

Abstract
Key points
There is assumed to be a monotonic association between melatonin suppression and circadian phase resetting induced by light exposure.
We tested the association between melatonin suppression and phase resetting in humans.
Sixteen young healthy participants received nocturnal bright light (∼9500 lux) exposure of continuous or intermittent patterns, and different durations ranging from 12 min to 6.5 h.
Intermittent exposure patterns showed significant phase shifts with disproportionately less melatonin suppression.
Each and every bright light stimulus in an intermittent exposure pattern induced a similar degree of melatonin suppression, but did not appear to cause an equal magnitude of phase shift.
These results suggest that phase shifts and melatonin suppression are functionally independent such that one cannot be used as a proxy measure of the other.
Abstract
Continuous experimental light exposures show that, in general, the conditions that produce greater melatonin suppression also produce greater phase shift, leading to the assumption that one can be used as a proxy for the other. We tested this association in 16 healthy individuals who participated in a 9‐day inpatient protocol by assessing melatonin suppression and phase resetting in response to a nocturnal light exposure (LE) of different patterns: (i) dim‐light control (<3 lux; n = 6) or (ii) two 12‐min intermittent bright light pulses (IBL) separated by 36 min of darkness (∼9500 lux; n = 10). We compared these results with historical data from additional LE patterns: (i) dim‐light control (<3 lux; n = 11); (ii) single continuous bright light exposure of 12 min (n = 9), 1.0 h (n = 10) or 6.5 h (n = 6); or (iii) an IBL light pattern consisting of six 15‐min pulses with 1.0 h dim‐light recovery intervals between them during a total of 6.5 h (n = 7). All light exposure groups had significantly greater phase‐delay shifts than the dim‐light control condition (P < 0.0001). While a monotonic association between melatonin suppression and circadian phase shift was observed, intermittent exposure patterns showed significant phase shifts with disproportionately less melatonin suppression. Each and every IBL stimulus induced a similar degree of melatonin suppression, but did not appear to cause an equal magnitude of phase shift. These results suggest unique specificities in how light‐induced phase shifts and melatonin suppression are mediated such that one cannot be used as a proxy measure of the other.
Keywords: melatonin, phase shift, light
Key points
There is assumed to be a monotonic association between melatonin suppression and circadian phase resetting induced by light exposure.
We tested the association between melatonin suppression and phase resetting in humans.
Sixteen young healthy participants received nocturnal bright light (∼9500 lux) exposure of continuous or intermittent patterns, and different durations ranging from 12 min to 6.5 h.
Intermittent exposure patterns showed significant phase shifts with disproportionately less melatonin suppression.
Each and every bright light stimulus in an intermittent exposure pattern induced a similar degree of melatonin suppression, but did not appear to cause an equal magnitude of phase shift.
These results suggest that phase shifts and melatonin suppression are functionally independent such that one cannot be used as a proxy measure of the other.
Introduction
Light is the strongest environmental time cue that resets the mammalian circadian pacemaker, located in the suprachiasmatic nucleus (SCN) (Czeisler et al. 1989; Meijer et al. 1992, 1998), with maximal phase shifts during the biological night (St Hilaire et al. 2012). Ocular light exposure during the biological night also acutely suppresses release of the pineal hormone melatonin (Lewy et al. 1980; Zeitzer et al. 2000). The light information required to entrain the SCN or suppress melatonin is detected by specialized retinal photoreceptors and transduced to the SCN via the retinohypothalamic tract (Klein & Moore, 1979; Johnson et al. 1998; Gooley et al. 2001). Subsequently, the SCN sends light information to the pineal via the retina–SCN–superior cervical ganglion (SCG)–pineal pathway, the canonical pathway by which light can suppress melatonin (Klein et al. 1971; Moore, 1978).
Light‐induced melatonin suppression shows sensitivities seemingly similar to those for phase resetting with respect to the duration (Chang et al. 2012), intensity (Boivin et al. 1996; Zeitzer et al. 2000) and spectral characteristics of the light exposure (Wright & Lack, 2001; Lockley et al. 2003; Gooley et al. 2010). The degree of melatonin suppression and the magnitude of phase resetting have been reported to be linearly correlated (Lockley et al. 2003) and, at least at the photoreceptor input level, have been proposed to be predictive of the strength of the photic stimulus on the SCN and therefore broader circadian function (Brainard et al. 2001; Rea et al. 2005, 2010). Given that melatonin suppression can only occur during the biological night (defined from the onset (SynOn) of melatonin until synthesis offset (SynOff)) (St Hilaire et al. 2007) and light can cause phase shifts at times when melatonin is not secreted (Jewett et al. 1997; Khalsa et al. 2003), the two responses can clearly be functionally independent.
In the present study we compared the magnitude and pattern of melatonin suppression with circadian phase resetting in humans during exposure to a range of continuous and intermittent light pulses in order to examine the extent to which melatonin suppression and phase resetting are functionally coupled.
Methods
Ethical approval
The study was approved by the Partners Human Research Committee, and participants provided written informed consent prior to study. The study conformed to the standards set by the latest revision of the Declaration of Helsinki and the study was registered as a clinical trial on http://www.clinicaltrials.gov (NCT no. 01330992).
The protocol for the 16 participants studied under the dim‐light control condition and the 1.0 h intermittent bright light (IBL) condition is reported here. The protocols for the 0.2, 1.0, 6.5 and 6.5 h IBL conditions have been reported previously (Gronfier et al. 2004; Chang et al. 2012; St Hilaire et al. 2012). Differences between the current and previous protocols are detailed below.
Participants and prestudy conditions
Sixteen young healthy individuals (9 females; mean age ± SD: 23.1 ± 2.8 years; range 19–29 years) participated in the current inpatient study at the Intensive Physiological Monitoring Unit in the Centre for Clinical Investigation (CCI) at Brigham and Women's Hospital. All were healthy as determined by physical, psychological and ophthalmologic exam. For at least 3 weeks prior to admission, participants maintained a self‐selected, constant 8 h sleep/rest/dark schedule confirmed with calls to a time‐ and date‐stamped voicemail at bedtime and wake time. Throughout screening, participants were asked to refrain from use of any prescription or non‐prescription medications, supplements, recreational drugs, caffeine, alcohol or nicotine. Compliance was verified by urine and blood toxicology during screening and urine toxicology upon entry to the unit. For at least 7 days prior to entering the unit, subjects’ schedules were monitored with actigraphy (Actiwatch‐L, Minimitter, Inc., Bend, OR, USA).
