

Abstract
Key points
The present study demonstrates, by in vitro and in vivo analyses, the novel concept that signal transmission between sympathetic neurons and the heart, underlying the physiological regulation of cardiac function, operates in a quasi‐synaptic fashion.
This is a result of the direct coupling between neurotransmitter releasing sites and effector cardiomyocyte membranes.
Abstract
Cardiac sympathetic neurons (SNs) finely tune the rate and strength of heart contractions to match blood demand, both at rest and during acute stress, through the release of noradrenaline (NE). Junctional sites at the interface between the two cell types have been observed, although whether direct neurocardiac coupling has a role in heart physiology has not been clearly demonstrated to date. We investigated the dynamics of SN/cardiomyocyte intercellular signalling, both by fluorescence resonance energy transfer‐based imaging of cAMP in co‐cultures, as a readout of cardiac β‐adrenergic receptor activation, and in vivo, using optogenetics in transgenic mice with SN‐specific expression of Channelrhodopsin‐2. We demonstrate that SNs and cardiomyocytes interact at specific sites in the human and rodent heart, as well as in co‐cultures. Accordingly, neuronal activation elicited intracellular cAMP increases only in directly contacted myocytes and cell–cell coupling utilized a junctional extracellular signalling domain with an elevated NE concentration. In the living mouse, optogenetic activation of cardiac SNs innervating the sino‐atrial node resulted in an instantaneous chronotropic effect, which shortened the heartbeat interval with single beat precision. Remarkably, inhibition of the optogenetically elicited chronotropic responses required a high dose of propranolol (20–50 mg kg–1), suggesting that sympathetic neurotransmission in the heart occurs at a locally elevated NE concentration. Our in vitro and in vivo data suggest that the control of cardiac function by SNs occurs via direct intercellular coupling as a result of the establishment of a specific junctional site.
Keywords: sympathetic neurons, optogenetics, neuro‐cardiac coupling
Key points
The present study demonstrates, by in vitro and in vivo analyses, the novel concept that signal transmission between sympathetic neurons and the heart, underlying the physiological regulation of cardiac function, operates in a quasi‐synaptic fashion.
This is a result of the direct coupling between neurotransmitter releasing sites and effector cardiomyocyte membranes.
Introduction
Intercellular communication between the sympathetic nervous system (SNS) and the heart provides the most important extrinsic regulation of heart function. Sympathetic neurons (SNs) mainly operate through the release of noradrenaline (NE), activating cardiomyocyte (CM) β‐adrenergic receptors (β‐ARs) with a subsequent increase in intracellular cAMP (Zaccolo & Pozzan, 2002; Rochais et al. 2004). In ventricular CMs, positive inotropic effects are mediated by cAMP activation of protein kinase A (PKA), which increases Ca2+ availability (Bers, 2008), whereas chronotropic effects result from cAMP or cAMP/PKA‐dependent amplification of the inward current underlying diastolic depolarization in sino‐atrial node (SAN) cells (Stieber et al. 2003). Cardiac sympathetic stimulation is engaged at the greatest degree during acute stress, commonly associated with the so‐called ‘fight‐or‐flight’ response (Jansen et al. 1995). At the same time, SNs continuously tune heart rhythm, contributing to the non‐random variation of the beat‐to‐beat intervals, referred to as physiological ‘heart rate variability’ (Lombardi et al. 1996). To accommodate such a wide effect range, neurons must deliver their signals to CMs rapidly (for the precise temporal control of cardiac responses), efficiently (to minimize neurotransmitter disposal) and, if needed, potently (to maximize the cardiac response). Neurotransmission based on direct coupling, similar to that occurring in neuromuscular junctions (NMJ) (Slater, 2003), would fulfill these requirements. Such a mode of communication has been proposed previously, mostly based on indirect morphologic and in vitro data, in a narrow group of studies (Choate et al. 1993; Hirst et al. 1996), although it has not been addressed in the intact innervated heart. Indeed, the ‘textbook’ explanation of the neurogenic control of heart function implies that NE is released into the myocardial interstitium and then diffuses to CM membranes to interact with β‐ARs. Abundant research has described the molecular details of adrenergic receptor signalling in the heart (Grimm et al. 2010), as well as the activation of adrenergic pathways by SNs and downstream changes in cardiac function (Levy & Martin, 1989; Wengrowski et al. 2015). Indeed, even the activation of intrinsic SNs in excised hearts causes profound increases in heart rate and developed force (Wengrowski et al. 2015). However, even though adrenergic signalling pathways are generally understood and the notion that cardiac SN dysfunction is central to the pathogenesis of multiple cardiovascular disorders (Gardner et al. 2016; Habecker et al. 2016) is generally accepted, the spatial arrangement and dynamics of intercellular communication between SNs and CMs is much less understood.
The present study aimed to investigate neurogenic control of cardiac function, with a specific focus on the interface between SNs and CMs. Accordingly, we studied in vitro co‐cultures of SNs and CMs and also investigated the response dynamics of cardiac SNs in vivo using optogenetics coupled with pharmacological β‐AR blockade. Live cell cAMP imaging of co‐cultures provided unique insight into the biophysics of neurocardiac spatial relationships and communication dynamics in vitro. Optogenetics enabled the spatiotemporal control of SNs innervating the SAN of living mice to provide new insights into the exquisite coupling of the neurocardiac interface.
Methods
All of the investigators involved in the present study understand the ethical principles under which the journal operates and the work conducted complies with the animal ethics checklist of the The Journal of Physiology (Grundy, 2015).
Human heart sample processing and immunofluorescence (IF)
We analysed postmortem heart samples archived in the historical collection of the Institute of Pathological Anatomy of the University of Padova, as had been acquired during routine postmortem clinical investigations. Samples were anonymous to the investigators and were used in accordance with the directives of the national committee of Bioethics and Raccomandazione (2006) della Commissione dei Ministri degli Stati Membri sull'utilizzo di campioni biologici di origine umana per scopi di ricerca.
Sample were analysed using the protocols described previously by Zaglia et al. (2016).
Ethical approval
All experimental procedures performed on rodents were approved by the local ethical committee (Authorization number C54) and communicated to the Ministry of Health (Ufficio VI), in compliance with Italian Animal Welfare Law (Law n 116/1992 and subsequent modifications). All procedures were performed by trained personnel with documented formal training and previous experience in experimental animal handling and care. All procedures were refined prior to starting the study, and the number of animals was calculated to use the least number of animals sufficient according to statistical sample power calculations.
Origin and source of the animals
Transgenic C57/BL6J mice expressing cre‐recombinase under the control of tyrosine hydroxylase (TOH) promoter B6.129 × 1‐Th<tm1(cre)Te>/Kieg (emma Laboratories, KCTT/EMMA/Johannes Wilbertz, Stockholm, Sweden) were bred with B6.Cg‐Gt(ROSA)26Sortm27.1(CAG‐COP4*H134R/td‐Tomato)Hze/J expressing mice (Jackson Laboratory, Bar Harbor, ME, USA). The resulting offspring had the STOP cassette deleted in dopaminergic and adrenergic neurons, thus expressing the hChR2(H134R)‐td‐Tomato fusion protein. TOH‐cre+/‐ lines were used to maintain the colonies and used as littermate controls. We also used C57BL/6J (Charles River Laboratories, Wilmington, MA, USA) and α‐MyHC/ChR2 mice in a C57/BL6J genetic background (Institutional Colony; Zaglia et al. 2015), as well as Sprague–Dawley rats (Charles River, Milan, Italy). In the present study, we used: P1–P3 TOH/ChR2 tg mice; P1–P3 rats; adult (3 months) male mice (24–28 g); and adult (3 months) male rats (250–350 g). Animals were maintained in individually ventilated cages in an Authorized Animal Facility (authorization number 175/2002A) under a 12:12 h light/dark cycle at a controlled temperature and had access to water and food available ad libitum.
