

Abstract
Key points
The medial entorhinal cortex (mEC) has an important role in initiation and propagation of seizure activity. Several anatomical relationships exist in neurophysiological properties of mEC neurons; however, in the context of hyperexcitability, previous studies often considered it as a homogeneous structure.
Using multi‐site extracellular recording techniques, ictal‐like activity was observed along the dorso‐ventral axis of the mEC in vitro in response to various ictogenic stimuli. This originated predominantly from ventral areas, spreading to dorsal mEC with a surprisingly slow velocity.
Modulation of inhibitory tone was capable of changing the slope of ictal initiation, suggesting seizure propagation behaviours are highly dependent on levels of GABAergic function in this region.
A distinct disinhibition model also showed, in the absence of inhibition, a prevalence for interictal‐like initiation in ventral mEC, reflecting the intrinsic differences in mEC neurons.
These findings suggest the ventral mEC is more prone to hyperexcitable discharge than the dorsal mEC, which may be relevant under pathological conditions.
Abstract
The medial entorhinal cortex (mEC) has an important role in the generation and propagation of seizure activity. The organization of the mEC is such that a number of dorso‐ventral relationships exist in neurophysiological properties of neurons. These range from intrinsic and synaptic properties to density of inhibitory connectivity. We examined the influence of these gradients on generation and propagation of epileptiform activity in the mEC. Using a 16‐shank silicon probe array to record along the dorso‐ventral axis of the mEC in vitro, we found 4‐aminopyridine application produces ictal‐like activity originating predominantly in ventral areas. This activity spreads to dorsal mEC at a surprisingly slow velocity (138 μm s−1), while cross‐site interictal‐like activity appeared relatively synchronous. We propose that ictal propagation is constrained by differential levels of GABAergic control since increasing (diazepam) or decreasing (Ro19‐4603) GABAA receptor activation, respectively, reduced or increased the slope of ictal initiation. The observation that ictal activity is predominately generated in ventral mEC was replicated using a separate 0‐Mg2+ model of epileptiform activity in vitro. By using a distinct disinhibition model (co‐application of kainate and picrotoxin) we show that additional physiological features (for example intrinsic properties of mEC neurons) still produce a prevalence for interictal‐like initiation in ventral mEC. These findings suggest that the ventral mEC is more likely to initiate hyperexcitable discharges than the dorsal mEC, and that seizure propagation is highly dependent on levels of GABAergic expression across the mEC.
Keywords: dorsal‐Ventral gradient, entorhinal cortex, hyperexcitability
Key points
The medial entorhinal cortex (mEC) has an important role in initiation and propagation of seizure activity. Several anatomical relationships exist in neurophysiological properties of mEC neurons; however, in the context of hyperexcitability, previous studies often considered it as a homogeneous structure.
Using multi‐site extracellular recording techniques, ictal‐like activity was observed along the dorso‐ventral axis of the mEC in vitro in response to various ictogenic stimuli. This originated predominantly from ventral areas, spreading to dorsal mEC with a surprisingly slow velocity.
Modulation of inhibitory tone was capable of changing the slope of ictal initiation, suggesting seizure propagation behaviours are highly dependent on levels of GABAergic function in this region.
A distinct disinhibition model also showed, in the absence of inhibition, a prevalence for interictal‐like initiation in ventral mEC, reflecting the intrinsic differences in mEC neurons.
These findings suggest the ventral mEC is more prone to hyperexcitable discharge than the dorsal mEC, which may be relevant under pathological conditions.
Introduction
The medial entorhinal cortex (mEC) occupies a pivotal anatomical position, acting as a gateway between the hippocampus proper and other cortical regions (Amaral & Witter, 1989; Canto et al. 2008). Functionally, it plays a key role in the encoding of spatial information, via grid, border, head direction and speed cells (Hafting et al. 2005; Solstad et al. 2008; Giocomo et al. 2014; Kropff et al. 2015), information which feeds onto, and is modulated by, hippocampal place cells (Brun et al. 2008; Bonnevie et al. 2013). Grid cells are organized in a highly topographically modular manner along the dorso‐ventral axis of the mEC. Thus, neurons of dorsal mEC have firing fields which are close together, while those in ventral mEC have firing fields further apart (Hafting et al. 2005; Stensola et al. 2012). This functional anatomical arrangement is mirrored by correlative dorso‐ventral anatomical relationships in the intrinsic and synaptic properties of layer 2 stellate neurons (Giocomo et al. 2007; Garden et al. 2008; Boehlen et al. 2010; Navratilova et al. 2012; Pastoll et al. 2012; Yoshida et al. 2013; Booth et al. 2016). For instance, stellate cells in the ventral mEC have higher input resistances, smaller medium afterhyperpolarizing potentials and a wider synaptic integration window than equivalent cells in the dorsal mEC. Many (although not all) of these dorso‐ventral variations in neurophysiology arise from a gradient in I h‐mediated sag potentials. For example, the sag time constant in mEC stellate cells varies considerably along the dorso‐ventral axis, particularly in juvenile/young adult mice (Boehlen et al. 2010), although these differences may diminish later in development (Boehlen et al. 2010; Booth et al. 2016). These differences in sag time constant likely arise from differences in I h amplitude (Garden et al. 2008) and/or activation time constants (Giocomo & Hasselmo, 2008).
In addition to these dorso‐ventral gradients in stellate cell intrinsic neurophysiology, an inhibitory gradient exists along the dorso‐ventral axis of the mEC, with stellate cells in the dorsal mEC receiving a greater number of inhibitory inputs (Beed et al. 2013). This variance in inhibitory synaptic inputs, combined with the gradients in intrinsic membrane properties, ultimately results in a gradient in physiological network rhythms, such as gamma band oscillations recorded in brain slices (Beed et al. 2013; Booth et al. 2016), in anaesthetized (Beed et al. 2013) and awake behaving (Booth et al. 2016) mice. Specifically, in the dorsal mEC, gamma oscillations are larger in amplitude (Beed et al. 2013; Booth et al. 2016) and may have a higher peak frequency (Booth et al. 2016).
Despite this wealth of recent research into the functional properties of subregional differences within the mEC, little is known about their impact on pathophysiological network activity. For example, bath application of the K+ channel blocker 4‐aminopyridine (4‐AP) is capable of inducing epileptiform network activity in brain slices prepared from various hippocampal and cortical regions, including the entorhinal cortex (D'Antuono et al. 2010; Berretta et al. 2012; Avoli et al. 2013b; Losi et al. 2015). This activity consists of brief (<1 s) interictal‐like events and more prolonged (>5 s) ictal‐like events (Avoli et al. 2013a). This type of activity provides a useful approach to model the network mechanisms underlying hyperactive neural activity associated with seizures.
In this study, we have used parasagittal slices of the mEC to examine the influence functional dorso‐ventral gradients may have on the generation and propagation of epileptiform activity in this brain area. We propose that the specific anatomical arrangement of physiological features that enables effective spatial information processing in the mEC may have implications for the pathological hyperexcitability observed under epileptic seizure conditions.
Methods
Ethical approval
All procedures were carried out in accordance with the UK Animals (Scientific Procedures) Act 1986 and were approved by the University of Exeter Animal Welfare and Ethical Review Body.