Historical data
Forty‐three individuals (13 females; mean age ± SD: 22.7 ± 3.5 years; range 18–31 years) participated in the previously reported protocols (Gronfier et al. 2004; Chang et al. 2012; St Hilaire et al. 2012). All prestudy conditions were identical.
Study protocols
Participants were studied for 9 or 10 days in an environment free of time cues (i.e. no access to windows, clocks, watches, live TV, radio, internet, telephones and current newspapers and continually supervised by staff trained not to reveal information about the time of day) (Fig. 1 A and B). The 9‐day schedule consisted of a 3‐day baseline (8 h:16 h sleep:wake cycle based on average sleep times in the 7 days prior to study entry), an initial ∼50 h constant routine (CR1, described below), a 16 h light exposure day, followed by 8 h of sleep. Following the subsequent 8 h sleep episode, participants completed a ∼30 h constant routine (CR2) followed by a final 8 h sleep episode, after which they were discharged (Fig. 1 A). The 10‐day protocol (Gronfier et al. 2004) differed in including a 26.2 h CR1 and a 64.0 h CR2. During the constant routine procedures, participants were asked to remain awake while supervised in constant dim light in a semi‐recumbent posture, with daily nutritional intake divided into hourly portions (150 mEq Na+/100 mEq K+ (±20%) controlled nutrient, isocaloric (basal energy expenditure × 1.3) diet, 2000–2500 mL fluids/24 h; Mills et al. 1978; Duffy & Dijk, 2002).
Figure 1. Study protocol to assess the functional coupling between melatonin suppression and phase resetting.
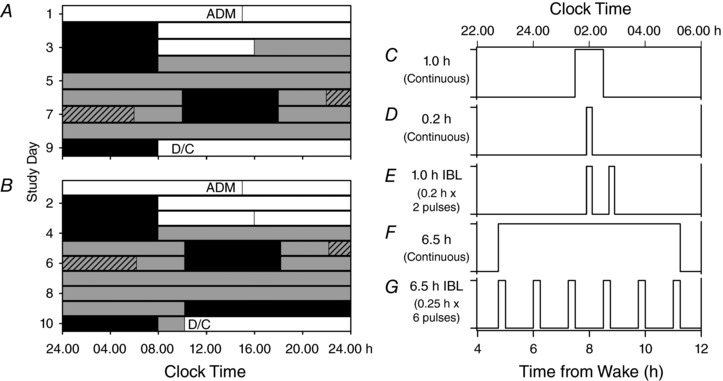
Participants were enrolled in a 9‐day (n = 39; A) or 10‐day (n = 20; B) inpatient protocol in an environment free of time cues. White bars indicate exposure to ambient room light (∼90 lux) and grey bars indicate exposure to dim ambient light (<3 lux). Black bars show scheduled sleep episodes in darkness that, for the first 3 baseline days, was scheduled the same time as the self‐selected sleep schedule maintained for 3 weeks prior to admission. The schedule consisted of a 3‐day baseline following admission (ADM), with 8:16 h sleep:wake cycle; an initial ∼50 h (A) or ∼26 h (B) constant routine, an 8 h sleep (sleep delayed by 10 h from pre‐admission self‐selected sleep schedule) and a 16 h wake episode in which the light exposure was centred; followed by 8 h sleep (sleep delayed by 10 h from pre‐admission self‐selected sleep schedule) and then a second ∼30 h (A) or ∼64 h (B) constant routine followed by an 8 h recovery sleep at the same time as the baseline days and then discharge (D/C), the 8 h interval during which the light exposure of different patterns and durations occurred is shown as a hatched bar. Participants received bright light exposure of different patterns and durations (C–G).
Lighting conditions
During the first 2.5 baseline days, maximum ambient light during scheduled wake was 48 μW cm−2 (∼190 lux) maximum when measured in the horizontal plane at a height of 187 cm and 23 μW cm−2 or (∼88 lux) when measured in the vertical plane (137 cm). In the 9‐day study, midway through day 3, maximum ambient light was decreased to <0.4 μW cm−2 [approximately 0.1 μW cm−2 (<3 lux; ∼1.5 lux)] when measured in the vertical plane at 137 cm and maintained at that level for the remainder of the study. In the 10‐day study, lighting conditions for day 3 were maintained at ∼90 lux until bedtime. Ambient room lighting was generated using ceiling‐mounted 4100K fluorescent lamps (F96T12/41U/HO/EW, 95W; F32T8/ADV841/A, 32W; F25T8/TL841, 25 W; Philips Lighting, The Netherlands) with digital ballasts (Hi‐Lume 1% and Eco‐10 ballasts, Lutron Electronics Co., Inc., Coopersburg, PA, USA) transmitted through a UV‐stable filter (Lexan 9030 with prismatic lens, GE Plastics, Pittsfield, MA, USA). Routine illuminance and irradiance measures were conducted using an IL1400 radiometer/powermeter with an SEL‐033/Y/W or SEL‐033/F/W detector, respectively (International Light, Inc., Newburyport, MA, USA).
Study groups and experimental light exposure
There were six light exposure (LE) conditions: (i) dim‐light control, n = 17; (ii) 1.0 h bright continuous light, n = 10 (Fig. 1 C); (iii) 0.2 h bright continuous light, n = 9 (Fig. 1 D); (iv) 1.0 h intermittent bright light (IBL), n = 10 (Fig. 1 E); (v) 6.5 h bright continuous light, n = 6 (Fig. 1 F); and (vi) 6.5 h bright IBL, n = 7; (Fig. 1 G). All LE occurred in constant posture conditions. The different LE conditions were aligned relative to the midpoints of LE timing, except for the 0.2 h LE, which occurred slightly earlier, with the midpoint 12 min earlier than the other groups. The 1.0 h IBL condition included two 12 min bright light pulses with a 36 min dark (lights off) interval in between. The 1.0 h IBL condition was timed so that the first pulse corresponded to the start of the single 0.2 h LE condition. The 6.5 h IBL condition included six 15 min bright light pulses with five 60 min dim light (<1 lux) intervals in between. The dim‐light control group received dim (<3 lux) ambient light during the constant posture. All participants in the 0.2 continuous, 1.0 h IBL, 1.0 h continuous and 10 participants in the control group completed the 9‐day inpatient study (Chang et al. 2012). All participants in the 6.5 h continuous, 6.5 h IBL and seven participants in the control group completed the 10‐day inpatient study (Gronfier et al. 2004). The phase shift and melatonin suppression results have been previously reported for the 0.2 h continuous (Chang et al. 2012), 1.0 h continuous (Chang et al. 2012), 6.5 h continuous (Gronfier et al. 2004; Chang et al. 2012), 6.5 h IBL (Gronfier et al. 2004) and 11 participants from the control group (Gronfier et al. 2004; St Hilaire et al. 2012). The LE conditions were centred within the 16 h wake episode and timed to occur at a circadian phase expected to induce melatonin suppression and phase delay shifts.
