

Abstract
Hepatocellular carcinoma (HCC) is the fifth most common primary cancer and second largest cause of cancer‐related death worldwide. The first‐line oral chemotherapeutic agent sorafenib only increases survival in patients with advanced HCC by less than 3 months. Most patients with advanced HCC have shown limited response rates and survival benefits with sorafenib. Although sorafenib is an inhibitor of multiple kinases, including serine/threonine‐protein kinase c‐Raf, serine/threonine‐protein kinase B‐Raf, vascular endothelial growth factor receptor (VEGFR)‐1, VEGFR‐2, VEGFR‐3, and platelet‐derived growth factor receptor β, HCC cells are able to escape from sorafenib treatment using other pathways that the drug insufficiently inhibits. The aim of this study was to identify and target survival and proliferation pathways that enable HCC to escape the antitumor activity of sorafenib. We found that insulin‐like growth factor 1 receptor (IGF1R) remains activated in HCC cells treated with sorafenib. Knockdown of IGF1R sensitizes HCC cells to sorafenib treatment and decreases protein kinase B (AKT) activation. Overexpression of constitutively activated AKT reverses the effect of knockdown of IGF1R in sensitizing HCC cells to treatment with sorafenib. Further, we found that ceritinib, a drug approved by the U.S. Food and Drug Administration for treatment of non‐small cell lung cancer, effectively inhibits the IGF1R/AKT pathway and enhances the inhibitory efficacy of sorafenib in human HCC cell growth and survival in vitro, in a xenograft mouse model and in the c‐Met/β‐catenin‐driven HCC mouse model. Conclusion: Our study provides a biochemical basis for evaluation of a new combination treatment that includes IGF1R inhibitors, such as ceritinib and sorafenib, in patients with HCC. (Hepatology Communications 2018;2:732‐746)
Abbreviations
- AKT
protein kinase B
- ALK
anaplastic lymphoma kinase
- CAT
Constitutively active β‐catenin
- c‐Raf
serine/threonine‐protein kinase Raf‐1
- CMV
cytomegalovirus
- ERK
extracellular signal‐regulated kinase
- FDA
U.S. Food and Drug Administration
- GFP
green fluorescent protein
- HCC
hepatocellular carcinoma
- HSB2
sleeping beauty transposase 2
- IGF1R
insulin‐like growth factor 1 receptor
- MET
c‐met
- p‐
phosphorylated
- PI3K
phosphatidylinositol 3‐kinase
- SCID
severe combined immunodeficiency mice
- shRNA
short hairpin RNA
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling
- VEGF
vascular endothelial growth factor
Hepatocellular carcinoma (HCC) is the primary malignancy of the liver and is now the second leading cause of cancer deaths worldwide.1 Low rates of early diagnosis coupled with high mortality rates make it critical to develop new treatment approaches to HCC.2 The multikinase inhibitor sorafenib is the first‐line treatment for advanced HCC, but the clinical impact of sorafenib is modest (2%‐3.3% objective partial response rate, 54%‐71% disease stabilization rate, and a nearly 3‐month survival advantage over placebo).3 Sorafenib suppresses tumor proliferation and angiogenesis by inhibiting multiple serine/threonine and receptor tyrosine kinases, including serine/threonine‐protein kinase Raf‐1 (or c‐Raf), wild‐type and mutant B‐Raf, vascular endothelial growth factor receptor (VEGFR)‐1, VEGFR‐2, VEGFR‐3, platelet‐derived growth factor receptor β, tyrosine‐protein kinase Kit (c‐KIT), FMS‐like tyrosine kinase 3 (FLT‐3), and proto‐oncogene tyrosine‐protein kinase receptor Ret (RET).4 However, other signaling pathways that sorafenib fails to inhibit can contribute to cell growth and survival in sorafenib‐acquired resistant cells, such as the phosphatidylinositol 3‐kinase (PI3K)/protein kinase B (AKT) signaling pathway.5 Therefore, combination drug treatment to inhibit the remaining active cell survival and growth pathways appears to be a promising approach to improve sorafenib efficacy.6, 7
Insulin‐like growth factor 1 receptor (IGF1R) is a receptor for IGF. IGF1R is activated through ligand‐induced phosphorylation and subsequently phosphorylates and activates both the PI3K/AKT and Ras/mitogen‐activated protein kinase pathways.8 Activation of IGF1R is crucial for malignant transformation and the survival of malignant cells.8, 9, 10 For example, aberrant expression and activation of IGF1R contributes to increased survival of pancreatic cancer cells,11 and knockdown of IGF1R led to inhibition of proliferation, migration, and invasiveness of prostate cancer cells.12 Overexpression of IGF1R was detected in 33% of human HCCs, and increased activation of IGF1R was observed in 52% of HCC tumors.13 Abrogation of IGF1R activation significantly but modestly decreases HCC cell viability and proliferation.14 Although several IGF1R inhibitors have been tested in clinical trials,9, 15, 16 none have been approved by the U.S. Food and Drug Administration (FDA). Intriguingly, ceritinib (Zykadia), a potent anaplastic lymphoma kinase (ALK) inhibitor that is FDA approved for treatment of non‐small cell lung cancer,17 has been reported to effectively inhibit IGF1R.18
In this study, we found that IGF1R remains activated in HCC cells after treatment with sorafenib. Moreover, knockdown of IGF1R sensitizes HCC cells to sorafenib by decreasing AKT activation. Overexpression of constitutively activated AKT reverses the effect of IGF1R knockdown in sensitizing HCC cells to sorafenib treatment. Furthermore, we found that ceritinib decreases phosphorylation of IGF1R and AKT and enhances the efficacy of inhibition by sorafenib in human HCC cell growth and survival in in vitro and in vivo models. Our study provides evidence that the combination of ceritinib and sorafenib has therapeutic potential for HCC and elucidates its possible mechanisms.
Materials and Methods
CELLS AND REAGENTS
Huh7 cells were purchased from the http://cellbank.nibiohn.go.jp/english/. Hep3B, HepG2, and 293T cells were purchased from the https://www.atcc.org/en/Products/Cells_and_Microorganisms/Human_Primary_Cells.aspx?gclid=Cj0KCQjw-uzVBRDkARIsALkZAdlA_a7Yjmfa99hen3E_Pfx7lsADENmu98W3cHqg8gQmC51kQqRloxwaAkTtEALw_wcB. All cells were cultured with Dulbecco's modified Eagle's medium (high glucose; Thermo Scientific, Waltham, MA), supplemented with 10% fetal bovine serum (Tissue Culture Biologicals) and penicillin and streptomycin (Sigma‐Aldrich, St. Louis, MO) in a humidified atmosphere of 5% CO2 at 37°C. Cells were plated in 12 or 6‐well plates at 30%‐40% density for 24 hours prior to treatment. Ceritinib was purchased from LC Laboratories (Cat#2086; Woburn, MA), and sorafenib was purchased from MedKoo Bioscience (Cat#100770a; Morrisville, NC).
