

Abstract
The platinum‐based DNA damaging agent cisplatin is used as a standard therapy for locally advanced head and neck squamous cell carcinoma (HNSCC). However, the mechanisms underpinning the cytotoxic effects of this compound are not entirely elucidated. Cisplatin produces anticancer effects primarily via activation of the DNA damage response, followed by inducing BCL‐2 family dependent mitochondrial apoptosis. We have previously demonstrated that cisplatin induces the expression of proapoptotic BCL‐2 family protein, Noxa, that can bind to the prosurvival BCL‐2 family protein, MCL‐1, to inactivate its function and induce cell death. Here, we show that the upregulation of Noxa is critical for cisplatin‐induced apoptosis in p53‐null HNSCC cells. This induction is regulated at the transcriptional level. With a series of Noxa promoter‐luciferase reporter assays, we find that the CRE (cAMP response element) in the promoter is critical for the Noxa induction by cisplatin treatment. Among the CREB/ATF transcription factors, ATF3 and ATF4 are induced by cisplatin, and downregulation of ATF3 or ATF4 reduced cisplatin‐induced Noxa. ATF3 and ATF4 bind to and cooperatively activate the Noxa promoter. Furthermore, ERK1 is involved in cisplatin‐induced ATF4 and Noxa induction. In conclusion, ATF3 and ATF4 are important regulators that induce Noxa by cisplatin treatment in a p53‐independent manner.
Keywords: apoptosis, ATF, cisplatin, ERK1, Noxa
Abbreviations
- ATF
activating transcription factor
- BCL‐2
B‐cell lymphoma‐2
- BH3
BCL‐2 homology 3
- CHOP
C/EBP‐homologous protein
- CREB
CRE binding protein
- CRE
cAMP response element
- DMEM
Dulbecco's modified Eagle's medium
- ERK
extracellular signal‐regulated kinase
- GAPDH
Glyceraldehyde 3‐phosphate dehydrogenase
- HNSCC
head and neck squamous cell carcinoma
- MCL‐1
myeloid cell leukemia‐1
- PARP
poly(ADP–ribose) polymerase
1. Introduction
Head and neck cancer is the sixth leading cancer worldwide. Head and neck squamous cell carcinoma (HNSCC) accounts for more than 90% of incident cases. Despite intense, multimodality treatment regimens for HNSCC including surgery, chemotherapy, and radiation, little progress has been made over the past 30 years in improving overall survival rates (Leemans et al., 2011; Rothenberg and Ellisen, 2012). Induction chemotherapy with platinum‐based compounds, taxanes, and 5‐fluorouracil is beneficial to head and neck cancer patients, but the prolonged use of chemotherapeutic drugs is limited by their toxicity and by the development of resistance (Schultz et al., 2010; Suh et al., 2014; Vermorken et al., 2007). Ultimately, cisplatin exerts anticancer effects via multiple mechanisms. Cisplatin is most prominent in the generation of DNA lesions, activation of DNA damage response, and the induction of p53 and BCL‐2 family dependent mitochondrial apoptosis. Tumor cell death induced by both conventional and targeted chemotherapy is often mediated by the BCL‐2 family dependent mitochondrial apoptotic pathway (Adams and Cory, 2007; Chipuk et al., 2010; Youle and Strasser, 2008). However, initiators of this apoptotic pathway, such as p53, are frequently mutated or deleted in HNSCC rendering the disease refractory to treatment (Poeta et al., 2007; Sano et al., 2011). The development of chemoresistance is often associated with cisplatin treatment, which leads to therapeutic failure. However, the mechanisms of the resistance by cisplatin remain unclear.
The BCL‐2 family consists of three main groups: prosurvival (e.g., BCL‐2, MCL‐1, BCL‐XL), multidomain prodeath (e.g., BAX, BAK), and BH3‐only prodeath (e.g., BAD, BID, BIM, Noxa). BH3‐only proteins cause cytochrome c release from the mitochondria by activating BAX and/or BAK, while the prosurvival BCL‐2 family of proteins prevents this process (Adams and Cory, 2007; Chipuk et al., 2010; Youle and Strasser, 2008). In the case of cisplatin‐induced apoptosis, a BH3‐only protein, Noxa, specifically binds to and recruits the prosurvival MCL‐1 from the cytosol to the mitochondria to inactivate the function of MCL‐1. Translocation of MCL‐1 initiates its phosphorylation and subsequent ubiquitination, which triggers proteasome‐mediated degradation (Nakajima et al., 2016). It has been demonstrated that the transcription of Noxa is induced by cisplatin, which is both p53‐dependent and p53‐independent mechanisms (Gutekunst et al., 2013; Sheridan et al., 2010; Zhu et al., 2013). Although, Noxa was originally identified as a p53 target gene, the p53‐independent mechanisms of the transcription of Noxa are not entirely elucidated. To investigate the mechanisms of cisplatin‐induced, p53‐independent apoptosis, we used HN8 and HN12 HNSCC cell lines in which p53 was inactive (Yeudall et al., 1997). Cisplatin treatment on these cells triggers the induction of Noxa and apoptosis. We found that the upregulation of Noxa was critical for cisplatin‐induced apoptosis in a p53‐independent manner. Therefore, we focused on the p53‐independent mechanism of Noxa induction by cisplatin treatment. We identified that transcription factors, ATF3 and ATF4, contribute to regulate Noxa mRNA induction by cisplatin treatment through CRE on the promoter. We further analyzed the signaling pathways to regulate ATF3 and ATF4 induction by cisplatin.
2. Materials and methods
2.1. Cell lines and cell culture
HN8 and HN12 cells were kindly provided by W. Andrew Yeudall (Augusta University). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Grand Island, NY, USA) supplemented with 10% heat‐inactivated fetal bovine serum (FBS) and 100 μg·mL−1 penicillin G/streptomycin at 37 °C in a humidified, 5% CO2 incubator.
