

Abstract
Tribbles pseudokinase 1 (Trib1) is a negative regulator of CCAAT/enhancer binding protein α (C/EBPα) and is known to induce granulopoiesis while suppressing monocyte differentiation. Loss of Trib1 was previously shown to increase the neutrophil population in the spleen but lead to M2‐like macrophage reduction. Because M2 macrophages are anti‐inflammatory and promote tissue repair by producing fibrogenic factors, we investigated liver fibrosis in Trib1‐deficient mice. Interestingly, loss of Trib1 suppressed fibrosis in the CCl4‐induced chronic liver injury model. Trib1 knockout increased neutrophils but had a minimal effect on the macrophage population in the liver. Hepatic expressions of neutrophil matrix metalloproteinases (Mmp)8 and Mmp9 were increased, but the production of fibrogenic factors, including transforming growth factor β1, was not affected by loss of Trib1. These results suggest that neutrophils are responsible for the suppression of fibrosis in Trib1‐deficient liver. Consistently, transplantation of Trib1‐deficient bone marrow cells into wild‐type mice alleviated CCl4‐induced fibrosis. Furthermore, expression of chemokine (C‐X‐C motif) ligand 1 (Cxcl1) by adeno‐associated viral vector in the normal liver recruited neutrophils and suppressed CCl4‐induced fibrosis; infusion of wild‐type neutrophils in CCl4‐treated mice also ameliorated fibrosis. Using recombinant adeno‐associated virus‐mediated expression of Mmp8 and Mmp9 alleviated liver fibrosis. Finally, neutrophil depletion by infusion of Ly6G antibody significantly enhanced CCl4‐induced fibrosis. Conclusion: While neutrophils are well known to exacerbate acute liver injury, our results demonstrate a beneficial role of neutrophils in chronic liver injury by promoting fibrolysis. (Hepatology Communications 2018;2:703‐717)
Abbreviations
- α‐SMA
α‐smooth muscle actin
- ALT
alanine aminotransferase
- C/EBPα
CCAAT‐enhancer‐binding protein α
- CD
clusters of differentiation
- cDNA
complementary DNA
- Cxcl1
chemokine (C‐X‐C motif) ligand 1
- Cyp2e1
cytochrome P450 2E1
- ECM
extracellular matrix
- FACS
fluorescence‐activated cell sorting
- HSC
hepatic stellate cell
- KO
knockout
- MMP
matrix metalloproteinase
- MSC
mesenchymal stem cell
- qRT‐PCR
quantitative reverse‐transcription polyacrylamide chain reaction
- rAAV
recombinant adeno‐associated virus
- TGF‐β
transforming growth factor β
- Trib1
Tribbles pseudokinase 1
Fibrosis is an essential process for tissue repair that encapsulates the damaged area. In the liver, chronic inflammation induced by a variety of causes, such as hepatitis virus, alcohol, drugs, and immune and metabolic disorders, accompanies fibrosis that often advances to cirrhosis.1 Damaged hepatocytes release factors that activate hepatic stellate cells (HSCs) directly and indirectly by nonparenchymal cells. Upon activation, HSCs become myofibroblasts and produce extracellular matrix (ECM). Progression of fibrosis is regulated by a balance between production and degradation of ECM. Transforming growth factor β (TGF‐β) produced by nonparenchymal cells in damaged liver plays a pivotal role in fibrogenesis by stimulating production of ECM from HSCs.2
Macrophages also play a central role for pathogenesis of chronic liver injury by phagocytosis of apoptotic liver cells, promoting inflammation, and producing matrix metalloproteinases (MMPs) that are responsible for the degradation of ECM.3 Hepatic macrophages are a heterogeneous cell population that includes the liver resident macrophages, Kupffer cells, and bone marrow‐derived macrophages.4 As they interact with a variety of cell types, they can be profibrotic as well as antifibrotic.5 While classification of macrophages has been a subject of debate, at least two macrophage subtypes have been recognized. M1 macrophages are proinflammatory, whereas M2 macrophages are anti‐inflammatory and promote tissue repair by producing fibrogenic factors.5 There are also many reports showing a scar‐resolving macrophage population that does not fit the M1 or M2 phenotype.6
Neutrophils are the most abundant type of granulocytes and are derived from the bone marrow progenitor cells that also give rise to monocytes. Neutrophils are a type of phagocyte and play a major role in defense against bacterial and fungal infection. They are rapidly recruited to the site of injury and are involved in pathogenesis by enhancing inflammation.7 In the liver, neutrophils are well known to exacerbate acute liver injury, such as ischemia/reperfusion and alcoholic injury.8 However, the role of neutrophils in chronic liver injury accompanying fibrosis remains obscure.
Tribbles pseudokinase 1 (Trib1) is one of the three members of the Tribbles family that is characterized by having pseudokinase domains that function as a scaffold.9 Trib1 plays a role in the degradation of CCAAT/enhancer binding protein α (C/EBPα), a crucial transcription factor for myeloid cell development, by recruiting it to E3 ubiquitin ligase. Liver‐specific knockout (KO) of Trib1 increases the hepatic expression of C/EBPα.10 Loss of C/EBPα expression inhibits the formation of granulocytes and leads to a concomitant increase in self‐renewal of hematopoietic stem cells. Expression of C/EBPα initiates with the commitment of multipotential precursors to the myeloid lineage. C/EBPα is specifically up‐regulated during granulocytic differentiation and is rapidly down‐regulated during the alternative monocytic pathway. Conditional expression of C/EBPα alone in stably transfected bipotential myeloid cells triggers neutrophilic differentiation.11 Thus, Trib1 deficiency may exhibit a phenotype similar to overexpression of C/EBPα. In fact, Satoh et al. showed that Trib1 KO increased the neutrophil population in the spleen whereas M2‐like macrophages and eosinophils were reduced.12 As M2 macrophages are anti‐inflammatory and promote tissue repair by producing fibrogenic factors, we were interested in evaluating liver fibrosis in the Trib1‐deficient mouse.