The target corneal light illuminance during the experimental light exposure (LE) was ∼9500 lux (∼2900 μW cm−2, ∼6700 melanopic lux; Lucas et al. 2014) and ∼1.5 lux (0.1 μW cm−2, ∼1.0 melanopic lux). The LE and dim‐light control conditions were generated using the same overhead fluorescent lamps as described previously. In the 9‐day inpatient study the constant posture for the light exposure session began ∼6 h after wake time on the LE day for 4.5 h. In the 10‐day inpatient study the constant posture for the LE session began ∼3 h after wake time on the LE day for ∼8 h. Participants remained seated in a specific location of the room for the entire constant posture. Participants wore clear unshaded UVEX glasses (UVEX Winter Optical, Smithfield, RI, USA) and were asked to maintain a ‘fixed gaze’ on a target on the wall directly in front of them for at least 4 min alternating with an equal interval ‘free gaze’ during which they were allowed to look elsewhere, as long as they did not close their eyes or shield them from the light. The 0.2 h continuous and 1.0 h continuous conditions used a 6 min fixed/free gaze interval; the 1.0 h IBL used a 4 min fixed/free gaze interval during the 1.0 h exposure interval and a 5 min fixed/free gaze interval during the remainder of the 4.5 h constant posture; the 6.5 h continuous and 6.5 h IBL conditions used a 5 min fixed/free gaze interval throughout the exposure interval. Light illuminance was measured at every change of gaze with the sensor placed next to the participant's eye and pointed at the target during ‘fixed gaze’ and in the angle of gaze during ‘free gaze’. A research technician was present during the LE session to administer the LE, measure and record light readings, and monitor adherence to study procedures.
Outcome measures of melatonin phase and melatonin suppression
In the current study (n = 16), blood samples were collected every 5–60 min throughout the protocol and assayed by radioimmunoassay for melatonin concentration. Blood collection frequency varied between protocols. Melatonin concentration was determined by double‐antibody radioimmunoassay (Vaughan, 1993) by a laboratory unaware of experimental conditions (Specialty Assay Research Core Laboratory, Brigham and Women's Hospital, Boston, MA, USA). The plasma melatonin intra‐assay coefficient of variation (%CV) was 8.5% at 5.85 pg mL−1 and 8.0% at 25.46 pg mL−1, and the interassay %CV was 10.8% at 5.98 pg mL−1; 14.5% at 23.07 pg mL−1. The previous studies had comparable %CV coefficients. In the 10‐day study (Gronfier et al. 2004), which included the 6.5 h continuous, 6.5 h IBL and dim‐light control conditions (n = 20), samples were assayed for melatonin by use of radioimmunoassay techniques (Diagnostech/Pharmasan, Osceola, WI, USA). The assay sensitivity was 2.5 pg mL−1. The average intra‐assay coefficients of variation were 6.4% below 50 pmol L−1 and 4.9% above. In the previous 9‐day studies (Chang et al. 2012; St Hilaire et al. 2012), which included the 0.2 h continuous, 1.0 h continuous conditions (n = 19) and the dim‐light control condition (n = 4), melatonin was assayed by radioimmunoassay (Pharmasan). Sensitivity of the assay was 2.5 pg mL−1; inter‐assay and intra‐assay coefficients of variance were 9.2–13.2% and 9.8–12.1%, respectively.
The dim light melatonin onset (DLMOn) was used as the endogenous phase marker and calculated as the time at which levels of melatonin went above the 25% threshold of the three harmonic fitted peak‐to‐trough amplitude of melatonin during CR1 (Lockley et al. 2003; Gronfier et al. 2004). Phase shifts were calculated as the difference in clock time between CR1 and CR2 phases. Due to the length of CR1 (∼50 h) in the 9‐day study, there were two DLMO phase measures determined for each participant during this constant routine; phase shifts were calculated using the DLMOn that occurred temporally closer (∼24 h prior) to the LE.
Melatonin suppression was determined in three ways: (i) the area under the curve (AUC) during the light exposure was compared to the AUC during the corresponding time window 24 h earlier during CR1, and the resulting percentage melatonin suppression due to the bright light was calculated using the formula [1 − (AUCLE/AUCCR1)] × 100 (Zeitzer et al. 2000; Lockley et al. 2003); (ii) the difference in melatonin levels at the start and end of each light pulse (interpolated linearly), expressed as a percentage; and (iii) AUC during the interval between DLMO and DLMOff (downward crossing of the 25% threshold) flanking the light‐exposure stimulus and the interval between DLMO and DLMOff 24 h earlier during CR1. Percentage melatonin suppression due to the bright light was calculated as described earlier.
Data analysis
Data from two participants in the current study were excluded from further analysis. One participant (1.0 h IBL condition) was excluded from analysis because post hoc analysis showed that the LE occurred 6.2 h after DLMOn, which is outside the interval (−3 to + 6 h around DLMOn) for inducing a phase delay (St Hilaire et al. 2012). The second individual (dim‐light control condition) showed considerable daytime melatonin secretion, which is unusual, and a reliable DLMO could not be determined. Due to differences in blood sampling frequency, melatonin values were linearly interpolated at the start and end of each light pulse and also at the start and end of the entire exposure window.
Melatonin phase shift and suppression data were compared among the six LE conditions using a general linear model one‐way ANOVA with LE condition as the main factor. If a significant main effect was observed, then data were subjected to Student–Newman–Keuls (SNK) post hoc analysis. The percentage change between the onset and offset of the light pulses was compared between groups using a general linear model with LE type as the main factor. If a significant main effect was observed then data were subjected to SNK post hoc analysis. All analyses were performed using SAS 9.3 (SAS Institute Inc., Cary, NC, USA).
Results
Melatonin suppression and phase resetting
Phase resetting increased with longer duration exposures, proportional to both the sum of the light exposure duration (Fig. 2 A) and the entire exposure window (start of the first pulse to the end of the last pulse in the IBL conditions; data not shown). All light exposure groups had significantly greater delay shifts than the dim‐light control condition. The phase shift in the 0.2 h continuous, 1.0 h IBL (0.4 h total light exposure) and 1.0 h continuous groups were not significantly different. The 6.5 h continuous and 6.5 h IBL (1.5 h total light exposure) groups were both significantly greater than the other three LE groups (P = 0.01) but not significantly different from each other (Fig. 2 A).