Primary culture of rat neonatal cardiomyocytes
P1–P3 Sprague–Dawley rats were killed by cervical dislocation (in accordance with Annex IV of European Directive 2010/63/EU). Hearts were harvested and processed as described previously (Castaldi et al. 2014). Cells were plated onto laminin‐coated (10 μg cm−2; Becton‐Dickinson Biosciences, Franklin Lakes, NJ, USA) coverslips, at a density of 110 000 cells cm−2, and maintained in a humidified atmosphere (5% CO2) at 37°C. Twenty‐four hours after seeding, CMs were transfected with the fluorescence resonance energy transfer (FRET)‐based sensor, GcAMPs (Nikolaev et al. 2004), or the PKA activity reporter, AKAR3 (A‐kinase activity reporter) (Allen & Zhang, 2006), using transfectin (Bio‐Rad, Hercules, CA, USA), in accordance with the manufacturer's instructions. In a subset of experiments, CMs were infected with the FRET‐based sensor, H187 (Nikolaev et al. 2004) for 24 h.
Isolation of superior cervical ganglia neuron and co‐culture with cardiomyocytes
P1–P3 Sprague Dawley rats or TOH/ChR2 mice were killed by cervical dislocation (in accordance with Annex IV of European Directive 2010/63/EU). Superior cervical ganglia neurons (SCGNs) were isolated by adapting a procedure described by (Zareen & Greene, 2009). SCGNs were minced in RPMI, supplemented with 10% horse serum and enzymatically dissociated in 0.25% trypsin with no EDTA (all from Thermo Fisher Scientific Inc., Waltham, MA, USA) for 30 min at 37°C. Rat SCSNs were seeded on CMs at a ratio of 1:50 (SN:CM). Cells were maintained in the CM culture medium, supplemented with 100 ng ml−1 NGF (Sigma, St Louis, MO, USA). ChR2 expressing SNs were cultured alone, on laminin‐coated dishes, at a density of 25000 cells cm−2.
In vitro IF
SCGN‐CM co‐cultures, as well as SCGNs cultured alone, were fixed with 3.7% formaldehyde for 30 min at 4°C, permeabilized with 1X PBS, supplemented with 0.1% Triton X‐100 for 5 min at room temperature and incubated with primary antibodies diluted in 1X PBS, supplemented with 1% BSA for 2 h at 37°C. Primary antibodies were revealed by a 30 min long incubation with secondary antibodies. Cells were analysed under a confocal microscope. The primary antibodies used for in vitro assays are listed in Table 1.
Table 1.
Primary antibodies used in the present study
| Antibody | Customer | Dilution | Analysis |
|---|---|---|---|
| Mouse anti‐α‐actinin | Sigma | 1:200 | IF |
| Rabbit anti‐cadherin | Abcam | 1:600 | IF |
| Mouse anti‐cardiac troponin I | Saggin et al. (1989) | 1:200 | IF |
| Rabbit anti‐β‐catenin | Abcam | 1:400 | IF |
| Mouse anti‐ChR2 | Progen Biotechnik (Germany) | 1:50 | IF |
| Rabbit anti‐dopamine β hydroxylase | Millipore | 1:200 | IF |
| Rabbit anti‐dystrophin | Abcam | 1:600 | IF |
| Rabbit anti‐HCN4 | Alomone | 1:200 | IF |
| Mouse anti‐SNAP25 | Sigma | 1:1000 | IF |
| Rabbit anti‐synapsin 1a | Millipore | 1:1000 | IF |
| Rabbit anti‐synaptotagmin | Abcam | 1:200 | IF |
| Rabbit anti‐TOH | Millipore | 1:500 | IF |
| Mouse anti‐TOH | Abcam | 1:400 | IF |
| Goat anti‐td‐Tomato | SicGene Antibodies | 1:200 | IF |
Processing of rodent heart samples and IF
Adult male mice and rats were killed by cervical dislocation. Hearts were harvested and processed as described previously (Zaglia et al. 2013). Sections (10 μm) were obtained with a cryostat (CM1860; Leica, Wetzlar, Germany) and processed for IF analysis. For identification and staining of the SAN cells, the heart base was fixed in 4% paraformaldehyde overnight at 4°C, dehydrated in sucrose gradient and processed as described above. Both ventricular and atrial heart sections were incubated with primary antibodies diluted in 1X PBS, supplemented with 1% BSA and 0.5–2% Triton X‐100 overnight at 4°C. 488‐ and Cy3‐conjugated secondary antibodies, all from Jackson Laboratory, were used to detect primary antibodies, by incubation at 37°C for 30 min. Heart sections were analysed under a confocal microscope (TCS SP5; Leica). The primary antibodies used in this study are listed in Table 1.
Ex vivo electron microscopy
Adult mice were anaesthetized with zoletil (20 mg g−1, i.p.) and the abdominal aorta was cannulated with a 22‐G needle. The inferior vena cava was cut to allow the outflow of the fixative. Hearts were retrogradely perfused with 1X Tyrode Solution (in mmol L–1): 136 NaCl, 5 Hepes, 0.33 NaH2PO4(H2O), 5.4 KCl, 1 MgCl2(6H2O) and 10 glucose (pH 7.4) at a rate of 60 ml h−1 and then fixed with 2.5% glutaraldehyde in 0.1 mol L−1 sodium cacodylate. Hearts were removed, with the right and left ventricles being dissected and then minced in 1 mm3 pieces and further fixed for 2 h at 4°C. Samples were then washed twice for 10 min with 0.2 mol L−1 sucrose in 0.1 mol L−1 sodium cacodylate. Post‐fixation was carried out in 1% osmium tetroxide in 0.1 mol L−1 sodium cacodylate for 2 h at 4°C. Samples were then treated with increasing ethanol concentrations, incubated with propylene oxide for 45 min and embedded in epoxy resin. Semi‐thin sections were cut, stained with uranyl acetate, 50% ethanol and Reynolds lead citrate, and then examined with a Tecnai 12 electron microscope (FEI, Hillsboro, OR, USA).
In vitro electron microscopy analysis
Transmission electron microscopy analysis was performed on co‐cultures. Cells were fixed with 2.5% glutaraldehyde in 0.1 m sodium cacodylate for 1 h at 4°C and washed twice for 30 min with 0.2 m sucrose and 0.1 m sodium cacodylate. Post‐fixation was carried out in 1% osmium tetroxide, 0.1 m sodium cacodylate for 2 h at 4°C. Samples were then treated with increasing ethanol concentrations, incubated with propylene oxide for 45 min and embedded in epoxy resin. The resin with the cells was extracted from the wells and semi‐thin sections were cut, stained with uranyl acetate, 50% ethanol and Reynolds lead citrate, and examined with a FEI Tecnai 12 electron microscope.
Electrophysiology on cultured neurons
Whole cell current clamp experiments were performed at room temperature (∼23°C). Data were recorded using a EPC‐7 amplifier (HEKA Electronic, Lambrecht, Germany) and pClamp10 software (Axon Instruments, Foster City, CA, USA). Signals were acquired at 10 kHz. Patch pipettes were prepared by pulling borosilicate glass capillaries (outer diameter 1.5 mm, inner diameter 1.16 mm; Harvard Apparatus Ltd, Cambridge, MA, USA) using a micropipette puller (Narishige, Tokyo, Japan). Pipette resistance was 2–4 MΩ when filled with intracellular solution. Extracellular solutions contained (in mmol L−1): 125 NaCl, 5 KCl, 1 Na3PO4, 1 MgSO4, 20 Hepes, 5.5 glucose and 1.8 CaCl2 (pH 7.4) with NaOH. Intracellular solution was (in mmol L−1): 100 K‐gluconate, 20 KCl, 10 Hepes, 10 phosphocreatine, 4 Mg‐ATP and 0.3 GTP (pH 7.3) with KOH. All membrane potential values were corrected for liquid junction potential, calculated as −14.5 mV. Blue light flashes were generated by a software‐controlled LED (Thorlabs, Newton, NJ, USA) and delivered through the microscope objective (60×; NA 1.4; Olympus, Tokyo, Japan).