2.2. Lentivirus production
The lentiviral short‐hairpin RNA (shRNA)‐expressing constructs were purchased from Sigma‐Aldrich (St. Louis, MO, USA). The target sequences for each shRNA are the following: Noxa 2: 5′‐CTTCCGGCAGAAACTTCTGAA‐3′, Noxa 4: 5′‐TGGAAGTCGAGTGTGCTACTC‐3′, ATF3‐1: 5′‐GCTGAACTGAAGGCTCAGATT‐3′, ATF3‐2: 5′‐CTTCATCGGCCCACGTGTATT‐3′, ATF4‐1: 5′‐GCCTAGGTCTCTTAGATGATT‐3′, ATF4‐2: 5′‐GCCAAGCACTTCAAACCTCAT‐3′, ERK1: 5′‐CCTGAATTGTATCATCAACAT‐3′, ERK2‐1: 5′‐CAAAGTTCGAGTAGCTATCAA‐3′, ERK2‐2: 5′‐TATCCATTCAGCTAACGTTCT‐3′, CREB: 5′‐ACAGCACCCACTAGCACTATT‐3′. The constructs were transfected into 293T packaging cells along with the packaging plasmids using EndoFectin Lenti (GeneCopoeia, Rockville, MD, USA) and the lentivirus‐containing supernatants were used to transduce the cells.
2.3. Luciferase assay
The sequences of p53 and CRE mutants on the Noxa promoter are the following: p53: 5′‐GAGAGTTTCCGGGAAGTTCGCG‐3′, CRE: 5′‐CTAAAAAA‐3′. Each promoter construct (−198 to +157 from the transcription start site) was cloned into KpnI‐BglII sites in PGV‐B2 (Toyo B‐Net, Tokyo, Japan). The ATF3 and ATF4 expression vectors were purchased from Addgene (Cambridge, MA, USA) (Wang et al., 2010). HN8 or HN12 cells (1 × 105 cells/12 well) were transfected with 0.5 μg of reporter plasmids, 0.4 μg of an ATF3 expression plasmid, an ATF4 expression plasmid (wild‐type or deletion mutants) or an empty vector, and 0.1 μg pRL‐SV40 Renilla luciferase plasmid (Promega, Madison, WI, USA) using EndoFectin Max (GeneCopoeia). Luciferase activity was measured using the Dual‐Luciferase Reporter System (Promega) and normalized to the Renilla luciferase activity expressed by pRL‐SV40.
2.4. Chemicals and antibodies
Cisplatin and SP600125 were purchased from ApexBio (Houston, TX, USA). SB203580 and PD184352 were purchased from LC Laboratories (Woburn, MA, USA). Cisplatin was dissolved in PBS and other reagents were dissolved in dimethyl sulfoxide (Hall et al., 2014). The following antibodies were used: cleaved‐PARP (D64E10), ATF4, CHOP and GAPDH from Cell Signaling Technology (Danvers, MA, USA); Noxa (114C307.1) from Thermo Fisher Scientific (Waltham, MA, USA); MCL‐1 from Enzo Life Sciences (Farmingdale, NY, USA); ATF3, ATF4, pERK, ERK1, and ERK2 from Santa Cruz Biotechnology (Santa Cruz, CA, USA); ATF4 from GeneTex (Irvine, CA, USA).
2.5. Western blot analyses
Whole cell lysates were prepared with CHAPS lysis buffer [20 mm Tris (pH 7.4), 137 mm NaCl, 1 mm DTT, 1% CHAPS, a protease inhibitor cocktail, and phosphatase inhibitor cocktails (Sigma‐Aldrich)]. Equal amounts of proteins were loaded on a SDS acrylamide gel, transferred to a nitrocellulose membrane, and analyzed by immunoblotting with ECL2 (Thermo Scientific, Rockford, IL, USA).
2.6. Cell viability assay
Cell death was quantified by Annexin V‐FITC (BD Biosciences, San Jose, CA, USA)‐propidium iodide (Sigma‐Aldrich) staining according to the manufacturer's protocol, followed by flow cytometric analysis using FACScan (BD Biosciences).
2.7. Quantitative real‐time PCR
Quantitative real‐time PCR analysis was performed as previously described. The primer and probe sets (GAPDH, Hs99999905_m1; Noxa, Hs00560402_m1) were purchased from Applied Biosystems (Carlsbad, CA, USA). Data were analyzed as mRNA expression levels relative to GAPDH according to the manufacturer's protocol.
2.8. Chromatin immunoprecipitation assay
The ChIP assay was performed using the kit (Cell Signaling Technology) according to the manufacture's instruction. To amplify the DNA, the following primers were used: Forward: 5′‐CCTACGTCACCAGGGAAGTT‐3′, Reverse: 5′‐GATGCTGGGATCGGGTGT‐3′.
2.9. Statistical analysis
Values represent the means ± SD for triplicates. The significance of differences between the experimental variables was determined using the Student's t‐test. Values were considered statistically significant at P < 0.05.
3. Results
3.1. Cisplatin‐induced apoptosis is Noxa‐dependent in HNSCC cells
It has been demonstrated that treatment with DNA‐damaging agents, such as cisplatin, induces Noxa expression and apoptosis in a variety of cell lines (Gutekunst et al., 2013; Lin et al., 2012; Nakajima et al., 2016; Sheridan et al., 2010; Zhu et al., 2013). To explore the significance of Noxa in cisplatin‐induced cell death in HNSCC, we first stably expressed the shRNA for Noxa or scrambled shRNA as control in HN8 (p53 deleted) and HN12 (p53 truncated and inactivated) cells (p53 expression is shown in Fig. S1) and then treated with cisplatin with the IC50 concentrations (50 μm for HN8 or 25 μm for HN12). In the control cells, Noxa and cleaved‐PARP (indicative of apoptosis) were induced starting at 8 h (Fig. 1A). Downregulation of Noxa resulted in reduction of cisplatin‐induced apoptosis, as judged by PARP cleavage and Annexin V staining (Fig. 1 and Fig. S2). These results suggest that Noxa is required for cisplatin‐induced apoptosis in HNSCC cells.
Figure 1.