MICE
All animals received humane care according to the Guide for the Care and Use of Laboratory Animals (http://oacu.od.nih.gov/ac_cbt/guide3.htm). The procedures for all animal experiments were approved by the Institutional Animal Care and Use Committee of Loyola University Chicago. The mice were housed in micro‐isolator cages in a room illuminated from 7:00 am to 7:00 pm (12‐hour:12‐hour light–dark cycle) and allowed access to water and chow ad libitum. C57BL/6 mice were obtained from the Jackson Laboratory, and severe combined immunodeficiency mice (SCID‐bg) were purchased from Charles River.
CELL PROLIFERATION/VIABILITY ANALYSIS BY ALAMARBLUE ASSAY
Hep3B, Huh7, and HepG2 cells were seeded into 96‐well plates (5 × 103 cells/well). After 24 hours, cells were treated with ceritinib (0.5‐1.5 μM), sorafenib (1.25‐5.0 μM), or both. At different time points, culture media was removed and alamarBlue (BUF012A; Bio‐Rad, Hercules, CA) solution (1:10 dilution in phosphate‐buffered saline) was added to the cells. After a 2‐hour incubation at 37°C, fluorescence values were measured with a fluorescent plate reader at 530‐560 nm excitation/590 nm emission.
LENTIVIRAL PARTICLE PREPARATION
For IGF1R knockdown experiments, Hep3B cells were infected with lentiviral plasmid (p)LKO.1 particles that contained IGF1R or scrambled short hairpin (sh) RNA and selected with 2 μg/mL puromycin for 5 days. Lentiviral pLKO.1 plasmids for shIGF1R (Table 1) or scrambled shRNA (SHC002; Sigma‐Aldrich) were packaged with the cytomegalovirus plasmid (pCMV)‐dr8.2 (Addgene) and pCMV‐VSVG (Addgene) in 293T cells to produce lentiviral particles as described.19, 20
Table 1.
shIGF1R Sequences
| Target | Sequences | RNA Interference Consortium Number | Company |
|---|---|---|---|
| Human shIGF1R‐1 | CCGGCGGCAACCTGAGTTACTACATCTCGAGATGTAGTAACTCAGGTTGCCGTTTTTG | TRCN0000121301 | Sigma‐Aldrich |
| Human shIGF1R‐2 | CCGGGCCGAAGATTTCACAGTCAAACTCGAGTTTGACTGTGAAATCTTCGGCTTTTTG | TRCN0000039675 | Sigma‐Aldrich |
For overexpression of constitutively active AKT experiments, Hep3B cells were infected with lentiviral particles packaged with FG12‐cmv‐green fluorescent protein (gfp)‐akt (constitutively active AKT) or FG12‐cmv‐gfp, pCMVΔ8.71, and pMD.G in 293T cells. Six days after infection, GFP‐positive cells expressing constitutively active AKT were sorted by flow cytometry (FACSAria Cell Sorter).
COLONY FORMATION ASSAY
Hep3B cells were seeded into 12‐well plates (6 × 103 cells/well) for 24 hours before treatment as described.21 After 48 hours of treatment with dimethyl sulfoxide, 1.25 μM sorafenib, 0.5 μM ceritinib, or a combination of both drugs, media was changed and the cells were cultured for 14 days. Cells were fixed in a mixture of 6.0% glutaraldehyde and stained with 0.5% crystal violet. Colonies were scored in at least four fields (magnification ×40) and reported as means ± SD.
XENOGRAFT MODEL
SCID‐bg mice were shaved following anesthesia using isoflurane, and 5 × 106 Hep3B cells in 100 μL of serum‐free media was injected into the left or right flanks of the mice. Two weeks postinjection, mice were treated with vehicle (30% captisol), ceritinib (25 mg/kg), sorafenib (25 mg/kg), or a combination of sorafenib (25 mg/kg) and ceritinib (25 mg/kg) by oral gavage daily. Tumor volumes were measured daily using a caliper until the day of sacrifice. Tumor volume was calculated using the ellipsoidal formula as follows: tumor volume (mm3) = 1/2(L × W2), where L is the greatest longitudinal distance of the tumor and W is the greatest transverse distance of the tumor. Tumors were harvested prior to volume growth beyond the humane threshold of 1,500 mm3.
MET/CAT‐DRIVEN HCC MODEL
For the c‐met (MET)/constitutively active β‐catenin (CAT)‐driven HCC model,19, 22, 23, 24 55 μg of total plasmids, encoding the sleeping beauty transposase (HSB2) and transposons with oncogenes MET/CAT and gaussia luciferase (Gluc) (22.5 μg pT3‐EF1a‐c‐MET [human], 22.5 μg pT3‐EF1a‐DN90‐β‐catenin [human], 5 μg pT3‐Gluc1, and 5 μg HSB2) were injected hydrodynamically into age‐ and sex‐matched mice. Six weeks after MET/CAT injection, mice were treated with vehicle (30% captisol), ceritinib (25 mg/kg), sorafenib (25 mg/kg), or a combination of ceritinib and sorafenib by oral gavage daily for 4 weeks prior to being sacrificed. All mice were maintained on the standard diet. Liver and body weights of each mouse were measured and recorded.
WESTERN BLOTTING
Western blotting was performed as described.19, 25 Primary antibodies, including those for IGF1R, phosphorylated IGF1R, caspase‐3, active caspase‐3, poly(adenosine diphosphate ribose) polymerase, phosphorylated (p‐)AKT (ser473), AKT, p‐extracellular signal‐regulated kinase (ERK), and ERK, were purchased from Cell Signaling Technology (Danvers, MA). Glyceraldehyde 3‐phosphate dehydrogenase and β‐actin antibodies were purchased from Sigma. More detailed information can be found in Table 2.
Table 2.
Antibodies Used in This Study
| Antibody | Catalog Number | Company |
|---|---|---|
| Phospho‐IGF1R (Tyr1131) | 3021 | Cell Signaling |
| IGF1R | 3027 | Cell Signaling |
| Phospho‐AKT (Ser473) | 4060 | Cell signaling |
| AKT | 9272 | Cell Signaling |
| Phospho‐ERK(Thr 202/Tyr 204) | 4370 | Cell Signaling |
| ERK | 4695 | Cell Signaling |
| Caspase 3 | 9662 | Cell Signaling |
| Cleavage caspase 3 | 9661 | Cell Signaling |
| PARP | 9532 | Cell Signaling |
| GAPDH | G8795 | Sigma‐Aldrich |
| β‐actin | A5441 | Sigma‐Aldrich |
| Ki67 | RM‐9106‐S0 | Fisher Scientific |
Abbreviations: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; PARP, poly(adenosine diphosphate ribose) polymerase; Phospho, phosphorylated.
Ki67 IMMUNOHISTOCHEMICAL STAINING
Immunohistochemistry was performed as described.25 Detailed antibody information can be found in Table 2. Cells with positive staining were scored in at least five fields (magnification ×400) and reported as means ± SD. Three mice were used in each group.