CCl4‐induced liver injury has been extensively used as a model of acute and chronic liver injury.13 A single injection of CCl4 induces centrilobular necrosis by a reactive free radical metabolite, which is generated by cytochrome P450 2E1 (Cyp2e1) in centrilobular hepatocytes, and causes transient activation of HSCs as a wound healing response. This is followed by rapid repair of the necrotic area by proliferation of the remaining parenchymal hepatocytes in a few days. However, repeated administration of CCl4 results in continuous activation of HSCs, leading to fibrosis. In this study, we investigated the role of Trib1 in fibrosis by using this model. Although the expression level of fibrogenic cytokine TGF‐β1 was comparable between wild‐type and Trib1 KO liver, the fibrotic area was significantly decreased in Trib1 KO liver. Extensive quantitative reverse‐transcription polyacrylamide chain reaction (qRT‐PCR) analysis revealed marked increases of the neutrophil MMPs Mmp8 and Mmp9 in Trib1 KO liver. Moreover, transplantation of Trib1 KO bone marrow cells suppressed liver fibrosis of wild‐type liver. Interestingly, recombinant adeno‐associated virus (rAAV)‐mediated expression of chemokine (C‐X‐C motif) ligand 1 (Cxcl1), a neutrophil chemoattractant, also suppressed liver fibrosis. Consistently, adoptive transfer of wild‐type neutrophils and rAAV‐mediated expression of Mmp8 and Mmp9 in CCl4‐induced fibrotic mice dramatically alleviated liver fibrosis. Immunodepletion of neutrophils significantly increased CCl4‐induced liver fibrosis. These results reveal an antifibrotic role for neutrophils in chronic liver injury.
Materials and Methods
MICE
C57BL/6 mice were obtained from CLEA Japan (Tokyo, Japan). Trib1‐deficient mice were provided by Dr. Akira Shizuo (Laboratory of Host Defense, World Premier International Immunology Frontier Research Center, Osaka University, Japan). Wild‐type littermates were used as controls. CD45.1 congenic C57BL/6 mice (RBRC 00144) were provided by the RIKEN BioResource Center through the National Bio‐Resource Project of the Ministry of Education, Culture, Sports, Science, and Technology, Japan. All mice were fed a standard chow diet, maintained on a 12‐hour light/dark cycle with free access to food and water unless otherwise indicated, and killed at 2:00 pm in the nonfasted state.
For bone marrow transplantation studies, pregnant Ly5.1 mice were injected subcutaneously with busulfan (15 mg/kg) at days 17 and 18 of gestation, a procedure that ablates hematopoietic stem cells in the neonates. The conditioned neonatal mice were injected intraperitoneally with 1 × 106 unmanipulated bone marrow cells obtained from femurs and tibias of Trib1‐deficient mice and littermate wild‐type mice within 24 hours of birth. All animal experiments were performed in accordance with our institutional guidelines.
INDUCTION OF ACUTE LIVER INJURY AND LIVER FIBROSIS
Acute liver injury was induced by a single intraperitoneal injection of CCl4. CCl4 (Wako Pure Chemical, Osaka, Japan) was diluted in corn oil (Wako) to 20% and injected into mice at a dose of 1 mL/kg. Liver fibrosis was induced by repeated injection of CCl4 2 times per week for 4 weeks unless otherwise indicated. Livers were harvested 3 days after the final CCl4 injection.
RECOMBINANT AAV VECTORS
rAAV vectors were produced in human embryonic kidney 293 cells as described.14 To create the rAAV expression vectors, the green fluorescent protein transgene was removed from plasmid liver‐specific promoter 1(pLSP1)‐enhanced green fluorescent protein (courtesy of Ian Alexander, Children's Medical Research Institute, Westmead, NSW, Australia) to obtain the rAAV vector backbone. Genes of interest (Cxcl1, Cre recombinase [Cre], Mmp8, Mmp9) were amplified by qPCR and cloned into the pLSP1 vector (primer sequences described below). All vectors were pseudoserotyped with adeno‐associated virus serotype 8 by calcium phosphate transfection with p5E18‐VD2/8 (courtesy of James M Wilson, University of Pennsylvania, Philadelphia, PA) and pXX6‐80 (courtesy of Jude Samulski, University of North Carolina, Chapel Hill, NC). The vectors generated were named as rAAV.Cxcl1, rAAV.Cre, rAAV.Mmp8, and rAAV.Mmp9, according to the encoded transgene.
INDUCED EXPRESSION OF Cxcl1, Mmp8, AND Mmp9 IN HEPATOCYTES
For hepatocyte‐specific gene delivery, mice were injected intraperitoneally with rAAV vectors encoding Cxcl1, Mmp8, or Mmp9 complementary DNA (cDNA). The primer pairs used for the expression vector are listed in Table 1.
Table 1.
Primer Sequences Used for This Study
| For qPCR: |
| Actb; 5′‐ccaaccgtgaaaagatgacc‐3′, 5′‐ccagaggcatacagggacag‐3′ |
| Cyp2e1; 5′‐ccaccagcacaactctgagata‐3′, 5′‐cccaataaccctgtcaatttctt‐3′ |
| Acta2; 5′‐gacaccacccacccagagt‐3′, 5′‐acatagctggagcagcgtct‐3′ |
| Tgfb1; 5′‐tcagacattcgggaagcagt‐3′, 5′‐acgccaggaattgttgctat‐3′ |
| Timp1; 5′‐gcaaagagctttctcaaagacc‐3′, 5′‐agggatagataaacagggaaacact‐3′ |
| Mmp8; 5′‐cactccgtggggagatttac‐3′, 5′‐aagaccgttgggtaggaagg‐3′ |
| Mmp9; 5′‐tgactacgataaggacggcaaa‐3′, 5′‐agatgaacgggaacacacagg‐3′ |
| Mmp13; 5′‐ctggcacacgcttttcctc‐3′, 5′‐atgggcagcaacaataaacaag‐3′ |
| Cxcl1; 5′‐ggattcacctcaagaacatccag‐3′, 5′‐atcttttggacaattttctgaacc‐3′ |
| Pdgfb; 5′‐cggcctgtgactagaagtcc‐3′, 5′‐gagcttgaggcgtcttgg ‐3′ |
| Il1b; 5′‐agttgacggaccccaaaag ‐3′, 5′‐agctggatgctctcatcagg ‐3′ |
| Il17a; 5′‐tgtgaaggtcaacctcaaagtc ‐3′, 5′‐gagggatatctatcagggtcttca ‐3′ |
| Cxcl2; 5′‐ccaaaagatactgaacaaaggcaag ‐3′, 5′‐ggcacatcaggtacgatcca ‐3′ |
| For expression vector construction: |
| Cre; 5′‐acgaattcgccgccaccatggtgcccaa ‐3′, 5′‐gtgatatctcagtccccatcctc ‐3′ |
| Cxcl1; 5′‐gaattcatgatcccagccacccgctcgc‐3′, 5′‐aagcttttacttggggacaccttttagc‐3′ |
| Mmp8;5′‐gaattcatgtttcgcctgaagacacttccattac‐3′, 5′‐cccgggctatgaacagttaagccataaattgc‐3′ |
| Mmp9;5′‐gaattcatgagtccctggcagcccctgctcctggc‐3′, 5′‐aagcttcaagggcactgcaggaggtcgtaggtc‐3′ |
HEMATOXYLIN AND EOSIN, SIRIUS RED, AND IMMUNOSTAINING
Liver cryosections (8 μm) were mounted on glass slides, and hematoxylin and eosin and sirius red staining were performed as described.15 Immunostaining was performed by cold acetone fixation. The fixed sections were incubated with ImmunoBlock (DS Pharma Biomedical) and then incubated with Ly6G (BioLegend) or F4/80 (BioLegend) and collagen I (Bio‐Rad, Hercules, CA) or Mmp9 (Abcam) antibodies, followed by Alexa555‐labeled anti‐rat and Alexa488‐labeled anti‐rabbit secondary antibodies (Molecular Probes). Images were captured using Observer Z1 with an AxioCam HRc (Zeiss, Oberkochen, Germany) except for quantification of sirius red‐positive areas, which were analyzed using cellSens software (Olympus, Tokyo, Japan). Ly6G‐positive (+) or F4/80+ cell numbers were counted using Image J software (for Figs. 2, 3, 4, 5). Ly6G and MMP9 double‐positive cell numbers were counted using the BZ‐X700 hybrid cell count software (Keyence, Osaka, Japan).