Slice preparation
Male C57/BL6 mice (aged 6–12 weeks) were bred at the University of Exeter and housed on a 12:12 h light–dark cycle with ad libitum access to food and water. Mice were killed by cervical dislocation and the brain rapidly extracted and placed in a cold (∼4°C), oxygenated sucrose‐based solution, comprising (in mm): sucrose (189), d‐glucose (10), NaHCO3 (26), KCl (3), MgSO4 (5), CaCl2 (0.1) and NaH2PO4 (1.25). The cerebellum was removed and the remaining brain tissue hemisected. Using a vibrating blade microtome (VT1200, Leica Microsystems, Wetzlar, Germany), parasagittal brain slices (400 μm thick), containing the mEC, were prepared whilst immersed in the sucrose‐based cutting solution. After cutting, the slices were immediately removed to a holding chamber containing oxygenated (95% O2–5% CO2) artificial cerebrospinal fluid (aCSF) comprising (in mm): NaCl (124), KCl (3), NaHCO3 (24), MgSO4 (1), d‐glucose (10) and CaCl2 (1.2). The slices were gradually warmed to ∼37°C (for 30 min) and then maintained at room temperature (∼20°C, for at least another 30 min) until ready for use. Whole slices were then transferred to an interface‐style recording chamber maintained at 34 ± 1°C and allowed to equilibrate for a further 30 min. Epileptiform activity was induced by bath application of either 4‐AP (100 μm; Sigma‐Aldrich, Poole, UK), picrotoxin (50 μm; Tocris Bioscience, Bristol, UK) and kainic acid (500 nm; Sigma‐Aldrich), or aCSF containing 0‐Mg2+, with each approach conducted on separate slice preparations. 4‐AP was followed by co‐application of GABAA receptor modulators acting at the benzodiazepine binding site, either diazepam (positive) or Ro19‐4603 (negative) (Wong & Skolnick, 1992; Tocris Bioscience). In a distinct subsection of slices used for kainate/picrotoxin experiments, a scalpel blade was used to make a cut in the intermediate mEC immediately after slice preparation, thus anatomically separating dorsal and ventral portions.
Data acquisition
Continuous extracellular recordings were made using one of two approaches: (1) a single 16‐channel silicone probe consisting of 16 individual shanks (55 μm wide, 100 μm apart), with a single electrode contact point at the end of each shank (Neuronexus, Ann Arbor, MI, USA; probe cat. no.: A16×1‐2mm‐100‐177), positioned in layer 2/3 (∼200 μm from surface), parallel to the dorso‐ventral axis of the mEC; or (2) pairs of glass micropipettes (filled with aCSF) positioned at the dorsal and ventral ends of the mEC. In a few experiments both a 16‐channel probe (in layer 5/6) and two glass electrodes (in layer 2) were used simultaneously. Dorsal recording electrodes were positioned 100–200 μm ventral to the entorhinal border (to ensure placement within the mEC proper), with silicon probe arrays spanning the subsequent 1.5 mm of the mEC. For glass electrode recordings, the ventral electrode was also positioned 100–200 μm from entorhinal border. The positions of these borders were estimated by comparison to the Allen Mouse Brain Atlas (2004). For the silicone probe recordings, data were recorded using a 32‐channel amplifier (RHD2132; Intan, Los Angeles, CA, USA) coupled to an open‐source acquisition board (Open Ephys Inc., Cambridge, MA, USA). These data were band‐pass filtered (1–500 Hz) and digitized at 2 kHz. For the glass electrode experiments, data were recorded using the two channels of a MultiClamp 700A (in I = 0 mode; Molecular Devices, Sunnyvale, CA, USA), band‐pass filtered at 1 Hz to 1 kHz and digitized at 5 kHz, using Clampex 10.4 software (Molecular Devices). All data were stored on the hard drive of a PC for off‐line analysis.
Data analysis
Data were analysed using built‐in and custom‐written functions in Matlab (The MathWorks Inc., Natick, MA, USA). Interictal and ictal bursts were identified using a threshold detection algorithm. Periods containing ictal‐like activity were identified manually. For each ictal‐like event, data were filtered (10–50 Hz or 50–250 Hz) and spectral power calculated for 0.5 s bins of data. Burst initiation in each channel was determined by the first time period over a threshold of 1.5–3 standard deviations from the mean. For interictal activity, data were filtered (0.5–10 Hz) and individual burst waveforms extracted (window size = 0.9 s) from each recording probe. The resulting waveforms were grouped using an unsupervised k‐means clustering algorithm (from the Matlab 2016a Statistics and Machine Learning Toolbox; distance measure was the sum of absolute differences). The most appropriate number of clusters (k) was the solution (where k > 1 and < 10) which resulted in the highest mean silhouette value.
Ictal burst start time for each electrode was plotted relative to the first recorded threshold crossing and slope of ictal propagation calculated in μm s−1 for each burst. We analysed two to six ictal discharges per slice and, since the slope value did not vary significantly over the course of these events, we calculated a mean slope value for each slice. For analysing within‐burst properties, cross‐correlation analysis was performed on 1 s time bins of data between the most ventral recording site and each subsequent dorsal electrode. Dorsal–ventral cross correlations were performed, meaning that positive peaks in the cross correlation correspond to waveforms that occur first in ventral mEC. Interictal bursts were also measured by a variable threshold search and their frequency expressed as number of bursts in each 60 s bin. Cross‐correlation analysis was also performed on time windows containing individual bursts.
Statistics
Experimental groups were compared using paired or unpaired Student's t tests or two‐way analysis of variance (ANOVA) for normally distributed data sets. For statistical analyses, each n refers to a slice. For some experiments more than one slice was used from a single animal; this is clearly stated in the text. Slopes of ictal initiation were produced by linear regression analysis of values relative to recording position on the probe. Results are displayed as means ± SEM in text and representative figures.
Results
While several studies have observed the effect of convulsant compounds on the mEC (Barbarosie & Avoli, 1997; Gnatkovsky et al. 2008; Berretta et al. 2012; Lévesque et al. 2016), differences in hyperexcitability across the anatomical extent of this cortical area are less well understood. We therefore cut parasagittal slices containing mouse mEC and recorded electrical activity from 16 sites across the dorso‐ventral axis, perfusing compounds commonly used to induce epileptiform activity. As reported previously (Nagao et al. 1996; Gulyás‐Kovács et al. 2002; Gonzalez‐Sulser et al. 2011), bath application of 4‐AP (100 μm) was shown to reliably induce both ictal‐ and interictal‐like bursting activity in mEC (Fig. 1). A wavelet transform‐based time–frequency analysis of individual bursts revealed that interictal‐like discharges consisted of waveforms which were readily apparent in the 1–10 Hz range. In contrast, ictal‐like activity comprised repetitive, large amplitude events which were apparent in the 1–10 Hz range on the wavelet scaleogram, but in addition these longer discharges were also associated with higher frequency activity (50–120 Hz) (Fig. 2 A).
Figure 1. 4‐AP induced ictal‐ and interictal‐like activity in mEC slices.

A, recording position of 16‐shank electrode array on parasagittal mEC slice, with scale depicting dorsal (D), ventral (V), rostral (R) and caudal (C) directions. B, example of ictal‐like bursting activity from dorsal (top) to ventral (bottom) mEC showing burst recorded first in most ventral electrode site (scale bar: 200 μV, 10 s) (a), with zoomed examples of interictal‐like (b) and ictal‐like (c) events (scale bars: 100 μV, 0.5 s and 200 μV, 2 s respectively).
Figure 2. 4‐AP‐induced ictal‐like activity in mEC is initiated in ventral recording sites.

A, example recording from a ventrally positioned electrode illustrating an ictal‐like burst (scale bar: 0.2 mV, 2 s) (a), with continuous wavelet transform scalogram illustrating the frequency components of the above recording (b). B, proportion of bursts starting at dorsal and ventral recording sites (n = 123 bursts from 10 slices slices from 8 animals). C, example trace showing 16 channels, filtered in low (10–50 Hz) frequency band (a), with normalized spectral power (b). D, average start time of burst relative to first channel to meet threshold for ictal activity using low frequency filter, showing linear increase with distance from ventral pole; linear regression: R 2 = 0.95, P < 0.001, slope = 138 μm s−1. E, example filtered in high (50–250 Hz) frequency band (a), with normalized spectral power (b). F, average burst start time; linear regression: R 2 = 0.95, P < 0.001, slope = 157 μm s−1.
Interictal‐like activity comprised brief (<1 s) paroxysmal discharges which appeared to be relatively synchronous along the dorso‐ventral axis of the mEC. We detected individual interictal‐like event traces using a threshold detection approach. Using an unsupervised k‐means clustering approach (see Methods) we grouped the waveforms based on the time of the peak of the waveforms. This approach usually resulted in two to three clusters of waveforms, which corresponded to bursts which were initiated at different sites. In the example in Fig. 3, there was an approximately even split between interictal waveforms travelling in a ventral‐to‐dorsal and a dorsal‐to‐ventral direction (Fig. 3 B), suggesting that these bursts were initiated at multiple sites along the dorso‐ventral axis of the mEC. The maximum time between interictal peaks across the 16 recording sites averaged 356 ± 30 ms (n = 10 slices from 8 animals).