Figure 2. Relationship between melatonin suppression and circadian phase resetting.
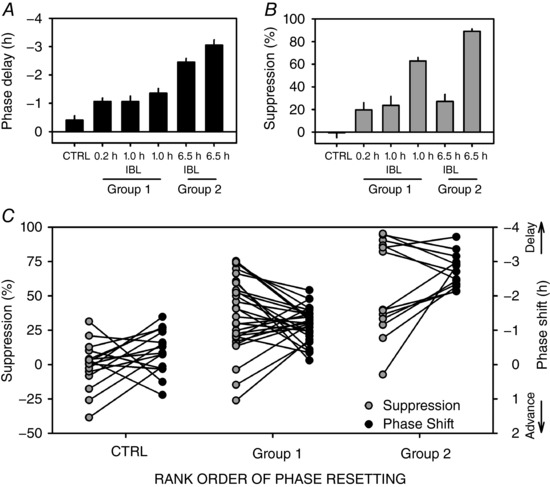
A and B, relationship between light pattern and (A) phase shift, assessed as the difference in circadian phase over 3 circadian cycles between the initial and final constant routines, and (B) melatonin suppression, assessed as the change in AUC over the interval within which the light exposure took place (0.2, 1.0 or 6.5 h, as appropriate) and the corresponding interval which occurred 24 h earlier. For IBL exposures, the entire exposure interval, including the intervening intervals of dim light, was used (e.g. for the 0.2 h condition, AUC was calculated over the entire 1 h interval). Three groups are apparent based on the rank order of the magnitude of phase shift: CTRL < Group 1 (0.2 h, 1.0 h IBL and 1.0 h continuous) < Group 2 (6.5 h IBL and 6.5 h continuous). C, individually matched phase shift and melatonin suppression values plotted for all individuals across the three groups as generated in A; data are shown as means ± SEM.
In contrast to phase resetting, melatonin suppression did not increase monotonically with summed LE duration (Fig. 2 B). All LE groups had significantly greater suppression than the control group but the 6.5 h IBL group (1.5 h total light exposure) had relatively less suppression than the 6.5 h continuous condition, and less than what could be expected from a 90 min exposure interval. The 0.2 h continuous and 6.5 h IBL (1.5 h total light exposure) conditions had similarly low melatonin suppression (∼25%) whereas the 1 h continuous and 6.5 h continuous conditions had similar higher suppression (∼60–80%). The rank order of the amount of melatonin suppression by condition did not correspond to the order observed for phase resetting magnitude. Therefore, the IBL conditions induced disproportionally larger phase shifts than melatonin suppression (Fig. 2 C).
To assess the relationship further, individually matched phase shift and melatonin suppression values were plotted for all individuals to examine whether the magnitudes of both responses were parallel. Figure 2 C shows that the phase‐shift magnitudes are highly variable and are not associated with the suppression response. Correlational analyses between melatonin suppression and phase shifts were not significant within any of the six different light exposure conditions (Table 1).
Table 1.
Correlation between phase shift and melatonin suppression within each light exposure condition
| Light exposure condition | Pearson's r | P |
|---|---|---|
| Dim‐light control | 0.26 | 0.34 |
| 1.0 h continuous | 0.41 | 0.24 |
| 0.2 h continuous | 0.47 | 0.20 |
| 1.0 h IBL | −0.37 | 0.32 |
| 6.5 h continuous | 0.18 | 0.74 |
| 6.5 h IBL | −0.42 | 0.35 |
Temporal dynamics of melatonin suppression
Melatonin levels decreased during each light pulse and started to recover shortly after the end of the light pulse, as expected. Melatonin levels remained low throughout the 1.0 h and 6.5 h continuous conditions (Fig. 3 A–G). Between the start and end of the 1.0 h and 6.5 h light pulses, melatonin changed 59.3 ± 5.8% and 78.0 ± 7.3%, respectively (Fig. 3 B–H). In the 6.5 h IBL condition, melatonin levels decreased during each 15 min pulse and recovered to pre‐pulse levels or higher by the end of each 60 min recovery interval (Fig. 3 I). Each of the six pulses reduced melatonin levels similarly (range 25–43%) without any evidence of desensitization in subsequent responses (Fig. 3 J).
Figure 3. Melatonin suppression and circadian phase resetting are not functionally coupled.
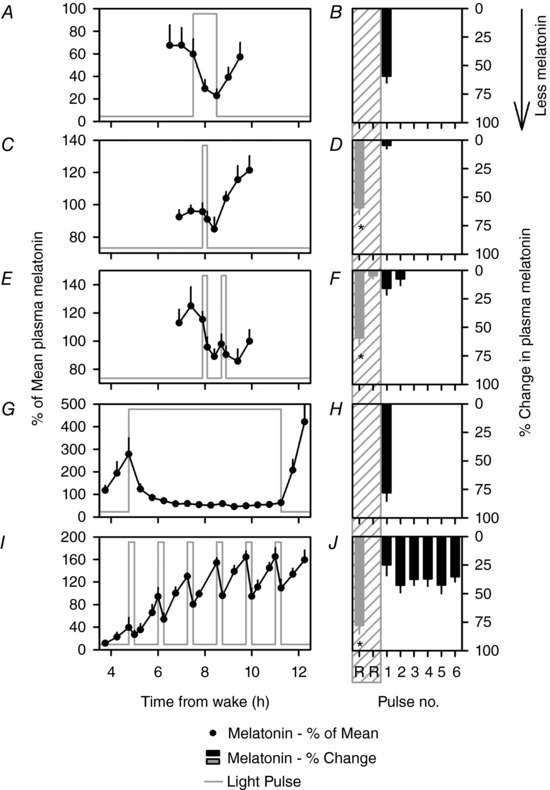
Melatonin levels are expressed as a percentage of the mean during the 8 h interval from 22.00 to 06.00 h shown by hatched bars in Fig. 1 A and B, which contains the light exposure for each individual, and then compared across individuals in the 1.0 h continuous (A), 0.2 h continuous (C), 1.0 h IBL (E), 6.5 h continuous (G) and 6.5 h IBL (I) conditions (mean ± SEM). Values were interpolated at the start and end of the light pulse and in 0.5 intervals starting 1 h before the pulse and ending 1.0 h after the pulse. The light pulse is shown as the grey line. The corresponding change in melatonin levels from each pulse calculated as the percentage change between the start and end of the light pulse is shown as black bars in B, D, F, H and J (mean ± SEM; *signifies P < 0.05). This method of calculating melatonin suppression is different from the AUC‐based method used for calculating suppression in Fig. 2, and allows for calculating the change in melatonin from the start to the end of each intermittent and continuous light pulse. The corresponding continuous conditions used as a comparative reference (denoted as Pulse no. R) are re‐plotted as grey bars in the grey hatched area. For the 1.0 h IBL condition, the comparator conditions include the 1.0 h and the 12 min continuous conditions. For the 6.5 h IBL condition, the comparator condition includes the 6.5 h continuous condition.