Real time cAMP imaging in SCGN‐CM co‐cultures
FRET imaging experiments were performed in 15 day co‐cultures. Cells were maintained in Hepes‐buffered Ringer modified saline (in mmol L−1): 125 NaCl, 5 KCl, 1 Na3PO4, 1 MgSO4, 5.5 glucose, 1.8 CaCl2 and 20 Hepes (pH 7.4) at room temperature and imaged on an inverted IX50 microscope (Olympus) coupled to a CCD camera (Sensicam QE; PCO, Kelheim, Germany) and a beam‐splitter optical device (Microimager, Optical Insight, Santa Fe, NM, USA). Images were acquired every 5 s, and processed using ImageJ (National Institutes of Health, Bethesda, MD, USA). Variations in FRET efficiencies were measured by drawing a region of interest in the cell cytosol and by monitoring changes in the 480 nm/545 nm fluorescence emission signals (R) on excitation at 430 nm after background subtraction. FRET values were expressed as ΔR/R 0, where R 0 is the ratio at t = 0 s and ΔR = R – R 0. The beginning of FRET variations, indicative of the intracellular cAMP concentration, was defined as the time of the first frame after which two consecutive values fell outside the SD of values under the basal condition. Membrane depolarization of SNs was obtained by applying high potassium solution (in mmol L−1) 80 NaCl, 50 KCl, 1 Na3PO4, 1 MgSO4, 5.5 glucose, 1.8 CaCl2 and 20 Hepes (pH 7.4) either via a perfusion system or via pulses generated by a software‐controlled pressurized micropipette (Eppendorf AG, Hamburg, Germany) within the proximity of the analysed cell.
Neuronal optogenetics in living mice
For surgical preparation, adult TOH/ChR2, C57BL/6J wild‐type and α‐MyHC/ChR2 male mice were anaesthetized with zoletil (15 mg g−1, i.p.), secured to the table in supine position and intubated with a 24‐G needle for ventilation (tidal volume 0.4 mL; 120 strokes min–1) from an artificial ventilator (SAR‐830; World Precision Instruments, Sarasota, FL, USA). Body temperature and heart rate (HR) were constantly monitored during the experiment. The skin was dissected by a lateral right subaxillary 1.5 cm cut, s.c. muscles were removed and a 0.5 cm incision was performed at the level of the first intercostal space. Self‐retaining microretractors separated the first and second ribs sufficiently to provide adequate exposure of the operating region.
For optogenetics, the right atrial surface was exposed and stimulated with a 1.5 mm diameter fibre optic (NA 0.27; Thorlabs), coupled to a LED (470 nm; 1.5 A; Thorlabs), controlled by the ECG acquisition hardware (Powerlab 8/30, Bioamp and LabChart, version 7.1; AD Instruments, Sydney, Australia), to generate power‐tunable light pulses. The fibre optic was moved to different epicardial areas using a micromanipulator (Narishige). Photostimulation was performed by delivering trains (from one to 50 flashes) of light pulses at frequencies ranging from 0.5 to 20 Hz. The surface ECG was recorded using Powerlab 8/30 and analysed in LabChart, version 7.1 (AD Instruments). In a subset of animals, dermographic ink was applied after functional optogenetic identification of the responsive atrial region, by drawing reference points with an inked tip positioned on the fibre optic side. After the death of the mice, the right atrium was excised and fixed and then a sample preparation was performed with the focus, during OCT inclusion and sectioning, on the ink‐marked myocardial area.
All procedures were terminal and animals were not reutilized after the experiments. Animals were eventually killed using authorized humane methods.
Estimation of blue light attenuation by the myocardium
To estimate the attenuation of blue light by the myocardial tissue, we used a method implemented and described previously (Zaglia et al. 2015). In detail, longitudinally cut ventricular slices of different thickness (0, 250, 400 and 800 μm) were placed with the endocardial side in contact with the light sensor of an optical power meter (PM100 D; Thorlabs). The emerging light intensity was measured with a dependency on the fibre output light intensity, fibre size diameter (from 100 to 1500 μm) and phantom thickness. Myocardial light attenuation was modelled by adapting previous studies performed in the brain (Dobrzynski et al. 2013). Briefly, values of light intensity experimentally determined using the tissue phantoms were fitted to a mono‐exponential decay curve to extrapolate light intensity as a function of the depth from the epicardial surface. The light intensity threshold for ChR2 activation was obtained from previous studies (Boyle et al. 2013; Nussinovitch et al. 2014). Based on these measurements, and on the evaluation of the SAN depth, we estimated that the light power irradiating the SAN region is in the order of 12 mW mm−2. Under these experimental conditions, we tested different pulse lengths (from 1 to 50 ms), which revealed that the minimum pulse duration required to reliably elicit positive chronotropic responses was 10 ms, whereas shorter lasting pulses activated responses with variable efficiency (∼50%).
Statistical analysis
All data are expressed as the mean ± SEM. Experimental groups were compared using unpaired (paired when appropriate) ANOVA tests and comparisons between multiple groups were performed using the Kruskal–Wallis test. P < 0.05 was considered statistically significant. Curve fitting was performed with Prism (GraphPad Software Inc., San Diego, CA, USA); NE dose/response analyses were fitted with a non‐linear regression sigmoidal dose‐response; and goodness of fits were assessed via the least squares method.
Results
Sympathetic neurons closely interact with cardiomyocytes both in vivo and in vitro
Cardiac sympathetic fibres, which display the typical pearl‐necklace morphology, highly innervate the human and rodent myocardium (Fig. 1 A–C) and express markers of neuroexocytosis (i.e. synapsin‐1a, SNAP25, syntaxin and synaptotagmin) (Fig. 1 D and E), specifically at the contact site with CMs (Fig. 2 A and B). Electron microscopy of mouse ventricular sections revealed sympathetic varicosities, in which NE vesicles were positioned close to the membrane on the side facing a juxtaposed CM and found at an inter‐membrane distance of 67 ± 16 nm (n = 18 contacts) (Fig. 2 C). These structural aspects are reminiscent of those of the well‐known NMJ (Slater, 2003). However, it remains unknown whether such organization has a functional role in intercellular neurocardiac communication (Franzoso et al. 2016). To analyse the dynamics of intercellular coupling between cultured SNs and CMs, at the single cell level, we used co‐cultures of SCGNs and neonatal rat CMs. SCGN were analysed in culture and had normal resting membrane potential and AP amplitude and duration (E m: −55.7 ± 1.6 mV; APamp: 59.1 ± 14.3 pA; APdur: 10.8 ± 8.1 ms; n = 17). In co‐culture, the majority of SCGNs were adrenergic, co‐expressing TOH (Fig. 2 D) and dopamine‐β‐hydroxylase (92 ± 4%, n = 240 neurons from three cultures). To further characterize the time‐dependency of the neurocardiac contact maturation, co‐cultures were analysed for the size of neuronal varicosities and the distribution of generic cell–cell adhesion molecules in pre‐ and post‐synaptic membranes (Anderson & Cohen, 1977; Okabe et al. 1999; Sharma et al. 2010). In particular, we focused on cadherins, which, in central synapses, connect the pre‐ and post‐synaptic membranes, and β‐catenin, which binds the cadherin‐complex to the actin cytoskeleton. Our IF staining demonstrated that varicosities increase in size with the time in co‐culture (5 days: 0.85 ± 0.23 μm2 vs. 15 days: 1.38 ± 0.10 μm2; n = 30 varicosities for each time point) (Fig. 2 E), accompanied by accumulation of post‐synaptic proteins, which suggest the progressive development of the neurocardiac contact site (Fig. 2 F and I). No significant differences were observed between 15‐ and 20‐day old co‐cultures, suggesting that 2 weeks was sufficient to reach the structural maturation of the intercellular contact.
