

Summary
Objective
Sympathetic predominance and ventricular repolarization abnormalities represent epilepsy‐associated cardiac alterations and may underlie seizure‐induced ventricular arrhythmias. Myocardial ion channel and electrical remodeling have been described early in epilepsy development and may contribute to ventricular repolarization abnormalities and excitability. Using the pilocarpine‐induced acquired epilepsy model we sought to examine whether altered myocardial ion channel levels and electrophysiological changes also occur in animals with long‐standing epilepsy.
Methods
We examined myocardial adrenergic receptor and ion channel protein levels of epileptic and age‐matched sham rats (9–20 months old) using western blotting. Cardiac electrical properties were examined using optical mapping ex vivo and electrophysiology in vivo. We investigated the propensity for ventricular tachycardia (VT) and the effects of β‐adrenergic blockade on ventricular electrical properties and excitability in vivo.
Results
In animals with long‐standing epilepsy, we observed decreased myocardial voltage‐gated K+ channels Kv4.2 and Kv4.3, which are known to underlie early ventricular repolarization in rodents. Decreased β1 and increased α1A adrenergic receptor protein levels occurred in the myocardium of chronically epileptic animals consistent with elevated sympathetic tone. These animals exhibited many cardiac electrophysiological abnormalities, represented by longer QRS and corrected QT (QTc) intervals in vivo, slower conduction velocity ex vivo, and stimulation‐induced VT. Administration of a β‐adrenergic antagonist late in epilepsy was beneficial, as the therapy shortened the QTc interval and decreased stimulation‐induced VT.
Significance
Our findings demonstrate that myocardial ion channel remodeling and sympathetic predominance, risk factors for increased ventricular excitability and arrhythmias, persist in chronic epilepsy. The beneficial effects of β‐adrenergic antagonist treatment late in the course of epilepsy suggest that attenuating elevated sympathetic tone may represent a therapeutic target for ameliorating epilepsy‐associated cardiac morbidity.
Keywords: Epilepsy, Cardiac remodeling, Ventricular tachycardia, Kv4.2, β‐Adrenergic blockade
Key Points.
Sympathetic predominance and myocardial ion channel remodeling are present in rats with chronic epilepsy
Altered cardiac electrophysiology is evident in a model of chronic epilepsy
Modulating sympathetic tone may represent a therapeutic target for cardiac protection in chronic epilepsy
Epilepsy is a common neurological condition affecting more than 50 million people worldwide.1 Clinical observations from individuals with epilepsy have demonstrated elevated resting heart rate (HR), decreased heart rate variability (HRV), and prolongation of the corrected QT interval (QTc), indicating cardiac sympathetic predominance and altered ventricular repolarization.2, 3, 4 Seizures can exacerbate these abnormalities with worsening of the QTc interval derangements, abnormal ventricular conduction, and ventricular arrhythmias.5, 6, 7, 8 Fatal and near‐fatal cases of seizure‐associated ventricular tachycardia (VT), ventricular fibrillation (VF), and asystole9, 10, 11 highlight the possibility that an increased ventricular arrhythmogenic risk may accompany epilepsy, which in some instances may contribute to early mortality including sudden unexpected death in epilepsy (SUDEP).12, 13
In models of status epilepticus–induced acquired epilepsy, in which there is an initial episode of status epilepticus that is followed by spontaneous recurrent seizures (epilepsy) later in life, molecular and electrophysiological alterations reflecting myocardial ion channel remodeling and sympathetic predominance have been described early in the development of epilepsy.14, 15, 16, 17, 18 Specifically, decreased myocardial voltage‐gated K+ channels (Kv4.2) and hyperpolarization‐activated cyclic nucleotide‐gated channel subunit 2 (HCN2), and the associated electrophysiological alterations have been reported in these epileptic animals.15, 16, 17 Furthermore, physiological interrogations of the autonomic nervous system's influence on HR and molecular examination of the myocardial β1‐adrenergic receptor levels revealed sympathetic predominance in the epileptic animals.14, 16, 18 Kv4.2 channels have predominant activity in early repolarization represented by the transient outward current (I TO), whereas HCN channels contribute to the automaticity of the cardiac pacemaker cells.19, 20 Alterations of the ion channels that contribute to cardiac action potential, in conjunction with increased sympathetic tone, may lead to cardiac electrical instability and increased excitability.
Accordingly, increased susceptibility to chemically induced VT/VF was observed early in the development of epilepsy.14, 15, 21 Attenuating sympathetic nervous system activity by administrating a β‐blocker (atenolol) prior to the induction of status epilepticus has been shown to decrease ventricular excitability, prevent QTc interval prolongation, and decrease VT/VF susceptibility following status epilepticus.15, 22 However, treatment with β‐adrenergic blockers before the onset of spontaneous recurrent seizures, as in these prior studies, has less translational significance than instituting therapy when epilepsy is fully established, which to our knowledge has not yet been evaluated. Furthermore, although chronically epileptic animals are known to exhibit periictal and interictal cardiac electrophysiological alterations,23, 24 the molecular underpinning of these alterations is not well understood. Thus, additional studies are needed to evaluate whether myocardial ion channel remodeling, sympathetic predominance, and the associated electrophysiological derangement persist in the chronically epileptic animals; and if so, whether treatment with β‐adrenergic blockers late in the course of epilepsy will modify the arrhythmogenic risk.
Here we build upon previous studies by further exploring cardiac molecular and electrophysiological alterations in chronically epileptic animals using the model of pilocarpine‐induced epilepsy in rats. We found in the chronically epileptic animals decreased myocardial Kv4.2 and Kv4.3 ion channel, as well as decreased β1 and increased α1A adrenergic receptor protein levels. Concurrently, the epileptic animals exhibited altered cardiac electrophysiology and stimulation‐induced ventricular excitability, which were ameliorated by β‐adrenergic antagonist. Our findings suggest that sympathetic predominance and myocardial ion channel remodeling, along with altered cardiac electrophysiology and ventricular excitability, remain a significant cardiac morbidity in a model of chronic epilepsy. The beneficial effects of the β‐adrenergic antagonist on the cardiac electrophysiology of the chronically epileptic animals suggest that modulating sympathetic tone represents a potential therapeutic target for cardiac protection in chronic epilepsy.