Figure 2.

Increased liver‐resident neutrophils in Trib1 KO mice. (A) Flow cytometric analysis of nonparenchymal cells from untreated liver. Numbers indicate the percentage of cells in the gate. The CD11b+ F4/80+ macrophage population was slightly reduced but the CD11b+ Ly6G+ neutrophil population was dramatically increased in Trib1 KO liver. (B) Immunohistochemical staining for Ly6G or F4/80 in untreated liver. Graphs show Ly6G+ or F4/80+ cell numbers in each field. Counting of Ly6G+ or F4/80+ cells was carried out with Image J analysis software (n = 4 per group; magnification ×10). (C,D) Mice were treated with CCl4 2 times per week for 4 weeks; this was followed by harvesting of the liver. Immunostaining for (C) Ly6G (red) or (D) F4/80 (red) and collagen type 1) (green) in liver. Graphs show Ly6G+ or F4/80+ cell numbers in each field (n = 7 per group; magnification ×10). Data are expressed as means ± SEM. *P < 0.05, ***P < 0.001. Abbreviations: ColI, collagen type 1; WT, wild type.
Figure 3.

Suppression of liver fibrosis by transplantation of Trib1 KO bone marrow cells. Ly5.1 mice received wild‐type or Trib1 KO bone marrow transplants following busulfan treatment. CCl4 was injected into each mouse 2 times per week for 4 weeks; this was followed by harvesting of the liver. (A) Immunostaining for Ly6G in liver sections. (B) Ly6G+ cell numbers in each field (WT BMT, n = 6; KO BMT, n = 5; magnification ×10). (C) Expression levels of Mmp8 and Mmp9 in whole liver (WT BMT, n = 6; KO BMT, n = 5). (D) Histochemical staining for collagen in liver sections. Photomicrographs illustrate hepatic collagen fibrils stained with sirius red. These pictures were represented by 21 serial tiling images (each magnification ×20). (E) Quantification of collagen‐positive area in liver sections as shown in (C) (WT BMT, n = 6; KO BMT, n = 5). Data are expressed as means ± SEM. **P < 0.01, ***P < 0.001. Abbreviations: BMT, bone marrow transplant; SR, sirius red; WT, wild type.
Figure 4.

Recruitment of neutrophils to the liver suppresses fibrosis. Cxcl1 was expressed in liver by administration of rAAV.Cxcl1 (1 × 1011 vg per mouse) 2 weeks before CCl4 administration; CCl4 was then administrated for 4 weeks. (A) Expression levels of Cxcl1, Mmp8, and Mmp9 in whole liver (n = 6 per group). (B) Collagen accumulation in the liver treated with CCl4 for 4 weeks. Photomicrographs illustrate hepatic collagen fibrils stained with sirius red. These pictures were represented by 21 serial tiling images (each magnification ×20). (C) Quantification of collagen‐positive area in liver sections as shown in (B) (n = 6 per group). (D) Immunostaining for Ly6G in liver sections treated with CCl4 for 4 weeks (magnification ×10). (E) Ly6G+ cell numbers in each field (n = 6 per group). Data are expressed as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviation: SR, sirius red.
Figure 5.

Infusion of wild‐type neutrophils suppresses CCl4‐induced liver fibrosis. Mice were treated with CCl4 2 times per week for 4 weeks; this was followed by injection of bone marrow‐derived neutrophils (5 × 106 cells/mouse) into the tail vein. PBS indicates control animals injected with PBS and Neu indicates neutrophil‐infused mice. (A) Sirius red staining of collagen in liver (left). Quantification of collagen‐positive area in liver sections (right) (n = 6 per group; ***P < 0.001). These pictures were represented by 21 serial tiling images (each magnification ×20). (B) Expression levels of Mmp8 and Mmp9 in whole liver (n = 6 per group; ***P < 0.001). (C) Mice were treated with CCl4 for 4 weeks followed by injection of 5 × 106 cells of bone marrow‐derived neutrophils. Neutrophil (red) in the liver at 1 hour after neutrophil infusion (magnification ×10). (D) Ly6G+ cell numbers in each field (PBS, n = 4; Neu, n = 5). Data are expressed as means ± SEM. **P < 0.01. Abbreviations: Neu, neutrophil; PBS, phosphate‐buffered saline; SR, sirius red.
WESTERN BLOTTING
Whole liver lysates were used for western blot analysis. Liver samples were sonicated in ice‐cold lysis buffer containing 10 mM CaCl2 and 0.2% Triton X‐100. We used liver microsomal fractions for Cyp2e1 analysis. The lysate was centrifuged at 10,000g for 10 minutes at 4°C, the supernatant was collected, and the protein concentration was determined by the Lowry assay (Bio‐Rad). Twenty micrograms of total protein extract was loaded onto a 10% polyacrylamide gel, separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and transferred to a Immobilon‐P polyvinylidene fluoride membrane (Millipore). After washing with trishydroxymethylaminomethane‐buffered saline containing 5% skimmed milk and 0.2% Tween 20, the membranes were incubated with antibody against α‐smooth muscle actin (αSMA; 1:400), actin (1:1,000), Mmp8 (1:2,000), Mmp9 (1:1,000), or Cyp2e1 (1:5, 000) overnight at 4°C.