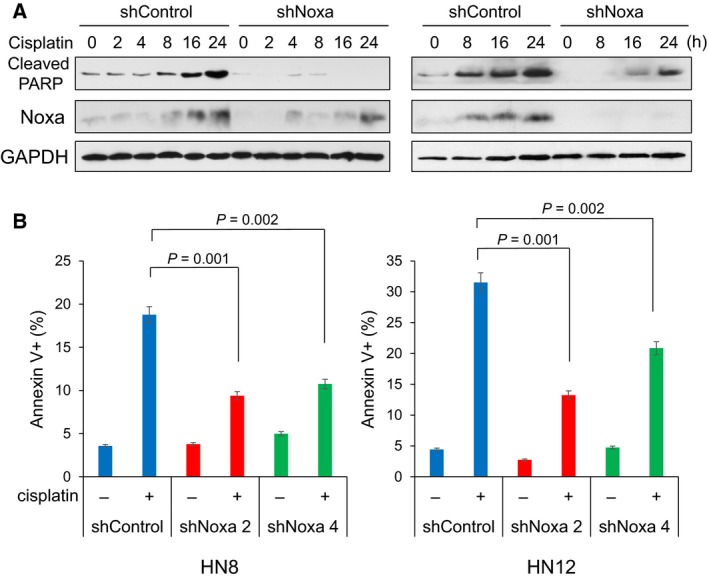
Noxa contributes to cisplatin‐induced apoptosis in a p53‐independent manner. (A) p53‐inactive HN8 and HN12 HNSCC cells were infected with lentiviruses encoding shRNA for nontargeting control or Noxa (shNoxa2). Cells were treated with cisplatin (50 μm for HN8 or 25 μm for HN12) with the indicated periods and equal amounts of the total extracts were used for immunoblot analysis with the indicated antibodies. (B) The cells in (A) were treated with cisplatin for 24 h and cell death was determined by Annexin V‐propidium iodide staining followed by FACS analyses. Another shNoxa construct, shNoxa4 was also introduced in each cell line, which was assayed similarly as shNoxa2. Values represent the mean ± SD of triplicates. The results are representative of at least two independent experiments and are reproducible.
3.2. The CRE on the Noxa promoter is essential for cisplatin‐mediated Noxa induction independent of p53
Although originally identified as a p53 target gene, Noxa is induced and required for cisplatin‐induced cell death even in p53‐null cell lines, such as HN8 and HN12 (Fig. 1). Thus, we explored the mechanisms of p53‐independent Noxa induction. In both HN8 and HN12 cells, gradual increases of Noxa mRNA were observed from 0 to 16 h, which correlated with Noxa protein induction (Fig. 2). This result suggests that the increase in Noxa expression is controlled by the mRNA level. Thus, our focus shifted to understanding the transcriptional regulation of Noxa.
Figure 2.
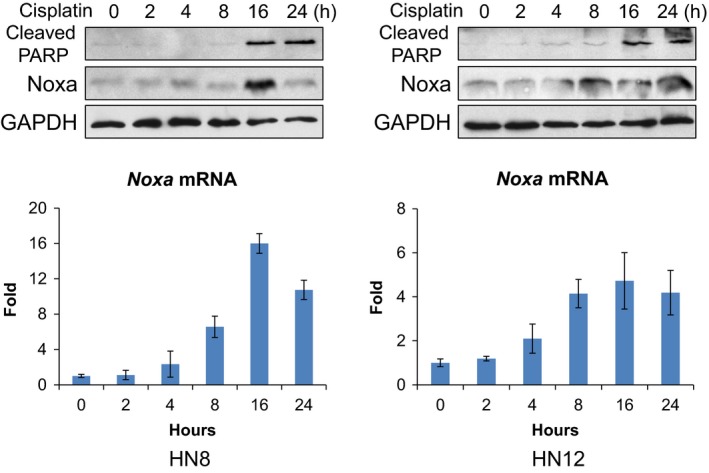
Noxa mRNA is induced by cisplatin in p53‐null HNSCC cells. Following cisplatin treatment (50 μm for HN8 or 25 μm for HN12), Noxa protein and mRNA expression at each time point were determined by western blots (upper panel) and quantitative real‐time PCR (lower panel), respectively. Values represent the mean ± SD of triplicates.
We next determined the promoter regions required for Noxa induction using a series of Noxa promoter‐luciferase constructs (Fig. 3A). In p53‐deleted HN8 cells, there was ~ 4‐fold induction with the −198 construct (−198 to +157 from the transcription start site). When the p53 binding site was mutated (−198 p53 mutant), similar levels of induction was still observed with cisplatin treatment. Consistently, there was ~ 4‐fold induction in the −171 construct in which the p53 binding site was deleted (Fig. 3B). The results from these constructs confirm that Noxa can be induced by cisplatin in a p53‐independent manner. When the CRE was mutated (−171 CRE mutant), cisplatin‐mediated Noxa induction was not detected (Fig. 3B). We further generated −131 to +21 construct and its CRE mutant (Fig. 3A). The intact −131 construct showed a ~ 4‐fold induction, whereas the −131 CRE mutant construct did not show the induction by cisplatin treatment (Fig. 3B). There was no induction with the −58 construct in which both the p53 binding site and CRE were deleted, but Myc and E2F binding sites were retained (Fig. 3B). Similar results were obtained using HN12 cells, confirming the requirement of CRE for cisplatin‐mediated Noxa induction (Fig. S3). Taken together, these data indicate that the CRE is critical for cisplatin‐mediated Noxa induction independent of the p53 binding site.
Figure 3.
