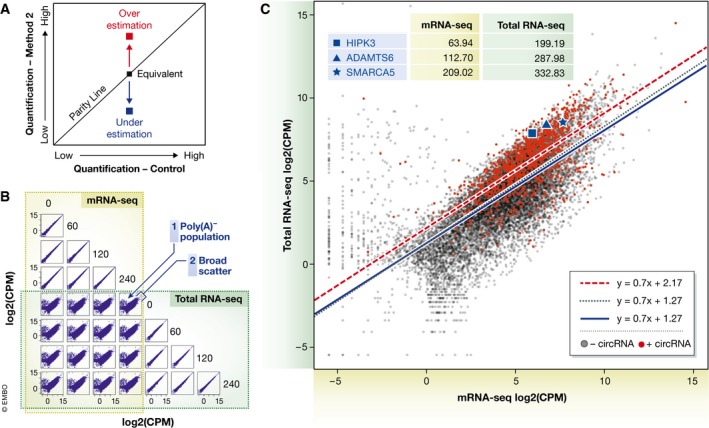
(A) Schematic scatterplot illustrating the interpretation of a transcript (square) that deviates from the parity line when comparing two methods of quantification from the same starting material. (B) Scatter matrix using data from Kelly et al (2015) comparing counts per million (CPM) at four time‐points after treatments with TGFβ (0, 60, 120 and 240 min). Unique poly(A)‐population and broad scatter highlighted for a prototypical mRNA‐seq versus total RNA‐seq plot. (C) Scatterplot of transcript abundance between mRNA‐seq and total RNA‐seq protocols for control HUVEC (0 min time‐point) libraries, with circRNA‐containing transcripts shown as red dots and circRNA‐deficient transcripts in black. Linear regression analysis for all transcripts (black dotted line), circRNA‐containing transcripts (red dashed line) and circRNA‐deficient transcripts (blue solid line). Three transcripts, each with a single prominent circRNA, annotated in blue (HIPK3—square, ADAMTS6—triangle, SMARCA5—star) along with their expression estimates in mRNA‐seq and total RNA‐seq (inset). All transcripts with CPM < 1 were removed from the analysis when calculating lines of best fit and intercepts (inset).
The presence of a population of RNAs, uniquely visible within total RNA‐seq libraries (poly(A)− population)
A distinct broadening of the scatterplot for mRNA transcripts (broad scatter)
The first anomaly, also observed and yet unexplained in a study by Zhao et al (2014), predominantly comprises the poly(A)− RNAs—specifically long non‐coding RNAs (lncRNAs), early termination transcripts and replication‐dependent histone transcripts (see Box 1 for more details on these species). While these transcripts distort the graphical comparison, it is primarily the contribution of circRNAs as “mRNA‐indistinguishable” transcripts that are responsible for the more significant broadening of the scatterplot.
CircRNA expression can affect quantification of transcript and exon levels
CircRNAs have attracted great interest as their elucidation through NGS studies has accelerated. Given that circRNAs lack poly(A) tails, they are rarely detected in mRNA‐seq, yet found abundantly in total RNA‐seq (~ 10,000 unique circRNAs per library; Kelly et al, 2015): as with random‐primed qRT–PCR, sequencing reads in total RNA‐seq arise from both the mRNA and the circRNA forms. Since calculations of gene expression from RNA‐seq data combine all reads mapping to a single transcript, circRNAs would artificially inflate read counts within coding regions of circRNA‐containing genes in the total RNA‐seq transcriptome. Similar issues with quantification of circRNA‐producing transcripts are expected for microarray experiments, the input of which is commonly total RNA.