Figure 1. Interaction between SNs and CMs in the mammalian heart.
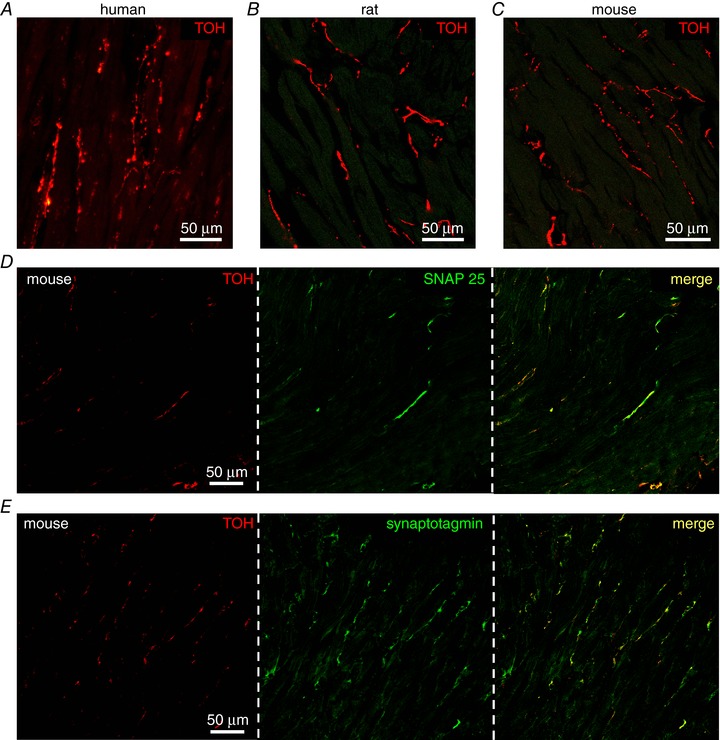
A–C, confocal IF analysis on human (A), rat (B) and mouse (C) heart sections stained with an antibody for TOH. D–E, confocal IF analysis on mouse heart sections stained with an antibody to TOH in combination with anti‐SNAP25 (D) or anti‐synaptotagmin (E).
Figure 2. Interaction between SNs and CMs ex vivo and in vitro .
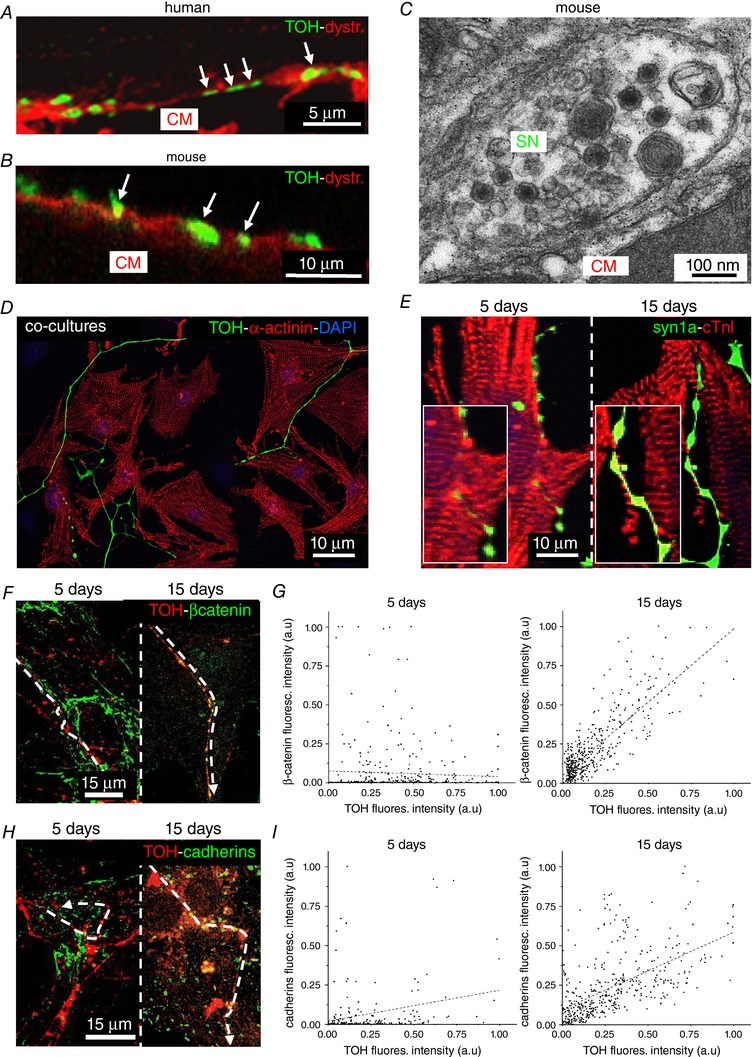
A and B, confocal double‐IF analysis on human (A) and mouse (B) hearts co‐stained with antibodies for dystrophin and TOH. Close contacts between SN varicosities and CM membrane are highlighted by arrows. C, electron microscopy on mouse heart sections. D, confocal IF on 15‐day SN‐CM co‐cultures, co‐stained with antibodies to sarcomeric actinin (α‐actinin) and TOH. Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole. E, confocal IF image of 5‐ and 15‐day SN‐CM co‐cultures, co‐stained with antibodies to cardiac troponin‐I (cTnI) (Saggin et al. 1989) and synapsin‐1a (syn1a). F and H, Confocal IF analysis on 5‐ (left) and 15‐day (right) co‐cultures co‐stained with antibodies to TOH and: β‐catenin (F) and pan‐cadherins (H). G and I, representative plot profiles showing time‐dependent β‐catenin and pan‐cadherin accumulation at the SN‐CM contact site.
In mature co‐cultures, the topology of interactions established between neuronal processes and CMs was classified into three categories: type (i) cells in direct contact with the neuronal varicosities, identified as round shaped enlargements of the axon within the CM area; type (ii) cells close to neuronal varicosities, which are found outside the CM area; and type (iii) CMs passed over by the neuronal processes devoid of varicosities (Fig. 3A). Remarkably, in 15‐day co‐cultures, the type (i) contacts showed morphological features similar to those observed in the intact heart, with reduced inter‐membrane distance between NE‐containing varicosities and target CMs (42.3 ± 7.1 nm; n = 35 contacts) (Fig. 3 B).
Figure 3. Selective signalling of SNs to target innervated cardiomyocytes.