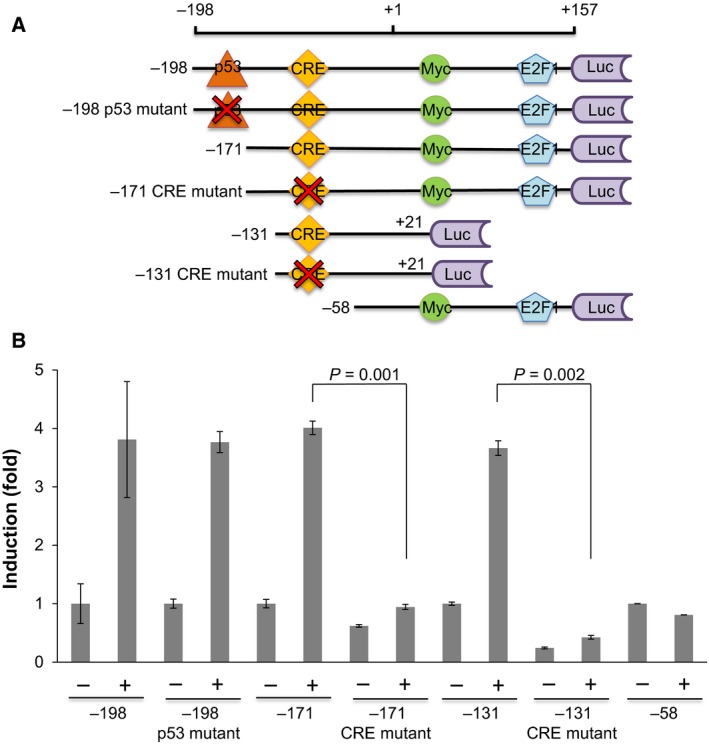
A CRE on the Noxa promoter is critical for cisplatin‐induced Noxa expression. (A) Schematic representation of the promoter region of Noxa. The mutations and deletions of the Noxa promoter were used for the promoter‐luciferase reporter fusion gene constructs. (B) A series of luciferase constructs shown in (A) were transfected in HN8 cells. On the next day, cells were treated with 50 μm of cisplatin for 16 h. Values represent the mean ± SD of triplicates. The results are representative of at least two independent experiments and are reproducible.
3.3. ATF3 and ATF4 regulate cisplatin‐mediated Noxa induction through binding to the CRE on the Noxa promoter
It has been demonstrated that CRE is regulated by the ATF/CREB family transcription factors such as ATF3, ATF4, and CREB (Ameri and Harris, 2008; Servillo et al., 2002). We first determined the expression of these transcription factors after cisplatin treatment in HN8 and HN12 cells. ATF3 showed the highest expression at 16 h with cisplatin treatment when Noxa and cleaved‐PARP were at the peak (Fig. 4A). ATF4 was slightly induced at 16 h. In contrast, CREB showed relatively constant expression (Fig. 4A).
Figure 4.
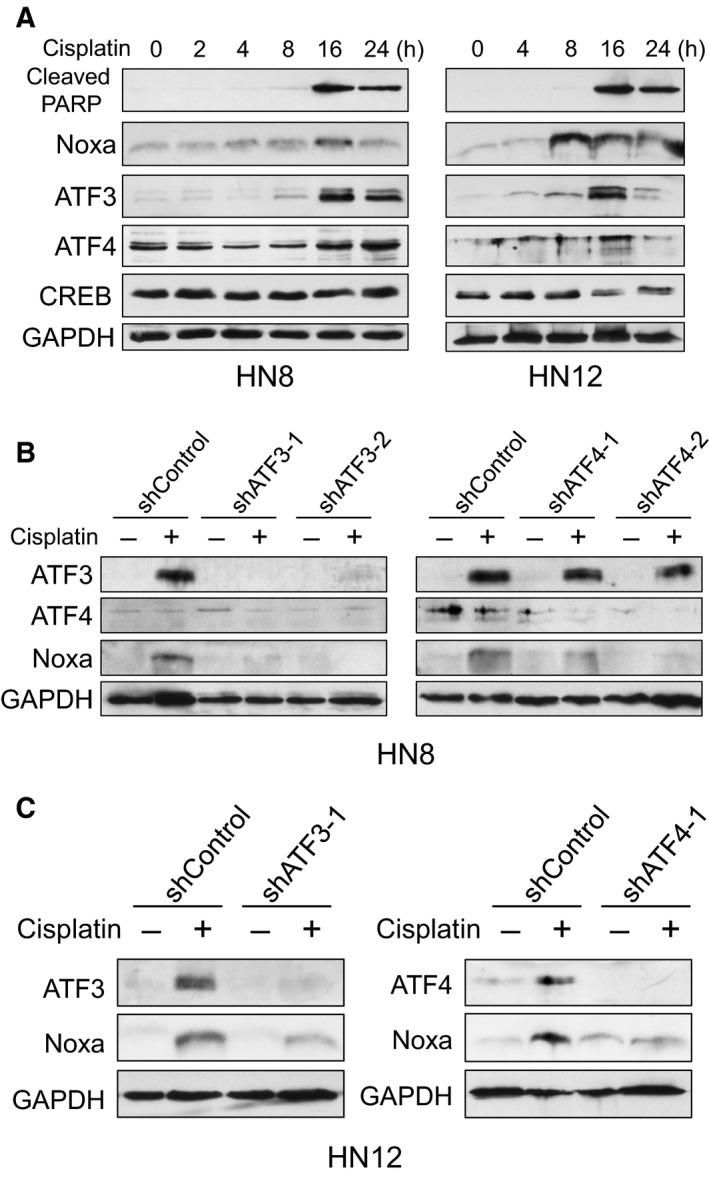
ATF3 and ATF4 contribute to Noxa induction by cisplatin treatment. (A) HN8 and HN12 cells were treated with cisplatin (50 μm for HN8 or 25 μm for HN12) for the indicated times. Equal amounts of the total extracts were used for immunoblot analysis with the indicated antibodies. (B, C) HN8 (B) or HN12 (C) cells were infected with lentiviruses encoding shRNA for nontargeting control, ATF3, or ATF4. Cells were treated with cisplatin for 16 h and equal amounts of the total extracts were used for immunoblot analysis with the indicated antibodies.
We then downregulated each transcription factor by specific shRNAs to examine which transcription factor was contributing to Noxa induction. The induction of Noxa was strongly reduced by shRNAs for ATF3 and ATF4 compared to shControl in both HN8 and HN12 cells (Fig. 4B,C). In contrast, downregulation of CREB by shRNA did not affect Noxa induction (Fig. S4). These results suggest that ATF3 and ATF4 control cisplatin‐mediated Noxa induction independent of p53.
Next, we examined whether endogenous levels of ATF3 and ATF4 bind to CRE in the Noxa promoter. We performed a ChIP assay using ATF3 or ATF4 specific antibodies and the primers that included CRE. Eight hours after cisplatin treatment, the binding of ATF3 and ATF4 to the DNA element containing CRE was clearly increased (Fig. 5A), suggesting that these transcription factors indeed bind to the CRE.
Figure 5.
