

Summary
We recently proposed that the maturation of a new epileptic focus (epileptogenesis) may explain late seizure recurrences, starting months to years after resective epilepsy surgery. We explore here the hypothesis that inherent transcriptomic changes may distinguish such “late relapsers.” An in‐depth clinical review of 2 patients with recurrent seizures starting years after surgery is contrasted to 4 controls who remained seizure‐free postoperatively. This clinical analysis is combined with RNA sequencing from the resected brain tissue, followed by unsupervised hierarchical clustering, independent pathway analysis, and multidimensional scaling analysis. Late‐recurrence patients clustered apart from seizure‐free patients, with late recurrence patients clustering together in the central space, whereas the seizure‐free patients clustered together in the periphery. We utilized RNA‐seq to identify differentially expressed genes between late‐recurrence and seizure‐free samples. We found 29 annotated genes with statistically significant differential expression (q < 0.05). The top canonical pathways identified as distinctly separating the late‐recurrence patients from the seizure‐free patients included the intrinsic prothrombin activation pathway (p = 1.55E‐06), the complement system (p = 4.57E‐05), and the atherosclerosis signaling pathway (p = 4.57E‐05). Our observations suggest that late recurrences after epilepsy surgery may be influenced partly by differences in gene expression in neuroinflammatory and brain healing/remodeling pathways. Such a hypothesis needs to be validated in the future.
Keywords: Epilepsy surgery, Epileptogenesis, Genetics, Neuroinflammation, Outcome research
Key Points.
We found 29 annotated genes with statistically significant differential expression in late‐recurrence versus seizure‐free patients after resective surgery
Intrinsic prothrombin activation pathway, complement system, and atherosclerosis signaling pathway differ in late‐recurrence from seizure‐free patients
Inherent, potentially genetic, differences in mechanisms of healing and neuroinflammation may contribute to seizure recurrence after epilepsy surgery
Epilepsy affects 50 million people worldwide and is resistant to medical therapy in up to 30%. The most effective option for this subgroup is resective brain surgery, but seizures recur in 45–50% of patients by 10 years after temporal lobectomy,1 and in 60–70% after extratemporal resections.2, 3 Traditional explanations of surgical failure revolve around the inability to localize or completely resect the epileptic tissue.4, 5 “Missing the epileptic focus” easily accounts for seizures recurring within the early or immediate postoperative period when epilepsy continues unabated despite surgery. However, “missing the epileptic focus” cannot explain later relapses, where patients initially respond to surgery and achieve a sustained seizure‐freedom, only to have seizures recur months to years after a resection. Difficulty attributing these late‐seizure recurrences to incomplete resection of an existing focus, together with the inability to predict postoperative seizure freedom solely based on clinical variables, we hypothesize that the mechanisms driving surgical outcomes may extend beyond traditional surgical variables and involve inherent biological patient characteristics that lower the threshold for epileptogenesis. Aiming to explore the hypothesis that such late recurrences may be partly influenced by differences in gene expression, we report a pilot study comparing the clinical and RNA sequencing data of 2 cases with “late seizure recurrence” after temporal lobectomy to 4 patients who were rendered seizure‐free with surgery.
Case Descriptions
Patient 1
This is a right‐handed man with no epilepsy risk factors and no family history of epilepsy. His seizures started at age 13 years with episodes of a sensation of “butterflies in the stomach,” a metallic taste, and a strange smell followed by loss of awareness, lip smacking, and fumbling with his hands, rarely progressing to convulsions. Seizures were initially controlled on carbamazepine, 200 mg/day, and he was seizure‐free until his senior high‐school year when seizures gradually worsened in frequency despite adjusting his anti‐epileptic drugs (AEDs). At age 27 years, resective epilepsy surgery was investigated. A detailed presurgical evaluation was consistent with right anterior temporal lobe epilepsy: scalp video–electroencephalography (EEG) showed right anterior temporal spikes and seizures; positron emission tomography (PET) showed right temporal hypometabolism, magnetic resonance imaging (MRI) showed right temporal pole blurring, and Wada test confirmed left‐hemisphere language dominance. The patient underwent a standard right anterior temporal lobectomy, including the mesial temporal structures. The pathology revealed mild gliosis in the hippocampus with no overt dysplastic abnormalities in the temporal cortex. He had rare postoperative olfactory auras, which resolved within 2 months, and no postoperative seizures. He returned to work as a computer analyst. Five years after being seizure‐free on levetiracetam and carbamazepine, he stopped levetiracetam. He remained seizure‐free for another 4 years, so he stopped carbamazepine. Seven months later, his family found him convulsing. Carbamazepine was reinitiated but seizures continued, initially at a frequency of 1 per month, gradually worsening over the subsequent 2 years to 1 per day. Postoperative seizures started with a “bad taste,” reminiscent of his preoperative auras, but with a distinctly different evolution of symptomatology to excessive salivation, hyperventilation, and tachycardia prior to losing awareness. Witnesses described him as scared, rolling around, and asking for help. Video‐EEG, PET scan, and stereo‐EEG evaluations confirmed that the recurrent seizures were in fact arising from the right orbitofrontal cortex. Repeat surgery revealed “nonspecific gliosis.” He was seizure‐free for 2 years. Seizures then recurred at a frequency of 1 every 3–4 months, increasing to 3 per month over the subsequent 2 years. Semiology now is a smell and taste aura followed by “seeing something on the right side of his visual field” and then convulsing. He has declined further surgical work‐up.