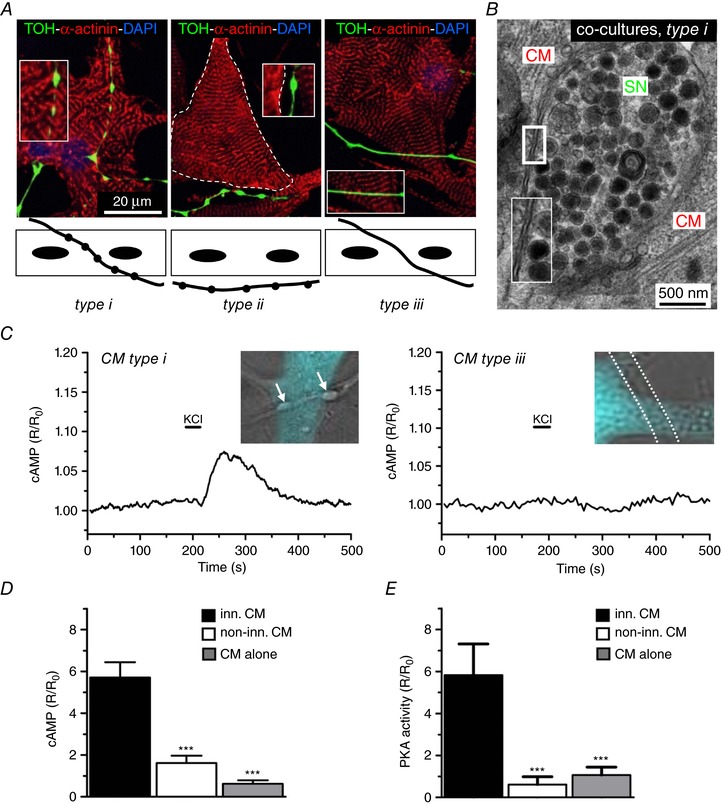
A, IF staining on 15 day co‐cultures stained with antibodies to: sarcomeric actinin (α‐actinin) and TOH. Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole. Confocal images (top) and schematic representations (bottom) of the different types of SN‐CM interactions observed in our experimental settings. B, electron microscopy on 15‐day co‐cultures. C, representative traces of cAMP changes in Epac1‐camps expressing CMs, upon SN depolarization, measured in innervated [type (i), left ], or non‐innervated [type (iii), right] CM (for details, see text). D, statistics of cAMP responses in corresponding groups. Bars indicate the SEM (*** P < 0.001; n = 20 CMs for each group). E, statistics of the PKA activity in response to KCl stimulation of SCGNs, in differently innervated CMs. Bars indicate the SEM (*** P < 0.001; n = 20 CMs for each group).
Cell–cell contact is required for neuronal‐dependent cardiomyocyte stimulation, in co‐cultures
Because β‐AR stimulation causes cardiac effects through cAMP‐dependent signalling, we used real‐time cAMP imaging in CMs expressing a FRET‐based camps biosensor Epac1‐camps (Nikolaev et al. 2004) in co‐cultures to analyse the CM responses to SN activation. Changes in intracellular [cAMP] were assessed at the single cell level by monitoring the variation of cyan fluorescent protein (CFP)/yellow fluorescent protein emission upon CFP illumination (also known as the sensitized emission ratio method). Depolarization of a single SN, as obtained by brief perfusion with 50 mmol L−1 KCl locally delivered with a pressurized micropipette, caused an increase in intracellular [cAMP] that was only detected in the innervated CMs [type (i)], whereas no changes were observed in non‐innervated cells [types (ii) and (iii)] or in KCl perfused CMs cultured alone (ΔR/R 0, innervated CMs: 5.71 ± 0.73%; non‐innervated CMs: 1.62 ± 0.35%; CMs alone: 0.62 ± 0.17%) (Fig. 3 C and D). All groups showed comparable cAMP responses to stimulation with NE. We then aimed to determine whether the cAMP rise, elicited solely in coupled CMs by neuronal depolarization, was paralleled by activation of downstream β‐AR/cAMP effectors. PKA activity was thus monitored with the fluorescence reporter AKAR3 (Allen & Zhang, 2006), under the experimental conditions used above. In line with the previous experiment, SN stimulation activated PKA only in directly contacted cells, whereas no effects were observed in non‐innervated CMs or in CMs cultured alone (ΔR/R 0, innervated CMs: 5.82 ± 1.48%; non‐innervated CMs: 0.62 ± 0.37%; CMs alone: 1.07 ± 0.38%) (Fig. 3 E). We excluded the possibility that the cAMP responses observed upon neuronal stimulation were a result of NE released in the culture medium. This was because administering the cell bathing solution, collected after KCl perfusion of all neurons in the dish, failed to induce any cAMP response in Epac1‐cAMPs expressing CMs (ΔR/R 0, CMs plus conditioned medium: 0.54 ± 0.16% vs. non‐treated CMs: 0.58 ± 0.12%). Altogether, our data firmly support the concept that the establishment of a structured and well‐organized intercellular contact is required for cSNs to elicit an efficient cAMP response in target CMs.
Sympathetic neurons communicate to cardiomyocytes in a restricted domain in vitro
To address the spatial selectivity of SN‐to‐CM communication, we first investigated cAMP responses in pairs of adjacent CMs, where only one of which was in direct contact with the stimulated SN. In these experiments, we transduced CMs with an adenoviral vector encoding the cAMP sensor H187 (Klarenbeek et al. 2015) to achieve higher efficiency and perform measurements with an elevated S/N ratio. Upon neuronal depolarization, activation of β‐ARs was limited to the innervated cell (ΔR/R 0, innervated CMs: 9.30 ± 0.92% vs. non‐innervated CMs: 0.18 ± 0.05%) (Fig. 4 A and B) and the amplitude of cAMP responses was unaffected by the rapid perfusion of the cells with normal saline solution. Altogether, these data indicate that SNs communicate to target CMs by discharging NE in a ‘diffusion‐restricted’ extracellular signalling domain, at the interface between the two cell types, and bounded by the close apposition of the two cell membranes (Fig. 3 B). Compared to distal sites, increases in cAMP elicited by neuronal depolarizations in CM regions proximal to the interaction site were consistently faster and greater, when normalized to the average response in the whole cell area (cAMP increaseproximal area/whole cell: 176.0 ± 26.9% vs. cAMP increasedistal area/whole cell: 97.9 ± 5.0%; speed of cAMP changeproximal area/whole cell: 88.0 ± 6.7 s vs. speed of cAMP change distal area/whole cell: 105.00 ± 3.79 s). By contrast, responses induced by diffuse NE addition to the culture medium were identical in all subcellular regions (cAMP increaseproximal area/whole cell: 106.0 ± 1.9% vs. cAMP increasedistal area/whole cell: 100 ± 1.5%; speed of cAMP changeproximal area/whole cell: 90 ± 7 s vs. speed of cAMP change distal area/whole cell: 100 ± 4 s) (Fig. 4 C–E). These data thus further support the concept that neuronally‐released NE activates only a confined pool of β‐ARs/adenylyl cyclases within the post‐synaptic CM membrane.
Figure 4. Local activation of CMs by innervating neurons.
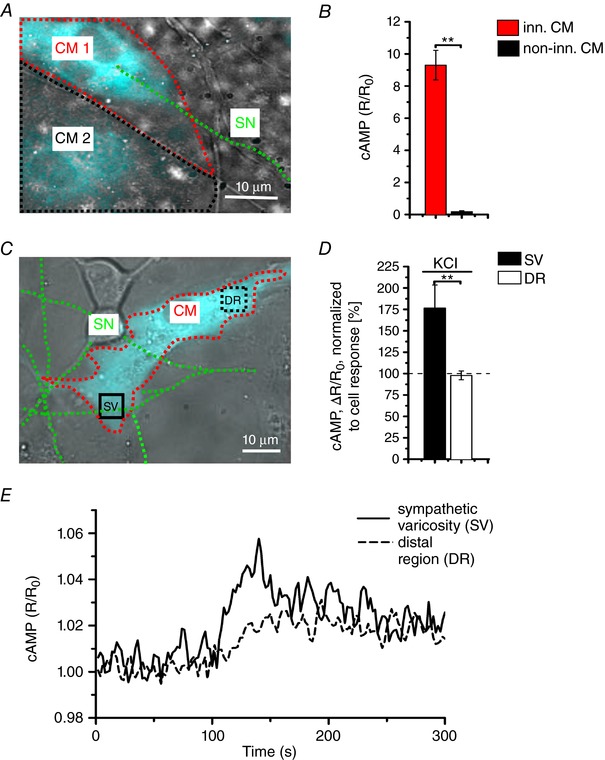
A, fluorescence image of adjacent H187 expressing CMs, one of which (CM1, red line) is in direct interaction with a SN (green line). CM2 decorated by a black line is not innervated by the depolarized SN. B, statistics of cAMP responses to KCl stimulation of SCGNs, in innervated vs. non‐innervated CMs. Bars indicate the SEM (** P < 0.01; n = 70 CM per group). C, Fluorescence image of a H187 expressing CM innervated by SN processes. cAMP variations were evaluated in the CM delineated by a red line, in regions close to (SV) or far from (DR, distal region) the neurocardiac interaction site. Green line highlights the innervating neuron. D, statistics of subcellular cAMP variations elicited by neuronal activation in the SV and DR of n = 36 CMs with similar neurocardiac arrangements as in (C); bars indicate the SEM (** P < 0.01). E, representative trace of cAMP changes calculated in the SV and DR regions of the CM shown in (C).