The first 12 min pulse of the 1.0 h IBL condition decreased melatonin levels by 15.7 ± 5.8%, which was not significantly different from the mean reduction in melatonin levels induced by the six 15 min pulses of the 6.5 h IBL condition (27.1 ± 6.3%). While the trend for suppression is apparent in the plots seen as a lower melatonin value at the end of the second pulse compared to the start of that pulse (Fig. 3 E), we did not detect a significant change in melatonin levels between the start and end of the light pulse for the 0.2 h continuous condition and the second pulse of the 1.0 h IBL condition, likely because the 36 min resetting interval was not sufficiently long to allow for full recovery of the melatonin levels, which would have allowed for observation of greater suppression.
Discussion
A positive relationship between melatonin suppression and phase delay resetting in response to nocturnal light exposure has been suggested (Boivin et al. 1996; Zeitzer et al. 2000; Lockley et al. 2003; Gooley et al. 2010; Chang et al. 2012), which may lead to the assumption that a light pulse that induces melatonin suppression may also induce circadian phase shifts, and that the responses are proportional such that melatonin suppression can be used as a proxy measure for phase resetting (Brainard et al. 2001; Thapan et al. 2001; Najjar et al. 2014; Rea et al. 2005, 2010). Poor association between melatonin suppression and circadian phase resetting has been previously reported, but a functional decoupling between the two responses was not elucidated (Zeitzer et al. 1997, 2011). Results from the current study show, however, that such close functional coupling does not exist, suggesting that any relationship is associative and inconsistent rather than causative. We present evidence that a similar degree of melatonin suppression occurs each and every time a light pulse is applied during the biological night (Fig. 3 J), which is not matched by a similar phase shifting response to each pulse, based on the overall phase shifting response (Fig. 2 A). The findings suggest that the phase shifting capacity of the SCN is diminished after an initial exposure and that this decreased response lasts for a specific interval during which additional photic stimuli have marginally additional phase resetting effects. Importantly, however, light continues to induce melatonin suppression during that interval. We hypothesize that the SCN appears to be able to relay the timing and ‘strength’ of the intermittent light exposures to the pineal while being insensitive to cumulative phase shifting effects of all pulses of light, most likely due to the topographical division of retinorecipient and circadian rhythm generation areas within the SCN. Notwithstanding the different temporal patterns of these two responses to light, these results further caution against using melatonin suppression as an indirect measure of the circadian resetting strength of a photic stimulus.
If melatonin suppression and phase resetting were functionally coupled, then the 0.2 h continuous, 1.0 h IBL and the 6.5 h IBL conditions could be expected to cause similar phase shifts since they induce similar levels of melatonin suppression. In contrast, the incongruent phase shift and melatonin suppression responses observed in the current study support the lack of a functional coupling between melatonin suppression and phase resetting. The results, therefore, show a divergence from the expected relationship between melatonin suppression and phase resetting, suggesting that melatonin suppression is not a reliable proxy for phase resetting.
The IBL data could be interpreted as indicating that most of the phase resetting effect is induced by the first few light pulses with subsequent light pulses adding less. The data do not support this hypothesis, however. First, the single 12 min (0.2 h) pulse induced the same phase shift of ∼1 h as the 60 min pulse. Second, when two 12 min pulses were administered within a 1 h exposure window, the same phase shift of ∼1 h was observed, suggesting that the second 12 min pulse did not induce additional phase resetting and that only the first pulse actively shifted the clock, despite both pulses inducing similar melatonin suppression.
Since melatonin levels returned to pre‐light exposure levels after 60 min, it could be hypothesized that SCN sensitivity to the phase resetting effects of light recovers within 60 min. If this were the case, the 6.5 h IBL exposure should induce a ∼6 h phase shift from the sum of six 15 min pulses administered with 60 min of recovery interval between pulses, given that one 15 min pulse induces a 1 h shift. The data show that only half of this phase resetting response is achieved following six 15 min pulses and therefore the pulses are not interpreted as six equal exposures by the SCN in terms of phase shifting. The 3 h shifts obtained are equivalent to approximately three individual 12 min shifts, consistent with the hypothesis that every other pulse induces a phase shift (pulses 1, 3 and 5) with the ‘even’ pulses contributing minimally to phase shifting, despite causing an equal amount of melatonin suppression each time. If this is the case, then the time interval during which there is reduced SCN phase resetting capacity starts as early as 15 min after the pulse, and remains low for ∼60 to ∼135 min after the pulse.
An alternate hypothesis is that the first three of the six pulses induced the entire phase resetting response and the latter three pulses were ineffective. In this case the interval of reduced sensitivity would be ∼36–120 min. Data from mechanistic studies in animal models do not support this conclusion, however. Multiple lines of evidence in animal models show that the SCN resolves individual pulses and returns to baseline between pulses. Light exposure‐induced c‐Fos expression in the SCN peaks 30 min after the pulse and is undetectable 60 min after the pulse (Butcher et al. 2003). Phosphorylated cAMP response element‐binding protein (pCREB) induction peaks ∼20 min after a pulse and returns to baseline within 60 min (Butcher et al. 2003). Light pulses delivered 60 and 120 min after the first 15 min pulse induce a second and third round of c‐Fos and pCREB expression in the SCN (Butcher et al. 2003). Importantly, even though the SCN can resolve individual pulses in 60 min intervals, additive phase resetting by multiple pulses may occur in ∼120 min intervals. In mice, two 15 min pulses administered 60 min apart did not induce significantly greater phase shift than the first pulse alone whereas when delivered 120 min apart, the two pulses cause significantly greater phase shift than the shift by one pulse alone (Best et al. 1999), consistent with our data.