After washing with trishydroxymethylaminomethane‐buffered saline Tween 20, the membranes were probed with a horseradish peroxidase‐conjugated secondary antibody (Amersham Biosciences, Little Chalfont, United Kingdom), and immunodetection was performed with a Western Lightning Plus enhanced chemiluminescence reagent (Perkin Elmer). Following immunodetection, membranes were incubated in Coomassie Brilliant blue as a loading control for protein quantification and signals were quantified with a luminoimage analyzer (LAS 4100; Fuji Film, Tokyo, Japan). Band intensity was measured using the Image J software.
GELATIN ZYMOGRAPHY
MMP9 activity of whole liver was detected with a gelatin‐zymography kit (Primary Cell, Sapporo, Japan) according to the manufacturer's instructions. In brief, each sample (20 μg), identical to that used for western blot analysis, was loaded for electrophoresis. Gels were washed and incubated 20 hours in incubation buffer (attached with kit) at 37°C, stained with Coomassie Brilliant blue, and scanned.
RT‐PCR AND REAL‐TIME PCR
Total RNA was isolated from mouse liver or blood cells using TRIzol reagent (Invitrogen). RT to cDNA templates was performed using PrimeScript RT Master Mix (Takara Bio, Kyoto, Japan). Real‐time RT‐PCR experiments were conducted with a LightCycler 96 system (Roche Diagnostics, Indianapolis, IN). The mouse actin beta gene assay was used as the normalization control. Sequence information for the primer pairs used is listed in Table 1.
CELL ISOLATION
A single‐cell suspension was obtained from livers by the collagenase perfusion method as described.15 Liver macrophages and neutrophils and bone marrow neutrophils were sorted by AutoMACS (Miltenyi Biotec) using the repeated Possel program.
QUANTITATIVE ANALYSIS OF LIVER SECTIONS STAINED WITH SIRIUS RED
Fibrotic areas detected by sirius red‐stained collagen fibers were quantified in the liver sections at 20× magnification. The mean value of 21 serial areas per sample was used as the percentage area of fibrosis (cellSens software; Olympus).
FLOW CYTOMETRY
Liver nonparenchymal cells were obtained by liver perfusion and dissociation to assess liver blood cell populations. For flow cytometric analysis, propydium iodide was used for staining dead cells and antibodies (BioLegend) were used to detect clusters of differentiation (CD)45, CD11b, F4/80, Ly6G, CD68, and CD206. To evaluate donor chimerism, peripheral blood samples were collected from the recipients after transplantation. After lysis of erythrocytes, cells were stained with propydium iodide and CD45.1 and CD45.2 antibodies. Donor contribution was calculated as follows: percentage of donor chimerism = (100 × CD45.1 cells [%]) / (CD45.1 cells [%] + CD45.2 cells [%]).
NEUTROPHIL DEPLETION BY Ly6G‐SPECIFIC ANTIBODY
Neutrophil depletion was performed as described16 with slight modifications. In brief, 500 μg of purified anti‐Ly6G rat monoclonal antibody (1A8; BioLegend) was administered to mice 6 hours before and 48 hours after the final CCl4 administration. Control animals received a similar dose of unlabeled rat immunoglobulin G (Beckman Coulter). Depletion of neutrophils in the peripheral blood was confirmed by fluorescence‐activated cell sorting (FACS) analysis.
MICROSOMAL PREPARATION
Liver microsomes were prepared as described.17 In brief, livers were homogenized in cold 0.15 M KCl and 10 mM ethylene diamine tetraacetic acid (pH 8.0). Following centrifugation of the homogenate at 15,000g for 15 minutes, microsomes were pelleted from the supernatant fraction by centrifugation at 105,000g for 60 minutes.
DETERMINATION OF Cyp2e1 ACTIVITY
Cyp2e1 activity was determined as described.18 We used 100 μg of microsomal samples per reaction individually.
SERUM LEVELS OF ALBUMIN, ASPARTATE AMINOTRANSFERASE, AND ALANINE AMINOTRANSFERASE
Albumin, aspartate aminotransferase, and alanine aminotransferase (ALT) in sera were measured by Oriental Yeast (Tokyo, Japan).
STATISTICAL ANALYSIS
Statistical analysis was performed using the unpaired two‐tailed Student t test. P < 0.05 was considered statistically significant.
Results
REDUCED FIBROSIS AND INCREASED EXPRESSION OF NEUTROPHIL MMPs IN Trib1 KO LIVER
Loss of Trib1 was previously shown to decrease tissue‐resident M2‐like macrophages and eosinophils and increase neutrophils by altering the expression level of C/EBPα in an E3‐ubiquitin ligase constitutive photomorphogenic 1‐dependent manner.12 Because macrophages have been implicated in liver fibrosis, we investigated liver fibrosis in Trib1‐deficient mice. After repeated administration of CCl4 for 4 weeks, sirius red staining of the liver sections revealed that collagen accumulation in Trib1 KO liver was significantly reduced compared with that of wild‐type littermates (Fig. 1A,B). Cyp2e1 is the major enzyme responsible for CCl4‐induced liver injury,19 but its expression level was not affected by loss of Trib1 (Fig. 1C). As continuous administration of CCl4 transforms quiescent HSCs to myofibroblasts that produce ECM, we examined expression of key fibrogenic cytokines Tgfb1 and actin α2 (Acta2), which are activation markers of myofibroblasts. However, there was no significant difference in the expression of these markers in wild‐type and Trib1 KO liver after 4 weeks of continuous CCl4 administration (Fig. 1C). Although Cyp2e1 protein levels and enzymatic activity in liver were slightly low in Trib1 KO liver (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full), necrotic area and expression levels of Tgfb1and Acta2/αSMA in Trib1 KO mice were comparable to those in wild‐type littermates (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full). Protein expression levels of αSMA were also comparable between wild‐type and Trib1 KO liver (Fig. 1D). Because lack of Trib1 did not cause changes to the expression of Acta2 and Tgfb1, we hypothesized that fibrolysis may be altered in Trib1‐deficient mice.
Figure 1.