Direct cell–cell interaction establishes potent and efficient neurocardiac coupling
The results reported above support the notion that neuronally‐released NE reaches a very high concentration within the synaptic cleft to elicit a cAMP response within the innervated CM. For this to be correct, we would expect that high doses of β‐blockers would be required to blunt the SN‐induced CM cAMP response. To test this hypothesis, we pre‐incubated co‐cultures with the selective β1‐AR blocker, metoprolol. Interestingly, although metoprolol significantly reduced elevations in cAMP caused by addition of NE to the culture medium, it was ineffective in counteracting the neuronal‐dependent CM response, which was comparable to that observed in the absence of β‐blockers (Fig. 5 A and B). Upon metoprolol washout, the neuronal‐dependent CM cAMP increase was totally preserved, and the response to diffuse NE was restored (Fig. 5 A and B). Based on this observation, we next estimated the [NE] generated by the activated neuron within the intercellular cleft. At plain comparison, neuronal activation elicited cAMP responses in CMs simliar to those elicited by 0.3 nm NE added diffusely to the culture dish (Fig. 5 C, red dot), therefore interacting with all the receptors on the cell membrane. However, it has to be taken into account that, given the tight spatial interaction, neuronally released NE only activates a restricted fraction of CM β‐ARs. We thus pursued a measurement of the effective agonist concentration independent of the number of activated receptors. Accordingly, we adopted the principle of competition binding assays and obtained inhibition curves by measuring CM cAMP responses to neuronal activation, after pre‐treating co‐cultures with increasing concentration of the non‐specific yet well‐characterized β‐AR blocker, propranolol. The dose‐inhibition kinetics were compared between responses to neuronal activation and those elicited by known amounts of NE (10, 100 and 1000 nmol L−1) (Fig. 5 C and D). The propranolol inhibition curve in neuronally‐stimulated CMs overlapped that obtained by direct stimulation with 100 nmol L−1 NE, indicating that [NE]cleft is within this order of magnitude (Fig. 5 D). Taken together, the spatial discrimination of target cell responses, the evidence that receptor‐dependent signalling is initiated at the contact site, and the high effective [NE]cleft all support the hypothesis that neurotransmission occurs in a quasi‐synaptic fashion within a restricted intermembrane domain. Based on the morphological evidence of our IF and EM analyses, we estimated the volume of the synaptic cleft to be in the order of 0.1 μm3.
Figure 5. In vitro assessment of [NE] in the synaptic cleft.
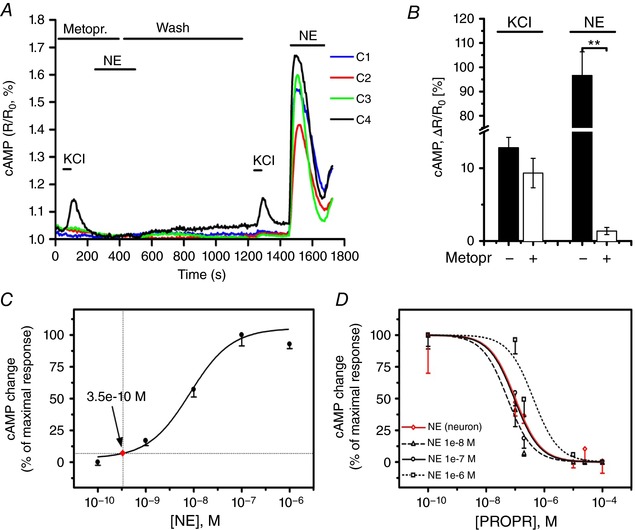
A and B, representative plot (A) and statistics (B) of cAMP changes in innervated vs. non‐innervated CMs upon neuronal depolarization, both in the absence and presence of the β1‐blocker metoprolol (100 μm). NE (100 nm) added to the culture medium was used to compare the effect of neuronal vs. diffuse stimulation. Bars indicate the SEM (** P < 0.01; n = 20 innervated, n = 63 non‐innervated CMs). C, dose‐effect curve of cAMP responses to NE directly applied to Epac1 expressing CMs. Arrow indicates the average cAMP response observed in co‐cultures. D, propranolol inhibition curve of cAMP responses elicited by neuronal stimulation (red trace), overlapped to that resulting from direct treatment of CMs with known [NE] (n = 6 measures for each point).
We next performed a series of experiments in living mice using sympathetic neuron optogenetics aiming to determine whether the properties of intercellular coupling observed in vitro were reflected by the physiology of cardiac autonomic control in vivo.
Optogenetic control of cardiac sympathetic neurons in vivo
The physiological requirements of autonomic control of HR demand that cardiac responses to neuronal activation be rapid and efficient. In line with this, morphological and ultrastructural evidence reveals that several neuronal processes innervate the SAN and that the single neuronal varicosities are found in close apposition with the membrane of target cells (Fig. 9 E and F) (Choate et al. 1993; Chow et al. 2001) Moreover, in SAN cells, β‐AR‐induced cAMP interacts directly with HCN4 channels, responsible for the amplification of I f current underlying diastolic pacemaker depolarization (DiFrancesco, 2010). We therefore used optogenetics to control SN activity at the same time as monitoring HR in anaesthetized mice to determine whether the effects of sympathetic activation reflect, in vivo, the direct cell–cell communication observed in the in vitro system.
Figure 9. Optogenetic control of cardiac CNs in vivo .
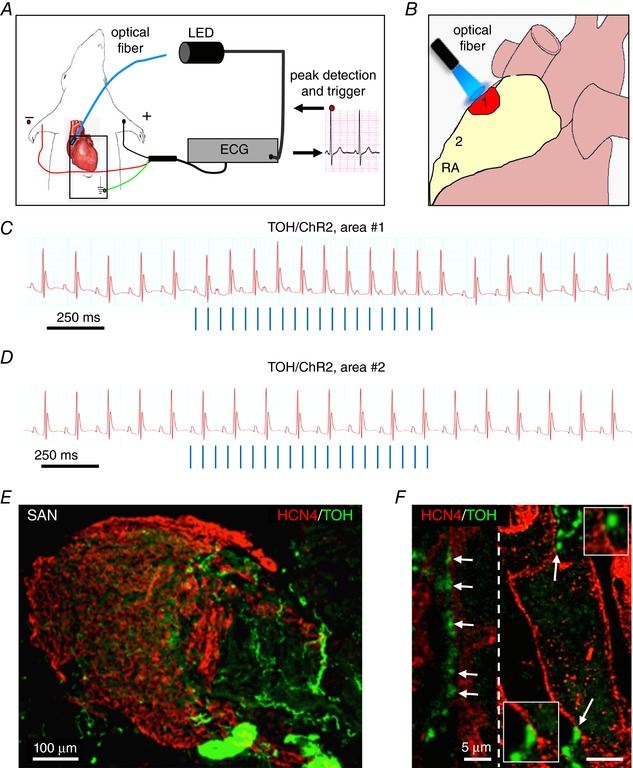
A, schematic illustration of the neuronal optogenetic set up used for right atrium illumination in open‐chest anaesthetized mice. B, representation of the different photostimulated atrial regions (areas#1–2). C, representative ECG trace of the optogenetic experiment, showing positive chronotropic response upon photoactivation (blue lines) of the right atrial area#1 in TOH/ChR2 mice. D, representative trace showing unchanged HR upon illumination of area#2. Behaviour of experiments (C and D) was observed in n = 15 mice. E and F, area#1 was analysed with double IF with SAN (HCN4) and SN (TOH) markers. The magnified image (F) highlights the close interaction between SN varicosities and SAN myocytes (left) and an example of multiple neuronal processes interacting with the same myocyte (right).