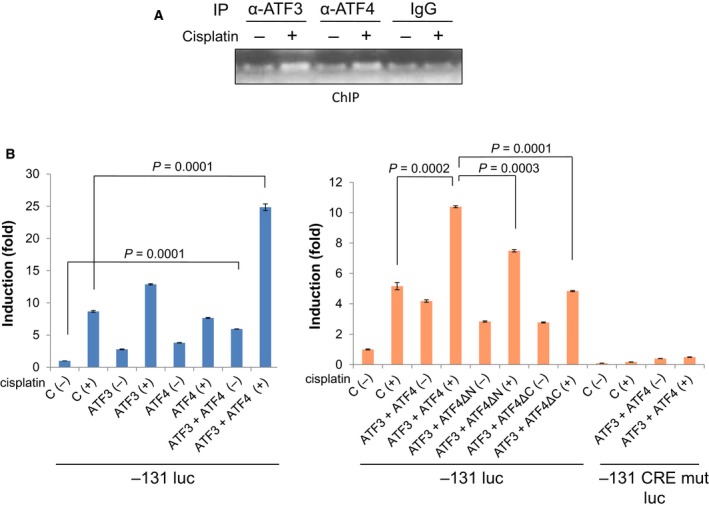
ATF3 and ATF4 bind to and activate the Noxa promoter through the CRE. (A) HN8 cells were treated with 50 μm of cisplatin for 8 h. Chromatin immunoprecipitation was performed with the indicated antibodies and DNA was analyzed by PCR using primers corresponding to −67 to +60 on the Noxa promoter. (B) HN12 cells were cotransfected with the −131 luc reporter construct and an ATF3 or ATF4 expression plasmid for 24 h. On the next day, cells were treated with 25 μm of cisplatin for 16 h. Values represent the mean ± SD of triplicates. The results are representative of at least two independent experiments and are reproducible.
Furthermore, in order to address whether ATF3 and ATF4 cooperatively activate the Noxa promoter, we transfected ATF3 and/or ATF4 expression vectors together with the −131 Noxa promoter‐luciferase construct that contains the CRE for induction (Fig. 3). ATF3 or ATF4 alone slightly activated the Noxa promoter, which was enhanced by cotransfection of ATF3 and ATF4 [cisplatin (–) in Fig. 5B and Fig. S5, left panels]. All these activities were augmented by cisplatin treatment. It has been demonstrated that ATF3 and ATF4 physically associate and activate the Noxa promoter in mantle cell lymphoma cells (Wang et al., 2010). This paper showed that the N‐terminal domain of ATF4 was dispensable for interaction with ATF3, and the C‐terminal DNA binding domain was required for interaction with ATF3. When deletion mutants of each domain of ATF4 were transfected, the activation of Noxa promoter was less compared to ATF4 wild‐type, but higher than the vector‐only control particularly without cisplatin (Fig. 5B and Fig. S5, right panels), suggesting that both DNA binding of ATF4 and interaction with ATF3 and ATF4 are required for cooperative activation of the Noxa promoter. The activation mediated by ATF3 and ATF4 was completely abrogated with a CRE mutant of the Noxa promoter. These results altogether suggest that ATF3 and ATF4 bind to the CRE of the Noxa promoter to cooperatively induce the Noxa expression.
3.4. ATF4 activation is regulated through ERK1 by cisplatin treatment
We next addressed the signaling pathways for the induction of ATF3 and ATF4 in cisplatin treatment. Noxa is known to be induced by ATFs with endoplasmic reticulum (ER) stress inducers, such as fenretinide and Eeyarestatin I (Qing et al., 2012; Wang et al., 2010). Fenretinide induced ATF3, ATF4, and Noxa similarly as cisplatin treatment. However, cisplatin did not induce an ER stress marker, CHOP, which was clearly induced by fenretinide (Fig. 6A), suggesting that ER stress was not induced by cisplatin treatment.
Figure 6.
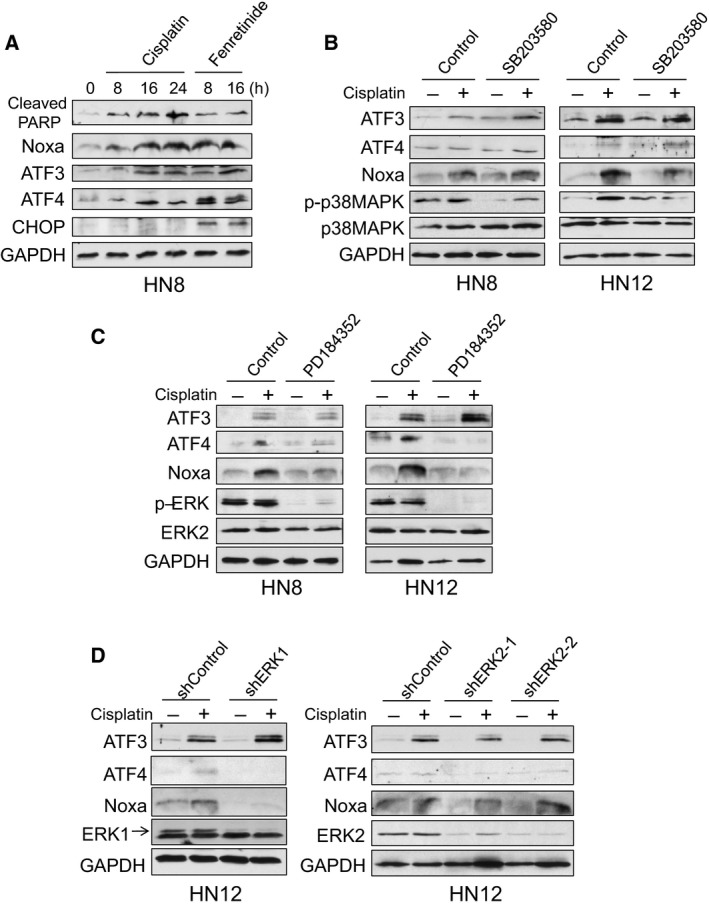
Cisplatin‐induced Noxa is regulated by ERK1 through ATF4 induction. (A) HN8 cells were treated with cisplatin (50 μm) or fenretinide (10 μm) for the indicated times. Equal amounts of the total extracts were used for immunoblot analysis with the indicated antibodies. (B) HN8 and HN12 cells were pretreated with a p38‐MAPK inhibitor, SB203580 (5 μm) for 30 min, and then treated with cisplatin for 16 h (50 μm for HN8 or 25 μm for HN12). Equal amounts of the total extracts were used for immunoblot analysis with the indicated antibodies. (C) HN8 and HN12 cells were pretreated with a MEK inhibitor, PD184352 (5 μm) for 30 min, and then treated with cisplatin for 16 h. Equal amounts of the total extracts were used for immunoblot analysis with the indicated antibodies. (D) HN12 cells were infected with lentiviruses encoding shRNA for nontargeting control, ERK1, or ERK2. Cells were treated with 25 μm of cisplatin for 16 h and equal amounts of the total extracts were used for immunoblot analysis with the indicated antibodies.