Materials and Methods
Generation of epileptic animals
This study was approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Status epilepticus was induced in 30‐ to 40‐day‐old male Sprague‐Dawley rats (120‐150 g) using pilocarpine (300 mg/kg, ip). To prevent pilocarpine from activating peripheral muscarinic receptors, animals were given scopolamine (1 mg/kg, ip) 30 min prior to pilocarpine administration. Behavioral seizures and status epilepticus were monitored using the Racine scale. Convulsive status epilepticus was defined as Racine stage 4 (rearing) or stage 5 (rearing and falling). Following 1 hour of status epilepticus, the animals were treated with pentobarbital (20 mg/kg, ip) to terminate the seizures. Subsequently, an observer monitored the animals in real‐time at random 4‐hour blocks for behavioral seizures from 2 to 5 months following status epilepticus. The animals were considered epileptic if they exhibited spontaneous behavioral seizures. Age‐matched sham animals received scopolamine (1 mg/kg, ip), normal saline (3 μl/kg), and pentobarbital (20 mg/kg).
Immunoblotting of the myocardial adrenergic receptors and ion channels
Myocardial adrenergic receptor and ion channel protein levels were examined in chronically epileptic (7‐18 months following pilocarpine induction) and age‐matched sham animals. Hearts were removed, placed in liquid nitrogen, and ground into fine powder. Whole heart powder was suspended (1 g/10 ml) and homogenized in a buffer containing HEPES (4 mm, pH 7.0), sucrose (320 mm), and protease inhibitor cocktail (Roche, Mannheim, Germany). The homogenate was centrifuged at 2,000×g for 10 minutes. The resulting pellets were suspended, homogenized, and centrifuged again at 2,000×g for 10 minutes. The supernatants from the 2 centrifugation steps were pooled as whole heart homogenates. Twenty‐five micrograms of proteins were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and probed with antibodies against the voltage‐gated Na+ channel: Nav1.5 (1:500); voltage‐gated K+ channels: Kv4.2 (1:500), Kv4.3 (1:500), Kv7.1 (1:200), and Kir2.1 (1:200); L‐type Ca2+ channel: Cav1.2 (1:500), and adrenergic receptors: β1‐ (1:500), β2‐ (1:500), and α1‐ (1:500). glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (1:5,000) was used as a lane loading control. After incubating with horseradish peroxidase–conjugated secondary antibody (1:5,000‐1:10,000) for one hour, the protein bands were visualized using chemiluminescent reagents and exposed to X‐ray film. The mean gray density of immunoreactive bands was obtained and analyzed using the Image J. The immunoreactive bands from the proteins of interest were normalized to the GAPDH levels for the corresponding protein. Kv4.2, Kv4.3 antibodies (mouse monoclonal) were purchased from NeuroMab (UC Davis, CA). Nav1.5, Cav1.2, and Kv7.1 antibodies (rabbit polyclonal) were purchased from Alomone Labs (Jerusalem, Israel). β1, β2, α1 (rabbit polyclonal), and GAPDH (mouse monoclonal) antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX).
Electrocardiography (ECG) measurements
Electrocardiography (ECG) recordings were performed in chronically epileptic animals (7‐18 months following pilocarpine induction, n = 15) and age‐matched shams (n = 21). Animals were anesthetized using ketamine/xylazine/acepromazine (per gram of body weight: 42.86 μg ketamine, 8.57 μg xylazine, 1.43 μg acepromazine, ip) and allowed 5 minutes of stabilization until ECG recordings were acquired. Single channel ECG in lead II configuration was recorded for 10 minutes using a VSM7 multiparameter monitor (VetSpecs, Canton, GA). HR, PR, and QRS and QT intervals were manually measured at 15 minutes from the induction of anesthesia. The QTc interval was calculated using Bazett's formula: QTc = QT/√RR.25
Ex vivo optical mapping
Age‐matched epileptic and sham animals (n = 5/group, 15 months following pilocarpine induction) were anticoagulated with heparin (1,000 U/kg, ip) and anesthetized using pentobarbital (50 mg/kg, ip). Hearts were rapidly removed and perfused in a retrograde fashion in a Langendorff apparatus with oxygenated Tyrode's solution (NaCl 136 mm, KCl 5.4 mm, CaCl2 1.8 mm, MgCl2 1.0 mm, NaH2PO4 0.33 mm, Hepes 10 mm, glucose 10 mm, pH 7.2), and kept at a constant perfusion pressure of 60‐65 mm Hg at 37°C. The perfusion pressure was monitored using an in‐line physiological pressure transducer that connected to an amplifier (AD Instruments, Colorado Springs, CO). The heart was stained with voltage‐sensitive dye RH237 (0.33 μm, Invitrogen, Carlsbad, CA) for 10 minutes, which binds to the outer layer of cell bilayer membranes of cardiomyocytes for measuring membrane potential. Optical mapping studies are based on the principle that voltage‐sensitive dyes alter their emission spectra in response to changes in transmembrane voltage. To prevent motion artifacts, blebbistatin at final concentration of 5 μm was applied as an excitation–contraction uncoupler. After staining, green light from both a 532 nm (Power Technology Inc, Alexander, AR) and a 520 ± 20 nm X‐Cite Series 120 system (Exfo, Ontario, Canada) laser light source was shone directly onto the heart. Emitted fluorescence was split by a 630 nm dichroic mirror and collected through a 710 nm long‐pass filter using two electron‐multiplying CCD cameras (Cascade 128+, Photometrics, Tucson, AZ). A grid was used to calibrate the locations in the field of view of these two CCD cameras. Simultaneous signals are recorded with synchronized CCD cameras operating at 500 frames per second with a spatial resolution of 0.14 × 0.14 mm2 per pixel. Unipolar stimuli were delivered to the ventricle at a 5 V output using platinum electrodes and a Grass stimulator (S88X, Grass Technologies, West Warwick, RI). Stimulations were computer‐controlled, and pacing sequences were the following: (1) steady state 3 Hz pacing, followed by (2) 2 s of pacing duration with progressively faster pacing rate from 200 msec cycle length to 70 msec cycle length. Each pacing protocol was performed in duplicate.