Figure 3. Interictal‐like bursts are generated in both dorsal and ventral portions of the mEC.
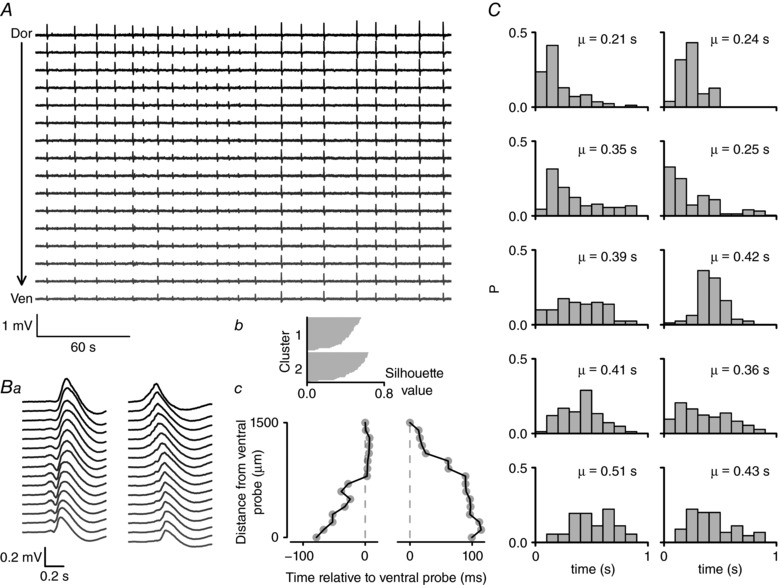
A, an example recording of interictal‐like bursts recorded using a 16‐shank electrode array on parasagittal mEC slice. This 4.5 min segment of data was recorded between 2 ictal‐like bursts (not shown). Numerous interictal‐like bursts were observed, visible on this time scale as brief vertical deflections on the recording. B, individual bursts were detected and clustered into groups according to the time of the waveform peak. In this recording, two groups were identified, the average waveforms of which are depicted in a. b, silhouette plot of the resulting k‐means clustering algorithm. c, the time of the average waveform peak (plotted relative to the time on the most ventral probe) for the two clusters. These data illustrate that interictal bursts are initiated at different points along the dorso‐ventral axis of the mEC. C, probability histograms showing the maximum difference in interictal peak times across all 16 probes for 10 different slices. The mean (μ) maximum difference in peak times is shown for each distribution. These data illustrate that, on average, interictal bursts take 0.2–0.5 s to spread along the dorso‐ventral axis of the mEC.
Ictal‐like discharges (>5 s) occurred in all the slices tested, appearing first after 1166 ± 148 s and continuing with an average interval of (259 ± 14 s). Interestingly, ictal activity was substantially more likely to be first detected in the most ventral mEC recording sites than those located in more dorsal aspects of mEC. In total 37/43 ictal bursts were first detected in ventral mEC compared to 6/43 in dorsal (Fig. 2 B). The propagation of activity from ventral to dorsal recording sites was shown to occur over a prolonged time frame (linear regression: R 2 = 0.95, P < 0.001, slope = 138 μm s−1), meaning that ictal activity in the most dorsal electrode occurred 14.7 ± 2.8 s (n = 10 slices from 8 animals) after the initiation of the event in the most ventral electrode on the recording array (Fig. 2 D). This pattern of activity was also observed using two glass electrodes placed at dorsal and ventral poles (paired t test; P = 0.03, n = 19 bursts/5 slices). Initially, this analysis was restricted to relatively low frequency components (10–50 Hz), but the same relationship was present when data were filtered in higher frequency bands (50–250 Hz), which more directly represent local neuronal firing (Fig. 2 F, linear regression: R 2 = 0.95, P < 0.001, slope = 157 μm s−1).
In several, but not all, cases, ictal‐like discharges were immediately preceded by an interictal‐like event (see Fig. 1 Bc). As with the interictal‐like discharges not associated with the ictal events (Fig. 3), there was no consistent directionality of these ictal‐associated interictal events. Thus, when we carefully examined the 5 s immediately preceding the ictal events we found that 6/30 ictal discharges were associated with interictal events travelling in a dorsal‐to‐ventral direction, 14/30 were associated with ventral‐to‐dorsal travelling interictal events and 10/30 were not associated with any preceding interictal event. Furthermore, there was no consistent relationship between the direction of the preceding interictal event and the subsequent ictal event (data not shown).
Closer examination of the burst waveforms within an ictal event indicated that the individual spike‐wave discharges were initiated in the ventral regions of the mEC. To quantify this, cross correlations were performed on data binned across time between the most ventral recording site and each of the subsequent dorsal electrodes. Figure 4 A and B shows recordings taken from dorsal and ventral poles of the electrode array, with dorsal–ventral cross‐correlation values for each time bin displayed in the colour ‘heatmap’ axis (Fig. 4 Ab). During the ictal bursting, activity across the dorsal and ventral electrodes became highly synchronous with largely positive lag time values, indicating that activity was largely led by the ventral mEC. The proportion of 1 s time bins with correlation peaks in the positive (ventral leading) was shown to be significantly greater during ictal events when compared to non‐ictal bins (Fig. 4 D, paired t test, P = 0.002, n = 10 slices from 8 animals). The lag times associated with the maximum correlation values were also observed to linearly increase with distance from the most ventral recording site. This indicates that within‐burst activity spread in the ventral to dorsal direction (Fig. 4 C, linear regression: R 2 = 0.93, P < 0.001, slope = (55.9 ± 5) × 103 μm s−1).
Figure 4. Intra‐ictal burst waveforms initiated in ventral mEC regions.
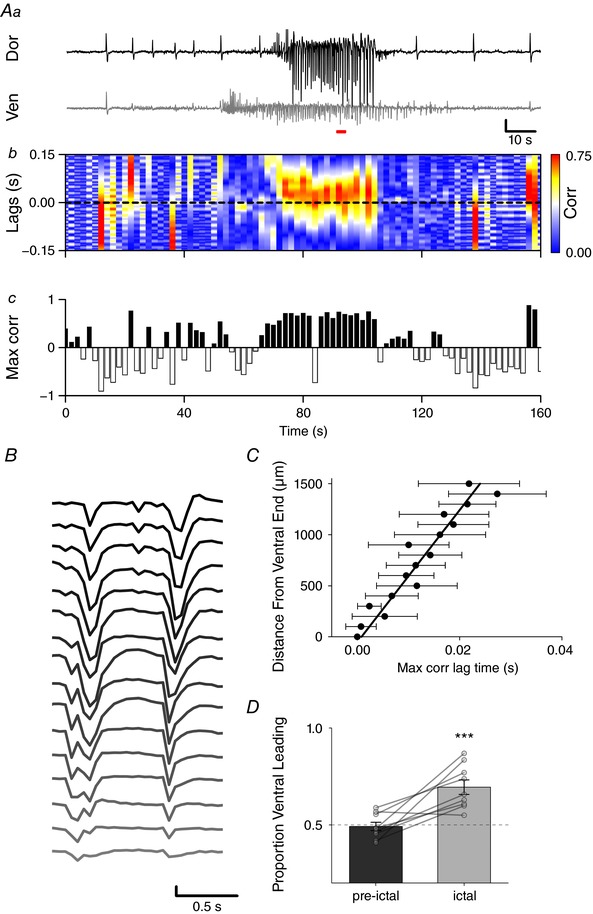
Aa, example traces from most dorsal (top) and ventral (bottom) recording sites of electrode array (scale bar: 100 μV, 10 s). b, binned cross correlations for every 1 s of data. c, correlation values are shown in the colour axis, with positive peaks indicating ventral‐leading activity and negative peaks dorsal‐leading. B, example of intra‐burst activity across 16‐shank electrode array initiating in ventral mEC during red bar in A (scale bar: 200 μV, 0.5 s). C, lag time associated with peak cross correlation between most ventral sites and each dorsal recording electrode, showing linear increase with distance from ventral pole (linear regression: R 2 = 0.93, P < 0.001, slope = 55.9 ± 5 mm s−1). D, proportion of 1 s time bins with correlation peaks in the positive (ventral leading) was greater during ictal events when compared to non‐ictal bins (paired t test, P = 0.002, n = 10 slices from 8 animals). *** P < 0.001. [Color figure can be viewed at http://wileyonlinelibrary.com]
Since ictal‐like bursting activity has also been shown to be initiated in deeper mEC layers, we next recorded along the dorso‐ventral axis of layer 5/6 mEC, with additional glass electrodes positioned in the superficial layers at either end of the recording array. Using this approach we found that, after application of 4‐AP, ictal‐like activity also occurred first in deep layer ventral recording sites, but there was no preference for bursting to be initiated in either deep or superficial mEC layers (Fig. 5 B, burst start time relative to onset (ventral L2/3: 1.431 ± 0.59 s; ventral L5/6: 0.514 ± 0.23 s; dorsal L2/3: 7.5 ± 1.2 s; dorsal L5/6: 7.14 ± 0.96 s; 2‐way repeated measures ANOVA, main effect, dorsal–ventral: F 1,10 = 56.5, P < 0.0001, layer: F 1,10 = 0.11, P = 0.75; n = 6) suggesting that ictal‐like activity may originate from either cortical layer.