It has been shown that the MEK‐ERK or p38‐MAPK pathways are involved in Noxa induction in cisplatin treatment (Sheridan et al., 2010; Zhu et al., 2013). Thus, we used a MEK inhibitor, PD184352, and a p38‐MAPK inhibitor, SB203580, to examine the effect on cisplatin‐induced Noxa expression. Pretreatment with SB203580 affected neither Noxa nor ATF3/ATF4 induction by cisplatin treatment (Fig. 6B). In contrast, pretreatment with PD184352 strongly inhibited Noxa induction in both HN8 and HN12 cells (Fig. 6C). The induction of ATF4, but not ATF3, was also strongly inhibited. It has been reported that cisplatin‐induced JNK activation is a key regulator of ATF3 induction in nonsmall cell lung carcinoma cells (Bar et al., 2016). Thus, we also tested the involvement of JNK for Noxa induction using a JNK inhibitor, SP600125. The inhibition of JNK affected neither ATF3 nor Noxa induction in HNSCC cells (Fig. S6). These results suggest that the MEK‐ERK pathway regulates Noxa induction through ATF4 activation independent of p53.
The MEK inhibitor, PD184352, equally inactivates ERK1 and ERK2 (pERK in Fig. 6C). ERK1 and ERK2 are thought as redundant isoforms due to high sequence homology. However, these proteins play a specific role in certain circumstances (Busca et al., 2016; Mehdizadeh et al., 2016). Thus, we performed specific downregulation by each shRNA to examine which ERK regulates ATF4 and Noxa by cisplatin. Downregulation of ERK1 clearly inhibited Noxa induction as well as ATF4 (Fig. 6D, Left panel). In contrast, downregulation of ERK2 did not reduce cisplatin‐induced ATF3, ATF4 or Noxa levels (Fig. 6D, Right panel). Taken together, ERK1 predominantly regulates cisplatin‐induced ATF4 and Noxa induction.
4. Discussion
DNA damaging agents, such as cisplatin, have been commonly used for chemotherapy in solid tumors for decades, but the molecular mechanisms of the action are still obscure. We and others have shown that a proapoptotic BCL‐2 family protein, Noxa plays a key role to induce cell death by cisplatin (Gutekunst et al., 2013; Lin et al., 2012; Nakajima et al., 2016; Sheridan et al., 2010; Zhu et al., 2013). Although Noxa was originally identified as a p53‐target gene, many cancers have p53 inactivation by deletion or mutations. Thus, it is important to identify p53‐independent regulatory mechanisms of Noxa in cisplatin treatment for overcoming the resistance. Using p53‐inactive HNSCC cells, we find that cisplatin‐induced Noxa is mainly regulated at the transcriptional level. Furthermore, the promoter analyses identified that CRE is crucial for the Noxa induction and transcription factors, ATF3 and ATF4 play an important role in this regulation. It has been shown that Noxa is regulated by a variety of signaling pathways through ATF3/ATF4 (Bagheri‐Yarmand et al., 2015; Qing et al., 2012; Wang et al., 2010; Yan et al., 2014). Thus, this study adds another pathway to induce Noxa through ATF3/ATF4. Although downregulation of Noxa by shRNA strongly inhibited cisplatin‐induced apoptosis, residual cell death activity was observed (Fig. 1), suggesting the involvement of other BH3‐only proteins. It has been shown that BIM is upregulated upon DNA‐damaging agent in a p53‐independent manner (Delbridge et al., 2016; Happo et al., 2010). Further studies are needed to clarify this point.
ATF3 and ATF4 are often activated by ER stress. However, cisplatin did not activate CHOP, an ER stress marker. In contrast, fenretinide induced ER stress, and ATF3, ATF4, and Noxa were induced as similar as those by cisplatin treatment (Fig. 6A). This result suggests that cisplatin and fenretinide induce Noxa mainly through ATF3/ATF4 in a p53‐independent manner, but the regulatory signaling pathways are distinct. A MEK inhibitor, PD184352 specifically inhibited cisplatin‐induced ATF4 followed by Noxa (Fig. 6C). Furthermore, ERK1, but not ERK2, plays a predominant role for this induction (Fig. 6D). The specific role of ERK1 in cisplatin induction has also been shown in hepatocellular carcinoma cells (Guegan et al., 2013). These results suggest that combination treatment with cisplatin and MEK inhibitors may not be effective for cancer therapies, since this combination may not synergize, but antagonize the effect of treatment. Cisplatin also activates p38‐MAPK (Zhu et al., 2013) or JNK (Bar et al., 2016), but treatment with neither a p38‐MAPK inhibitor SB203580 nor a JNK inhibitor SP600125 affected cisplatin‐induced Noxa or ATF3/ATF4 (Fig. 6B and Fig. S6), suggesting that p38‐MAPK and JNK play alternative roles in cisplatin‐induced apoptosis. Thus, it is still unclear how ATF3 is induced by cisplatin. Once the signaling pathways of ATF3 activation become elucidated, the activators of these pathways could be alternative strategies for targeting Noxa to efficiently induce cell death for cancer treatment.
Our results indicate that Noxa can be efficiently induced by not only cisplatin, but also by an ER stress inducer, fenretinide, through ATF3/ATF4 activation (Fig. 6A). The induction of Noxa mainly and solely targets MCL‐1 to inactivate its function. Thus, combining the two treatments to inhibit MCL‐1 together with BCL‐2/BCL‐XL should effectively induce apoptosis in cancer cells. Indeed, when HN8 and HN12 cells were treated with fenretinide and ABT‐263 (navitoclax), we observed synergistic induction of cell death (unpublished results). It has been demonstrated that combination of fenretinide and ABT‐737, a preclinical compound of ABT‐263, shows enhanced apoptosis in neuroblastoma, melanoma, and B‐cell chronic lymphocytic leukemia models (Bruno et al., 2012; Fang et al., 2011; Mukherjee et al., 2015). Since fenretinide and navitoclax are used in clinical trials in a variety of cancers, this combination might become an alternative strategy to treat HNSCC.