In vivo cardiac electrophysiology
Cardiac electrophysiological studies in vivo were performed to evaluate the propensity for ventricular arrhythmias in chronically epileptic (7‐12 months following pilocarpine induction) and age‐matched sham animals. To evaluate whether β‐adrenergic blockade was protective against ventricular arrhythmias in the epileptic compared to sham rats, a subset of animals was treated with atenolol (10 mg/kg/dose, ip)26 or saline twice daily for 3 days prior to the studies (sham + vehicle = 13, sham + atenolol = 4, epileptic + vehicle = 17, epileptic + atenolol = 9). To investigate whether atenolol could reverse ECG abnormalities, a subset of animals (n = 3 for sham + vehicle, sham + atenolol, and epileptic + vehicle; n = 6 for epileptic + atenolol) underwent ECG recording prior to and following atenolol treatment. Animals were anesthetized using 2% isoflurane in 1 L/min O2. A 6‐lead body‐surface ECG and 4 intracardiac electrograms were recorded using a computer‐based acquisition system (Emka Technologies). Animals’ body temperatures were maintained at 36°C using a heating pad. We measured HRV from the surface ECG recordings prior to the electrophysiological (EP) studies as an additional surrogate marker for cardiac autonomic control. Standard deviation of the NN intervals (SDNN) and square root of the mean squared differences of successive NN intervals (RMSSD) represented time domain variables. Frequency domain variables included low frequency (LF: 0.05–0.75 Hz) and high frequency (HF: 0.75–2.5 Hz).24 Atrial and ventricular intracardiac electrograms were recorded using a 1.6F octopolar catheter (EPR‐802, Millar Instruments, Houston, TX) inserted into the right external jugular vein. Bipolar right atrial pacing and right ventricular pacing were performed using 2‐msec current pulses delivered by an external stimulator (STG‐3008; Multi Channel Systems, Reutlingen, Germany). To induce ventricular arrhythmias, programmed electrical stimulation was performed at cycle lengths of 100 msec for 8 beats followed by 1 extra‐stimulus (S2) at shorter coupling intervals. Ventricular tachycardia was defined as a sequence of >3 spontaneous ventricular beats following program electrical stimulation. Each animal underwent 2 or 3 programmed electrical stimulation (PES) trials. An animal was considered susceptible to PES‐induced VT if 2 of the 3 trials resulted in VT.
Statistical analysis
Survival curves were compared using the Mantel‐Cox test. Continuous variables were analyzed using either 2‐tailed Student t‐test or analysis of variance (ANOVA) with post hoc Tukey. Categorical variables were analyzed using χ2 test with post hoc Fisher's exact test. Optical mapping data were analyzed with custom software using spatial and temporal filtering. Conduction velocities were calculated using single vector method by the line of conduction from the stimulus site (apex of the ventricle) to the homogeneous spread distally (base of the ventricle). All results were expressed as mean ± standard error of the mean (SEM).
Results
Chronically epileptic animals exhibited altered levels of myocardial adrenergic receptors and Kv4.x potassium channels
Starting at around 12 months of age, the epileptic animals began to exhibit increased mortality compared with the age‐matched shams, with log‐rank hazard ratio analysis demonstrating double the death rate in the epileptic animals (95% confidence interval: 1.40–3.86, p < 0.01; Method S1, Figure S1) Because these death events were unexpected, we sought to investigate whether myocardial changes in animals with chronic epilepsy may provide a plausible mechanism contributing the early mortality.
We evaluated changes in myocardial adrenergic receptor levels as a surrogate marker for persistently increased sympathetic tone in chronically epileptic animals between 9 and 20 months age. We found decreased protein levels of β1‐adrenergic receptors (100.1 ± 13.4% vs. 53.3 ± 6.9%, sham vs. epileptic, n = 10/group, p < 0.01), increased levels of α1A‐adrenergic receptors (100.0 ± 3.4% vs. 136.0 ± 4.6%, sham vs. epileptic, n = 5/group, p < 0.001), and no difference in the β2‐receptor protein levels (100.0 ± 21.6% vs. 110.7 ± 35.4%, sham vs. epileptic, n = 6/group) in the epileptic compared with sham animals (Figure 1A). We surveyed representative myocardial ion channels in each phase of ventricular action potential and found no changes in the protein levels of Nav1.5 (upstroke), Cav1.2 (plateau), and Kv7.1 (late repolarization) (Figure S2). Compared with the sham animals, the myocardium of epileptic animals exhibited decreased protein levels of Kv4.2 (100.0 ± 18.6% vs. 52.0 ± 10.3% sham vs. epileptic, n = 12–14/group, p < 0.05) and Kv4.3 (100.0 ± 12.7% vs. 34.5 ± 10.1% sham vs. epileptic, n = 12–14/group, p < 0.01), which have predominant activity during early repolarization (Figure 1B).
Figure 1.
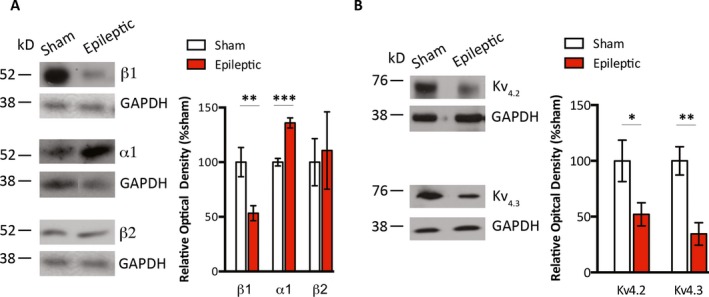
Altered myocardial protein levels of adrenergic receptors and Kv4.x ion channels in epileptic animals. A, Whole heart homogenates that were obtained from the hearts of sham and epileptic animals were probed with antibodies against β1‐, β2‐, and α1‐adrenergic receptors. Myocardial homogenates from epileptic compared to that from sham animals had significantly decreased immunoreactivity for β1‐adrenergic receptors, increased immunoreactivity for α1A‐adrenergic receptors, and no significant difference in immunoreactivity for β2‐receptors. B, Whole heart homogenates were probed with antibodies against Kv4.2 and Kv4.3 ion channels. Myocardial homogenates from epileptic compared with sham animals had significantly decreased immunoreactivity for Kv4.2 and Kv4.3 channels. *p < 0.05, **p < 0.01, ***p < 0.001
Myocardium from chronically epileptic animals exhibited altered electrical properties and increased excitability
Compared with age‐matched sham animals, chronically epileptic animals (7‐18 months following status epilepticus) exhibited a myriad of ECG alterations. These changes included higher resting HR (244 ± 3 bpm vs. 270 ± 10 bpm, sham vs. epileptic, n = 15–21/group, p < 0.05) (Figure 2A), longer QTc intervals (272 ± 10 msec vs. 336 ± 11 msec sham vs. epileptic, n = 15–21/group, p < 0.001) (Fig. 2B), and longer QRS intervals (67 ± 4 msec vs. 89 ± 5 msec sham vs. epileptic, n = 15–21/group, p < 0.01) (Fig. 2C). There were no differences in HRV parameters between sham and chronically epileptic animals (Method S1, Table S1).