Figure 5. Ictal‐like bursts show similar propagation in deep cortical layers.
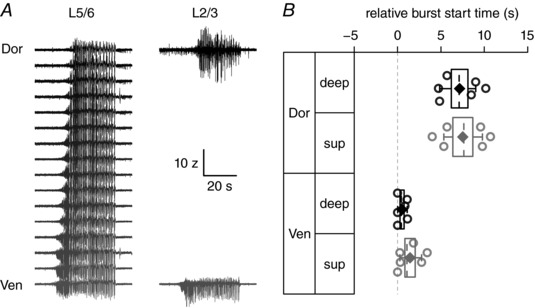
A, example silicon probe recording of ictal‐like bursting activity from deep layers (L5/6) of mEC, dorsal (top) to ventral (bottom) (scale bar: 10 z, 20 s), with simultaneous glass electrode recordings from superficial layers (L2/3). B, start time of each recording location relative to initiation of first burst.
The relatively slow spread of 4‐AP‐induced ictal‐like waveforms from ventral to dorsal recording sites suggests that some process regulates and dampens spike‐wave propagation from the ventral to dorsal pole of the mEC. Since there is a gradient in GABAerigc inhibition along the dorso‐ventral axis of the mEC (Beed et al. 2013; Booth et al. 2016), we reasoned that a greater inhibitory drive onto principal cells in the dorsal mEC may be responsible for the slow spread of ictal‐like discharge activity (Fig. 2). To examine this hypothesis, we pharmacologically modulated GABAA receptors during pre‐established 4‐AP‐induced ictal‐like activity. Application of diazepam, a positive allosteric modulator of GABAA receptors, significantly decreased the speed of ictal propagation by ∼2‐fold (Fig. 6 Aa and B), from 147.5 ± 23 to 64 ± 14 μm s−1 (Fig. 6 Cb, paired t test, P < 0.001, n = 6 slices from 6 animals), with no significant difference in the extent of ictal propagation (4‐AP: 907 ± 183 μm; 4‐AP+diazepam: 682 ± 145 μm; unpaired t test: P = 0.3, n = 6 slices from 6 animals, data not shown). Conversely, the application of GABAA receptor inverse agonist, Ro19‐4603, significantly increased propagation speed by ∼7.5‐fold (from 170.3 ± 45 to 1272.7 ± 117 μm s−1) when compared to paired baseline, such that burst initiation was almost instantaneous along the ventral to dorsal axis of the mEC (Fig. 6 Db, paired t test, P < 0.001, n = 6 slices from 6 animals).
Figure 6. Modulation of GABAergic transmission changes rate of ictal‐like propagation in mEC slices.

A and B, example traces of ictal‐like events (top) with normalized power (bottom) on 16‐shank recording array after application of 4‐AP (scale bar: 500 μV, 5 s) (Aa and Ba) and subsequent application of diazepam (DZP; 30 μm; Ab) or Ro19‐4603 (RO; 10 nm; Bb). Ac and Bc show time course of ictal burst slope before and after manipulation of GABAergic transmission. C, decreased ictal slope in an example slice after diazepam application (white) compared to 4‐AP alone (grey); 3 ictal bursts shown pre‐ (1800–2400 s) and post‐ (3000–3600 s) drug, with mean slope decreasing ∼2‐fold (b); paired t test, P < 0.001, n = 6 slices from 6 animals. D, ictal propagation is faster after application of Ro19‐4603 (a); paired t test, P < 0.001, n = 6 slices from 6 animals (b). *** P < 0.001.
In order to determine whether the increased excitability of ventral mEC was specific to factors dependent on 4‐AP application, we continued with alternative chemoconvulsant strategies. Another common approach for inducing epileptiform activity in vitro is the removal of Mg2+ from the extracellular medium, producing hyperexcitability mediated by enhanced NMDA receptor activation (Jones & Heinemann, 1988; Jones, 1989; Bragdon et al. 1992; Traub et al. 1994; Whittington et al. 1995; Armand et al. 2000). Bathing slices in aCSF containing 0‐Mg2+ was also capable of producing large, ictal‐like waveforms. Similar to our findings with 4‐AP application, discharges were almost always observed first in ventral recording sites (Fig. 7 A; 24/26 ventral vs. 2/26 dorsal, n = 6 slices from 6 animals). Since the slope of ictal propagation has been shown to increase with continued 0‐Mg2+ exposure (Trevelyan et al. 2007b), we restricted our observations to relatively early periods after Mg2+ washout (30 min). Nevertheless, this approach was sufficient to see regular periods of ictal‐like activity being initiated 1352 ± 66 s after removal of Mg2+ and continuing to occur with an interval of 141 ± 32 s. The slope of ictal initiation was also shown to occur in the ventral to dorsal direction (Fig. 7 C; 55.7 ± 9.4 μm s−1; 1‐way ANOVA, F 1,7 = 8.6, P = 0.02, n = 6 slices from 6 animals). Compared to 4‐AP‐induced seizure activity, the directionality of within‐burst discharges was more variable under 0‐Mg2+. However across the duration of the ictal burst, there was a trend towards an increased proportion of time bins with correlation peaks in the positive (ventral leading) compared to non‐ictal bins (Fig. 7 D–F, paired t test, P = 0.06, n = 6 slices from 6 animals). The lag time associated with the maximum correlation values were also observed to linearly increase with distance from the most ventral recording site. This indicates that within‐burst activity spread in the ventral to dorsal direction (Fig. 7 E, linear regression: R 2 = 0.81, P < 0.001, slope = (35.8 ± 2) × 103 μm s−1). Trevelyan et al. (2007a) reported that in neocortical slices from juvenile mice, late ‘intra‐ictal’ discharges induced by removal of Mg2+ switched direction from the main ictal wavefront. In contrast, we found that in the majority of parasagittal mEC slices (5/6 slices), the ventral‐to‐dorsal directionality of individual discharges was maintained throughout the ictal burst (see Fig. 7 D for a representative example). In one slice we did observe a switch from ventral‐leading to dorsal‐leading discharges approximately halfway through the ictal events (data not shown).
Figure 7. Ictal‐like activity from slices in 0‐Mg2+ aCSF is also initiated in ventral mEC recording sites.