Trib1‐deficient mice exhibit reduced liver fibrosis and increased expression of neutrophil Mmps. (A) Histochemical staining for collagen. CCl4 was injected into each mouse 2 times per week for 4 weeks; the liver was harvested 72 hours after the final CCl4 administration. Sirius red staining of liver sections after CCl4 treatment. These pictures were represented by 21 serial tiling images (each magnification ×20). (B) Quantification of collagen‐positive area in liver sections as shown in (A) (WT, n = 10, Trib1 KO, n = 7 per group). (C) Real‐time RT‐PCR analysis of Cyp2e1, Acta2, and Tgfb1 in whole liver cDNA (n = 7 per group). (D) Western blot analysis for αSMA of extracts from wild‐type littermates and Trib1 KO liver. Actin was used for the loading control. (E) Real‐time RT‐PCR analysis of Mmp8, Mmp9, Mmp13, and Timp1 in whole liver cDNA (n = 7 per group). (F) Gelatin‐zymography of extracts from wild‐type and Trib1 KO liver after chronic CCl4 administration. (G) Real‐time RT‐PCR analysis of Mmp8 and Mmp9 in macrophages or neutrophils. Macrophages were isolated from normal liver by collagenase perfusion followed by magnetic bead sorting with F4/80 antibody. Neutrophils were isolated from bone marrow by magnetic bead sorting with Ly6G antibody (n = 3 per group). Data are expressed as means ± SEM. **P < 0.01, ***P < 0.001. Abbreviations: Mφ, macrophage; Neu, neutrophil; pro hMMP, pro‐form human MMP; SR, sirius red; WT, wild type.
As MMPs are critically involved in the resolution of fibrosis,1 we examined the expression of Mmps and found that Trib1‐deficient liver exhibited a marked increase of Mmp8 (collagenase) and Mmp9 (gelatinase B) whereas the expression level of Mmp13, an interstitial collagenase, was unchanged (Fig. 1E). Furthermore, expression of tissue inhibitor of MMP‐1 (Timp1), a negative regulator of MMPs, was comparable between wild‐type and Trib1 KO liver. Protein levels of MMP8 and MMP9 were also increased in Trib1 KO liver lysate (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full). Increased MMP9 enzymatic activity in Trib1 KO liver was confirmed by zymography (Fig. 1F). These results strongly suggest that increased MMP activity in Trib1 KO liver is responsible for the reduced fibrosis. MMP8 is a known neutrophil collagenase, and MMP9 is produced by myeloid cells.20 To identify the cell source of MMPs, we sorted Ly6G+ neutrophils and F4/80+ macrophages and examined expression levels of Mmps by qPCR (Fig. 1G). Results showed that the major source of Mmp8 and Mmp9 was neutrophils rather than macrophages. We also performed co‐immunofluorescence staining for Ly6G and MMP9 to identify localization of the cells and matrix‐degrading activity (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full). Although Ly6G+ cell numbers were significantly different between wild‐type and Trib1 KO liver (Fig. 2), the MMP9 signal was colocalized with Ly6G in both genotypes. These results indicate that loss of Trib1 increases the expression of neutrophil‐derived MMPs in the liver.
NEUTROPHILS ARE ABUNDANT IN Trib1 KO LIVER
To address whether the up‐regulation of Mmps in Trib1 KO liver is due to the increased number of neutrophils in the mutant liver, we examined Ly6G+ cells and F4/80+ cells by FACS. Even before the administration of CCl4, Ly6G+ neutrophils were clearly increased in Trib1 KO liver whereas F4/80+ macrophages were slightly reduced, consistent with a previous report12 (Fig. 2A; http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full). Immunohistochemical analysis also showed that the Ly6G+ neutrophil population was increased in Trib1 KO liver whereas the F4/80+ macrophage population was slightly decreased in Trib1 KO liver (Fig. 2B). After 4 weeks of CCl4 administration, the Ly6G+ cells were more abundant in Trib1 KO liver compared with wild‐type littermates whereas F4/80+ cells were not increased in Trib1 KO liver (Fig. 2C,D). These results indicate that neutrophils are abundant in Trib1 KO liver before and after CCl4 administration.
TRANSPLANTATION OF Trib1 KO BONE MARROW CELLS ALLEVIATES LIVER FIBROSIS
To rule out the possible influence of altered expression of hepatocytic metabolic enzymes on the Trib1 KO phenotype, we investigated whether blood cells, including neutrophils, are responsible for the suppression of liver fibrosis. For transplantation of Trib1 KO bone marrow cells, busulfan was administered to pregnant Ly5.1 congenic mice to deplete bone marrow cells. Busulfan‐treated neonatal mice were transplanted with wild‐type or Trib1 KO bone marrow cells. We used chimeric mice that showed at least 70% donor chimerism in the peripheral blood (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full). Transplantation of Trib1 KO bone marrow cells but not wild‐type littermate cells increased Ly6G+ cells in the liver (Fig. 3A,B). Consistently, Trib1 KO bone marrow cells increased Mmps expression in liver after CCl4 treatment (Fig. 3C). Furthermore, sirius red staining showed that collagen deposition was significantly decreased by transplantation of Trib1 KO bone marrow cells (Fig. 3D,E). These data demonstrate that blood cells derived from Trib1 KO bone marrow cells alleviated CCl4‐induced liver fibrosis.
rAAV‐MEDIATED EXPRESSION OF Cxcl1 IN WILD‐TYPE LIVER RECRUITS NEUTROPHILS AND SUPPRESSES LIVER FIBROSIS
To address whether wild‐type neutrophils are capable of suppressing fibrosis as effectively as Trib1 KO neutrophils, we expressed Cxcl1 (also known as KC), a neutrophil chemoattractant chemokine, by rAAV vector under the control of a liver‐specific promoter.14 Adeno‐associated virus serotype 8 was chosen as it can efficiently transduce hepatocytes in mice. Induction of Cxcl1 expression in the wild‐type liver resulted in a marked increase in the number of neutrophils in the liver without inducing liver injury as evidenced by unchanged levels aspartate aminotransferase/ALT (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full).
We then administered CCl4 8 times after transduction with rAAV.Cxcl1. Following CCl4 administration, the expression of Cxcl1 in the liver was confirmed (Fig. 4A). Consistent with increased expression of Cxcl1, expressions of Mmp8 and Mmp9 were significantly increased in liver that received rAAV.Cxcl1 (Fig. 4A). The number of Ly6G+ cells was also significantly increased in rAAV.Cxcl1 transduced liver compared to those that received the control vector rAAV.Cre (Fig. 4D,E), and sirius red staining was reduced by Cxcl1 expression (Fig. 4B,C), indicating that accumulation of neutrophils in the liver alleviates liver fibrosis.