Figure 2.
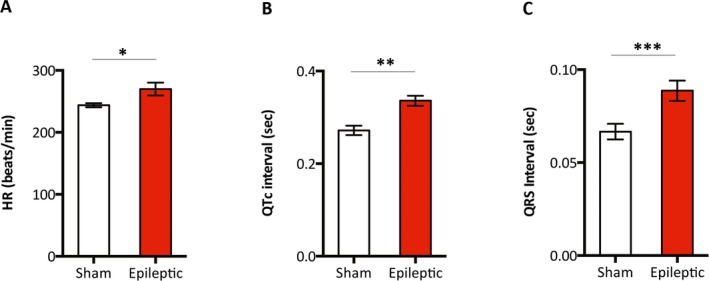
Epileptic animals exhibited higher HR and longer QRS and QTc intervals. Single channel ECG recordings in the lead II configuration were obtained in anesthetized animals. Compared with the age‐matched sham animals, chronically epileptic animals (7‐18 months following status epilepticus) had a higher HR (A), and longer QTc (B) and QRS (C) intervals. n = 15–21/group. *p < 0.05, **p < 0.01, ***p < 0.001
Observed ECG alterations were not associated with structural or functional changes in the hearts of chronically epileptic animals. Cardiac magnetic resonance imaging studies in the epileptic animals at 4 months of age demonstrated no significant differences in the left and right ventricular ejection fraction, end diastolic volume, and left ventricular mass compared with sham animals (Method S1, Table S2). Similarly, echocardiographic studies in the epileptic animals performed at 9 months of age demonstrated no significant differences in shortening fraction, interventricular septal thickness, posterior wall thickness, and internal dimension of the left ventricle during systole and diastole compared with the sham animals (Method S1, Table S3). Furthermore, a veterinary pathologist who was blinded to the group assignment reviewed the hematoxylin and eosin (H&E) staining at 9‐14 months of age and concluded that sham and epileptic animals exhibited comparable myocardial histology (Method S1, Figure S3).
To determine whether the observed ECG abnormalities in chronically epileptic animals reflected changes in the intrinsic cardiac electrical properties and contributed to increased ventricular excitability, we mapped action potential propagation across the ventricles using an ex vivo (Langendorff) heart preparation with denervated hearts, a voltage‐sensitive dye, and electrical stimulation of the ventricles. The cardiac optical mapping ex vivo demonstrated significantly slower conduction velocity in the hearts of chronically epileptic animals compared with sham animals at all diastolic intervals (n = 5/group, p < 0.05, unpaired Student t‐test, Figure 3A). Ventricular ectopic foci were observed in 2 of 5 epileptic animals during electrical stimulation, whereas no ectopic foci were evident in ventricles from sham animals (Figure 3B). In sham animals, all the action potentials were elicited by the electrical stimulation. These action potentials always originated at the site of the electrical stimulation and each action potential propagated in the same direction (Figure 3B). In contrast, in the epileptic animals, each electrical stimulus elicited additional ectopic action potentials in a 1:2 ratio (paced‐to‐ectopic beats). The ectopic action potentials arose at locations different from the site of electrical stimulation and propagated in various directions (Figure 3B). Together, these findings suggest alterations in the intrinsic electrical properties of the myocardium of chronically epileptic animals, which are associated with increased ventricular excitability.
Figure 3.
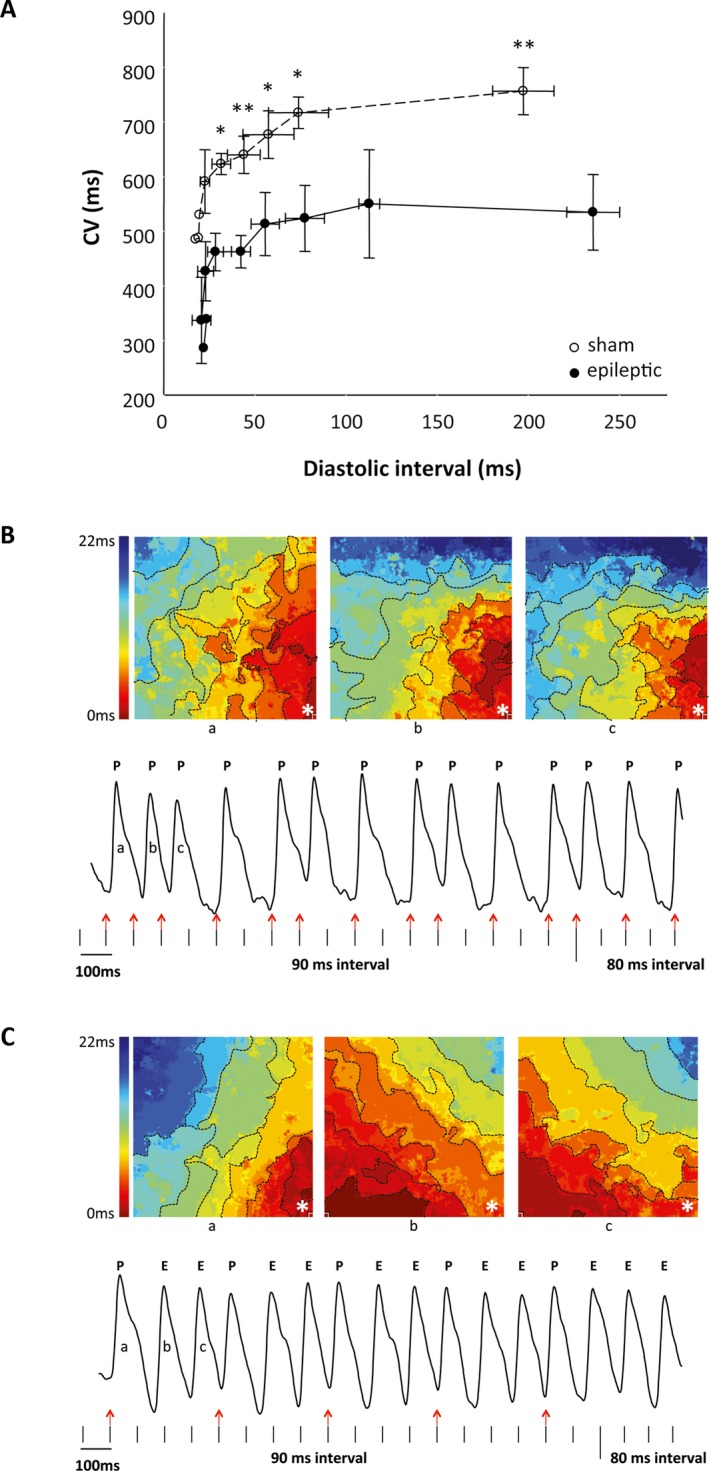
Epileptic animals exhibited altered cardiac electrical properties and increased ventricular excitability. A, Ex vivo optical mapping using voltage‐sensitive dye RH237 and incrementally faster electrical pacing revealed slower conduction velocity (CV) in epileptic (open circles) compared to sham animals (closed circles) at all cycle lengths. n = 5/group. * p < 0.05. ** p < 0.01. B, C, Sample tracings of the isochronal maps (top) and corresponding voltage‐dependent fluorescence intensity (bottom) are shown from a sham (B) and an epileptic animal (C). Vertical marks represent electrical stimulation. B, In the sham animals, all the action potentials (P) were elicited by the electrical stimulation (red arrows). The isochronal maps for the first 3 action potentials (beats a, b, c) in the sham demonstrate that they originated at the site of the electrical stimulation (*) and propagated in the same direction. C, In the epileptic animals, electrical stimulations that elicited action potentials (red arrows) also triggered additional ectopic action potentials (E). The corresponding isochronal maps for the ectopic beats (beats b, c) demonstrated different origin and direction of propagation away from the site of the electrical stimulation (*).