TERMINAL DEOXYNUCLEOTIDYL TRANSFERASE–MEDIATED DEOXYURIDINE TRIPHOSPHATE NICK‐END LABELING
Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) staining was performed as described26 using a kit purchased from Millipore (Cat#S7101). The TUNEL‐positive cell numbers were scored in at least five fields (magnification ×400) per mouse and are reported as means ± SD. Three mice were used in each group.
STATISTICAL ANALYSIS
Statistical analysis was carried out using GraphPad Prism V software. Data are presented as means ± SD (shown in the figures where applicable). Statistical significance was calculated with the Student t test, and P < 0.05 was considered significant.
Results
KNOCKDOWN OF IGF1R ENHANCES THE INHIBITION EFFICACY OF SORAFENIB ON HCC CELL GROWTH BY INHIBITING AKT
Although sorafenib inhibits multiple kinase activities to suppress tumor angiogenesis and proliferation, other signaling pathways, such as PI3K/AKT, which sorafenib does not inhibit at low or even higher doses, contribute to cell growth and survival in sorafenib‐acquired resistant cells.5 The IGF1R signal pathway has been reported to be enriched in sorafenib‐acquired resistant tumor cells.27 Therefore, we examined whether IGF1R remains active after sorafenib treatment in HCC cells. We found that sorafenib failed to inhibit IGF1R phosphorylation at Tyr1131, which is required for activation of IGF1R's kinase,28 in three HCC cell lines (Fig. 1A). Phosphorylation of AKT was also not decreased by sorafenib (Fig. 1A; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1181/full). However, phosphorylation of ERK, a downstream target of RAF/mitogen‐activated protein kinase/ERK kinase (MEK),29 was decreased after sorafenib treatment, indicating sorafenib had activity against ERK in all three HCC cell lines (Fig. 1A). To determine whether IGF1R activity remains contributory to HCC cell growth and survival after sorafenib treatment, we used lentiviral shRNAs to knock down IGF1R expression in Hep3B cells. We found that knockdown of IGF1R enhanced the inhibitory efficacy of sorafenib on Hep3B cell growth (Fig. 1B,C). Further, knockdown of IGF1R decreased the phosphorylation of AKT but not ERK in Hep3B cells (Fig. 1B). To determine whether knockdown of IGF1R enhances the inhibitory efficacy of sorafenib on HCC cell growth by inhibiting AKT activity, we overexpressed constitutively active AKT in IGF1R knockdown cells (Fig. 1D). We found that overexpression of constitutively active AKT abrogated the ability of IGF1R knockdown to sensitize HCC cells to sorafenib (Fig. 1E). In general, these data indicate that knockdown of IGF1R sensitizes HCC cells to sorafenib treatment by inhibiting AKT activity.
Figure 1.
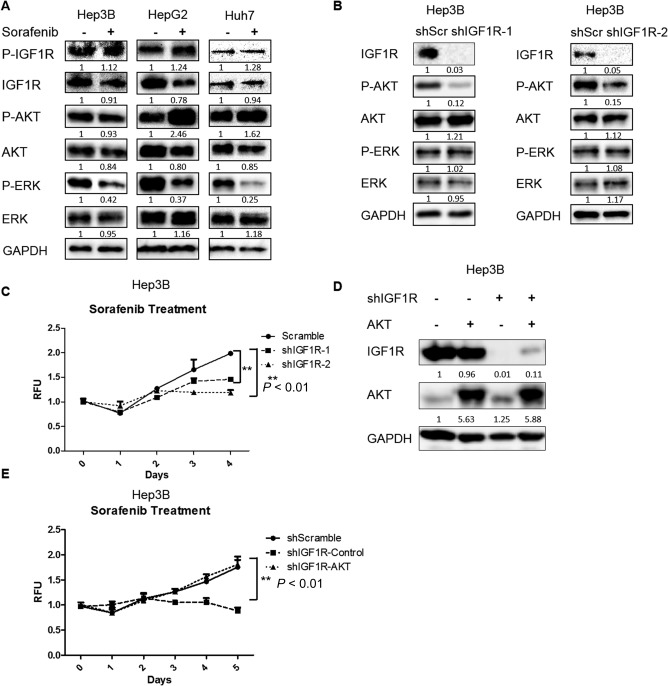
Knockdown of IGF1R enhanced the inhibitory efficacy of sorafenib in HCC cells by inhibiting AKT. (A) HCC cells were treated with sorafenib (1.25 μM for Hep3B, 2.5 μM for HepG2, and 5 μM for Huh7) for 24 hours. Expressions of p‐IGF1R, IGF1R, p‐AKT (ser473), AKT, p‐ERK, ERK, and GAPDH proteins were examined by western blotting. (B) Expressions of IGF1R, p‐AKT (ser473), AKT, p‐ERK, ERK, and GAPDH proteins were examined by western blotting in Hep3B cells infected with IGF1R shRNAs and scrambled shRNA lentiviral particles. (C) Cell proliferation was analyzed by the alamarBlue assay in Hep3B cells infected with IGF1R shRNAs and scrambled shRNA lentiviral particles and then treated with 1.25 μM sorafenib. (D) Expressions of IGF1R, AKT, and GAPDH proteins were examined by western blotting in Hep3B cells infected with scrambled shRNA, IGF1R shRNA, constitutively active AKT, or both lentiviral particles. (E) Cell proliferation was analyzed by the alamarBlue assay in IGF1R knockdown Hep3B cells infected with control or constitutively active AKT lentiviral particles and then treated with 1.25 μM sorafenib. Each experiment was repeated at least 3 times. Abbreviations: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; RFU, relative fluorescence unit; shScr, short hairpin scrambled. Values in C and E were mean ± SD (n = 3 in each group).
CERITINIB ENHANCES THE EFFICACY OF SORAFENIB IN INHIBITING HCC CELL GROWTH AND SURVIVAL
To determine whether the use of a combination treatment using IGF1R inhibitors and sorafenib is a viable clinical translational strategy to treat HCC, we examined whether ceritinib, an FDA‐approved dual ALK and IGF1R inhibitor for the treatment of non‐small cell lung cancer,17, 18 further sensitizes HCC cells to sorafenib treatment. To do so, we first examined the effect of ceritinib on HCC cell growth, which has not been reported. We found that ceritinib effectively inhibited IGF1R activity, which was marked by the phosphorylation of IGF1R and AKT, in HCC cells. Interestingly, ERK phosphorylation was not affected by ceritinib (Fig. 2A). As ceritinib is also an ALK inhibitor, we examined whether ceritinib inhibits ALK activity in a number of HCC cell lines, including Hep3B, Huh7, and HepG2. We found that ALK expression was too low to be detected in these cell lines (Fig. 2A), suggesting that ALK does not play an important role in the growth of these HCC cells. We also found that ceritinib inhibited the growth of HCC cells (Fig. 2B), which phenocopies the effect of knockdown of IGF1R in HCC cells (Fig. 2C). In addition, overexpression of constitutively active AKT abrogated the inhibitory effect on HCC cell growth by either ceritinib or knockdown of IGF1R (Fig. 2D,E). These results suggest that ceritinib suppresses HCC cell growth by inhibiting the IGF1R/AKT pathway.