To illustrate the average global impact of circRNAs on gene expression quantification using total RNA‐seq relative to the more focused mRNA‐seq, we have annotated the aforementioned dataset from Kelly et al (2015) comparing mRNA‐seq and total RNA‐seq data (0 min time‐point in the TGFβ dataset; Fig 2C). Transcripts were identified as circRNA‐containing if they expressed > 5 reads across their back‐splice junction (definitive for circRNAs), which is a modest, yet high‐confidence circRNA expression level. Fitting linear regression lines to these data revealed an almost twofold overestimation of mRNA expression for circRNA‐containing genes over either (i) all genes or (ii) circRNA‐deficient genes (Δlog2 = 0.9, Fig 2C) based on the linear regression intercepts (Fig 2C). Three exemplary circRNA‐producing genes—HIPK3, ADAMTS6 (Jeck et al, 2013) and SMARCA5 (Conn et al, 2015)—contain an abundant circRNA and exhibit an observable amplification of mRNA expression in total RNA‐seq of 1.6‐fold to 3.2‐fold relative to the value obtained by mRNA‐seq (Fig 2C). By quantifying the expression level of individual exons from the gene ADAMTS6, it is clear that there is a significantly higher abundance in total RNA‐seq for those exons which contain circRNAs compared to those that do not have circRNAs (Fig 3A). This also manifests on coverage plots where the peak height in circRNA‐containing exons is significantly higher than non‐circRNA‐containing exons in total RNA‐seq (Fig 3B, green track). However for mRNA‐seq (Fig 3B, gold track), the exon read height is fairly uniform across the entire gene. This shows a global impact of circRNAs on expression levels of the parent mRNA in total RNA‐seq.
Figure 3. CircRNAs confounding gene quantification at the exon level.
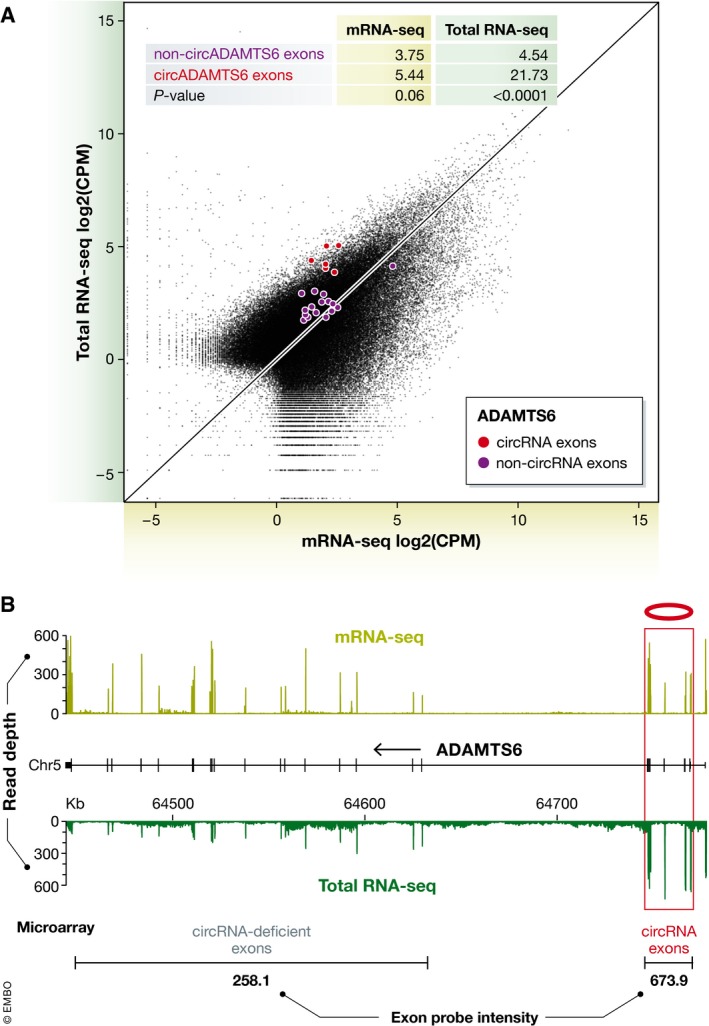
(A) Scatterplot of exon abundance (black points) between mRNA‐seq and total RNA‐seq protocols for TGFβ 0 min time‐point libraries highlighting the expression levels of exons in ADAMTS6 and their corresponding CPM (inset table) for circRNA‐containing exons (red) and non‐circRNA exons (blue). (B) Upper: Read coverage plot for mRNA‐seq (blue) and total RNA‐seq (red) protocols for ADAMTS6 expressing a prominent circRNA from exons 2–6 (shaded grey). Lower: Average microarray exon probe signal over circRNA exons (673.9) and non‐circRNA exons (258.1).