The method is based on the expression of Channelrhodopsin‐2 (ChR2), a microbial‐derived light‐activated ion channel, permeable to Na+ (Nagel et al. 2003; Boyden et al. 2005), in SNs. ChR2 has been used for cardiac pacing (Arrenberg et al. 2010; Bruegmann et al. 2010) and, recently, it was used to modulate SN activity in isolated perfused hearts (Wengrowski et al. 2015). Illumination of ChR2‐expressing neurons generates an inward depolarizing current that results in neurotransmitter release within milliseconds (Boyden et al. 2005; Zhang et al. 2007; Wengrowski et al. 2015). We thus generated transgenic mice expressing the ChR2‐H134R‐td‐Tomato gene under control of the TOH promoter. These mice had structurally and functionally normal hearts with ChR2 expression that was negligible in SAN cells and working CMs. Expression was specifically restricted to the SNs that innervate both the atria and the ventricles, with a morphology and a topology identical to those of control hearts (Figs 6 and 7). In addition, SCGNs isolated from TOH/ChR2 mice expressed ChR2 within the plasma membrane (Fig. 8 A and B). During whole cell voltage clamp measurements, photoactivation of cultured ChR2‐expressing SNs with blue light activated the expected inward photocurrent and 10 ms flashes were sufficient to trigger APs that were indistinguishable from spontaneous APs (Fig. 8 C and D). These results confirmed that we could achieve control of the activity of TOH/ChR2 neurons in vivo by delivering blue light to the neuronal efferents with a fibre‐optic probe.
Figure 6. Heart morphology and function in TOH/ChR2 mice.
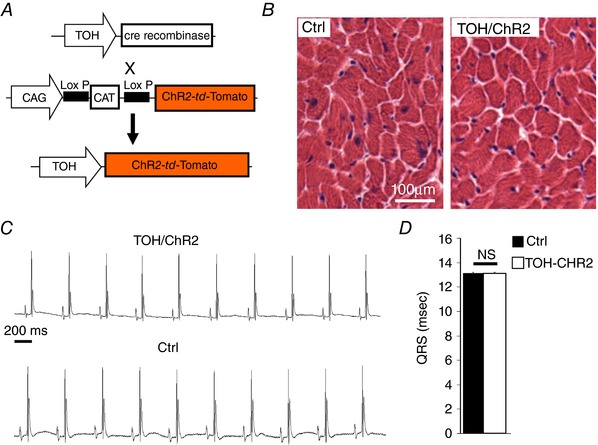
A, generation of transgenic mice expressing the fused protein ChR2‐td‐Tomato under the control of the TOH promoter (TOH/ChR2). B, haematoxylin and eosin staining in heart cryosections from control and TOH/ChR2 mice. C, ECG traces from TOH/ChR2 and control mice. D, measurement of the QRS interval in transgenic mice and littermate controls (n = 6 mice for each group; NS, not significant).
Figure 7. Characterization of cardiac autonomic innervation in TOH/ChR2 mice.
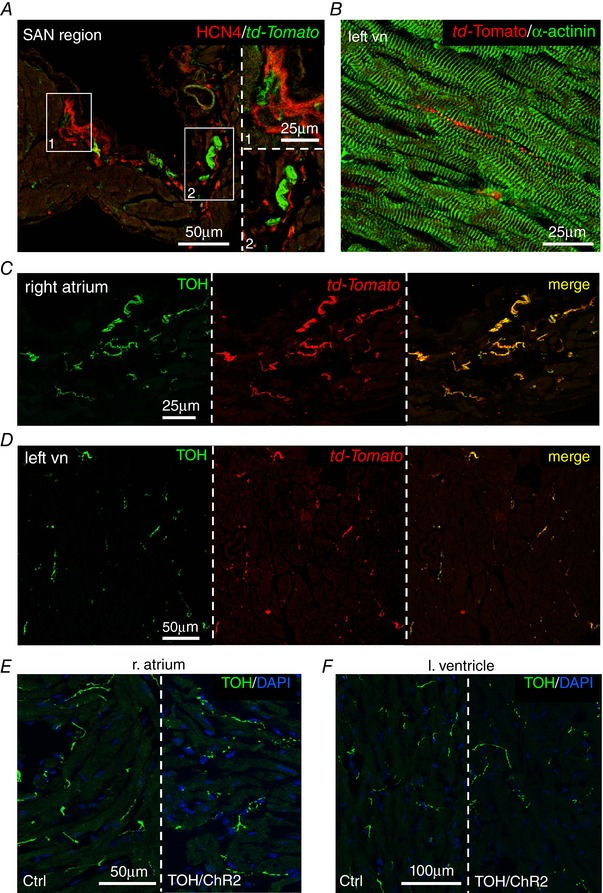
A, confocal IF analysis on right atrium sections co‐stained with antibodies to HCN4 and td‐Tomato. B, confocal IF analysis on left ventricular sections co‐stained with antibodies to td‐Tomato and α‐actinin. C and D, confocal IF analysis on right atrium (C) and left ventricle (D) heart sections, co‐stained with antibodies to td‐Tomato and TOH. E and F, confocal IF analysis on heart sections from the right atrium (E) and left ventricle (F) of control and TOH/ChR2 mice, stained with an antibody to TOH. Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole.
Figure 8. Optogenetic modulation of cultured SNs.
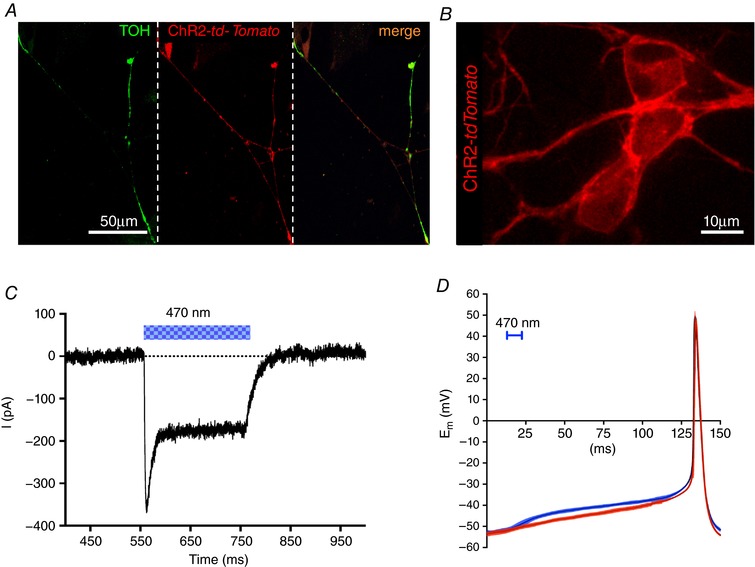
A, confocal IF analysis on cultured SNs from TOH/ChR2 mice stained with an antibody to TOH (left). Middle: fluorescence image of td‐Tomato. Right: merged image of the two signals. B, confocal image analysis on cultured TOH/ChR2 SNs. C, patch clamp recording of ChR2 photocurrent elicited by light stimulation (blue bar) in cultured TOH/ChR2 SNs. D, comparison of spontaneous (red) and light‐induced (blue) (10 ms, 470 nm light pulse) APs in cultured TOH/ChR2 SNs (n = 12 per group). Colour filled area indicates SD.