Figure 2.
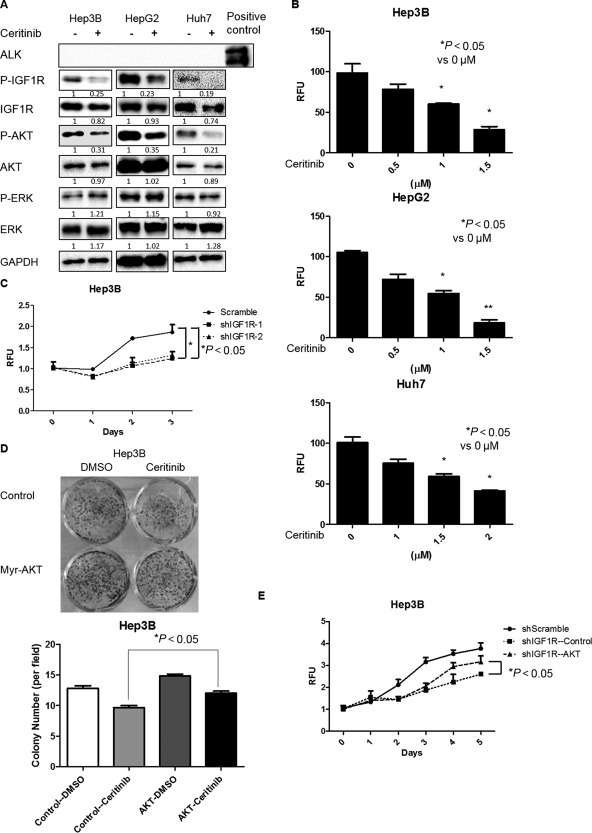
Ceritinib suppressed HCC cell growth by inhibiting the IGF1R/AKT pathway. (A) HCC cells were treated with ceritinib (0.5 μM for Hep3B, 1 μM for HepG2, and 2 μM for Huh7) for 24 hours. Expressions of p‐IGF1R, IGF1R, p‐AKT (ser473), AKT, p‐ERK, ERK, and GAPDH proteins were examined by western blotting. (B) HCC cells were treated with ceritinib at different doses for 48 hours. Cell proliferation was analyzed by the alamarBlue assay. (C) Cell proliferation was analyzed by the alamarBlue assay in Hep3B cells infected with IGF1R shRNAs and scrambled shRNA lentiviral particles. (D) Hep3B cells infected with control or constitutively active AKT lentiviral particles were treated with 0.5 μM ceritinib for 48 hours. Cells were then cultured for 14 days and stained with 0.5% crystal violet. (E) Cell proliferation was analyzed by the alamarBlue assay in IGF1R knockdown Hep3B cells infected with control or constitutively active AKT lentiviral particles. Each experiment was repeated at least 3 times. Abbreviations: DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; RFU, relative fluorescence unit. Values in B, C, D, and E were mean ± SD (n = 3 in each group).
We next examined whether ceritinib enhances the inhibitory efficacy of sorafenib on HCC cell growth and survival. Indeed, ceritinib enhanced the efficacy of sorafenib in inhibiting HCC cell growth in a dose‐ and time‐dependent manner (Fig. 3A,B). Further, we found that ceritinib increased sorafenib‐induced cellular apoptosis, marked by cleavage of caspase‐3 and poly(adenosine diphosphate ribose) polymerase (Fig. 3C). Collectively, our data indicate that ceritinib enhances the inhibitory efficacy of sorafenib on HCC cell growth and survival.
Figure 3.
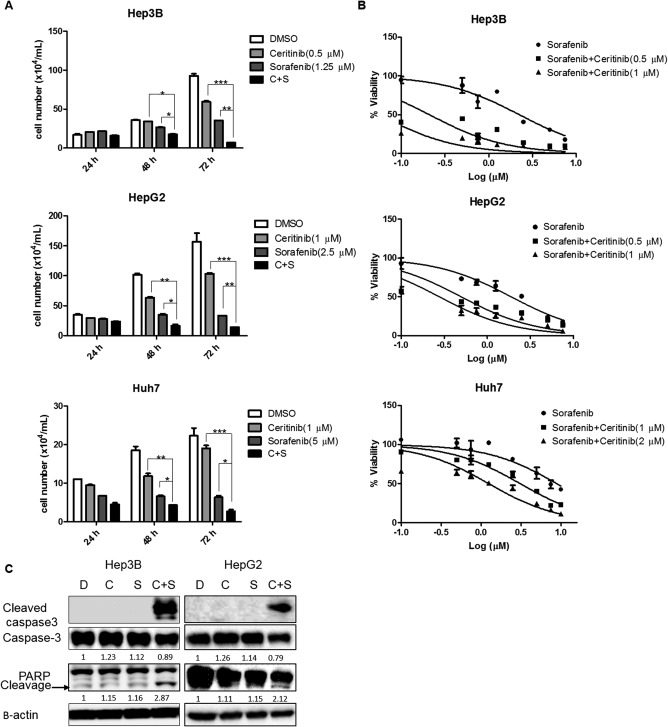
Ceritinib enhanced the efficacy of sorafenib in inhibiting HCC cell growth and survival in vitro. (A) Hep3B, HepG2, or Huh7 cell numbers were counted following treatment with sorafenib or a combination of sorafenib and ceritinib for varying lengths of time. *P < 0.05; **P < 0.01; ***P < 0.001. (B) Viability of Hep3B, HepG2, and Huh7 cells was analyzed by the alamarBlue assay 48 hours following treatment with sorafenib or a combination of sorafenib and ceritinib. (C) Expressions of cleaved caspase‐3, caspase‐3, PARP, and β‐actin proteins were examined by western blotting in Hep3B and HepG2 cells treated with vehicle, sorafenib, ceritinib, or a combination of both. Each experiment was repeated at least 3 times. Abbreviations: C, ceritinib; D, dimethyl sulfoxide; DMSO, dimethyl sulfoxide; PARP, poly(adenosine diphosphate ribose) polymerase; S, sorafenib. Values in A and B were mean ± SD (n = 3 in each group).
CERITINIB ENHANCES THE EFFICACY OF SORAFENIB IN INHIBITING HCC CELL GROWTH AND SURVIVAL BY INHIBITING THE IGF1R/AKT PATHWAY
To determine whether ceritinib enhances the efficacy of sorafenib in inhibiting HCC cell growth and survival by inhibiting the IGF1R/AKT pathway, we first examined the levels of phosphorylated IGF1R and AKT by western blotting. We found decreased phosphorylation of IGF1R in HCC cells treated with a combination of ceritinib and sorafenib compared to such cells treated with sorafenib alone (Fig. 4A). The level of phosphorylated AKT was also decreased in HCC cells treated with a combination of ceritinib and sorafenib compared to such cells treated with sorafenib alone (Fig. 4A), even when phosphorylation of AKT was increased by sorafenib treatment in HepG2 and Huh7 cells (Fig. 4A); this is consistent with results reported by other groups.5 Notably, the level of phosphorylated ERK was not further decreased by ceritinib in HCC cells treated with a combination of ceritinib and sorafenib compared to such cells treated with sorafenib alone (Fig. 4A). Importantly, overexpression of constitutively active AKT rescued the ability of ceritinib to sensitize HCC cells to sorafenib treatment (Fig. 4B,C), suggesting that ceritinib enhances the efficacy of sorafenib in inhibiting HCC cell growth and survival by inhibiting the IGF1R/AKT pathway.