Microarray
Predating NGS technologies and arguably more widely used, microarrays quantify expression levels for thousands of genes simultaneously by hybridisation of labelled RNA to probes or probe sets custom designed to capture selected coding and, in some cases, non‐coding regions. The use of probe sets can increase the accuracy of expression estimates as well as inform on isoform usage. However, no single statistically solid method exists for combining this information, and inconsistencies between probes within a probe set have been reported, but not explained, by many groups (Schneider et al, 2012; Marakhonov et al, 2014). Again, we argue that circRNAs contribute to these inconsistencies.
To illustrate the confounding effect of “non‐mRNA species” in microarrays, we analysed expression levels of ADAMTS6 in two publicly available human microarray datasets (GSE25979 and GSE36837; Appleby et al, 2012; Weigand et al, 2012), which resulted in the same phenomenon observed in total RNA‐seq (Fig 3B). Specifically, circRNAs indiscriminately hybridise to complementary exon probes resulting in an elevated intensity signal in circRNA‐containing exons compared with non‐circRNA‐containing exons (Fig 3B). Unlike RNA‐seq data (processing of which cumulates read counts mapping to coding sequences of a transcript), microarray data processing allows for many options when summarising probe sets: selection of the probe(s) with the highest normalised intensity, lowest P‐value, likelihood to cross‐hybridise or taking the median intensity of all probes. Critically, no single option will compensate for the inherent variations introduced by circRNAs and the wrong choice will, on occasion, return misleading results. Again, mRNAs‐resembling poly(A)+ RNA species including truncated and bimorphic transcripts may impose the same effect or synergistically elevate the confounding beyond that of the circRNAs described here. Of course, prior purification of poly(A)+ RNA will mitigate the bulk of the problem.
Potential impact on alternative splicing predictions
Beyond the impact on gene expression quantification, circRNAs also alter reads that cross splice junctions of a given transcript (junction counts). The most abundant class of circRNAs are two exons in length, and they could therefore contribute additional junction counts that do not represent real alternatively spliced mRNAs. The consequence are skewed predictions in differential alternative splicing (dAS) of the cognate mRNA. Currently, a small number of methods exist to assess dAS from NGS, each relying on the accurate estimation of either exon–exon junction counts or exon coverage (or both). As a result, circRNAs and other “mRNA‐indistinguishable” factors that are visible in total RNA‐seq will likewise artificially inflate estimations of abundance and may increase the rate of false‐positive dAS candidates.
Cautionary points for RNA quantification experiments
An awareness of the confounding effects that can be caused by ncRNAs lacking poly(A) tails can help ensure that gene expression quantification remains an important and reliable research tool. To enhance the confidence in the conclusions drawn when circRNAs are confounding gene quantification, we suggest three approaches. Firstly, identify circRNAs within a gene of interest by cross‐referencing with online circRNA databases, for example CircNet (http://circnet.mbc.nctu.edu.tw) or circBase (http://circbase.org). This will permit design of qRT–PCR primers outside the circRNA regions, and for NGS, an awareness of the significant impact circRNAs may have on splice site junction counts and global gene expression. Secondly, whenever possible, design qRT–PCR primers towards the 3′ end of the transcript, with the reverse primer anchored in the UTR as this provides greater specificity, and circRNAs comprising the 3′ UTR represent a very minor fraction of the circRNA population. Finally, to mitigate cross‐talk between sense and antisense transcripts, we suggest using strand‐specific NGS protocols.
Concluding remarks
As we have explained, gene expression quantification can be unreliable. Therefore, all new RNA types that fall in the category of “mRNA indistinguishable” should be disseminated among the research community to ensure these are considered in quantification strategies. This is of particular importance as genome‐wide expression profiling studies are commonly employed as a primary screen to inform future research. We support further evolution of the reference genome annotation and approaches to account for gene expression estimates of circRNA‐containing genes. This will support the continued rise in popularity of total RNA‐seq compared with mRNA‐seq.
The circRNAs are exciting transcripts in their own right and straightforward to identify from existing total RNA‐seq experiments. Far from being troublesome molecules, the presence of circRNAs offers us not only a wake‐up call to reflect on experimental design for gene expression profiling, but to study their expression and regulation in any system where total RNA‐seq is utilised and to draw novel and exciting conclusions.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This publication was supported by National Health and Medical Research Council (GNT1144250) and Ray & Shirl Norman Cancer Research Trust project grants awarded to SJC. Fellowship support was provided by the Australian Research Council Future Fellowship to SJC (FT160100318).