5. Conclusions
We have previously demonstrated that Noxa‐mediated MCL‐1 phosphorylation followed by MCL‐1 degradation is critical for apoptosis induced by DNA damaging agents through regulation of the Noxa/MCL‐1/CDK2 complex (Nakajima et al., 2016). In the current study, Noxa induction is mediated by ATF3/ATF4 transcription factors, which are regulated specifically by ERK1 in the upstream signaling pathway via cisplatin treatment. Taken together, we propose one of the signaling pathways that cisplatin takes to induce apoptosis, as shown in Fig. 7. We expect some of these proposed molecules in the figure could be used as biomarkers and targets to enhance the cisplatin sensitivity to overcome resistance.
Figure 7.
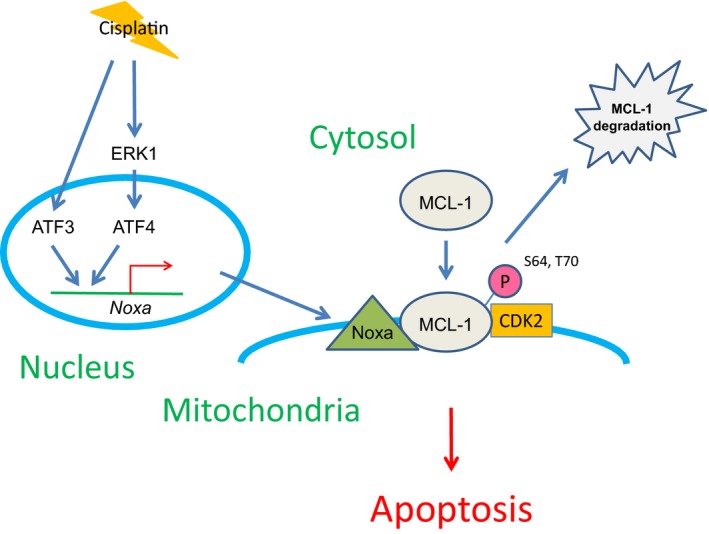
A schematic representation of the signaling pathway from cisplatin treatment to the induction of apoptosis.
Author contributions
KS and HH conceived and designed the experiments. WN provided a series of luciferase constructs. KS, T‐TV, WC, MN, and KZ carried out the experiments. KS and HH wrote the manuscript.
Supporting information
Fig. S1. The expression of p53 in HNSCC cell lines.
Fig. S2. Noxa contributes to cisplatin‐induced apoptosis.
Fig. S3. A CRE on the Noxa promoter is critical for cisplatin‐induced Noxa expression.
Fig. S4. The involvement of CREB in cisplatin‐mediated Noxa induction.
Fig. S5. The expression of ATF3 and ATF4 transfected in HN12 cells.
Fig. S6. The involvement of JNK in cisplatin‐mediated Noxa induction.
Acknowledgements
We thank Andrew Yeudall for cell lines and valuable comments and Steven Grossman, Simitra Deb, Swati Deb and Bradford Windle for valuable comments. This work was supported in part by VCU Presidential Research Quest Fund (to HH). Services in support of this research project were performed by the VCU Massey Cancer Center core laboratories, which is supported in part by funding from NIH‐NCI Cancer Center Support Grant P30 CA016059.
References
- Adams JM and Cory S (2007) The Bcl‐2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameri K and Harris AL (2008) Activating transcription factor 4. Int J Biochem Cell Biol 40, 14–21. [DOI] [PubMed] [Google Scholar]
- Bagheri‐Yarmand R, Sinha KM, Gururaj AE, Ahmed Z, Rizvi YQ, Huang SC, Ladbury JE, Bogler O, Williams MD, Cote GJ et al (2015) A novel dual kinase function of the RET proto‐oncogene negatively regulates activating transcription factor 4‐mediated apoptosis. J Biol Chem 290, 11749–11761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar J, Hasim MS, Baghai T, Niknejad N, Perkins TJ, Stewart DJ, Sekhon HS, Villeneuve PJ and Dimitroulakos J (2016) Induction of activating transcription factor 3 is associated with cisplatin responsiveness in non‐small cell lung carcinoma cells. Neoplasia 18, 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno S, Ghiotto F, Tenca C, Mazzarello AN, Bono M, Luzzi P, Casciaro S, Recchia A, Decensi A, Morabito F et al (2012) N‐(4‐hydroxyphenyl)retinamide promotes apoptosis of resting and proliferating B‐cell chronic lymphocytic leukemia cells and potentiates fludarabine and ABT‐737 cytotoxicity. Leukemia 26, 2260–2268. [DOI] [PubMed] [Google Scholar]
- Busca R, Pouyssegur J and Lenormand P (2016) ERK1 and ERK2 map kinases: specific roles or functional redundancy? Front Cell Dev Biol 4, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ and Green DR (2010) The BCL‐2 family reunion. Mol Cell 37, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbridge AR, Pang SH, Vandenberg CJ, Grabow S, Aubrey BJ, Tai L, Herold MJ and Strasser A (2016) RAG‐induced DNA lesions activate proapoptotic BIM to suppress lymphomagenesis in p53‐deficient mice. J Exp Med 213, 2039–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Harned TM, Kalous O, Maldonado V, DeClerck YA and Reynolds CP (2011) Synergistic activity of fenretinide and the Bcl‐2 family protein inhibitor ABT‐737 against human neuroblastoma. Clin Cancer Res 17, 7093–7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guegan JP, Ezan F, Theret N, Langouet S and Baffet G (2013) MAPK signaling in cisplatin‐induced death: predominant role of ERK1 over ERK2 in human hepatocellular carcinoma cells. Carcinogenesis 34, 38–47. [DOI] [PubMed] [Google Scholar]
- Gutekunst M, Mueller T, Weilbacher A, Dengler MA, Bedke J, Kruck S, Oren M, Aulitzky WE and van der Kuip H (2013) Cisplatin hypersensitivity of testicular germ cell tumors is determined by high constitutive Noxa levels mediated by Oct‐4. Cancer Res 73, 1460–1469. [DOI] [PubMed] [Google Scholar]
- Hall MD, Telma KA, Chang KE, Lee TD, Madigan JP, Lloyd JR, Goldlust IS, Hoeschele JD and Gottesman MM (2014) Say no to DMSO: dimethylsulfoxide inactivates cisplatin, carboplatin, and other platinum complexes. Cancer Res 74, 3913–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happo L, Cragg MS, Phipson B, Haga JM, Jansen ES, Herold MJ, Dewson G, Michalak EM, Vandenberg CJ, Smyth GK et al (2010) Maximal killing of lymphoma cells by DNA damage‐inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood 116, 5256–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemans CR, Braakhuis BJ and Brakenhoff RH (2011) The molecular biology of head and neck cancer. Nat Rev Cancer 11, 9–22. [DOI] [PubMed] [Google Scholar]