Figure 4.
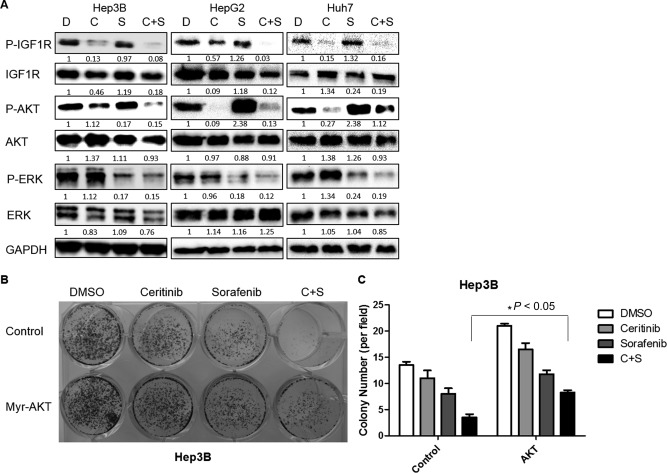
The combination of ceritinib and sorafenib inhibited HCC cell growth by inhibiting the IGF1R/AKT pathway. (A) Expressions of p‐IGF1R, IGF1R, p‐AKT, AKT, and GAPDH proteins in Hep3B, HepG2, and Huh7 cells following treatment with DMSO, sorafenib, ceritinib, or a combination of both drugs for 24 hours were examined by western blotting. (B) Hep3B cells infected with control or constitutively active AKT lentiviral particles were treated with DMSO, ceritinib, sorafenib, or a combination of both drugs for 48 hours. Cells were then cultured for 14 days and stained with 0.5% crystal violet. (C) Colonies from (B) were quantified. Each experiment was repeated at least 3 times. Abbreviations: C, ceritinib; D, dimethyl sulfoxide; DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; S, sorafenib. Values in C were mean ± SD (n = 3 in each group).
CERITINIB ENHANCES THE EFFICACY OF SORAFENIB IN INHIBITING HCC TUMOR GROWTH IN VIVO
To further investigate the efficacy of ceritinib in sensitizing HCC cells to sorafenib treatment in vivo, we first examined the effect of the combination of ceritinib and sorafenib in a xenograft model. We found that treatment with either ceritinib or sorafenib alone was able to inhibit tumor growth while combination treatment using ceritinib and sorafenib had the best overall effectiveness (Fig. 5A‐C). Importantly, mouse weight was not significantly affected by the combination treatment, suggesting minimal toxicity for the combination treatment regimen (Fig. 5D). We further confirmed that ceritinib treatment inhibited IGF1R and AKT activities in xenografted tumors (Fig. 5E). Additionally, we found that combination treatment with ceritinib and sorafenib further decreased tumor cell proliferation (Fig. 5F) and increased tumor cell apoptosis (Fig. 5G; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1181/full) compared to sorafenib or ceritinib treatment alone. Overall, our xenograft model results demonstrate that ceritinib enhances the efficacy of sorafenib in inhibiting human HCC tumor growth.
Figure 5.
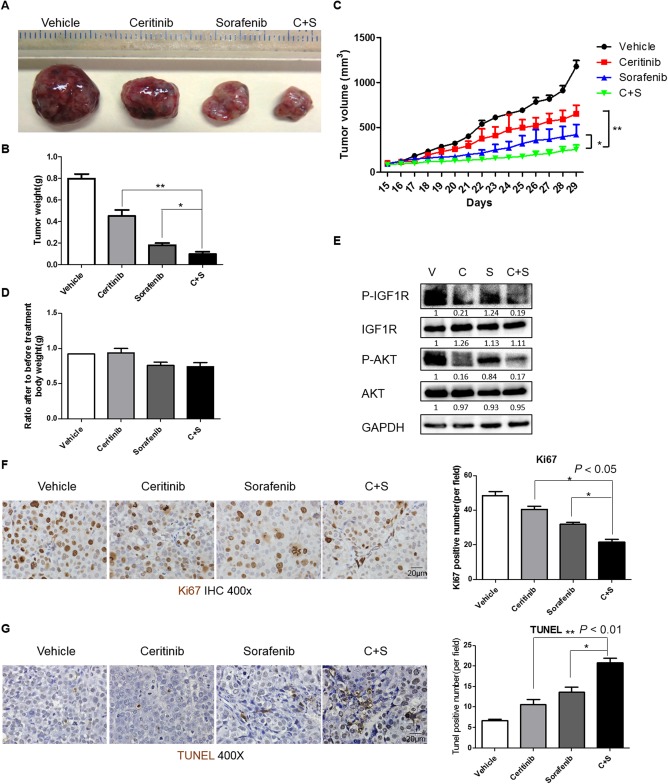
Ceritinib enhanced the efficacy of sorafenib‐mediated inhibition of HCC tumor growth in a xenograft model. (A) Hep3B cells were injected into SCID‐bg mice subcutaneously to construct a xenograft model. Two weeks after injection, five mice were treated with vehicle (30% captisol), ceritinib (25 mg/kg), sorafenib (25 mg/kg), or combination of ceritinib and sorafenib daily by oral gavage for two weeks. Tumor volumes were measured daily using external calipers until the day of sacrifice. Gross tumors were photographed after harvesting (magnification, 1x). (B) Tumor weights of the mice from (A). (C) Tumor volumes from the mice from (A). Mice were weighed before and after treatment. (D) Mouse weight ratios (after/prior to treatment) of the mice from (A). (E) Expressions of p‐IGF1R, IGF1R, p‐AKT, AKT, and GAPDH proteins in tumors from (A) were examined by western blotting. (F) Proliferation in tumors from (A) was examined by immunohistochemistry for Ki67. (G) Apoptosis in tumors from (A) was examined by TUNEL staining. Each experiment was repeated at least 3 times. Abbreviations: C, ceritinib; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; IHC, immunohistochemistry; S, sorafenib; V, vehicle. Values in B, C, D, F, and G were mean ± SD (n = 5 for B‐D in each group, and n = 3 for F and G in each group).