Chronically epileptic animals exhibited stimulation‐induced ventricular tachycardia in vivo that was rescued with β‐adrenergic blockade
To assess arrhythmogenic potential in chronically epileptic animals and to evaluate the contribution of increased sympathetic drive, in vivo cardiac EP studies were conducted. Atenolol‐treated epileptic animals exhibited lower HR and shorter QTc intervals after 3 days of drug administration (HR: 343 ± 11 bpm vs. 298 ± 8 bpm, pre‐atenolol vs post‐atenolol, n = 6/group, p < 0.01; QTc: 239.8 ± 19.4 msec vs 208.6 ± 16.4 msec, n = 6/group, p < 0.01; paired Student t‐test, Figure 4A,B). We did not observe HR differences in sham animals before and after atenolol treatment; however, QTc intervals were decreased after 3 days of atenolol treatment (Figure S4). We found that 12 of 17 vehicle‐treated epileptic animals were susceptible to PES‐induced VT (Figure 4C). In contrast, fewer atenolol‐treated epileptic animals (2 of 9) were susceptible to PES‐induced VT as compared with vehicle‐treated epileptic animals (12 of 17) (Fisher's exact test, p < 0.05, Figure 4C). By comparison, 4 of 13 vehicle‐treated and 0 of 4 atenolol‐treated sham animals were susceptible to PES‐induced VT, which was not significantly different from the vehicle‐treated epileptic animals (Figure 4C). There were 3 deaths each in vehicle‐treated sham and epileptic animals that resulted from prolonged VT that degenerated to VF and asystole. In contrast, there were no deaths in atenolol‐treated sham and epilepsy animals. Of interest, 2 atenolol‐treated epileptic animals survived after having prolonged stimulation‐induced VT (>1 min) that spontaneously returned to sinus rhythm (Figure 4D).
Figure 4.
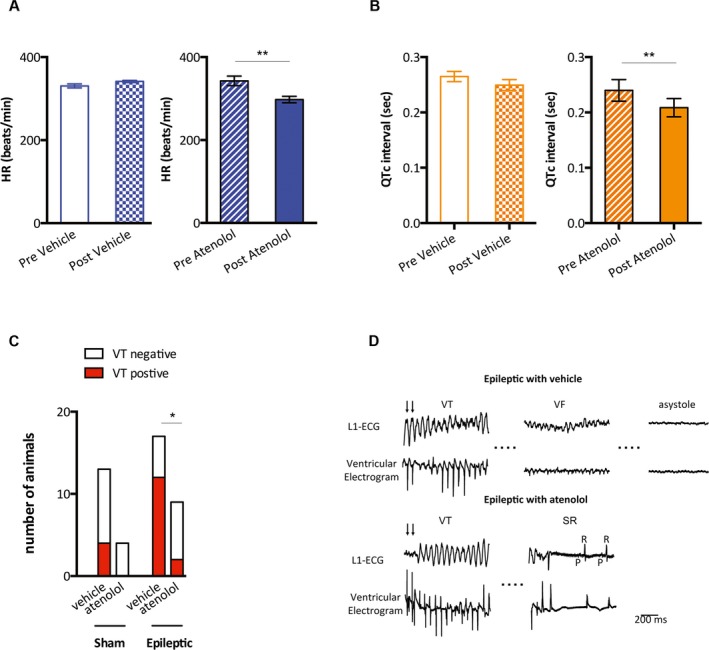
Atenolol slowed HR, shortened the QTc interval, and decreased the incidence of VT in the epileptic animals. A, Epileptic animals that were treated with normal saline (epileptic + vehicle, n = 3) exhibited no changes in HR (pre‐vehicle vs post‐vehicle). In contrast, atenolol‐treated epileptic animals (epileptic + atenolol, n = 6), exhibited lower HR following atenolol administration (pre‐atenolol vs post‐atenolol). B, Saline‐treated epileptic animals exhibited no changes in QTc intervals (pre‐vehicle vs post‐vehicle). Epileptic animals that received atenolol exhibited decreased QTc intervals following atenolol administration (pre‐atenolol vs post‐atenolol, n = 6). C, Four of 13 vehicle‐treated and 0 of 4 atenolol‐treated sham animals had PES‐induced VT. By comparison, 12 of 17 vehicle‐treated epileptic animals had PES‐induced VT. Significantly fewer atenolol‐treated epileptic animals (2 of 9) exhibited PES‐induced VT as compared with vehicle‐treated epileptic animals. D, Surface (L1) and intracardiac tracings (ventricular electrogram) demonstrate a prolonged VT episode degenerating into ventricular fibrillation (VF) and ultimately asystole in the vehicle‐treated epileptic animals. Traces are shown from an atenolol‐treated epileptic animal that exhibited spontaneous conversion from prolonged VT to sinus rhythm (SR). VT is shown as marked and conversion to SR is indicated by a P wave (P) that is followed by the QRS (R). Arrows indicate the last electrical stimulations. *p < 0.05, **p < 0.01
Discussion
We found in animals with chronic epilepsy, molecular and electrophysiological alterations that suggest persistent sympathetic predominance and myocardial ion remodeling. These changes may contribute to ventricular excitability and stimulation‐induced ventricular arrhythmias. Although cardiac sympathetic predominance with increased HR, a prolonged QTc interval, and increased susceptibility to ventricular arrhythmia have been described early following experimental status epilepticus,15, 18, 21 cardiac electrophysiological and molecular alterations associated with chronic epilepsy are not well understood. There have been conflicting electrophysiological observations in chronic epilepsy models, with a higher HR but without QTc interval prolongation being described in chronically epileptic rats in one study,24 and a slower HR with QTc interval prolongation being reported in another.17 Our finding of decreased β1 and increased α1 myocardial receptor levels provide a molecular correlate that is consistent with increased sympathetic tone.27, 28, 29 However, decreased parasympathetic input has been reported in chronic epilepsy,24 which can also account for autonomic nervous system imbalance favoring sympathetic overdrive. Therefore, whether sympathetic predominance is due to a decrease in parasympathetic or an increase in sympathetic input in chronic epilepsy remains to be an area of active investigation.