A, example traces of ictal‐like events (top) with normalized power (bottom) on 16‐shank recording array after Mg2+ washout (scale bar: 500 μV, 5 s). B, example burst start times along dorso‐ventral axis of the mEC normalized to start of burst for 3 ictal‐like events recorded after Mg2+ washout. C, average slope of ictal initiation under 0‐Mg2+ conditions shows bursts occur in the ventral to dorsal direction (55.7 ± 9.4 μm s−1; 1‐way ANOVA, F 1,7 = 8.6, P = 0.02, n = 6 slices from 6 animals). Da, example traces from most dorsal (top) and ventral (bottom) recording sites (scale bar: 100 μV, 10 s); b, 1 s binned cross correlations. c, values shown in colour axis, with positive peaks indicating ventral‐leading activity and negative peaks dorsal‐leading. E, lag time associated with peak cross correlation between most ventral sites and each dorsal recording electrode (linear regression R 2 = 0.81, P < 0.001, slope = (35.8 ± 2) × 103 μm s−1). F, proportion of time bins with correlation peaks in the positive (ventral leading) compared to non‐ictal bins (paired t test, P = 0.06, n = 6 slices from 6 animals). [Color figure can be viewed at http://wileyonlinelibrary.com]
Our data suggest that local inhibitory networks provide tight control of epileptiform activity within the mEC. However, there are also strong gradients in the intrinsic electrical properties of excitatory stellate neurons within the mEC (Garden et al. 2008; Giocomo & Hasselmo, 2009; Boehlen et al. 2010; Booth et al. 2016). To determine the contribution of dorso‐ventral gradients in properties other than GABAergic inhibition to the generation of hyper‐excitability, we used an additional pharmacological model, namely combined application of kainate (500 nm) and picrotoxin (50 μm). This approach both increased excitation via the excitatory (i.e. depolarizing) action of kainate on GluK receptor subtypes and produced a complete blockade of GABAA receptor‐mediated inhibition in the mEC. This produced slow, interictal‐like events at both dorsal and ventral recording sites (Fig. 8 A and B). Under these conditions, we observed that interictal‐like bursting activity was led by ventral mEC, such that individual bursts were almost always initiated at the ventral end of the mEC (Fig. 8 B–E). Furthermore, the onset of bursting activity was also seen to appear first at ventral recording sites (434 ± 56 s after kainate/picrotoxin application) compared to those in the dorsal aspect of mEC (635 ± 98 s) (Fig. 8 D, paired t test, P = 0.01, n = 8 from 5 animals), though upon reaching equilibrium bursting occurred uniformly across the mEC (Fig. 8 C).
Figure 8. Application of 500 nm kainate and 50uM picrotoxin produces interictal‐like events which originate in ventral mEC.
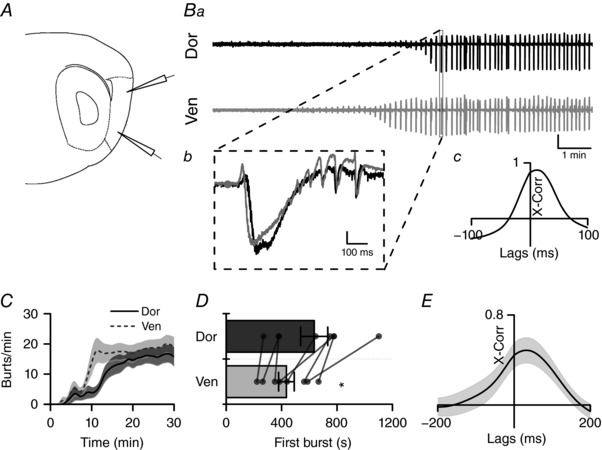
A, illustration of relative position of glass recording electrodes in dorsal (top) and ventral (bottom) mEC. B, example trace after application of picrotoxin (50 μm; a); box represents one interictal event (b) with cross correlation (c) showing peak occurring in ventral mEC before dorsal (scale bar: 0.1 mV). C, average time‐pooled data showing the development of burst frequency (min−1) in dorsal (black) and ventral (grey) mEC (n = 8 slices from 5 animals). Continuous line represents mean (± SEM shown by shaded areas). D, mean (±SEM) time until first epileptic event is shorter in ventral than dorsal mEC (paired t test P = 0.013, n = 8 slices from 5 animals). E, average cross correlation between dorsal and ventral events (n = 8 slices from 5 animals) showing peak lag time >0 s. * P < 0.05.
To further test the hypothesis that the ventral mEC is more excitable than the dorsal, we anatomically separated the two ends of the mEC with a scalpel cut (Fig. 9 A). This allowed us to observe whether the dorsal mEC would produce interictal bursting independently, rather than as a result of ventral mEC hyper‐excitability. Cut slices produced interictal bursting in both ventral and dorsal recording sites. Similar to control (uncut) slices, bursting activity was first recorded in the ventral mEC after kainate/picrotoxin application (Fig. 9 D; dorsal: 907 ± 184 s; ventral: 682 ± 145 s; paired t test, P = 0.025, n = 4, from 4 animals). However, in contrast to observations in intact control mEC slices, it was evident that events in cut dorsal mEC slices occurred at a slower rate when compared to ventral (Fig. 9 B and C). At ventral mEC recording sites, burst frequency was similar between cut and control slices (Fig. 9 D). Conversely, bursts in the cut dorsal mEC occurred at a lower frequency than those in intact mEC slices (Fig. 9 E). As expected, the cross correlation between dorsal and ventral electrodes was largely absent following anatomical separation of the dorsal and ventral mEC, illustrating that the two regions had become desynchronized (Fig. 9 G and H, unpaired t test, P = 0.03, n = 4/8 slices from 4/5 animals). Taken together these findings suggest that the dorsal mEC is less likely to produce epileptiform activity in the absence of the ventral mEC.
Figure 9. Separation of dorsal and ventral mEC produces preferential decrease in epileptic events in the dorsal mEC.
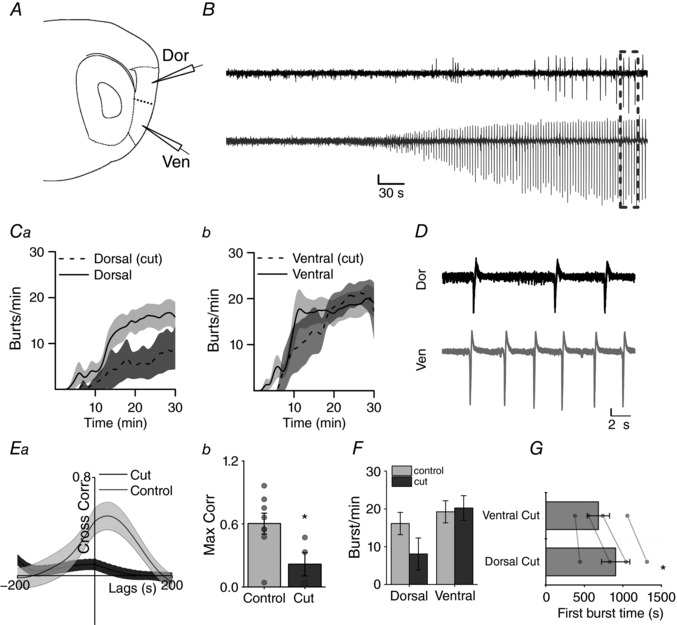
A, relative position of dorsal (top) and ventral (bottom) recording electrodes and scalpel cut (dotted line) between electrodes. B, example trace (scale bar 0.2 mV, 30 s). C, averaged time‐pooled data showing the development of burst frequency in dorsal (a) and ventral (b) cut slices compared to control (n = 4). D, zoomed example trace (box) showing desynchronized bursting in dorsal and ventral mEC (scale bar 0.2 mV, 2 s). E, average cross correlation of cut slices (n = 4 slices from 4 animals) compared to controls (n = 8 slices from 5 animals) (a) shows significant decrease in correlation of epileptic bursts (b) (unpaired t test P = 0.031). * P < 0.05. F, decreased average burst frequency in dorsal mEC in cut slices compared to control. G, bar graph showing mean (±SEM) time in seconds until first recording epileptic event is also shorter in ventral than dorsal mEC when ends are separated (*paired t test P = 0.026) (n = 4 slices from 4 animals).
Discussion
This study is the first to highlight the differential role of dorsal and ventral mEC in the generation of epileptiform events in vitro. In essence, we suggest that previously reported gradients in inhibitory networks (Beed et al. 2013) and intrinsic membrane properties (Garden et al. 2008; Giocomo & Hasselmo, 2009; Boehlen et al. 2010; Dodson et al. 2011; Pastoll et al. 2012; O'Reilly et al. 2015; Booth et al. 2016) combine to make the ventral mEC more prone than the dorsal mEC to the generation of pathological hyperexcitability.