To further test the efficacy of ceritinib in sensitizing HCC cells to sorafenib treatment in a mouse model with intact immune responses, we used the MET/CAT‐driven HCC model. Co‐activation of MET and β‐catenin often occurs in HCC.22 Codelivery of both MET and CAT (ΔN90‐β‐catenin, exon 3 deleted), but neither MET nor CAT alone, into mouse livers using the sleeping beauty transposon system efficiently induces HCC within several weeks.22, 23, 24 Therefore, this model (referred to here as MET/CAT) is useful for studying the functions of genes in hepatocarcinogenesis because of its clinical relevance and efficiency of HCC induction. We found that both IGF1R and AKT are activated in MET/CAT‐induced HCCs (Fig. 6A). We injected C57B6/J mice with MET/CAT to induce HCC and then treated them with vehicle, ceritinib, sorafenib, or a combination of both drugs for 4 weeks. Consistent with the results from our xenograft model, either ceritinib or sorafenib when used alone was able to inhibit HCC development, but the combination treatment had the best efficacy in tumor inhibition (Fig. 6B,C; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1181/full). Mouse weights also were not significantly changed by the combination treatment, suggesting tolerable toxicity of the combination treatment in mice with intact immune responses (Fig. 6D). Phosphorylation of both IGF1R and AKT was also inhibited by ceritinib treatment in MET/CAT‐induced tumors (Fig. 6E). Furthermore, combination treatment with ceritinib and sorafenib decreased tumor cell proliferation (Fig. 6F) and increased tumor cell apoptosis (Fig. 6G) compared to ceritinib or sorafenib monotherapy in this model. Overall, our data demonstrate that ceritinib enhances the efficacy of sorafenib in inhibiting HCC tumor growth in preclinical mouse models with intact immune responses.
Figure 6.
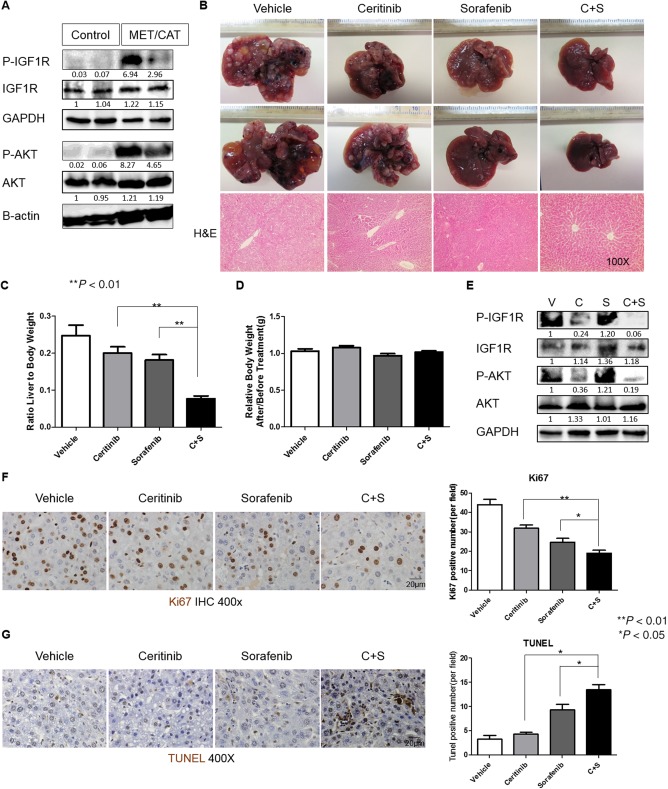
Ceritinib enhanced the efficacy of sorafenib in inhibiting tumor growth in the MET/CAT‐driven HCC model. (A) Expressions of p‐IGF1R, IGF1R, p‐AKT, AKT, and GAPDH proteins were detected by western blotting in the livers of five C57B6/J mice 8 weeks after hydrodynamic injection of MET/CAT or pT3 control. (B) Photographs (magnification, 0.5x) and H&E staining of livers of C57B6/J mice 6 weeks after injection of MET/CAT followed by treatment with vehicle (30% captisol), ceritinib (25 mg/kg), sorafenib (25 mg/kg), or a combination of ceritinib and sorafenib for 4 weeks. (C) Liver weight/body weight ratios were analyzed in mice from (B) (n = 5). (D) Mouse weight ratios (after/prior to treatment) of mice from (B) (n = 5). (E) Expressions of p‐IGF1R, IGF1R, p‐AKT, AKT, and GAPDH proteins in mice from (B) were examined by western blotting. (F) Proliferation in liver tumors from (B) was examined by immunohistochemistry for Ki67. (G) Apoptosis in liver tumors from (B) was examined by TUNEL staining. Each experiment was repeated at least 3 times. Abbreviations: C, ceritinib; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; H&E, hematoxylin and eosin; IHC, immunohistochemistry; S, sorafenib; V, vehicle. Values in C, D, F and G were mean ± SD (n = 5 for C and D in each group, and n = 3 for F and G in each group).
Discussion
HCC is a common and fatal malignancy of the liver and is the second leading cause of cancer‐related mortality in the world.1, 30 Sorafenib, the first‐line oral therapeutic agent for advanced HCC increases patient survival by less than 3 months. Regorafenib was recently approved by the FDA for treatment of patients with HCC who do not or no longer respond to sorafenib, but it only increases survival by about 3 months.31 Nivolumab, a programmed cell death protein 1 inhibitor, also was approved to treat sorafenib‐treatment failures, but the overall response rate was only 14.3% (22/154) (http://www.opdivohcp.com/advanced-hcc/efficacy/clinical-trial-results). Therefore, there is still an urgent need to develop new and more effective therapeutic agents and strategies to treat HCC. In this study, we demonstrate that inhibition of IGF1R efficiently increases sensitivity of HCC cells to sorafenib treatment in in vitro and in vivo models. The findings of the current study provide a basis to investigate a new combination of sorafenib with an IGF1R inhibitor, such as ceritinib, for treatment of HCC.
IGFIR overactivation is one of the hallmarks of HCC and can be mediated by increased levels of IGFIR protein and/or an excess of IGF ligands.32 Healthy mature hepatocytes do not express IGFIR.16 In HCC samples, up‐regulation of IGFIR is one of the most common alterations, occurring in 30% of patients.33 Activation of IGFIR signaling in HCC was significantly associated with AKT and mammalian target of rapamycin signaling.16 In vitro studies showed that abrogation of IGF1R activation and downstream signaling by the monoclonal antibody A12 significantly decreased cell viability and proliferation.14 In vivo, A12 delayed tumor growth and prolonged survival, reducing proliferation rates and inducing apoptosis.14 Although several IGF1R inhibitors or blocking antibodies have been tested in preclinical models or clinical trials for patients with HCC,9, 14, 15, 16 none has been approved by the FDA, possibly because inhibition of IGF1R alone may not be sufficient to effectively inhibit HCC cell growth and survival. Our data support this hypothesis as inhibition of IGF1R by shRNA or ceritinib has only a modest suppression on HCC proliferation and survival (Figs. 1 and 2). However, we show for the first time that the combination of sorafenib and the IGF1R inhibitor ceritinib is more effective against HCC in in vitro and in vivo models compared to sorafenib or inhibition of IGF1R alone. The addition of an IGF1R inhibitor is advantageous because sorafenib is insufficient to inhibit IGF1R and downstream AKT activation in HCC cells and IGF1R is critical for AKT activation, which promotes HCC cell proliferation and survival.34 Combination treatment using sorafenib and IGF1R inhibitors not only suppresses platelet‐derived growth factor receptor, VEGFR, and RAF phosphorylation but also inhibits the IGF1R/AKT axis, resulting in a more efficacious anti‐HCC effect (Fig. 7).