INFUSION OF WILD‐TYPE NEUTROPHILS ALLEVIATES CCl4‐INDUCED LIVER FIBROSIS
To further examine the beneficial effect of neutrophils in injured liver, we adoptively transferred 5 × 106 normal bone marrow neutrophils into mice that received CCl4 for 4 weeks. Neutrophils were isolated using magnetic activated cell sorting with 14 kinds of antibodies to deplete blood cells except for neutrophils. Consistent with the above findings, infusion of neutrophils significantly reduced collagen accumulation in the liver within 2 days after infusion (Fig. 5A). In contrast, infusion of the Ly6G‐negative cell population did not reduce liver fibrosis (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full). These results indicate that infused neutrophils effectively reduced fibrosis. Notably, as the average lifespan of mouse neutrophils is estimated to be 1.5‐12.5 hours in tissue,7 transferred neutrophils were not detected at the time of sacrifice. Thus, to verify whether transferred neutrophils reached the liver, we killed mice 1 hour after infusion. Immunohistochemical staining of liver sections revealed that the Ly6G+ cells were significantly increased in mice that had been infused with neutrophils (Fig. 5C,D). Furthermore, the expression levels of Mmp8 and Mmp9 were also increased (Fig. 5B). These results suggest that adoptively transferred neutrophils rapidly migrate to the liver and resolve collagen fibers.
EXPRESSION OF Mmp8 AND Mmp9 AMELIORATES CCl4‐INDUCED LIVER FIBROSIS
We next investigated the mechanisms of neutrophil‐mediated resolution of fibrosis. Because the expression of Mmp8 and Mmp9 is a characteristic feature of neutrophils, we transduced both Mmp8 and Mmp9 cDNA into the liver by rAAV vector after repeated treatments of CCl4 for 6 weeks. The expression levels of Mmps were verified by qRT‐PCR (Fig. 6A). Because we fixed the total dose to 1 × 1011 vector genomes (vg) of rAAV per mouse, for the combinatorial expression of both Mmps, mice received a dose of 5 × 1010 vg for each vector. Expression of Mmp8 or Mmp9 resulted in a marked decrease in collagen staining by sirius red (Fig. 6B,C). Serum ALT levels showed no significant hepatic damage by overexpression of Mmp8 and Mmp9 (Fig. 6D). Thus, these results suggest that the resolution of liver fibrosis by neutrophils can be, at least in part, due to expressions of Mmp8 and Mmp9. Taken together, these findings indicate that neutrophils play a beneficial role in the resolution of liver fibrosis.
Figure 6.

Hepatic expression of Mmp8 and Mmp9 reduces CCl4‐induced liver fibrosis. Mice were treated with CCl4 2 times per week for 6 weeks; this was followed by injection with rAAV.Mmp8 or rAAV.Mmp9 to express Mmp8 and Mmp9 for 2 weeks. For expression of Mmp8 and Mmp9 alone, a dose of 1011 vg of rAAV was injected intraperitoneally into each mouse. For combined expression of Mmp8 and Mmp9, a dose of 5 ± 1010 vg was used for each vector, resulting in a combined dose of 1011 vg of rAAV per mouse. (A) Expression levels of Mmp8 and Mmp9 in whole liver (n = 6 per group). (B) Sirius red staining for collagen in liver 2 weeks after rAAV transduction. These pictures were represented by 21 serial tiling images (each magnification ×20). (C) Quantification of collagen‐positive area in murine liver sections as shown in (B) (n = 6 per group). (D) Evaluation of serum ALT levels at 2 weeks after rAAV vector administration (n = 6 per group). Data are expressed as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviation: SR, sirius red.
NEUTROPHIL IMMUNE DEPLETION EXACERBATES CCl4‐INDUCED LIVER FIBROSIS
We depleted neutrophils by Ly6G antibody (1A8) to further prove that neutrophils play a role for resolution of liver fibrosis. According to the protocol described,21, 22, 23 we administrated 500 μg of antibody twice (Fig. 7A). FACS analysis showed that the Ly6G+CD11b+ cells were markedly decreased at 72 hours after the final CCl4 administration when the scar resolution was most active (Fig. 7B).6 Consistently, immunostaining of liver sections demonstrated a significant decrease of Ly6G+ cells in liver at 96 hours after the final CCl4 administration (Fig. 7C‐F). Moreover, sirius red staining analysis clearly showed that fibrosis resolution was significantly delayed by anti‐Ly6G antibody treatment (Fig. 7G,H). These findings indicate that neutrophils are involved in natural fibrosis resolution in liver.
Figure 7.
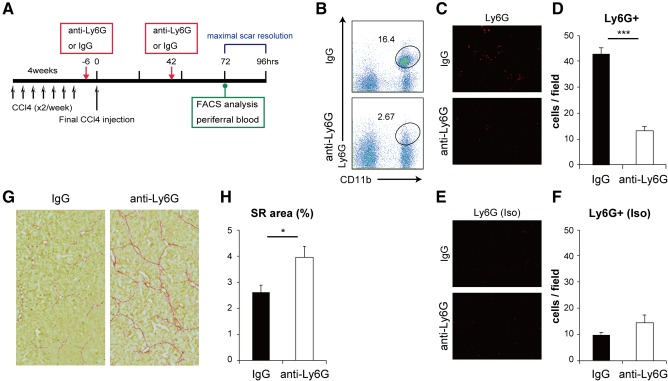
Immunodepletion of neutrophils enhances CCl4‐induced liver fibrosis. (A) Experimental design. Hepatic fibrosis was induced in C57BL/6 mice by intraperitoneal injection of CCl4 2 times per week for 4 weeks. Next, 500 μg of Ly6G (1A8) antibody or control IgG was administrated intraperitoneally 6 hours before the final injection and 42 hours after the final injection of CCl4. (B) Flow cytometric analysis of the peripheral blood at 72 hours after the final CCl4 injection. Numbers indicate the percentage of cells in the gate. The CD11b+Ly6G+ neutrophil population was dramatically reduced by immunodepletion. (C,D) Immunohistochemical staining for Ly6G in liver. Ly6G+ cell numbers in each field were determined using Image J analysis software (IgG, n = 7; anti‐Ly6G, n = 6; magnification ×10). (E,F) Isotype control of (C,D). (G) Collagen accumulation in the liver treated with CCl4 for 4 weeks. Tissues were harvested at 96 hours after the final CCl4 injection. Photomicrographs illustrate hepatic collagen fibrils stained with sirius red. These pictures were represented by 21 serial tiling images (each magnification ×20). (H) Quantification of collagen‐positive area in liver sections shown in (G) (IgG, n = 7; anti‐Ly6G, n = 6). Data are expressed as means ± SEM. *P < 0.05, ***P < 0.001. Abbreviations: IgG, immunoglobulin G; SR, sirius red.