Bath application of 4‐AP resulted in complex neuronal network behaviours in parasagittal slices of the mEC, consisting of both ictal‐ and interictal‐like spike‐wave discharges (Fig. 1). This combination of brief and prolonged epileptiform activity has been extensively studied previously, both in the entorhinal cortex (D'Antuono et al. 2010; Avoli et al. 2013b; Lévesque et al. 2016) and in other brain regions such as the hippocampus (Nagao et al. 1996; Gonzalez‐Sulser et al. 2011; Berretta et al. 2012). Nevertheless, the propagation of this activity within the entorhinal cortex has not previously been studied. Indeed, many of these previous studies have often considered the entorhinal cortex as a homogeneous structure. Using multi‐site extracellular recording techniques, we first studied the interictal‐like events which generally propagated along the full extent of the dorso‐ventral axis of the mEC. By detecting individual bursts and statistically grouping them on the basis of the relative time of the waveform peak, we established that interictal‐like discharges could be generated at multiple points along the dorso‐ventral axis (Fig. 3). Furthermore, we found that these bursts propagated from the site of origin to the furthest extent of our recording probes (maximum distance 1.5 mm) within a few tenths of a second.
In contrast, the slow time frame of the spread of the ictal‐like activity was surprising. On average ictal‐like events initiated in the ventral mEC spread dorsally with a velocity of ∼160 μm s−1, taking ∼15 s to propagate to the most dorsal aspects of mEC. Similar results were obtained using Mg2+‐free aCSF. Since interictal events appear to show no preference for directionality in this model, this may suggest that these different facets of epileptiform activity are governed by different mechanisms. Nevertheless, it is unclear from our results whether the precise timing of interictal events along the sagittal plane influenced the initiation of seizure activity under these conditions. The stereotyped propagation of ictal‐like events from ventral to dorsal mEC was evident in both the low (10–50 Hz) and the high (50–250 Hz) frequency components of ictal waveforms (Fig. 2 D–F). The presence of high frequency activity along the entire dorso‐ventral axis suggests active recruitment of these areas to the ictal core, since this activity is more likely to represent the direct firing of a substantial population of mEC neurons in response to hyperexcitability (Schevon et al. 2012; Weiss et al. 2013, 2015). Once ictal‐like behaviour was apparent in both dorsal and ventral poles of the mEC, spike‐wave discharges became tightly synchronized, albeit with the ventral burst generally preceding the dorsal by a few milliseconds. This was the case for both 4‐AP‐ and 0‐Mg2+‐induced seizures, although the latter showed slightly more variability with respect to within‐burst directionality. Specifically, in 1/6 slices we did observe a late switch from ventral‐leading to dorsal‐leading ‘intra‐ictal’ discharges as has been reported previously in neocortical slices prepared from juvenile mice (Trevelyan et al. 2007a), although this was not the case in the majority of our recordings. The ictal‐like events recorded by Trevelyan et al. (2007a) do not appear to display any common directionality, so, as with the initiation of seizure events, it may be that the specific functional anatomical organization of the mEC lends itself to intra‐ictal events generally maintaining a ventral‐to‐dorsal directionality throughout an ictal discharge. It is also possible that differences in the age of the mice from which slices were prepared may play a role in the discrepancy between our data (aged 6–12 weeks) and those of Trevelyan et al. (aged 11–18 days), as dorsal and ventral mEC neurons undergo differential functional developmental changes during this period (Boehlen et al. 2010), which may affect the dynamics of burst propagation.
Given that axonal action potential conduction velocity and synaptic transmission are several orders of magnitude faster than the ictal propagation speed, we reasoned that some other physiological process must constrain the spread of activity. We consequently hypothesized that ictal propagation is constrained by differential levels of GABAergic control along the dorso‐ventral axis of the mEC (Beed et al. 2013; Booth et al. 2016). In support of this, we found that application of pharmacological agents that increased (diazepam) or decreased (Ro19‐4603) postsynaptic GABAA receptor activation, respectively, reduced or increased the slope of ictal initiation (Fig. 6). In this context, it is pertinent to note that mEC stellate cells are unlikely to form large numbers of recurrent excitatory connections, with less than 1 in 500 pairs of stellate cells being synaptically coupled (Couey et al. 2013; Pastoll et al. 2013). Fast spiking GABAergic interneurons, in contrast, form a powerful recurrent inhibitory circuit, with stellate cells connecting primarily to interneurons which in turn predominantly project back onto other stellate cells (Couey et al. 2013; Buetfering et al. 2014). In this situation, the anatomical arrangement of such inhibitory connections will have strong implications for the generation of epileptiform events. Dorsal mEC stellate cells receive a greater number of inhibitory inputs than those in ventral mEC, but perhaps more significantly, they receive a greater proportion of their inputs from more distal inhibitory neurons (Beed et al. 2013). This would therefore suggest that ictal events would need to overcome an increasing level of feed‐forward inhibition as they travel from ventral to dorsal mEC.
Numerous reports suggest the activity of GABAergic interneurons regulates seizure‐like activity (Dichter & Spencer, 1969; Prince & Wong, 1981; Schwartz & Bonhoeffer, 2001; Trevelyan et al. 2006, 2007b; Trevelyan, 2009). The period immediately before ictal events can be characterized by an increased interneuron firing that reaches its peak at ictal onset, while the activity of principal cells does not change until after initiation (Ziburkus et al. 2006; Lévesque et al. 2016). The activation of PV‐positive GABAergic interneurons is capable of suppressing ictal seizure propagation in cortico‐hippocampal circuits, through feed‐forward inhibition (Trevelyan et al. 2006; Schevon et al. 2012; Cammarota et al. 2013; Sessolo et al. 2015; Lu et al. 2016). However, under certain conditions activation of these cells may favour seizure initiation (Avoli & de Curtis, 2011; Sessolo et al. 2015). These reports may suggest, in contrast to our findings, that ictal‐like activity would be more likely to originate from areas where inhibition is more dominant. However, it is important to distinguish between the spread, or recruitment, of ictal‐like activity and the direct trigger for seizure initiation, which may still occur upstream of the recording location. Furthermore, it is unclear whether relative differences in inhibitory expression would be relevant in these conditions. Since there is still considerable inhibition in the ventral mEC (Beed et al. 2013), the dorso‐ventral organization of the mEC does not necessarily preclude the interneuron‐mediated initiation of ictal events in ventral areas, which may be more vulnerable due to the higher levels of intrinsic neuronal excitability in this region (Garden et al. 2008; Giocomo & Hasselmo, 2009; Boehlen et al. 2010; Booth et al. 2016). Nevertheless, taken together, our findings suggest that GABAergic systems act to control the propagation of seizure‐like events. Coupled with the high density of PV‐positive staining in the dorsal mEC, this may suggest that the dorsal mEC would be less likely to initiate an ictal bursts than the ventral mEC.
Epileptiform discharges have been shown to originate in both deep and superficial areas (Avoli et al. 2002). Our data suggest that, while ictal‐like activity consistently originates from ventral regions, there was no preference for bursting to initiate in either the deep or superficial layers of mEC. While this was the case, the location of burst initiation did not affect the dorsal–ventral propagation of ictal activity (Fig. 5). Inhibitory gradients have previously been reported only in superficial mEC layers (L2/3), with limited PV staining in layer 5 (Beed et al. 2013; Booth et al. 2016). It is therefore possible that it is the inhibitory organization of layer 2/3 that is largely responsible for the slow propagation across the dorso‐ventral axis of the mEC.
The intrinsic properties of mEC stellate cells are also likely to play a role in the organization of hyperexcitable activity. In this regard, it has been widely reported that ventral mEC stellate cells exhibit a higher input resistance, a slower membrane time constant and a lower action potential threshold compared to dorsal mEC stellate cells (Garden et al. 2008; Giocomo & Hasselmo, 2009; Boehlen et al. 2010; Booth et al. 2016). There are also numerous reports of gradients in I h along the dorso‐ventral axis of the mEC, such that I h is more prominent in the dorsal mEC stellate cells (Garden et al. 2008; Giocomo & Hasselmo, 2008; Boehlen et al. 2010). Combined, these cell‐intrinsic properties will produce higher levels of excitability in the ventral mEC, with less current required to produce action potential firing and greater levels of synaptic integration, in part due to the differences in I h‐mediated potentials (Garden et al. 2008). For example, seizure‐induced plastic reductions in I h in entorhinal cortex neurons result in substantial increases in neuronal excitability (Shah et al. 2004). Consequently, even in the absence of GABAergic inhibition, one might expect to observe an increased propensity for epileptiform bursting in the ventral compared to the dorsal mEC. We tested this hypothesis by incubating mEC slices in a blocker of GABAA receptors (picrotoxin) along with a glutamate receptor agonist (kainate) (Fig. 8). The treatment resulted in interictal‐like, but not ictal‐like, discharges. We found that not only did the disinhibition‐mediated interictal‐like discharges develop first in the ventral mEC, but that once bursts were established in both dorsal and ventral ends of the mEC, a cross‐correlation analysis of individual bursts revealed that the ventral bursts almost always preceded the dorsal bursts. Furthermore, when the dorsal and ventral poles of the mEC were physically separated with a scalpel cut, we found that bursts recorded from the ventral mEC were of a similar frequency to those in uncut slices, whilst bursts in the dorsal mEC were significantly less frequent than those in the uncut dorsal mEC (Fig. 9). Presumably, in the uncut slices, the dorsal mEC is entrained to the more frequent disinhibition‐mediated epileptiform bursts in the ventral mEC. Taken together, these data suggest that the intrinsic properties and/or excitatory synaptic transmission properties (which are intimately linked; Garden et al. 2008) of ventral mEC neurons predispose this region to seizure‐like activity, when compared to the dorsal mEC.