Patient 2
This is a right‐handed girl who started having seizures at 9 years of age. She has no epilepsy risk factors, and no family history of seizures. Her seizures were staring episodes associated with manual and oral automatisms. By 14 years of age, she had already tried multiple AEDs without success, so surgery was considered. At the time, she was receiving zonisamide monotherapy (600 mg/day) and was having 1–7 seizures per month. Scalp video‐EEG, PET scan, and ictal single‐proton emission computerized tomography (SPECT) results suggested a mesial left temporal epilepsy localization, which was subsequently confirmed with invasive EEG recordings, so she underwent a standard left anterior temporal lobectomy. Pathology showed “mild” cortical dysplasia. A 6‐month postoperative scalp EEG showed no epileptiform activity. She was seizure‐free and aura‐free for 2 years on a reduced dose of zonisamide (300 mg/day) until a breakthrough seizure in the context of sleep deprivation in college. Zonisamide was increased, but seizures still gradually increased in frequency from 3 per year initially to 1 per month within the subsequent 4 years. Postoperative seizure semiology differed from her preoperative events: she has sudden onset tachycardia followed by overall body numbness/tingling and blurring of vision. Witnesses describe confusion and eye‐blinking for a couple of minutes. Scalp EEG showed left temporal spiking. She has declined work‐up for re‐operation.
Materials and Methods
Patient accrual and sample collection
Patients 1 and 2 were enrolled into an existing repository of fresh frozen tissue from epilepsy patients. Four seizure‐free patients who underwent temporal lobe resection within a similar timeframe were selected as a comparison for the transcriptomic analysis (Table 1). This study was approved by the Cleveland Clinic Institutional Review Board.
Table 1.
Detailed clinical characteristics of 2 cases of late seizure relapse and 4 seizure‐free patients used as controls in the RNA‐sequencing studies
| Patient number | Sex | Hand | Age at surgery in years | Seizure outcome | Age at onset in years | Epilepsy duration in years | Pathology | AEDs at surgery | Surgery side | Seizure frequency at time of surgery/month | Comorbidities at the time of surgery | Invasive EEG |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | Right | 28 | Relapse 9 years after surgery | 13 | 14 | Non‐specific with gliosis | CBZ, LEV | Right | 40 | None | None |
| 2 | F | Right | 14 | Relapse <1 year after surgery | 9 | 5 | Cortical dysplasia | ZNS | Left | 4 | None | SDE |
| A | M | Right | 46 | Seizure‐ free 3 years after surgery | 38 | 8 | Cortical dysplasia | CBZ | Right | 80 | None | SDE |
| B | F | Ambi‐dextrous | 48 | Seizure‐free 11 years after surgery | 1 | 47 | Cortical dysplasia and hippocampal sclerosis | LEV | Left | 1 | Hypertension, asthma, fibromyalgia | SDE |
| C | M | Right | 23 | Seizure‐free 16 years after surgery | 15 | 8 | Hippocampal sclerosis | LEV, LTG | Right | 3 | None | None |
| D | F | Left | 14 | Seizure‐ free 9 years after surgery | 9 | 5 | Cortical dysplasia | OXC | Left | 90 | None | SDE |
AEDs, antiepileptic drugs; CBZ, carbamazepine; LEV, levetiracetam; ZNS, zonisamide; LTG, lamotrigine; OXC, oxcarbazepine; SDE, subdural electrodes.