Discussion
In this study, we showed that loss of Trib1 results in the accumulation of neutrophils in the liver, leading to reduced fibrosis in the chronic liver injury model induced by CCl4. We also showed that recruiting neutrophils by rAAV‐mediated expression of Cxcl1 in the liver as well as infusion of neutrophils alleviated fibrosis. Trib1‐deficient liver exhibited increased expression of the neutrophil proteases Mmp8 and Mmp9, and their rAAV‐mediated expression in liver reduced CCl4‐induced fibrosis. Neutrophil depletion by Ly6G antibody delayed liver fibrosis resolution (Fig. 7). Taken together, these results demonstrate that neutrophils play a beneficial role for CCl4‐induced liver fibrosis by producing these MMPs.
Because M2 macrophages have been considered to be profibrotic by producing TGF‐β and lack of Trib1 was previously shown to decrease tissue resident M2‐like macrophages and increase neutrophils,12 we hypothesized that liver fibrosis would be altered in Trib1 KO mice. Although macrophages in Trib1 KO liver were slightly decreased, no significant change was observed in the expression of fibrogenic TGF‐β at 48 hours after either a single dose or continuous administration of CCl4 (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full; Fig 1C). Instead we found a significant increase of neutrophils in Trib1 KO liver. As Trib1 is a negative regulator of C/EBPα and C/EBPα is required for neutrophil differentiation from myeloid progenitors, the increase of neutrophils in Trib1 KO mice was likely due to a consequence of up‐regulation of C/EBPα, consistent with reports showing that overexpression of C/EBPα increased the neutrophil population.11 There was no statistically significant difference in expression of neutrophil cytokines in Trib1 KO liver (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full), suggesting that accumulation of neutrophils in the liver was due to increased neutrophil production rather than increased neutrophil chemoattractants in the liver.
Neutrophils are the most abundant leukocytes in the circulatory system and have been considered to play key roles in acute inflammatory responses, whereas the roles of neutrophils in chronic inflammation have not been intensively studied. In many tissues, neutrophils are recruited to the injury site and promote inflammatory reactions. Likewise, it has been well established that neutrophils are rapidly recruited to the liver in acute liver injury models and exacerbate inflammatory responses, leading to occasional liver failure.8 However, the role of neutrophils in chronic hepatitis remains uncertain. The most interesting finding of our study is that neutrophils play an antifibrotic role in chronic liver injury. As fibrosis was ameliorated by recruiting wild‐type neutrophils by rAAV‐mediated expression of Cxcl1 in the liver and also by infusion of wild‐type neutrophils, it is likely that the effect of Trib1 KO was due to the increased number but not functional alteration of neutrophils by the mutation.
Neutrophils reportedly have a minimal impact on wound repair and regeneration,22 which contradicts our findings. However, the experimental setting of Moles et al. differed from ours in that they used S100A9 KO mice in which inflammatory cytokines were reduced and neutrophil levels in the liver remained 50%‐60% of the normal level.22 In our system, neutrophil levels were reduced to 14% in the peripheral blood and 30% in the liver by Ly6G antibody. Thus, we think that neutrophil levels in the liver could be a reason for the difference.
The beneficial role of neutrophils for liver injuries is not completely unexpected. Infusion of bone marrow‐derived macrophages has been shown to reduce hepatic fibrosis by recruiting endogenous macrophages and neutrophils.3 Administration of granulocyte colony‐stimulating factor enhanced migration of bone marrow cells into fibrotic liver and accelerated the regression of liver fibrosis.24 It was also reported that depletion of neutrophils by polymorphonuclear cell‐specific antibody treatment blocked degradation of collagen during the recovery process of reversible biliary obstruction in rats.25 In a transgenic mouse (CD11b‐DTR) study, depletion of macrophages during the recovery phase after CCl4‐induced injury impaired matrix degradation26; however, because neutrophils also express CD11b, neutrophils could have been depleted in the experimental setting, suggesting that the impaired degradation of matrix could be attributed to neutrophil depletion. Although those reports suggest a positive role for neutrophils in suppressing fibrosis, none provided direct evidence for the antifibrotic role of neutrophils in liver injuries. As described above, the results of our study provide comprehensive evidence showing a beneficial role for neutrophils in liver fibrosis. Furthermore, ectopic expression of Mmp8 and Mmp9 suppressed liver fibrosis in wild‐type liver (Fig. 6) and immune depletion of neutrophils delayed liver fibrosis resolution (Fig. 7); both of these findings provide a foundation for the antifibrotic function of neutrophils. Furthermore, neutrophil transfusion into wild‐type animals after establishing fibrosis induced rapid resolution of fibrosis (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full), suggesting that the antifibrotic function of neutrophils is likely due to fibrosis resolution by expression of Mmp8 and Mmp9.
Infusion of autologous bone marrow cells has been shown to alleviate cirrhosis not only in animal models but also in patients.27 Mesenchymal stem cells (MSCs) and macrophages have been considered the effective cell types in bone marrow cells. In addition to MSCs from bone marrow, MSCs from other tissues, such as adipose tissue, have been shown to be effective.28 However, the mechanism underlying the positive effect of MSCs or macrophages on cirrhosis remains unknown, although several possibilities have been considered; these include transdifferentiation of MSCs into hepatocyte‐like cells, secretion of factors that suppress activated stellate cells, modulation of immunity, resolution of fibrosis, and stimulation of hepatocyte proliferation. On the other hand, it has been shown that bone marrow‐derived macrophage delivery induced up‐regulation of a chemokine that recruits endogenous macrophages and neutrophils in the liver, improving fibrosis.3 Furthermore, hepatic macrophages have been reported to promote the neutrophil‐dependent resolution of fibrosis in cholestatic rat liver.29
Taken together with our results, it is strongly suggested that neutrophils may be the cell type that executes the antifibrotic function of bone marrow‐derived macrophages. Although the neutrophil has a short lifespan and is rapidly eliminated from the body after infusion, neutrophil infusion quickly ameliorates fibrosis. Thus, if a sufficient number of neutrophils could be prepared, allogenic neutrophils could be a potential source of cell therapy for cirrhosis. Development of neutrophil differentiation from human‐induced pluripotent stem cells30, 31 would be supportive of such a strategy. Furthermore, delivery of chemoattractive chemokines for neutrophils as well as MMP8 and MMP9 in the liver might be an alternative strategy for the treatment of liver fibrosis.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full.