Dorso‐ventral gradients in mEC activity appear to be important for effective spatial information processing, but the anatomical organization of the mEC may leave it vulnerable to disease pathology. For example, we have previously reported that cellular and network properties of the dorsal mEC are preferentially disrupted in a mouse model of dementia (Booth et al. 2016). Equivalent changes to mEC physiology could also be present under prolonged epileptic conditions, this time with pathology most likely disrupting ventral mEC function. It remains to be seen whether results seen here in parasagittal slices are also relevant in the temporal lobe in vivo, either in rodent models or human patients. However, this study suggests that, within the entorhinal cortex, the ventral portion of the mEC is more likely to initiate seizure activity. Furthermore, investigating means to perturb communication between ventral and dorsal regions might disrupt seizure propagation in vivo, although this may also generate consequences for spatial navigation. Indeed, the relatively slow ventral– dorsal propagation of ictal activity potentially presents an opportunity to intervene in seizureogenisis. For example, it is possible to conceive of a scenario where hyperactivity is detected within the most ventral aspects of mEC and interventions activated, for example optogenetically, that serve to block dorsal‐wards spread and subsequent pan‐entorhinal hypersynchrony.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
Conception and design of the work: J.T.B, A.D.R, K.G.P, and T.R. Acquisition, analysis, and interpretation of data: T.R, P.M, J.T.B. Drafting the manuscript: T.R, J.T.B. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by a University of Exeter and Eli Lilly studentship (T.R). P.M. was supported by an MRC Proximity to Discovery award in partnership with AstraZeneca. K.G.P. was an employee of Eli Lilly. A.D.R. was part funded by a Royal Society Industrial Fellowship. J.T.B. was an Alzheimer's Research UK Senior Research Fellow (ARUK‐SRF2012‐6).
Biography
Tom Ridler received an undergraduate masters (MSci) in ‘Neuroscience with study in industry’ from the University of Bristol, during which time he spent a year working at Eli Lilly. He moved to Exeter in 2013 as a PhD student under the supervision of Dr Jon Brown and Prof Andrew Randall and is now employed as a post‐doctoral researcher in the Exeter Applied Neurophysiology group. He is interested in how neurological disorders such as epilepsy and dementia produce changes to neuronal networks that ultimately affect cognition. His current work focuses on the medial entorhinal cortex, which is essential for processing spatial information.

Edited by: Ole Paulsen & Matthew Nolan
Linked articles This article is highlighted by a Perspective by Parrish & Trevelyan. To read this Perspective, visit https://doi.org/10.1113/JP276184.
References
- Amaral D & Witter M (1989). The three‐dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience 31, 571–591. [DOI] [PubMed] [Google Scholar]
- Armand V, Rundfeldt C & Heinemann U (2000). Effects of retigabine (D‐23129) on different patterns of epileptiform activity induced by low magnesium in rat entorhinal cortex hippocampal slices. Epilepsia 41, 28–33. [DOI] [PubMed] [Google Scholar]
- Avoli M, D'Antuono M, Louvel J, Köhling R, Biagini G, Pumain R, D'Arcangelo G & Tancredi V (2002). Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol 68, 167–207. [DOI] [PubMed] [Google Scholar]
- Avoli M & de Curtis M (2011). GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog Neurobiol 95, 104–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, de Curtis M & Köhling R (2013a). Does interictal synchronization influence ictogenesis? Neuropharmacology 69, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, Panuccio G, Herrington R, D'Antuono M, de Guzman P & Lévesque M (2013b). Two different interictal spike patterns anticipate ictal activity in vitro. Neurobiol Dis 52, 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbarosie M & Avoli M (1997). CA3‐driven hippocampal‐entorhinal loop controls rather than sustains in vitro limbic seizures. J Neurosci 17, 9308–9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beed P, Gundlfinger A, Schneiderbauer S, Song J, Böhm C, Burgalossi A, Brecht M, Vida I & Schmitz D (2013). Inhibitory gradient along the dorsoventral axis in the medial entorhinal cortex. Neuron 79, 1197–1207. [DOI] [PubMed] [Google Scholar]
- Berretta N, Ledonne A, Mango D, Bernardi G & Mercuri NB (2012). Hippocampus versus entorhinal cortex decoupling by an NR2 subunit‐specific block of NMDA receptors in a rat in vitro model of temporal lobe epilepsy. Epilepsia 53, e80–e84. [DOI] [PubMed] [Google Scholar]
- Boehlen A, Heinemann U & Erchova I (2010). The range of intrinsic frequencies represented by medial entorhinal cortex stellate cells extends with age. J Neurosci 30, 4585–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnevie T, Dunn B, Fyhn M, Hafting T, Derdikman D, Kubie JL, Roudi Y, Moser EI & Moser M‐B (2013). Grid cells require excitatory drive from the hippocampus. Nat Neurosci 16, 309–317. [DOI] [PubMed] [Google Scholar]
- Booth CA, Ridler T, Murray TK, Ward MA, de Groot E, Goodfellow M, Phillips KG, Randall AD & Brown JT (2016). Electrical and network neuronal properties are preferentially disrupted in dorsal, but not ventral, medial entorhinal cortex in a mouse model of tauopathy. J Neurosci 36, 312–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragdon AC, Kojima H & Wilson WA (1992). Suppression of interictal bursting in hippocampus unleashes seizures in entorhinal cortex: a proepileptic effect of lowering [K+]0 and raising [Ca2+]0 . Brain Res 590, 128–135. [DOI] [PubMed] [Google Scholar]
- Brun VH, Leutgeb S, Wu H‐Q, Schwarcz R, Witter MP, Moser EI & Moser M‐B (2008). Impaired spatial representation in CA1 after lesion of direct input from entorhinal cortex. Neuron 57, 290–302. [DOI] [PubMed] [Google Scholar]
- Buetfering C, Allen K & Monyer H (2014). Parvalbumin interneurons provide grid cell‐driven recurrent inhibition in the medial entorhinal cortex. Nat Neurosci 17, 710–718. [DOI] [PubMed] [Google Scholar]
- Cammarota M, Losi G, Chiavegato A, Zonta M & Carmignoto G (2013). Fast spiking interneuron control of seizure propagation in a cortical slice model of focal epilepsy. J Physiol 591, 807–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto CB, Wouterlood FG & Witter MP (2008). What does the anatomical organization of the entorhinal cortex tell us? Neural Plast 2008, 381243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couey JJ, Witoelar A, Zhang S‐J, Zheng K, Ye J, Dunn B, Czajkowski R, Moser M‐B, Moser EI, Roudi Y & Witter MP (2013). Recurrent inhibitory circuitry as a mechanism for grid formation. Nat Neurosci 16, 318–324. [DOI] [PubMed] [Google Scholar]
- D'Antuono M, Köhling R, Ricalzone S, Gotman J, Biagini G & Avoli M (2010). Antiepileptic drugs abolish ictal but not interictal epileptiform discharges in vitro. Epilepsia 51, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichter M & Spencer WA (1969). Penicillin‐induced interictal discharges from the cat hippocampus. I. Characteristics and topographical features. J Neurophysiol 32, 649–662. [DOI] [PubMed] [Google Scholar]
- Dodson PD, Pastoll H & Nolan MF (2011). Dorsal‐ventral organization of theta‐like activity intrinsic to entorhinal stellate neurons is mediated by differences in stochastic current fluctuations. J Physiol 589, 2993–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden DLF, Dodson PD, O'Donnell C, White MD & Nolan MF (2008). Tuning of synaptic integration in the medial entorhinal cortex to the organization of grid cell firing fields. Neuron 60, 875–889. [DOI] [PubMed] [Google Scholar]