The EMBO Journal (2018) 37: e97945
References
- Adiconis X, Borges‐Rivera D, Satija R, DeLuca DS, Busby MA, Berlin AM, Sivachenko A, Thompson DA, Wysoker A, Fennell T, Gnirke A, Pochet N, Regev A, Levin JZ (2013) Comparative analysis of RNA sequencing methods for degraded or low‐input samples. Nat Methods 10: 623–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleby SL, Cockshell MP, Pippal JB, Thompson EJ, Barrett JM, Tooley K, Sen S, Sun WY, Grose R, Nicholson I, Levina V, Cooke I, Talbo G, Lopez AF, Bonder CS (2012) Characterization of a distinct population of circulating human non‐adherent endothelial forming cells and their recruitment via intercellular adhesion molecule‐3. PLoS One 7: e46996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwal‐Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S (2014) circRNA biogenesis competes with pre‐mRNA splicing. Mol Cell 56: 55–66 [DOI] [PubMed] [Google Scholar]
- Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ (2015) The RNA binding protein quaking regulates formation of circRNAs. Cell 160: 1125–1134 [DOI] [PubMed] [Google Scholar]
- Gallego Romero I, Pai AA, Tung J, Gilad Y (2014) RNA‐seq: impact of RNA degradation on transcript quantification. BMC Biol 12: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19: 141–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S, Greenman C, Cook PR, Papantonis A (2015) Exon skipping is correlated with exon circularization. J Mol Biol 427: 2414–2417 [DOI] [PubMed] [Google Scholar]
- Li W, You B, Hoque M, Zheng D, Luo W, Ji Z, Park JY, Gunderson SI, Kalsotra A, Manley JL, Tian B (2015) Systematic profiling of poly(A)+ transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet 11: e1005166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y‐C, Li J‐R, Sun C‐H, Andrews E, Chao R‐F, Lin F‐M, Weng S‐L, Hsu S‐D, Huang C‐C, Cheng C, Liu C‐C, Huang H‐D (2016) CircNet: a database of circular RNAs derived from transcriptome sequencing data. Nucleic Acids Res 44: D209–D215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marakhonov A, Sadovskaya N, Antonov I, Baranova A, Skoblov M (2014) Analysis of discordant Affymetrix probesets casts serious doubt on idea of microarray data reutilization. BMC Genom 15(Suppl 12): S8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzluff WF, Wagner EJ, Duronio RJ (2008) Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat Rev Genet 9: 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir P, Li S, Lou S, Wang D, Spakowicz DJ, Salichos L, Zhang J, Weinstock GM, Isaacs F, Rozowsky J, Gerstein M (2016) The real cost of sequencing: scaling computation to keep pace with data generation. Genome Biol 17: 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Brown GR, Hiatt SM, Thibaud‐Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM, Murphy MR, O'Leary NA, Pujar S, Rajput B, Rangwala SH, Riddick LD, Shkeda A, Sun H, Tamez P, Tully RE et al (2014) RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 42: D756–D763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Mason CE, Smith TM (2012) Limitations of the human reference genome for personalized genomics. PLoS One 7: e40294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO (2013) Cell‐type specific features of circular RNA expression. PLoS Genet 9: e1003777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO (2012) Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7: e30733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S, Smith T, Hansen U (2012) SCOREM: statistical consolidation of redundant expression measures. Nucleic Acids Res 40: e46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Pan Z, Lee JY (2007) Widespread mRNA polyadenylation events in introns indicate dynamic interplay between polyadenylation and splicing. Genome Res 17: 156–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigand JE, Boeckel J‐N, Gellert P, Dimmeler S (2012) Hypoxia‐induced alternative splicing in endothelial cells. PLoS One 7: e42697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Duff MO, Graveley BR, Carmichael GG, Chen L‐L (2011) Genomewide characterization of non‐polyadenylated RNAs. Genome Biol 12: R16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, He X, Hoadley KA, Parker JS, Hayes DN, Perou CM (2014) Comparison of RNA‐Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genom 15: 419 [DOI] [PMC free article] [PubMed] [Google Scholar]