Supporting Information 1
Acknowledgment
We thank N. Miyata for cell sorting, N. Imaizumi for animal care, and the members of the Miyajima laboratory for helpful discussions and advice. We also thank the University of Tokyo Institute of Molecular and Cellular Biosciences Olympus Bioimaging Center for help with microscopy and image acquisition.
Potential conflict of interest: Nothing to report.
Supported, in part, by the Japan Society for the Promotion of Science KAKENHI (grant number 26253023), The Grant for National Center for Global Health and Medicine (29‐1005) and the Japan Agency for Medical Research and Development.
REFERENCES
- 1. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol 2014;14:181‐194. [DOI] [PubMed] [Google Scholar]
- 2. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon‐Walker TT, Hartland S, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology 2011;53:2003‐2015. [DOI] [PubMed] [Google Scholar]
- 4. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 2017;17:306‐321. [DOI] [PubMed] [Google Scholar]
- 5. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011;11:723‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly‐6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A 2012;109:E3186‐3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 2013;13:159‐175. [DOI] [PubMed] [Google Scholar]
- 8. Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol 2007;35:757‐766. [DOI] [PubMed] [Google Scholar]
- 9. Eyers PA, Keeshan K, Kannan N. Tribbles in the 21st century: the evolving roles of Tribbles pseudokinases in biology and disease. Trends Cell Biol 2017;27:284‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bauer RC, Sasaki M, Cohen DM, Cui J, Smith MA, Yenilmez BO, et al. Tribbles‐1 regulates hepatic lipogenesis through posttranscriptional regulation of C/EBPalpha. J Clin Invest 2015;125:3809‐3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol 1998;18:4301‐4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Satoh T, Kidoya H, Naito H, Yamamoto M, Takemura N, Nakagawa K, Tolba R, Trautwein C, Trebicka J, Weiskirchen R. Critical role of Trib1 in differentiation of tissue‐resident M2‐like macrophages. Nature 2013;495:524‐528. [DOI] [PubMed] [Google Scholar]
- 13. Liedtke C, Luedde T, Sauerbruch T, Scholten D, Streetz K, Tacke F, et al. Experimental liver fibrosis research: update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair 2013;6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cunningham SC, Dane AP, Spinoulas A, Alexander IE. Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol Ther 2008;16:1081‐1088. [DOI] [PubMed] [Google Scholar]
- 15. Yagai T, Miyajima A, Tanaka M. Semaphorin 3E secreted by damaged hepatocytes regulates the sinusoidal regeneration and liver fibrosis during liver regeneration. Am J Pathol 2014;184:2250‐2259. [DOI] [PubMed] [Google Scholar]
- 16. Tate MD, Deng YM, Jones JE, Anderson GP, Brooks AG, Reading PC. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J Immunol 2009;183:7441‐7450. [DOI] [PubMed] [Google Scholar]
- 17. Lang MA, Nebert DW. Structural gene products of the Ah locus. Evidence for many unique P‐450‐mediated monooxygenase activities reconstituted from 3‐methylcholanthrene‐treated C57BL/6N mouse liver microsomes. J Biol Chem 1981;256:12058‐12067. [PubMed] [Google Scholar]
- 18. Chang TKH Crespi CL, Waxman DJ. Spectrophotometric analysis of human CYP2E1‐catalyzed p‐nitrophenol hydroxylation In: Phillips IR, Shephard EA, eds. Cytochrome P450 Protocols. Methods in Molecular Biology. 2nd ed Totowa, NJ:Humana Press; 2006:364. [Google Scholar]
- 19. Wong FW, Chan WY, Lee SS. Resistance to carbon tetrachloride‐induced hepatotoxicity in mice which lack CYP2E1 expression. Toxicol Appl Pharmacol 1998;153:109‐118. [DOI] [PubMed] [Google Scholar]
- 20. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2002;2:161‐174. [DOI] [PubMed] [Google Scholar]
- 21. Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G‐specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol 2008;83:64‐70. [DOI] [PubMed] [Google Scholar]
- 22. Moles A, Murphy L, Wilson CL, Chakraborty JB, Fox C, Park EJ, et al. A TLR2/S100A9/CXCL‐2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol 2014;60:782‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bruhn KW, Dekitani K, Nielsen TB, Pantapalangkoor P, Spellberg B. Ly6G‐mediated depletion of neutrophils is dependent on macrophages. Results Immunol 2015;6:5‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Higashiyama R, Inagaki Y, Hong YY, Kushida M, Nakao S, Niioka M, et al. Bone marrow‐derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology 2007;45:213‐222. [DOI] [PubMed] [Google Scholar]
- 25. Harty MW, Muratore CS, Papa EF, Gart MS, Ramm GA, Gregory SH, et al. Neutrophil depletion blocks early collagen degradation in repairing cholestatic rat livers. Am J Pathol 2010;176:1271‐1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 2005;115:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Terai S, Ishikawa T, Omori K, Aoyama K, Marumoto Y, Urata Y, et al. Improved liver function in patients with liver cirrhosis after autologous bone marrow cell infusion therapy. Stem Cells 2006;24:2292‐2298. [DOI] [PubMed] [Google Scholar]
- 28. Berardis S, Dwisthi Sattwika P, Najimi M, Sokal EM. Use of mesenchymal stem cells to treat liver fibrosis: current situation and future prospects. World J Gastroenterol 2015;21:742‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harty MW, Papa EF, Huddleston HM, Young E, Nazareth S, Riley CA, et al. Hepatic macrophages promote the neutrophil‐dependent resolution of fibrosis in repairing cholestatic rat livers. Surgery 2008;143:667‐678. [DOI] [PubMed] [Google Scholar]
- 30. Choi KD, Vodyanik MA, Slukvin II. Generation of mature human myelomonocytic cells through expansion and differentiation of pluripotent stem cell‐derived lin‐CD34+CD43+CD45+ progenitors. J Clin Invest 2009;119:2818‐2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morishima T, Watanabe K, Niwa A, Fujino H, Matsubara H, Adachi S, et al. Neutrophil differentiation from human‐induced pluripotent stem cells. J Cell Physiol 2011;226:1283‐1291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1178/full.
Supporting Information 1