Figure 7.
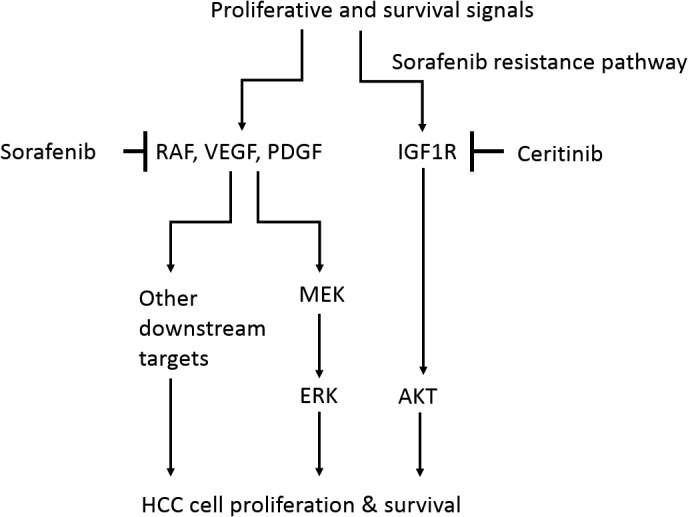
Schematic model. Sorafenib inhibits its downstream targets RAF, VEGF, and PDGF, resulting in inhibition of MEK/ERK activity, which leads to inhibition of HCC cell proliferation and survival. However, HCC cells are able to escape from sorafenib's effects using other resistance pathways that sorafenib insufficiently inhibits. IGF1R/AKT is such a sorafenib‐resistant pathway. IGF1R remains activated after sorafenib treatment in HCC cells and its activity leads to AKT activation, which is critical for HCC cell proliferation and survival. Ceritinib is a potent IGF1R inhibitor. Ceritinib effectively inhibits the IGF1R/AKT pathway but fails to suppress ERK activity. Therefore, ceritinib alone has a modest effect on HCC cell proliferation and survival. The combination of sorafenib and ceritinib effectively inhibits both the MEK/ERK and IGF1R/AKT pathways, which results in more effective inhibition of HCC cell growth compared to either sorafenib or ceritinib used alone. Abbreviations: MEK, mitogen‐activated protein kinase/ERK kinase; PDGF, platelet‐derived growth factor.
Ceritinib is well known as an adenosine triphosphate‐competitive tyrosine kinase inhibitor of ALK. Intriguingly, the expression of both ALK (Fig. 2A) and p‐ALK in the HCC cell lines (Hep3B, Huh7, and HepG2) is very low (data not shown). Therefore, it is unlikely that ceritinib sensitizes these HCC cells to sorafenib by inhibiting ALK. Two recent studies indicate that ALK is overexpressed in 13%‐44% human HCC and that this overexpression is correlated with poor prognosis.35, 36 ALK can be activated by different ligands (e.g., growth factors pleiotrophin or midkine) and various pathways.37 Therefore, it is likely that ceritinib inhibits ALK activity in some patients with HCC. We found that overexpression of ALK decreases the sensitivity of HCC cells to sorafenib (data not shown), suggesting inhibition of ALK might sensitize HCC cells to sorafenib. Therefore, combination therapy consisting of ceritinib plus sorafenib might be suitable not only for patients with HCC with activated IGF1R but also for a subset of patients with HCC with ALK activation as well as for patients showing both IGF1R and ALK activation.
It has been shown that IGF1R activation leads to both PI3/AKT and RAS/RAF/ERK activation.8 However, we found that knockdown of IGF1R or ceritinib inhibits only the activation of AKT but not ERK in HCC cells (Fig. 2A). The failure to inhibit ERK activity may explain why knockdown of IGF1R or the use of ceritinib alone has a modest effect on HCC cell growth and survival. On the other hand, sorafenib treatment is sufficient to effectively inhibit ERK activity but not AKT activity in HCC cells (Fig. 1A). Therefore, the combined use of sorafenib and ceritinib leads to inhibition of both ERK and AKT activities, which results in profound inhibition of HCC cell growth and ultimately cell death (Fig. 7). The mechanisms by which IGF1R inhibition fails to abrogate ERK activity in HCC cells remain unclear. It has been shown that A12, a monoclonal antibody against IGF1R, failed to inhibit ERK in leukemic cells treated with interleukin‐3.38 Therefore, it is possible that ERK can be activated by other upstream factors, such as interleukin‐3, and that IGF1R is not a major regulator of ERK activation in HCC cells.
In conclusion, our study shows that IGF1R inhibition effectively sensitizes HCC cells to sorafenib treatment by inhibiting AKT activity. The combined inhibition of IGF1R using ceritinib and sorafenib may offer an improved strategy to treat HCC. As ceritinib is already FDA approved, a clinical trial using combination treatment with sorafenib is warranted. A number of IGF1R inhibitors have been developed and are being studied in clinical trials for multiple types of solid tumors. A combination of one of these inhibitors plus sorafenib might provide a new direction in HCC therapy, especially in patients with activated IGF1R.
Authors names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1181/full.
Supporting Information 1
Potential conflict of interest: Nothing to report.
Supported in part by an American Association for the Study of Liver Diseases Liver Scholar Award (to W.Q.) and National Institutes of Health awards R03CA195183, R03CA184652, and R01CA197128 (to W.Q.).