The molecular mechanisms of phase resetting explored in animal models, in conjunction with the current results, support longer duration (>15 min) pulses being resolved into discrete pulses. For example, exposure to a 120 min continuous pulse leads to extracellular‐signal‐regulated kinase (ERK) activation in the SCN, which is necessary and influences the magnitude of phase resetting (Butcher et al. 2002, 2003; Dziema et al. 2003) within 15 min of the pulse. ERK activation then decreases to near basal levels (∼30% of the initial induction) by 60 min and remains at basal levels until the end of the light pulse (Butcher et al. 2003). In contrast, the G‐protein Ras, which is upstream of ERK in the mitogen‐activated protein kinase pathway, remains in an activated state throughout the 120 min light pulse. Taken together, these results suggest that the SCN is responsive to a continuous light pulse but that the resetting capacity becomes minimal ∼15 min after the start of the pulse. This hypothesis is further supported by the sustained firing activity of SCN neurons to light pulses longer than 15 min and the discrete sustained response of SCN neuronal firing to a train of intermittent 15 min pulses but a lack of an additive increase in phase resetting in response to multiple pulses (Meijer et al. 1992). This ability of the SCN to continue receiving the photic stimulus likely explains the sustained melatonin suppression observed with the 1.0 h and 6.5 h continuous conditions. In addition, all six pulses within the 6.5 h IBL induced the same level of melatonin suppression.
Additionally, exogenous melatonin inhibits SCN firing (Mason & Brooks, 1988; Zhou et al. 2000), a marker of SCN activation in response to light and monotonically related to the magnitude of phase resetting (Meijer et al. 1992). Therefore, there is a likely impact of melatonin on the differential phase resetting observed under continuous and intermittent light exposure patterns. Robust and sustained melatonin suppression during continuous bright light exposure may have allowed the SCN to fully phase shift in response to light, whereas high melatonin levels (low melatonin suppression) during intermittent bright light exposure may have partially inhibited SCN phase shifting in response to light. This may explain the slightly reduced phase resetting, albeit not significantly, observed in the 6.5 h IBL condition compared to the 6.5 h continuous light exposure patterns, but requires additional studies to be confirmed.
Overall, our data suggest that melatonin suppression and phase resetting are sometimes correlated, but ultimately are generated by distinct processes downstream of retinal signal transduction (Paul et al. 2003). A limitation of the current study was the limited sample size in each light exposure condition, however. Additionally, the blood sampling frequency was different between protocols, which was accounted for post hoc by linearly interpolating melatonin values. Future studies with additional participants in the IBL conditions, and the same sampling frequency for melatonin are necessary to confirm the decoupled phase resetting and melatonin suppression under intermittent light exposure conditions. We hypothesize that the photic signal received at the retinorecipient core of the SCN is transmitted to the pineal, given that the light pulse induces immediate and continuous melatonin suppression for the entire light exposure duration, each and every time a light pulse is given between SynOn and SynOff. In contrast, the same photic stimulus induces an initial phase resetting response within the SCN, although if the following pulse occurs within 36–135 min, the SCN appears to be considerably less sensitive to additional phase resetting. After the interval of reduced phase shifting capacity ends, the system appears to respond fully to a second phase shifting pulse.
The ability to reliably predict and estimate phase resetting capacity of a given photic stimulus from the melatonin suppression induced by the same stimulus appears attractive in applied settings primarily due to less resource requirements for assessing melatonin suppression. Our work shows clearly, however, that melatonin suppression is not a reliable surrogate for phase resetting, often the major chronobiotic and chronotherapeutic outcome of interest. Therefore, melatonin suppression is a reliable marker of the SCN's ability to detect light and transduce that information to the pineal gland but cannot be used as a surrogate measure of the stimulus strength that induces circadian phase resetting in humans.
Additional information
Competing interests
Competing interests are reported for the last two years. A.M.C., N.S. and C.G. have no conflicts of interest to declare.
S.A.R. holds a patent for prevention of circadian rhythm disruption by using optical filters and improving sleep performance in subject exposed to light at night; he owns equity in Melcort Inc.; he is a co‐investigator on studies sponsored by Biological Illuminations, LLC and Vanda Pharmaceuticals Inc. He has provided paid consulting services to Sultan & Knight Ltd t/a Circadia.
M.S.H. has provided paid consulting services to The MathWorks, Inc. (Natick, MA, USA) and the CRC for Alertness, Safety, and Productivity (Melbourne, Australia).
C.A.C. reports having received consulting fees from or served as a paid member of scientific advisory boards for: Bose Corporation; Boston Celtics; Boston Red Sox; Columbia River Bar Pilots; Institute of Digital Media and Child Development; Klarman Family Foundation; Novartis; Samsung Electronics; Quest Diagnostics, Inc.; Vanda Pharmaceuticals and V‐Watch/PPRS. He has also received education/research support from Cephalon Inc., Mary Ann & Stanley Snider via Combined Jewish Philanthropies, Optum, Philips Respironics, Inc., ResMed Foundation, San Francisco Bar Pilots, Schneider Inc., and Sysco. He has received lecture fees from American Academy of Sleep Medicine (AADSM), CurtCo Media Labs LLC, Global Council on Brain Health/AARP, Hawaii Sleep Health and Wellness Foundation, Harvard School of Public Health, Maryland Sleep Society, National Sleep Foundation, University of Michigan, University of Washington, and Zurich Insurance Company, Ltd. The Sleep and Health Education Program of the Harvard Medical School Division of Sleep Medicine (which C.A.C. directs) has received Educational Grant funding from Cephalon, Inc., Jazz Pharmaceuticals, Takeda Pharmaceuticals, Teva Pharmaceuticals Industries Ltd, Sanofi‐Aventis, Inc., Sepracor, Inc. and Wake Up Narcolepsy. He is the incumbent of an endowed professorship provided to Harvard University by Cephalon, Inc. and holds a number of process patents in the field of sleep/circadian rhythms (e.g. photic resetting of the human circadian pacemaker). Since 1985, he has also served as an expert on various legal and technical cases related to sleep and/or circadian rhythms including those involving the following commercial entities: Bombardier, Inc.; Continental Airlines; FedEx; Greyhound; and United Parcel Service (UPS). He owns or owned an equity interest in Somnus Therapeutics, Inc., and Vanda Pharmaceuticals. He received royalties from McGraw Hill, Houghton Mifflin Harcourt and Koninklijke Philips Electronics, N.V. for the Actiwatch‐2 and Actiwatch‐Spectrum devices. His interests were reviewed and managed by Brigham and Women's Hospital and Partners HealthCare in accordance with their conflict of interest policies.
E.B.K. reports non‐university income from consulting for a law firm on a transportation‐related case.