Isolation and purification of RNA from brain tissue
Total RNA was isolated from approximately 50 mg of flash frozen brain tissues as we have reported previously.6 Briefly, tissues were homogenized in 1 ml Trizol (Life Technologies, Carlsbad, CA, U.S.A.) using 5 mm stainless steel beads for 2 minutes at 25 Hz with a TissueLyserII (Qiagen, Valencia, CA, U.S.A.). Total RNA was then extracted using Trizol standard extraction protocol. We utilized the Ambion PureLink RNA Mini Kit (Life Technologies) for RNA clean‐up and DNase treatment. We assessed RNA quality using a 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, U.S.A.), with all RNA samples yielding acceptable RIN quality scores >7.
Library preparation and RNA sequencing
Sequencing libraries were prepared using the Illumina TruSeq Stranded Total RNA with Ribo Zero Sample Prep Kit (Illumina, San Diego, CA, U.S.A.). Libraries were sequenced (100 bp, paired‐end) using the Illumina HiSeq2000 platform at Génome Québec (Montréal, Canada). RNA and sequencing quality control metrics are summarized in Table S1. Raw sequencing FASTQ files were quality controlled using FastQC analysis (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to identify base quality profiles, inefficient removal of ribosomal RNA (rRNA), k‐mer analysis to identify problems such as adapter contamination, and abundance of fragments that are shorter than target read length (100 bp). Finally, adapter trimming was performed prior to alignment and downstream analysis to remove adaptor sequences used during library preparation.
Read mapping and differential gene expression analysis
Paired‐end FASTQ files were aligned to human genome assembly (hg19) using TopHat (release 2.0.11).7 Cufflinks (release 2.2.0) was used to assemble the aligned transcripts.7 Cuffdiff2 (release 2.2.0) was used to quantitate differential expression between groups of samples.7 Gene transcripts with a false discovery rate (q‐value) <0.05 were considered to be differentially expressed. To visualize the differential expression output from Cuffdiff2, we employed CummeRbund (release 2.6.7).7 We performed pathway analysis using Ingenuity Pathway Analysis (IPA; QIAGEN Bioinformatics, Redwood City, CA, U.S.A.). p‐values derived from IPA indicate significance after Benjamini‐Hochberg multiple testing correction.
Results
Clustering of samples
With unsupervised hierarchical clustering of RNA expression in epileptic foci, using Jensen‐Shannon distance within CummeRbund, late‐recurrence patients (Fig. 1A, Patients 1 and 2) cluster apart from seizure‐free patients (Fig. 1A, Patients A‐D). Similarly, with multidimensional scaling (MDS) analysis performed to reduce the overall differential expression to a single point in 2‐dimensional space, late‐recurrence patients cluster together in the central space, whereas the seizure‐free patients cluster together in the periphery (Fig. 1B).
Figure 1.
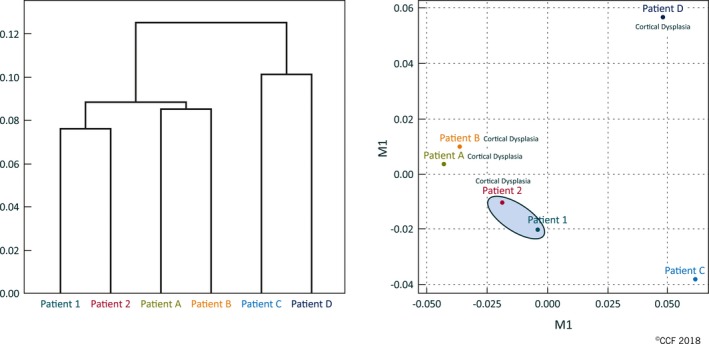
RNA‐seq expression differences cluster late‐recurrence patient apart from seizure‐free patients. Panel A shows a dendrogram reflecting the relatedness of the expression profiles between patients. Panel B shows a multidimensional scaling (MDS) analysis, which reduces the overall differential expression of all the regions of the genome for a patient to a single point in 2‐dimensional space. The gray box highlights the late recurrence patients. We annotated patients with cortical dysplasia to show lack of clustering due to this pathology.
Identification of differentially expressed genes
Using Cuffdiff 2 to distinguish differentially expressed genes from late‐recurrence and seizure‐free patient samples, we found 29 annotated genes with statistically significant (q < 0.05) differential expression (Fig. S1 and Table S2).