- Giocomo LM & Hasselmo ME (2008). Time constants of h current in layer II stellate cells differ along the dorsal to ventral axis of medial entorhinal cortex. J Neurosci 28, 9414–9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giocomo LM & Hasselmo ME (2009). Knock‐out of HCN1 subunit flattens dorsal‐ventral frequency gradient of medial entorhinal neurons in adult mice. J Neurosci 29, 7625–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giocomo LM, Stensola T, Bonnevie T, Van Cauter T, Moser M‐B & Moser EI (2014). Topography of head direction cells in medial entorhinal cortex. Curr Biol 24, 252–262. [DOI] [PubMed] [Google Scholar]
- Giocomo LM, Zilli EA, Fransén E & Hasselmo ME (2007). Temporal frequency of subthreshold oscillations scales with entorhinal grid cell field spacing. Science 315, 1719–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnatkovsky V, Librizzi L, Trombin F & de Curtis M (2008). Fast activity at seizure onset is mediated by inhibitory circuits in the entorhinal cortex in vitro. Ann Neurol 64, 674–686. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Sulser A, Wang J, Motamedi GK, Avoli M, Vicini S & Dzakpasu R (2011). The 4‐aminopyridine in vitro epilepsy model analyzed with a perforated multi‐electrode array. Neuropharmacology 60, 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás‐Kovács A, Dóczi J, Tarnawa I, Détári L, Banczerowski‐Pelyhe I & Világi I (2002). Comparison of spontaneous and evoked epileptiform activity in three in vitro epilepsy models. Brain Res 945, 174–180. [DOI] [PubMed] [Google Scholar]
- Hafting T, Fyhn M, Molden S, Moser M‐BB & Moser EI (2005). Microstructure of a spatial map in the entorhinal cortex. Nature 436, 801–806. [DOI] [PubMed] [Google Scholar]
- Jones RSG (1989). Ictal epileptiform events induced by removal of extracellular magnesium in slices of entorhinal cortex are blocked by baclofen. Exp Neurol 104, 155–161. [DOI] [PubMed] [Google Scholar]
- Jones RS & Heinemann U (1988). Synaptic and intrinsic responses of medical entorhinal cortical cells in normal and magnesium‐free medium in vitro. J Neurophysiol 59, 1476–1496. [DOI] [PubMed] [Google Scholar]
- Kropff E, Carmichael JE, Moser M‐B & Moser EI (2015). Speed cells in the medial entorhinal cortex. Nature 523, 419–424. [DOI] [PubMed] [Google Scholar]
- Lévesque M, Herrington R, Hamidi S & Avoli M (2016). Interneurons spark seizure‐like activity in the entorhinal cortex. Neurobiol Dis 87, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losi G, Marcon I, Mariotti L, Sessolo M, Chiavegato A & Carmignoto G (2015). A brain slice experimental model to study the generation and the propagation of focally‐induced epileptiform activity. J Neurosci Methods 260, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhong C, Wang L, Wei P, He W, Huang K, Zhang Y, Zhan Y, Feng G & Wang L (2016). Optogenetic dissection of ictal propagation in the hippocampal–entorhinal cortex structures. Nat Commun 7, 10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao T, Alonso A & Avoli M (1996). Epileptiform activity induced by pilocarpine in the rat hippocampal‐entorhinal slice preparation. Neuroscience 72, 399–408. [DOI] [PubMed] [Google Scholar]
- Navratilova Z, Giocomo LM, Fellous J‐M, Hasselmo ME & McNaughton BL (2012). Phase precession and variable spatial scaling in a periodic attractor map model of medial entorhinal grid cells with realistic after‐spike dynamics. Hippocampus 22, 772–789. [DOI] [PubMed] [Google Scholar]
- O'Reilly KC, Flatberg A, Islam S, Olsen LC, Kruge IU & Witter MP (2015). Identification of dorsal–ventral hippocampal differentiation in neonatal rats. Brain Struct Funct 220, 2873–2893. [DOI] [PubMed] [Google Scholar]
- Pastoll H, Ramsden HL & Nolan MF (2012). Intrinsic electrophysiological properties of entorhinal cortex stellate cells and their contribution to grid cell firing fields. Front Neural Circuits 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastoll H, Solanka L, van Rossum MCW & Nolan MF (2013). Feedback inhibition enables theta‐nested gamma oscillations and grid firing fields. Neuron 77, 141–154. [DOI] [PubMed] [Google Scholar]
- Prince D & Wong R (1981). Human epileptic neurons studied in vitro. Brain Res 210, 323–333. [DOI] [PubMed] [Google Scholar]
- Schevon CA, Weiss SA, McKhann G, Goodman RR, Yuste R, Emerson RG & Trevelyan AJ (2012). Evidence of an inhibitory restraint of seizure activity in humans. Nat Commun 3, 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TH & Bonhoeffer T (2001). In vivo optical mapping of epileptic foci and surround inhibition in ferret cerebral cortex. Nat Med 7, 1063–1067. [DOI] [PubMed] [Google Scholar]
- Sessolo M, Marcon I, Bovetti S, Losi G, Cammarota M, Ratto GM, Fellin T & Carmignoto G (2015). Parvalbumin‐positive inhibitory interneurons oppose propagation but favor generation of focal epileptiform activity. J Neurosci 35, 9544–9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X & Johnston D (2004). Seizure‐induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron 44, 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solstad T, Boccara CN, Kropff E, Moser M‐B & Moser EI (2008). Representation of geometric borders in the entorhinal cortex. Science 322, 1865–1868. [DOI] [PubMed] [Google Scholar]
- Stensola H, Stensola T, Solstad T, Frøland K, Moser M‐B & Moser EI (2012). The entorhinal grid map is discretized. Nature 492, 72–78. [DOI] [PubMed] [Google Scholar]
- Traub RD, Jefferys JG & Whittington MA (1994). Enhanced NMDA conductance can account for epileptiform activity induced by low Mg2+ in the rat hippocampal slice. J Physiol 478, 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ (2009). The direct relationship between inhibitory currents and local field potentials. J Neurosci 29, 15299–15307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Baldeweg T, van Drongelen W, Yuste R & Whittington M (2007a). The source of afterdischarge activity in neocortical tonic‐clonic epilepsy. J Neurosci 27, 13513–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Watson BO & Yuste R (2006). Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci 26, 12447–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D & Yuste R (2007b). Feedforward inhibition contributes to the control of epileptiform propagation speed. J Neurosci 27, 3383–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss SA, Banks GP, McKhann GM, Goodman RR, Emerson RG, Trevelyan AJ & Schevon CA (2013). Ictal high frequency oscillations distinguish two types of seizure territories in humans. Brain 136, 3796–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss SA, Lemesiou A, Connors R, Banks GP, McKhann GM, Goodman RR, Zhao B, Filippi CG, Nowell M, Rodionov R, Diehl B, McEvoy AW, Walker MC, Trevelyan AJ, Bateman LM, Emerson RG & Schevon CA (2015). Seizure localization using ictal phase‐locked high gamma: A retrospective surgical outcome study. Neurology 84, 2320–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington MA, Traub RD & Jefferys JG (1995). Erosion of inhibition contributes to the progression of low magnesium bursts in rat hippocampal slices. J Physiol 486, 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong G & Skolnick P (1992). High affinity ligands for “diazepam‐insensitive” benzodiazepine receptors. Eur J Pharmacol 225, 63–68. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Jochems A & Hasselmo ME (2013). Comparison of properties of medial entorhinal cortex layer II neurons in two anatomical dimensions with and without cholinergic activation. PLoS One 8, e73904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziburkus J, Cressman JR, Barreto E & Schiff SJ (2006). Interneuron and pyramidal cell interplay during in vitro seizure‐like events. J Neurophysiol 95, 3948–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]