Under physiological conditions, β‐adrenergic receptors (primarily β1) mediate intracellular signal transduction in the myocardium in response to sympathetic nervous system stimulation. However, persistent sympathetic stimulation can adversely affect myocardial health through several mechanisms including increased arrhythmogenic potential, myocardial death, and pathological cardiac hypertrophy with fibrosis.30, 31 Desensitization of β1 adrenergic receptors through either decreased receptor levels or uncoupling of the intracellular signal cascades may represent an adaptive response to persistently elevated sympathetic tone at the expense of decreased catecholamine sensitivity.27 Therefore, under these pathological conditions, increased α1 adrenergic receptor levels play a greater role in maintaining cardiac function by maintaining myocardial catecholamine sensitivity and inducing physiological myocardial hypertrophy.29, 32, 33 Our observations of altered myocardial adrenergic receptor levels and electrophysiological properties, as well as ventricular hyperexcitability, suggest that myocardial adrenergic receptor remodeling may be deleterious to cardiac electrical properties in epilepsy. Additional studies will be needed to further elucidate the roles of individual adrenergic receptor subtypes on epilepsy‐associated cardiac electrical remodeling.
Somatic ion channel mutations have been shown to manifest in seizures, altered cerebral and cardiac electrophysiology, and arrhythmias.34, 35, 36, 37 These observations suggest that myocardial ion channelopathies may contribute to the cardiac electrophysiological alterations associated with epilepsy. Accordingly, decreased expression of HCN2, an ion channel that contributes to the cardiac pacemaker activity, was observed in the epileptic rats early following kainate‐induced status epilepticus.17 Furthermore, we have previously reported decreased myocardial Kv4.2 protein levels 2 weeks following pilocarpine‐ and in kainate‐induced status epilepticus.15, 16 In the studies reported here, we surveyed representative ion channels that contribute to the ventricular action potential in chronically epileptic rats (7‐18 months following pilocarpine induction) and found significantly decreased protein levels of Kv4.2 and Kv4.3 ion channels. Our findings indicate that changes in these ion channels previously reported as downregulated early in the development of epilepsy persist until late in the course of chronic epilepsy. These voltage‐dependent K+ channels contribute to the transient outward potassium current (I TO) in early myocardial repolarization in rodents. A decrease in Kv4.x channels and a subsequent loss of I TO is a finding seen in diverse cardiac pathologies and is known to confer an increased arrhythmogenic risk.38 Furthermore, these changes in Kv4.x channels and I TO are seen with chronic β‐adrenergic stimulation and tachycardia,31, 39 similar to that described by our findings. Therefore, decreased myocardial Kv4.2 protein levels observed in this study, coupled with previous reports of decreased myocardial Kv4.2 levels during epileptogenesis,15, 16 suggest that decreased Kv4.2 ion channel levels may represent one candidate mechanism of potentially many that contributes to cardiac hyperexcitability in this model of epilepsy. In our studies, significant changes in the other ion channels evaluated were not seen. Other studies have also implicated altered Na+ currents in the cardiac remodeling associated with kainate‐induced status epilepticus and acquired epilepsy.40 Decreased Na+ currents could manifest in slower conduction velocity, as we have seen in the myocardium from epileptic animals. Although we did not observe decreased protein levels of myocardial Nav1.5 in the epileptic animals, we cannot exclude the possibility of altered Nav1.5 electrophysiological properties in the myocardium from epileptic animals due to post‐translational or other alterations in the channels. In addition, we did not investigate in this study region‐specific ion channel changes that may contribute to the observed alterations in cardiac electrophysiological properties. Further molecular and electrophysiological studies that examine region‐specific ion channel changes and in single, isolated cardiomyocytes are important future directions for identifying additional molecular candidates contributing to cardiac electrical remodeling.
Our data also revealed altered intrinsic electrical properties of the myocardium in chronically epileptic animals, represented by the decreased conduction velocity of the action potential propagation through the ventricles of the denervated hearts. In addition, experiments ex vivo demonstrated stimulation‐induced ventricular ectopy, suggesting that altered intrinsic electrical properties may be associated with increased ventricular excitability in the myocardium of epileptic animals. These cardiac electrical alterations occurred in the absence of obvious structural changes in the myocardium as reflected in normal myocardial histology, cardiac magnetic resonance imaging, and echocardiography in the epileptic animals. In this regard, our findings are consistent with the prevailing literature, which demonstrates a lack of clinically significant heart disease in individuals with epilepsy and risk factors for SUDEP.12, 13 In contrast, left ventricular hypertrophy and impaired cardiac performance have been reported previously in rats with audiogenic seizures and in a model of acquired epilepsy.23, 24 Of interest, impaired cardiac performance in chronically epileptic animals has been shown to occur following a seizure episode,24 which supports seizure‐induced acute myocardial failure as a potential etiology for sudden death in epilepsy.13 These changes may be model‐specific, or an alternative explanation for the discrepancy is that in the absence of an acute seizure these parameters may be normal in epilepsy. Recent unpublished studies (Schreiber J et al. Abstract Number 3.082, American Epilepsy Society Annual Meeting, 2016) have shown changes consistent with cardiac injury in chronically epileptic children using Echo speckle tracking. Thus, broader application of this technique in epilepsy may be warranted.