Pathway analysis
Pathway analysis was performed on differentially expressed genes with fragments per kilobase of transcript per million mapped reads (FPKM) ≥5 and top significant pathways identified after Benjamini‐Hochberg multiple testing correction of p‐values. Such a filtration approach resulted in 19 differentially expressed genes with FPKM ≥5. The top canonical pathways identified as distinctly separating the late recurrence patients from the seizure‐free patients included the intrinsic prothrombin activation pathway (p = 1.55E‐06), the complement system (p = 4.57E‐05), and the atherosclerosis signaling pathway (p = 4.57E‐05). Notably, all pathways presented include extensive overlap with respect to the prioritized genes within each pathway (Table 2). We validated expression changes of genes (n = 9) belonging to the prioritized canonical pathways using quantitative real‐time reverse transcription PCR (Figs. S2 and S3).
Table 2.
Ingenuity Pathway Analysis (IPA) top canonical pathways
| Ingenuity canonical pathwaysa | −log(B‐H p‐value) | p‐value | Ratio | Differentially expressed genesb |
|---|---|---|---|---|
| Intrinsic prothrombin activation pathway | 5.81 | 1.549E‐06 | 0.0976 | COL1A2, COL1A1, F13A1, COL3A1 |
| Complement system | 4.34 | 4.571E‐05 | 0.0833 | C1R, C7, CFI |
| Atherosclerosis signaling | 4.34 | 4.571E‐05 | 0.0323 | COL1A2, COL1A1, LYZ, COL3A1 |
| GP6 signaling pathway | 2.79 | 1.622E‐03 | 0.0229 | COL1A2, COL1A1, COL3A1 |
| Hepatic fibrosis/hepatic stellate cell activation | 2.53 | 2.951E‐03 | 0.0164 | COL1A2, COL1A1, COL3A1 |
| Dendritic cell maturation | 2.53 | 2.951E‐03 | 0.0162 | COL1A2, COL1A1, COL3A1 |
| Neuroprotective role of THOP1 in Alzheimer's disease | 1.73 | 1.862E‐02 | 0.0179 | C1R, SERPINA3 |
| Acute phase response signaling | 1.45 | 3.548E‐02 | 0.0118 | C1R, SERPINA3 |
| Extrinsic prothrombin activation pathway | 1.37 | 4.266E‐02 | 0.0625 | F13A1 |
Significant pathways after Benjamini‐Hochberg multiple testing correction.
Derived from an input of n = 19 differentially expressed genes with FPKM ≥5.
Discussion
It is clear that epilepsy surgery is far from curative, as seizures recur in more than half the patients,1, 2, 3 and less than one‐fourth successfully stop AEDs.8 Longitudinal seizure outcomes following temporal and extratemporal epilepsy surgery reveal that regardless of the type of resection, half of all postoperative seizure recurrences start within 2–6 postoperative months, whereas the remaining half start within the subsequent 10–15 years.1, 2, 3 This consistent observation of 2 separate “early” and “late” phases of seizure recurrence suggests 2 distinct mechanisms of seizure recurrence.9 In fact, “early” postoperative recurrences (<6 postoperative months) are very likely to be drug‐resistant from their onset, and to arise from foci distant to the surgical bed,10, 11 suggesting that they indeed represent a persistence of an inaccurately localized or incompletely resected epileptogenic zone. The predictors of such early recurrences support markers of diffuse and poorly localized epilepsy like: (1) bilateral abnormalities on brain MRI, (2) the need to obtain ictal and functional mapping through invasive EEG recordings for surgical planning, and (3) interictal spikes on postoperative EEG.1
The two “late‐recurrence” patients reported here clearly deviate from this traditional profile of surgical failures: their EEG, clinical, and functional neuroimaging pre‐operative testing yielded concordant information, so preoperative epilepsy localization was not challenging. Their postoperative EEG did not show any residual epileptogenicity, and, in fact, their surgery was initially “successful”: both were having almost daily seizures prior to resection and became seizure‐free for 2–9 years after surgery, a phenomenon that is difficult to attribute to chance alone, but rather suggests adequate response to surgery, followed by de novo development of epilepsy. For both patients, the recurrent seizures behaved more like a “new‐onset,” epilepsy as they started in the context of a provoking factor (stopping AEDs in Patient 1 and sleep deprivation in Patient 2), represented a “new” seizure semiology that was not present preoperatively, and were infrequent at their onset (1 per month in Patient 1 and 1 per 4 months in Patient 2), gradually developing drug‐resistance over the subsequent several years. Patient 1 repeated this course of prolonged postoperative seizure‐freedom followed by relapse twice. These observations raise the alternative hypothesis that a different mechanism—beyond a missed epileptic focus—may contribute to seizure recurrence in these special situations of late relapse.