- Lin C, Zhao XY, Li L, Liu HY, Cao K, Wan Y, Liu XY, Nie CL, Liu L, Tong AP et al (2012) NOXA‐induced alterations in the Bax/Smac axis enhance sensitivity of ovarian cancer cells to cisplatin. PLoS One 7, e36722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehdizadeh A, Somi MH, Darabi M and Jabbarpour‐Bonyadi M (2016) Extracellular signal‐regulated kinase 1 and 2 in cancer therapy: a focus on hepatocellular carcinoma. Mol Biol Rep 43, 107–116. [DOI] [PubMed] [Google Scholar]
- Mukherjee N, Reuland SN, Lu Y, Luo Y, Lambert K, Fujita M, Robinson WA, Robinson SE, Norris DA and Shellman YG (2015) Combining a BCL2 inhibitor with the retinoid derivative fenretinide targets melanoma cells including melanoma initiating cells. J Invest Dermatol 135, 842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima W, Sharma K, Lee JY, Maxim NT, Hicks MA, Vu TT, Luu A, Yeudall WA, Tanaka N and Harada H (2016) DNA damaging agent‐induced apoptosis is regulated by MCL‐1 phosphorylation and degradation mediated by the Noxa/MCL‐1/CDK2 complex. Oncotarget 7, 36353–36365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, Ridge JA, Goodwin J, Kenady D, Saunders J et al (2007) TP53 mutations and survival in squamous‐cell carcinoma of the head and neck. N Engl J Med 357, 2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X, Mayes PA, Wise DR, Thompson CB, Maris JM et al (2012) ATF4 regulates MYC‐mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 22, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg SM and Ellisen LW (2012) The molecular pathogenesis of head and neck squamous cell carcinoma. J Clin Invest 122, 1951–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano D, Xie TX, Ow TJ, Zhao M, Pickering CR, Zhou G, Sandulache VC, Wheeler DA, Gibbs RA, Caulin C et al (2011) Disruptive TP53 mutation is associated with aggressive disease characteristics in an orthotopic murine model of oral tongue cancer. Clin Cancer Res 17, 6658–6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz JD, Bran G, Anders C, Sadick H, Faber A, Hormann K and Sauter A (2010) Induction chemotherapy with TPF (docetaxel, carboplatin and fluorouracil) in the treatment of locally advanced squamous cell carcinoma of the head and neck. Oncol Rep 24, 1213–1216. [DOI] [PubMed] [Google Scholar]
- Servillo G, Della Fazia MA and Sassone‐Corsi P (2002) Coupling cAMP signaling to transcription in the liver: pivotal role of CREB and CREM. Exp Cell Res 275, 143–154. [DOI] [PubMed] [Google Scholar]
- Sheridan C, Brumatti G, Elgendy M, Brunet M and Martin SJ (2010) An ERK‐dependent pathway to Noxa expression regulates apoptosis by platinum‐based chemotherapeutic drugs. Oncogene 29, 6428–6441. [DOI] [PubMed] [Google Scholar]
- Suh Y, Amelio I, Guerrero Urbano T and Tavassoli M (2014) Clinical update on cancer: molecular oncology of head and neck cancer. Cell Death Dis 5, e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermorken JB, Remenar E, van Herpen C, Gorlia T, Mesia R, Degardin M, Stewart JS, Jelic S, Betka J, Preiss JH et al (2007) Cisplatin, fluorouracil, and docetaxel in unresectable head and neck cancer. N Engl J Med 357, 1695–1704. [DOI] [PubMed] [Google Scholar]
- Wang Q, Shinkre BA, Lee JG, Weniger MA, Liu Y, Chen W, Wiestner A, Trenkle WC and Ye Y (2010) The ERAD inhibitor Eeyarestatin I is a bifunctional compound with a membrane‐binding domain and a p97/VCP inhibitory group. PLoS One 5, e15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Zhong N, Liu G, Chen K, Liu X, Su L and Singhal S (2014) Usp9x‐ and Noxa‐mediated Mcl‐1 downregulation contributes to pemetrexed‐induced apoptosis in human non‐small‐cell lung cancer cells. Cell Death Dis 5, e1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeudall WA, Jakus J, Ensley JF and Robbins KC (1997) Functional characterization of p53 molecules expressed in human squamous cell carcinomas of the head and neck. Mol Carcinog 18, 89–96. [DOI] [PubMed] [Google Scholar]
- Youle RJ and Strasser A (2008) The BCL‐2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9, 47–59. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Regunath K, Jacq X and Prives C (2013) Cisplatin causes cell death via TAB 1 regulation of p53/MDM2/MDMX circuitry. Genes Dev 27, 1739–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The expression of p53 in HNSCC cell lines.
Fig. S2. Noxa contributes to cisplatin‐induced apoptosis.
Fig. S3. A CRE on the Noxa promoter is critical for cisplatin‐induced Noxa expression.
Fig. S4. The involvement of CREB in cisplatin‐mediated Noxa induction.
Fig. S5. The expression of ATF3 and ATF4 transfected in HN12 cells.
Fig. S6. The involvement of JNK in cisplatin‐mediated Noxa induction.