We have previously described decreased β1 receptor protein levels and activation of downstream intracellular signaling cascades 2 weeks following kainate‐induced status epilepticus suggesting early sympathetic predominance in a model of acquired epilepsy.16 Our findings in this study suggest cardiac sympathetic predominance in the chronically epileptic animals. Together, these observations indicate that persistent sympathetic predominance may represent a significant pathological feature associated with epilepsy. Therefore, reducing sympathetic input through β‐adrenergic blockade may represent a potential cardioprotective strategy. Indeed, attenuating increased sympathetic nervous system activity by administering atenolol prior to the induction of status epilepticus has been shown to prevent QTc prolongation and decrease VT/VF susceptibility 24 h following status epilepticus.15 However, treatment with β‐adrenergic blockers before the onset of spontaneous recurrent seizures as in these prior studies has less translational significance than instituting therapy when epilepsy is fully established. Here we have demonstrated that atenolol treatment in chronically epileptic animals at the time when cardiac alterations have become evident is protective. In primary cardiac pathology, blunting persistent sympathetic stimulation using a β‐blocker has been shown to ameliorate adrenergic receptor remodeling and other adverse cardiac effects.30 Because persistent sympathetic predominance may represent a significant pathological feature throughout epilepsy development, whether early initiation of β‐blockers may prevent or reverse epilepsy‐associated cardiac remodeling represents an important and clinically relevant area of future investigation.
There are additional factors that may influence the interpretation of our findings. First, ECG measurements were obtained under sedation/anesthesia. Furthermore, different anesthetic agents may exhibit varying effects on HR and ventricular repolarization. Therefore, the effects of sedation/anesthesia represent a significant confounder and our observations may not be readily translatable to awake, freely moving animals. Only male rats were used, which is a significant limitation of our study. Because gender is known to play an important role in cardiovascular health, our findings may not be applicable to the female gender.
In a model of chronic epilepsy. we have observed molecular correlates supporting persistently increased cardiac sympathetic tone and identified the remodeling of myocardial Kv4.2/Kv4.3 ion channels as a candidate mechanism contributing to increased ventricular excitability. These myocardial changes and susceptibility to stimulation‐induced VT occurred at the time when the epileptic animals exhibited an increased mortality rate. Future studies are needed to investigate whether fatal cardiac arrhythmias as a potential sequela of these myocardial changes contribute to early mortality in the epileptic animals, and whether modulating sympathetic tone will prevent long‐term cardiac remodeling and reduce the risk of arrhythmias later in the course of chronic epilepsy.
Disclosure
The authors do not have any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Figure S1. Epileptic animals have decreased survival.
Figure S2. Epileptic animals have myocardial Nav1.5, Cav1.2, Kv7.1, and Kir2.1 protein levels comparable to sham animals.
Figure S3. Epileptic animals have normal cardiac histology.
Figure S4. Atenolol shortens QTc intervals in sham animals.
Table S1. Comparable heart rate variability between sham and epileptic animals.
Table S2. Epileptic animals exhibited cardiac structure and function comparable to sham animals by MRI.
Table S3. Epileptic animals exhibited cardiac structure and function comparable to sham animals by echocardiography.
Method S1. Supplemental methods.
Acknowledgments
Funding support: NIH/NINDS: R01NS039943, R01NS081053, R21NS077028, K08NS063117; Citizens United for Research in Epilepsy (CURE) Christopher Donalty and Kyle Coggins Memorial Award for Interdisciplinary Research; Emma Bursick Memorial Fund.
Biography
Dr. Yi‐Chen Lai's research focuses on early and chronic cardiac alterations associated with epilepsy.

References
- 1. Ngugi AK, Bottomley C, Kleinschmidt I, et al. Estimation of the burden of active and life‐time epilepsy: a meta‐analytic approach. Epilepsia 2010;51:883–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nei M, Ho RT, Abou‐Khalil BW, et al. EEG and ECG in sudden unexplained death in epilepsy. Epilepsia 2004;45:338–345. [DOI] [PubMed] [Google Scholar]
- 3. Neufeld G, Lazar JM, Chari G, et al. Cardiac repolarization indices in epilepsy patients. Cardiology 2009;114:255–260. [DOI] [PubMed] [Google Scholar]
- 4. Poh MZ, Loddenkemper T, Reinsberger C, et al. Autonomic changes with seizures correlate with postictal EEG suppression. Neurology 2012;78:1868–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boggs JG, Painter JA, DeLorenzo RJ. Analysis of electrocardiographic changes in status epilepticus. Epilepsy Res 1993;14:87–94. [DOI] [PubMed] [Google Scholar]
- 6. Brotherstone R, Blackhall B, McLellan A. Lengthening of corrected QT during epileptic seizures. Epilepsia 2010;51:221–232. [DOI] [PubMed] [Google Scholar]
- 7. Nei M, Ho RT, Sperling MR. EKG abnormalities during partial seizures in refractory epilepsy. Epilepsia 2000;41:542–548. [DOI] [PubMed] [Google Scholar]
- 8. Surges R, Adjei P, Kallis C, et al. Pathologic cardiac repolarization in pharmacoresistant epilepsy and its potential role in sudden unexpected death in epilepsy: a case‐control study. Epilepsia 2010;51:233–242. [DOI] [PubMed] [Google Scholar]
- 9. Espinosa PS, Lee JW, Tedrow UB, et al. Sudden unexpected near death in epilepsy: malignant arrhythmia from a partial seizure. Neurology 2009;72:1702–1703. [DOI] [PubMed] [Google Scholar]
- 10. Ferlisi M, Tomei R, Carletti M, et al. Seizure induced ventricular fibrillation: a case of near‐SUDEP. Seizure 2013;22:249–251. [DOI] [PubMed] [Google Scholar]
- 11. Lanz M, Oehl B, Brandt A, et al. Seizure induced cardiac asystole in epilepsy patients undergoing long term video‐EEG monitoring. Seizure 2011;20:167–172. [DOI] [PubMed] [Google Scholar]