REFERENCES
- 1. Zhu RX, Seto WK, Lai CL, Yuen MF. Epidemiology of hepatocellular carcinoma in the Asia‐Pacific region. Gut Liver 2016;10:332‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wild AT, Gandhi N, Chettiar ST, Aziz K, Gajula RP, Williams RD, et al. Concurrent versus sequential sorafenib therapy in combination with radiation for hepatocellular carcinoma. Plos One 2013;8:e65726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li J, Wang L, Cong N, Shi C, Bu W, Song J, et al. Efficacy of sorafenib for advanced hepatocellular carcinoma and prognostic factors. Hepatogastroenterology 2014;61:954‐957. [PubMed] [Google Scholar]
- 4. Keating GM, Santoro A. Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs 2009;69:223‐240. [DOI] [PubMed] [Google Scholar]
- 5. Chen KF, Chen HL, Tai WT, Feng WC, Hsu CH, Chen PJ, et al. Activation of phosphatidylinositol 3‐kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J Pharmacol Exp Ther 2011;337:155‐161. [DOI] [PubMed] [Google Scholar]
- 6. Gao JJ, Shi ZY, Xia JF, Inagaki Y, Tang W. Sorafenib‐based combined molecule targeting in treatment of hepatocellular carcinoma. World J Gastroenterol 2015;21:12059‐12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Siegel AB, Olsen SK, Magun A, Brown RS Jr. Sorafenib: where do we go from here? Hepatology 2010;52:360‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Denduluri SK, Idowu O, Wang Z, Liao Z, Yan Z, Mohammed MK, et al. Insulin‐like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis 2015;2:13‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen HX, Sharon E. IGF‐1R as an anti‐cancer target‐‐trials and tribulations. Chin J Cancer 2013;32:242‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tognon CE, Sorensen PH. Targeting the insulin‐like growth factor 1 receptor (IGF1R) signaling pathway for cancer therapy. Expert Opin Ther Targets 2012;16:33‐48. [DOI] [PubMed] [Google Scholar]
- 11. Nair PN, De Armond DT, Adamo ML, Strodel WE, Freeman JW. Aberrant expression and activation of insulin‐like growth factor‐1 receptor (IGF‐1R) are mediated by an induction of IGF‐1R promoter activity and stabilization of IGF‐1R mRNA and contributes to growth factor independence and increased survival of the pancreatic cancer cell line MIA PaCa‐2. Oncogene 2001;20:8203‐8214. [DOI] [PubMed] [Google Scholar]
- 12. Kato H, Sekine Y, Furuya Y, Miyazawa Y, Koike H, Suzuki K. Metformin inhibits the proliferation of human prostate cancer PC‐3 cells via the downregulation of insulin‐like growth factor 1 receptor. Biochem Biophys Res Commun 2015;461:115‐121. [DOI] [PubMed] [Google Scholar]
- 13. Desbois‐Mouthon C, Baron A, Blivet‐Van Eggelpoel MJ, Fartoux L, Venot C, Bladt F, et al. Insulin‐like growth factor‐1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin‐like growth factor‐1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin Cancer Res 2009;15:5445‐5456. [DOI] [PubMed] [Google Scholar]
- 14. Tovar V, Alsinet C, Villanueva A, Hoshida Y, Chiang DY, Sole M, et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre‐clinical efficacy of IGF‐1R blockage. J Hepatol 2010;52:550‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu J, Zhu AX. Targeting insulin‐like growth factor axis in hepatocellular carcinoma. J Hematol Oncol 2011;4:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Enguita‐German M, Fortes P. Targeting the insulin‐like growth factor pathway in hepatocellular carcinoma. World J Hepatol 2014;6:716‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang LL, Li GB, Ma S, Zou C, Zhou S, Sun QZ, et al. Structure‐activity relationship studies of pyrazolo[3,4‐d]pyrimidine derivatives leading to the discovery of a novel multikinase inhibitor that potently inhibits FLT3 and VEGFR2 and evaluation of its activity against acute myeloid leukemia in vitro and in vivo. J Med Chem 2013;56:1641‐1655. [DOI] [PubMed] [Google Scholar]
- 18. Lovly CM, McDonald NT, Chen H, Ortiz‐Cuaran S, Heukamp LC, Yan Y, et al. Rationale for co‐targeting IGF‐1R and ALK in ALK fusion‐positive lung cancer. Nat Med 2014;20:1027‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shang N, Arteaga M, Zaidi A, Stauffer J, Cotler SJ, Zeleznik‐Le NJ, et al. FAK is required for c‐Met/beta‐catenin‐driven hepatocarcinogenesis. Hepatology 2015;61:214‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arteaga M, Shang N, Ding X, Yong S, Cotler SJ, Denning MF, et al. Inhibition of SIRT2 suppresses hepatic fibrosis. Am J Physiol Gastrointest Liver Physiol 2016;310:G1155‐G1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006;1:2315‐2319. [DOI] [PubMed] [Google Scholar]
- 22. Tward AD, Jones KD, Yant S, Cheung ST, Fan ST, Chen X, et al. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci U S A 2007;104:14771‐14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patil MA, Lee SA, Macias E, Lam ET, Xu C, Jones KD, et al. Role of cyclin D1 as a mediator of c‐Met‐ and beta‐catenin‐induced hepatocarcinogenesis. Cancer Res 2009;69:253‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stauffer JK, Scarzello AJ, Andersen JB, De Kluyver RL, Back TC, Weiss JM, et al. Coactivation of AKT and beta‐catenin in mice rapidly induces formation of lipogenic liver tumors. Cancer Res 2011;71:2718‐2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shang N, Arteaga M, Chitsike L, Wang F, Viswakarma N, Breslin P, et al. FAK deletion accelerates liver regeneration after two‐thirds partial hepatectomy. Sci Rep 2016;6:34316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shang N, Arteaga M, Zaidi A, Cotler SJ, Breslin P, Ding X, et al. FAK kinase activity is required for the progression of c‐MET/beta‐catenin‐driven hepataocellular carcinoma. Gene Expr 2016;17:79‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tovar V, Cornella H, Moeini A, Vidal S, Hoshida Y, Sia D, et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut 2017;66:530‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hernandez‐Sanchez C, Blakesley V, Kalebic T, Helman L, LeRoith D. The role of the tyrosine kinase domain of the insulin‐like growth factor‐I receptor in intracellular signaling, cellular proliferation, and tumorigenesis. J Biol Chem 1995;270:29176‐29181. [DOI] [PubMed] [Google Scholar]
- 29. Kalathil SG, Lugade AA, Iyer R, Miller A, Thanavala Y. Endothelial progenitor cell number and ERK phosphorylation serve as predictive and prognostic biomarkers in advanced hepatocellular carcinoma patients treated with sorafenib. Oncoimmunology 2016;5:e1226718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 31. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, et al.; RESORCE Investigators . Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2017;389:56‐66. [DOI] [PubMed] [Google Scholar]
- 32. Aleem E, Nehrbass D, Klimek F, Mayer D, Bannasch P. Upregulation of the insulin receptor and type I insulin‐like growth factor receptor are early events in hepatocarcinogenesis. Toxicol Pathol 2011;39:524‐543. [DOI] [PubMed] [Google Scholar]
- 33. Faivre S, Bouattour M, Raymond E. Novel molecular therapies in hepatocellular carcinoma. Liver Int 2011;31(Suppl 1.):151‐160. [DOI] [PubMed] [Google Scholar]
- 34. Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased lipogenesis, induced by AKT‐mTORC1‐RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011;140:1071‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jia SW, Fu S, Wang F, Shao Q, Huang HB, Shao JY. ALK gene copy number gain and its clinical significance in hepatocellular carcinoma. World J Gastroenterol 2014;20:183‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu J, Jin H, Tian H, Lian G, Chen S, Li J, et al. Anaplastic lymphoma kinase protein expression predicts micrometastases and prognosis for patients with hepatocellular carcinoma. Oncol Lett 2016;11:213‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wellstein A. ALK receptor activation, ligands and therapeutic targeting in glioblastoma and in other cancers. Front Oncol 2012;2:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bertrand FE, Steelman LS, Chappell WH, Abrams SL, Shelton JG, White ER, et al. Synergy between an IGF‐1R antibody and Raf/MEK/ERK and PI3K/Akt/mTOR pathway inhibitors in suppressing IGF‐1R‐mediated growth in hematopoietic cells. Leukemia 2006;20:1254‐1260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1181/full.
Supporting Information 1