S.W.L. reports receiving consulting fees from Atlanta Falcons, Atlanta Hawks, Carbon Limiting Technologies Ltd on behalf of PhotoStar LED, Perceptive Advisors, PlanLED, Serrado Capital, Slingshot Insights, and has ongoing consulting contracts with Akili Interactive, Consumer Sleep Solutions, Delos Living LLC, Environmental Light Sciences LLC, Focal Point LLC, Headwaters Inc., Hintsa Performance AG, Light Cognitive, Mental Workout, OpTerra Energy Services Inc., Pegasus Capital Advisors LP, Wyle Integrated Science and Engineering. He has received unrestricted equipment gifts from Biological Illuminations LLC; Bionetics Corporation; and F. Lux Software LLC; royalties from Oxford University Press; honoraria plus travel, accommodation or meals for invited seminars, conference presentations or teaching from Estee Lauder, Informa Exhibitions, and Teague; travel, accommodation and/or meals only (no honoraria) for invited seminars, conference presentations or teaching from Hintsa Performance AG, Lightfair and USGBC; ongoing investigator‐initiated research grants from Biological Illumination LLC and F. Lux Software LLC; has completed service agreements with Rio Tinto Iron Ore and Vanda Pharmaceuticals Inc.; and has completed three sponsor‐initiated clinical research contracts with Vanda Pharmaceuticals Inc. He also holds a process patent for the use of short‐wavelength light for resetting the human circadian pacemaker and improving alertness and performance which is assigned to the Brigham and Women's Hospital per Hospital policy. He has also served as a paid expert witness in two cases related to sleep, circadian rhythms, work hours and/or light. He also serves as a Program Leader in the Cooperative Research Centre for Alertness, Safety and Productivity.
Author contributions
This work was completed at the Intensive Physiological Monitoring Unit at the Centre for Clinical Investigation at Brigham and Women's Hospital, Boston, MA, USA. Authors responsible for conception or design of the work: S.A.R., M.S.H., C.G., A.M.C., N.S., C.A.C., E.B.K. and S.W.L.; acquisition, analysis or interpretation of data for the work: S.A.R., M.S.H., C.G., A.M.C., N.S., C.A.C., E.B.K. and S.W.L.; drafting the work or revising it critically for important intellectual content: S.A.R., M.S.H., C.G., A.M.C., N.S., C.A.C., E.B.K. and S.W.L. All authors approved the final version of the manuscript, agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by NASA NAG 5‐3952, NSBRI HFP02801, HFP02802, and NIH P01‐AG09975, R01‐HL114088 (E.B.K.), RC2‐HL101340‐0 (E.B.K., S.W.L., S.A.R.), K02‐HD045459 (E.B.K.), K24‐HL105664 (E.B.K.), T32‐HL07901 (M.S.H., S.A.R.), HL094654 (C.A.C.). The project described was supported by grants from the National Centre for Research Resources to the General Clinical Research Centre GCRC‐M01‐RR02635, NIH Grant Number 1UL1 TR001102‐01, 8UL1TR000170‐05, UL1 RR 025758, Harvard Clinical and Translational Science Centre, from the National Centre for Advancing Translational Science. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Centre for Research Resources, the National Centre for Advancing Translational Science or the National Institutes of Health.
Acknowledgements
We thank the research staff and research participants at the Division of Sleep Medicine, BWH; and the technical, dietary, nursing and medical staff at the Centre for Clinical Investigation at the Brigham and Women's Hospital.
Biography
Shadab Rahman's research interests are in sleep and circadian neurophysiology. His primary research interest is in basic and applied circadian photobiology. He has been studying how the spectral, duration and pattern characteristics of light modulate behavioural, endocrine and molecular outputs that are under the control of the biological clock. His secondary interest is in peripheral rhythms including metabolic, immune and reproductive rhythms. He is exploring the endogenous circadian regulation of these rhythms and the effects of light exposure on these rhythms. His goal is to combine the findings from these two research streams to develop effective photobiological countermeasures for circadian disruption.

Edited by: Ole Paulsen & Kim Barrett
Linked articles This article is highlighted by a Perspective by Zeitzer. To read this Perspective, visit https://doi.org/10.1113/JP276076.
This is an Editor's Choice article from the 1 June 2018 issue.
References
- Best JD, Maywood ES, Smith KL & Hastings MH (1999). Rapid resetting of the mammalian circadian clock. J Neurosci 19, 828–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin DB, Duffy JF, Kronauer RE & Czeisler CA (1996). Dose‐response relationships for resetting of human circadian clock by light. Nature 379, 540–542. [DOI] [PubMed] [Google Scholar]
- Brainard GC, Hanifin JP, Greeson JM, Byrne B, Glickman G, Gerner E & Rollag MD (2001). Action spectrum for melatonin regulation in humans: Evidence for a novel circadian photoreceptor. J Neurosci 21, 6405–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher GQ, Doner J, Dziema H, Collamore M, Burgoon PW & Obrietan K (2002). The p42/44 mitogen‐activated protein kinase pathway couples photic input to circadian clock entrainment. J Biol Chem 277, 29519–29525. [DOI] [PubMed] [Google Scholar]
- Butcher GQ, Lee B & Obrietan K (2003). Temporal regulation of light‐induced extracellular signal‐regulated kinase activation in the suprachiasmatic nucleus. J Neurophysiol 90, 3854–3863. [DOI] [PubMed] [Google Scholar]
- Chang AM, Santhi N, St Hilaire M, Gronfier C, Bradstreet DS, Duffy JF, Lockley SW, Kronauer RE & Czeisler CA (2012). Human responses to bright light of different durations. J Physiol 590, 3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeisler CA, Kronauer RE, Allan JS, Duffy JF, Jewett ME, Brown EN & Ronda JM (1989). Bright light induction of strong (type 0) resetting of the human circadian pacemaker. Science 244, 1328–1333. [DOI] [PubMed] [Google Scholar]
- Duffy JF & Dijk DJ (2002). Getting through to circadian oscillators: Why use constant routines? J Biol Rhythms 17, 4–13. [DOI] [PubMed] [Google Scholar]
- Dziema H, Oatis B, Butcher GQ, Yates R, Hoyt KR & Obrietan K (2003). The ERK/MAP kinase pathway couples light to immediate‐early gene expression in the suprachiasmatic nucleus. Eur J Neurosci 17, 1617–1627. [DOI] [PubMed] [Google Scholar]