- 12. Devinsky O. Sudden, unexpected death in epilepsy. N Engl J Med 2011;365:1801–1811. [DOI] [PubMed] [Google Scholar]
- 13. Surges R, Thijs RD, Tan HL, et al. Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol 2009;5:492–504. [DOI] [PubMed] [Google Scholar]
- 14. Read MI, McCann DM, Millen RN, et al. Progressive development of cardiomyopathy following altered autonomic activity in status epilepticus. Am J Physiol Heart Circ Physiol 2015;309:H1554–H1564. [DOI] [PubMed] [Google Scholar]
- 15. Bealer SL, Little JG, Metcalf CS, et al. Autonomic and cellular mechanisms mediating detrimental cardiac effects of status epilepticus. Epilepsy Res 2010;91:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brewster AL, Marzec K, Hairston A, et al. Early cardiac electrographic and molecular remodeling in a model of status epilepticus and acquired epilepsy. Epilepsia 2016;57:1907–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Powell KL, Jones NC, Kennard JT, et al. HCN channelopathy and cardiac electrophysiologic dysfunction in genetic and acquired rat epilepsy models. Epilepsia 2014;55:609–620. [DOI] [PubMed] [Google Scholar]
- 18. Metcalf CS, Radwanski PB, Bealer SL. Status epilepticus produces chronic alterations in cardiac sympathovagal balance. Epilepsia 2009;50:747–754. [DOI] [PubMed] [Google Scholar]
- 19. Baruscotti M, Barbuti A, Bucchi A. The cardiac pacemaker current. J Mol Cell Cardiol 2010;48:55–64. [DOI] [PubMed] [Google Scholar]
- 20. Birnbaum SG, Varga AW, Yuan LL, et al. Structure and function of Kv4‐family transient potassium channels. Physiol Rev 2004;84:803–833. [DOI] [PubMed] [Google Scholar]
- 21. Metcalf CS, Poelzing S, Little JG, et al. Status epilepticus induces cardiac myofilament damage and increased susceptibility to arrhythmias in rats. Am J Physiol Heart Circ Physiol 2009;297:H2120–H2127. [DOI] [PubMed] [Google Scholar]
- 22. Read MI, Harrison JC, Kerr DS, et al. Atenolol offers superior protection against cardiac injury in kainic acid‐induced status epilepticus. Br J Pharmacol 2015;172:4626–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fazan R Jr, Silva CA, Oliveira JA, et al. Evaluation of cardiovascular risk factors in the wistar audiogenic rat (WAR) strain. PLoS ONE 2015;10:e0129574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Naggar I, Lazar J, Kamran H, et al. Relation of autonomic and cardiac abnormalities to ventricular fibrillation in a rat model of epilepsy. Epilepsy Res 2014;108:44–56. [DOI] [PubMed] [Google Scholar]
- 25. Bazett HC. An analysis of the time‐relations of electrocardiograms. Heart 1920;7:353–370. [Google Scholar]
- 26. Mehvar R, Gross ME, Kreamer RN. Pharmacokinetics of atenolol enantiomers in humans and rats. J Pharm Sci 1990;79:881–885. [DOI] [PubMed] [Google Scholar]
- 27. Bristow MR, Ginsburg R, Minobe W, et al. Decreased catecholamine sensitivity and beta‐adrenergic‐receptor density in failing human hearts. N Engl J Med 1982;307:205–211. [DOI] [PubMed] [Google Scholar]
- 28. Hwang KC, Gray CD, Sweet WE, et al. Alpha 1‐adrenergic receptor coupling with Gh in the failing human heart. Circulation 1996;94:718–726. [DOI] [PubMed] [Google Scholar]
- 29. Skomedal T, Borthne K, Aass H, et al. Comparison between alpha‐1 adrenoceptor‐mediated and beta adrenoceptor‐mediated inotropic components elicited by norepinephrine in failing human ventricular muscle. J Pharmacol Exp Ther 1997;280:721–729. [PubMed] [Google Scholar]
- 30. El‐Armouche A, Eschenhagen T. Beta‐adrenergic stimulation and myocardial function in the failing heart. Heart Fail Rev 2009;14:225–241. [DOI] [PubMed] [Google Scholar]
- 31. Rossow CF, Dilly KW, Yuan C, et al. NFATc3‐dependent loss of I(to) gradient across the left ventricular wall during chronic beta adrenergic stimulation. J Mol Cell Cardiol 2009;46:249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang B, Qin D, El‐Sherif N. Spatial alterations of Kv channels expression and K(+) currents in post‐MI remodeled rat heart. Cardiovasc Res 2001;52:246–254. [DOI] [PubMed] [Google Scholar]
- 33. Jensen BC, O'Connell TD, Simpson PC. Alpha‐1‐adrenergic receptors in heart failure: the adaptive arm of the cardiac response to chronic catecholamine stimulation. J Cardiovasc Pharmacol 2014;63:291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Auerbach DS, Jones J, Clawson BC, et al. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS ONE 2013;8:e77843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Glasscock E, Yoo JW, Chen TT, et al. Kv1.1 potassium channel deficiency reveals brain‐driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci 2010;30:5167–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goldman AM, Glasscock E, Yoo J, et al. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med 2009;1:2ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ludwig A, Budde T, Stieber J, et al. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J 2003;22:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nattel S, Maguy A, Le Bouter S, et al. Arrhythmogenic ion‐channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 2007;87:425–456. [DOI] [PubMed] [Google Scholar]
- 39. Xiao L, Coutu P, Villeneuve LR, et al. Mechanisms underlying rate‐dependent remodeling of transient outward potassium current in canine ventricular myocytes. Circ Res 2008;103:733–742. [DOI] [PubMed] [Google Scholar]
- 40. Biet M, Morin N, Lessard‐Beaudoin M, et al. Prolongation of action potential duration and QT interval during epilepsy linked to increased contribution of neuronal sodium channels to cardiac late Na+ current: potential mechanism for sudden death in epilepsy. Circ Arrhythm Electrophysiol 2015;8:912–920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Epileptic animals have decreased survival.
Figure S2. Epileptic animals have myocardial Nav1.5, Cav1.2, Kv7.1, and Kir2.1 protein levels comparable to sham animals.
Figure S3. Epileptic animals have normal cardiac histology.
Figure S4. Atenolol shortens QTc intervals in sham animals.
Table S1. Comparable heart rate variability between sham and epileptic animals.
Table S2. Epileptic animals exhibited cardiac structure and function comparable to sham animals by MRI.
Table S3. Epileptic animals exhibited cardiac structure and function comparable to sham animals by echocardiography.
Method S1. Supplemental methods.