Finding a credible biological mechanism for postoperative seizure recurrence after prolonged seizure‐freedom has been difficult,9 as most occur in patients with no clear histopathological substrate for their operated epilepsy12 (as in our cases). If inherent predisposing factors contribute to a proepileptic tendency, then examining the resected tissue may give useful predictive insights, even though the epilepsy is now arising from a different localization. We therefore performed RNA sequencing of the resected brain tissue to investigate variation in gene expression that may make these patients particularly more vulnerable to de novo developing epilepsy—the process of epileptogenesis.
Comparing RNA‐sequencing findings in our two “late recurrence” cases to those of the seizure‐free controls helps with filtering out common alterations due to having epilepsy per se, and with preferentially detecting “signals” of higher risk of late relapse. The comparable AED usage, epilepsy duration, and seizure frequencies in the controls help homogenize the impact of these variables on the genetic makeup of the tissue.
The genes and pathways we identified represent integral components of epileptogenesis. ATP diphosphohydrolase and 5’‐nucleotidase involved in atherosclerosis signaling are critical in maintaining an adequate ATP (excitatory) to adenosine (neuroprotective) balance, have been implicated in kindling of temporal lobe epilepsy after status epilepticus in pentylenetetrazol animal models, and are altered in the dentate gyrus of human hippocampal epilepsy.13 Abundant literature supports the role of neuroinflammation and complement system in epileptogenesis.14, 15 With recent data linking single nucleotide polymorphism (SNP) interleukin (IL)–1β variability with higher risks of posttraumatic epilepsy,16 it is interesting to note that IL‐1 receptor type II gene, IL‐7R, and IL‐8 genes, among other inflammation markers, were differentially expressed in our late relapsers, suggesting that genetic variability in neuroinflammatory pathways implicated in brain healing after head trauma (or brain surgery) may affect long‐term postoperative seizure outcomes. Involvement of the intrinsic prothrombin pathway suggested by our data further supports this hypothesis. It is notable that Table 1 documents the long duration of follow‐up in our seizure‐free patients, validating an appropriate choice for controls, and the relative lack of conditions predisposing to the prothrombin/atherosclerosis/complement pathway differences supporting the potential to attribute our findings to differences in seizure outcomes.
Limitations
This pilot study is underpowered to make definitive conclusions. The goal is exploratory at this stage, however, and we fully acknowledge in the context of this preliminary report that our findings need to be validated. Gene expression studies, as done here, can be influenced by several complex variables that we cannot account for in these retrospectively collected human samples. However, stringent pathway analysis including only differentially expressed genes with FPKM ≥5 and p‐value correction resulted in the same prioritized canonical pathways. These data suggest a strong biological signature that warrants validation in larger cohorts or by other groups.
Conclusion
So far, approaches at improving surgical outcomes have focused exclusively on improving epilepsy localization. Our observations suggest that for half of these surgical failures, the “late‐relapsers,” a genomic predisposition to epileptogenesis may also be partly relevant. This pilot series is hypothesis‐generating and requires hypothesis‐testing in larger cohorts. If validated, this may open a new approach to epilepsy surgery incorporating a more focused patient selection and a modified perioperative medical therapy to mitigate epileptogenesis.
Disclosure
No author has any relevant conflicts of interest. The study was approved by Cleveland Clinic Institutional Review Board. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Author Contributions
Lara Jehi: design of the study, interpretation of the results, drafting of the manuscript; Lamis Yehia: reanalysis and experimentation for the revised manuscript, critical revision of the manuscript; Charissa Peterson: analysis of the data, interpretation of the results; Farshad Niazi: analysis of the data; Robyn Busch, Richard Prayson, Zhong Ying, William Bingaman, Imad Najm: revision of the manuscript; Charis Eng: design and conceptualization of the study, interpretation of the data, revision of the manuscript.
Supporting information
Figure S1. Dendrogram and heatmap representing log2(FPKM) values of 29 significant differentially expressed genes.
Figure S2. Fold changes of the differentially expressed genes belonging to IPA top signaling pathways as determined through transcriptomic analysis.
Figure S3. Validation of the differentially expressed genes belonging to IPA top signaling pathways using quantitative reverse transcription polymerase chain reaction.
Table S1. RNA and sequencing quality control metrics.