- Gooley JJ, Lu J, Chou TC, Scammell TE & Saper CB (2001). Melanopsin in cells of origin of the retinohypothalamic tract. Nat Neurosci 4, 1165. [DOI] [PubMed] [Google Scholar]
- Gooley JJ, Rajaratnam SMW, Brainard GC, Kronauer RE, Czeisler CA & Lockley SW (2010). Spectral responses of the human circadian system depend on the irradiance and duration of exposure to light. Sci Transl Med 2, 31ra33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronfier C, Wright KP Jr, Kronauer RE, Jewett ME & Czeisler CA (2004). Efficacy of a single sequence of intermittent bright light pulses for delaying circadian phase in humans. Am J Physiol Endocrinol Metab 287, E174–E181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett ME, Rimmer DW, Duffy JF, Klerman EB, Kronauer RE & Czeisler CA (1997). Human circadian pacemaker is sensitive to light throughout subjective day without evidence of transients. Am J Physiol Regul Integr Comp Physiol 273, R1800–R1809. [DOI] [PubMed] [Google Scholar]
- Johnson RF, Moore RY & Morin LP (1998). Loss of entrainment and anatomical plasticity after lesions of the hamster retinohypothalamic tract. Brain Res 460, 293–313. [DOI] [PubMed] [Google Scholar]
- Khalsa SBS, Jewett ME, Cajochen C & Czeisler CA (2003). A phase response curve to single bright light pulses in human subjects. J Physiol 549, 945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DC & Moore RY (1979). Pineal N‐acetyltransferase and hydroxyindole‐O‐methyltransferase: Control by the retinohypothalamic tract and the suprachiasmatic nucleus. Brain Res 174, 245–262. [DOI] [PubMed] [Google Scholar]
- Klein DC, Weller JL & Moore RY (1971). Melatonin metabolism: neural regulation of pineal serotonin: acetyl coenzyme A N‐acetyltransferase activity. Proc Natl Acad Sci USA 68, 3107–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewy AJ, Wehr TA, Goodwin FK, Newsome DA & Markey SP (1980). Light suppresses melatonin secretion in humans. Science 210, 1267–1269. [DOI] [PubMed] [Google Scholar]
- Lockley SW, Brainard GC & Czeisler CA (2003). High sensitivity of the human circadian melatonin rhythm to resetting by short wavelength light. J Clin Endocrinol Metab 88, 4502–4505. [DOI] [PubMed] [Google Scholar]
- Lucas RJ, Peirson SN, Berson DM, Brown TM, Cooper HM, Czeisler CA, Figueiro MG, Gamlin PD, Lockley SW, O'Hagan JB, Price LL, Provencio I, Skene DJ & Brainard GC (2014). Measuring and using light in the melanopsin age. Trends Neurosci 37, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason R & Brooks A (1988). The electrophysiological effects of melatonin and a putative melatonin antagonist (N‐acetyltryptamine) on rat suprachiasmatic neurones in vitro. Neurosci Lett 95, 296–301. [DOI] [PubMed] [Google Scholar]
- Meijer JH, Rusak B & Ganshirt G (1992). The relation between light‐induced discharge in the suprachiasmatic nucleus and phase shifts of hamster circadian rhythms. Brain Res 598, 257–263. [DOI] [PubMed] [Google Scholar]
- Meijer JH, Watanabe K, Schaap J, Albus H & Detari L (1998). Light responsiveness of the suprachiasmatic nucleus: long‐term multiunit and single‐unit recordings in freely moving rats. J Neurosci 18, 9078–9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JN, Minors DS & Waterhouse JM (1978). Adaptation to abrupt time shifts of the oscillator[s] controlling human circadian rhythms. J Physiol 285, 455–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RY (1978). The innervation of the mammalian pineal gland. Biology 4, 1–29. [Google Scholar]
- Najjar RP, Chiquet C, Teikari P, Cornut PL, Claustrat B, Denis P, Cooper HM & Gronfier C (2014). Aging of non‐visual spectral sensitivity to light in humans: compensatory mechanisms? PLoS One 23, e85837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul KN, Fukuhara C, Tosini G & Albers HE (2003). Transduction of light in the suprachiasmatic nucleus: evidence for two different neurochemical cascades regulating the levels of Per1 mRNA and pineal melatonin. Neuroscience 119, 137–144. [DOI] [PubMed] [Google Scholar]
- Rea MS, Figueiro MG, Bierman A & Bullough JD (2010). Circadian light. J Circadian Rhythms 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea MS, Figueiro MG, Bullough JD & Bierman A (2005). A model of phototransduction by the human circadian system. Brain Res Brain Res Rev 50, 213–228. [DOI] [PubMed] [Google Scholar]
- St Hilaire MA, Gooley JJ, Khalsa SB, Kronauer RE, Czeisler CA & Lockley SW (2012). Human phase response curve to a 1 h pulse of bright white light. J Physiol 590, 3035–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Hilaire MA, Gronfier C, Zeitzer JM & Klerman EB (2007). A physiologically‐based mathematical model of melatonin including ocular light suppression and interactions with the circadian pacemaker. J Pineal Res 43, 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapan K, Arendt J & Skene DJ (2001). An action spectrum for melatonin suppression: Evidence for a novel non‐rod, non‐cone photoreceptor system in humans. J Physiol 535, 261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan GM (1993). New sensitive serum melatonin radioimmunoassay employing the Kennaway G280 antibody: Syrian hamster morning adrenergic response. J Pineal Res 15, 88–103. [DOI] [PubMed] [Google Scholar]
- Wright HR & Lack LC (2001). Effect of light wavelength on suppression and phase delay of the melatonin rhythm. Chronobiol Int 18, 801–808. [DOI] [PubMed] [Google Scholar]
- Zeitzer JM, Kronauer RE & Czeisler CA (1997). Photopic transduction implicated in human circadian entrainment. Neurosci Lett 5, 135–138. [DOI] [PubMed] [Google Scholar]
- Zeitzer JM, Dijk DJ, Kronauer RE, Brown EN & Czeisler CA (2000). Sensitivity of the human circadian pacemaker to nocturnal light: Melatonin phase resetting and suppression. J Physiol 526, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitzer JM, Ruby NF, Fisicaro RA & Heller HC (2011). Response of the human circadian system to millisecond flashes of light. PLoS One 6, e22078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XJ, Jiang XH, Yu GD & Yin QZ (2000). Modulation of circadian rhythm of discharges of suprachiasmatic nucleus neurons in rat hypothalamic slices by melatonin. Sheng Li Xue Bao 52, 215–219. [PubMed] [Google Scholar]