Table S2. Statistically significant differentially expressed annotated genes.
Acknowledgments
We thank Ann Warbel, NP, for maintaining the Cleveland Clinic Epilepsy Center's Outcomes database. We also thank Sanaa Jehi, PhD, for help in preparing the figures. C.E. is the Sondra J. and Stephen R. Hardis Endowed Chair at the Cleveland Clinic, and an ACS Clinical Research Professor. L.J. is supported by NINDS R21 NS099734.
Biography
Dr. Lara Jehi is an epileptologist, and the director of research at the Cleveland Clinic Epilepsy Center.

References
- 1. Jehi L, Yardi R, Chagin K, et al. Development and validation of nomograms to provide individualised predictions of seizure outcomes after epilepsy surgery: a retrospective analysis. Lancet Neurol 2015;14:283–290. [DOI] [PubMed] [Google Scholar]
- 2. Simasathien T, Vadera S, Najm I, et al. Improved outcomes with earlier surgery for intractable frontal lobe epilepsy. Ann Neurol 2013;73:646–654. [DOI] [PubMed] [Google Scholar]
- 3. See SJ, Jehi LE, Vadera S, et al. Surgical outcomes in patients with extratemporal epilepsy and subtle or normal magnetic resonance imaging findings. Neurosurgery 2013;73:68–77. [DOI] [PubMed] [Google Scholar]
- 4. Reed CM, Dewar S, Fried I, et al. Failed epilepsy surgery deserves a second chance. Clin Neurol Neurosurg 2017;163:110–115. [DOI] [PubMed] [Google Scholar]
- 5. Englot DJ, Raygor KP, Molinaro AM, et al. Factors associated with failed focal neocortical epilepsy surgery. Neurosurgery 2014;75:648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tilot AK, Bebek G, Niazi F, et al. Neural transcriptome of constitutional Pten dysfunction in mice and its relevance to human idiopathic autism spectrum disorder. Mol Psychiatry 2016;21:118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc 2012;7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yardi R, Irwin H, Gupta A. Reducing versus stopping antiepileptic medications after temporal lobe surgery. Ann Clin Transl Neurol 2014;1:115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Najm I, Jehi L, Palmini A, et al. Temporal patterns and mechanisms of epilepsy surgery failure. Epilepsia 2013;54:772–782. [DOI] [PubMed] [Google Scholar]
- 10. Jehi L, Sarkis R, Bingaman W, et al. When is a postoperative seizure equivalent to “epilepsy recurrence” after epilepsy surgery? Epilepsia 2010;51:994–1003. [DOI] [PubMed] [Google Scholar]
- 11. Jehi LE, Silveira DC, Bingaman W, et al. Temporal lobe epilepsy surgery failures: predictors of seizure recurrence, yield of reevaluation, and outcome following reoperation. J Neurosurg 2010;113:1186–1194. [DOI] [PubMed] [Google Scholar]
- 12. Schwartz TH, Jeha L, Tanner A, et al. Late seizures in patients initially seizure free after epilepsy surgery. Epilepsia 2006;47:567–573. [DOI] [PubMed] [Google Scholar]
- 13. Schetinger MR, Morsch VM, Bonan CD, et al. NTPDase and 5’‐nucleotidase activities in physiological and disease conditions: new perspectives for human health. BioFactors 2007;31:77–98. [DOI] [PubMed] [Google Scholar]
- 14. Klang A, Schmidt P, Kneissl S, et al. IgG and complement deposition and neuronal loss in cats and humans with epilepsy and voltage‐gated potassium channel complex antibodies. J Neuropathol Exp Neurol 2014;73:403–413. [DOI] [PubMed] [Google Scholar]
- 15. Vezzani A, Friedman A, Dingledine RJ. The role of inflammation in epileptogenesis. Neuropharmacology 2013;69:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diamond ML, Ritter AC, Failla MD, et al. IL‐1β associations with posttraumatic epilepsy development: a genetics and biomarker cohort study. Epilepsia 2015;56:991–1001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dendrogram and heatmap representing log2(FPKM) values of 29 significant differentially expressed genes.
Figure S2. Fold changes of the differentially expressed genes belonging to IPA top signaling pathways as determined through transcriptomic analysis.
Figure S3. Validation of the differentially expressed genes belonging to IPA top signaling pathways using quantitative reverse transcription polymerase chain reaction.
Table S1. RNA and sequencing quality control metrics.
Table S2. Statistically significant differentially expressed annotated genes.