Mice were anaesthesized and the heart exposed during ECG monitoring. During the procedure, basal HR dropped by 22 ± 8% as a result of anaesthesia. We then assessed the effect of SN activation on HR (Fig. 9 A), using the hardware and protocols for in vivo optogenetics described previously (Zaglia et al. 2015) and scanned the surface of the right atrium with the tip of a fibre optic probe, at the same time as delivering trains of light flashes. The rate and light intensity were modulated to achieve different tissue irradiation volumes (see Methods). Photoactivation of the right atrial epicardium reproducibly induced sinus tachycardia, which was triggered when illuminating a very specific region of approximately 1–2 mm in diameter (area#1), whereas no HR increase was elicited by illumination of the adjacent surrounding or remote atrial areas (area#2) (Fig. 9 B–D). The responsive area of the atria was marked with dermographic ink and analysed by IF (see Methods), which revealed CMs expressing the bona fide marker of SAN cells, HCN4 (Liu et al. 2007), interacting closely with SNs (Fig. 9 E and F). Atrial responses were identical at photoactivation pulse lengths between 2 and 10 ms, in line with the observation that within the AP duration, each photostimulating pulse evokes one neurotransmitter release event. A single neuronal photoactivation pulse only shortened the inter‐beat interval of the same or immediately following R‐R cycle (Fig. 10 A), and the average delay from the neuronal AP and R‐R shortening was 166 ± 38 ms [n = 18 trials, in five different mice; the 95% confidence interval (CI) is shown]. The maximal effect on the single interval shortening was typically achieved with two consecutive photoactivation pulses and only affected the rate of up to two beats. As a consequence of very fast and short‐acting HR response kinetics, the effect of multiple photostimulation pulses on the average heart acceleration depended on the repetition frequency with respect to the resting HR. Indeed, the chronotropic responses were progressively higher when the frequency of light pulses was increased, reaching the maximal effect when the pulse frequency was approximately three times that of the underlying HR (Fig. 10 B). Photostimulation did not alter the morphology of ECG parameters (i.e. p wave, PR interval and QRS duration) (Table 2), even at the highest pulse frequencies.
Figure 10. Optogenetic control of cardiac neurogenic responses in vivo .
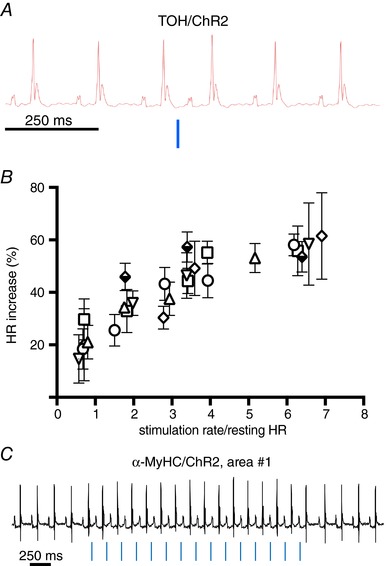
A, chronotropic effect of single neuronal photoactivation pulse (10 ms, blue line) delivered to area#1. B, relationship between neuronal photostimulation rate and chronotropic effect (% HR increase). Each point is the average of 10 repeats, and error bars show the 95% confidence interval (CI). Experiments were repeated in n = 6 different mice, each indicated by a unique symbol. Multiple comparison was performed with two‐way ANOVA followed by Tukey's test. C, representative ECG trace of sympathetic optogenetic experiment, showing atrial pacing (chronotropic stimulation at fixed light/p‐wave delay) upon photoactivation (blue lines) of the right atrial area#1 in α‐MyHC/ChR2 mice. The same behaviour was observed in all α‐MyHC/ChR2 mice analysed (n = 6) (Zaglia et al. 2015).
Table 2.
ECG parameters evaluated before and during 20 Hz photostimulation
| ECG parameter (ms) | Before photostimulation | During photostimulation | Statistics |
|---|---|---|---|
| PR | 57.04 ± 0.09 | 58.64 ± 1.10 | NS |
| P duration | 15.14 ± 0.09 | 18.74 ± 2.96 | NS |
| QRS | 13.18 ± 0.19 | 13.11 ± 0.11 | NS |
The analysis was performed in 10 mice and tested with paired ANOVA. NS, not significant.
Altogether, our optogenetic data demonstrate that rapid, selective and efficient modulation of pacemaker activity is achieved by the activation of a subset of cardiac SNs that directly target the SAN myocytes. Based on our morphological and ultrastructural evidence (Fig. 2 A–C), all these properties probably depend on the direct interaction between SNs and CMs, and in vitro experiments suggest that such an interaction motivates intercellular communication through a restricted domain with high [NE].
Validation of the specificity of sympathetic neuron optogenetics
Given that local increase in tissue temperature may potentially enhance neuronal firing rate (Acker et al. 2012), independently from opsin expression, we performed a series of control experiments to exclude the effect of photoactivation on HR could be a result of tissue heating. Accordingly, we first applied our phostimulation protocols in wild‐type mice, not expressing ChR2. As expected, photostimulation of area#1 of the right atrium, at light power up to 24 mW mm−2 and pulse duration from 1 ms to 20 s (n = 6 mice), did not have any effect on HR (HR: before photostimulation, 430 ± 1.2 beats min–1 vs. after photostimulation, 428 ± 2.2 beats min–1). Furthermore, brief stimulation trains (10 ms pulse−1, 10 Hz, for 1 s) of the same atrial region (area#1), of TOH/ChR2 mice, were repeated with a short inter‐train interval (10 s). There was no difference between the effect of the first vs. the tenth stimulation train (n = 20 repeats/mouse in n = 6 mice) and, at the end of each photoactivation train, HR returned promptly to the same baseline value. In addition, the chronotropic effect of a photoactivation train delivered prior to 20 s of continuous illumination was identical to that of the same stimulation delivered afterwards. Finally, at the end of the photostimulation experiments, hearts were harvested and the atria analysed by standard histological methods, which excluded signs of tissue damage, confirming the results previously obtained with cardiac optogenetics (Zaglia et al. 2015). Altogether, these results exclude artefacts as a result of tissue heat and support well the specific effect of light on neuronal ChR2 activation. Additional control experiments were performed to confirm that light‐induced HR acceleration was exclusively a result of sympathetic stimulation of the sinus node, and not atrial pacing through non‐specific ChR2 expression in CMs. During photoactivated sinus tachycardia, the timing of light pulses and P‐waves were unrelated (Fig. 9 C), consistent with the physiology of cardiac sympathetic regulation. This was different from the results of identical illumination protocols in α‐MyHC/ChR2 mice (Zaglia et al. 2015), which expressed ChR2 in CMs but not in SNs. In these animals, paced beats were synchronized with the light flashes applied across the atrial surface (flash/p‐wave delay: 8.00 ± 0.46 ms, n = 8 mice) (Fig. 10 C). Illumination of the SAN in TOH/ChR2 mice with long‐lasting light pulses (100–500 ms) also had no effect on HR, thus excluding the possibility that the observed chronotropic responses were a result of activation of inward Na+ entry through ChR2 mistargeted to SAN cells, which would contribute to directly increasing their diastolic depolarization rate.
Optogenetic assessment of neurocardiac communication in vivo
To assess whether the dynamics of neurocardiac signalling are similar between our co‐cultures and the intact heart, we performed neuronal photostimulation at the same time as gradually increasing the dose of the β‐AR antagonist propranolol (Fig. 11). We used trains of 20 light pulses at 20 Hz (10 ms pulse−1) which elicited, in the absence of propranolol, a significant increase in HR (see above). Treatment with propranolol (5 mg kg−1) decreased resting HR [ΔHRbpm, −77.8 (95% CI: −57.5 to –98.1)] but did not have any effect on the neurogenic increase in HR [ΔHRbpm, photoactivation: 126.5 (106.2–146.9) vs. photoactivation+: 158.5 (95% CI: 138.1–178.8)] (Fig. 11 A). Indeed, a much higher dosage of propranolol (20–50 mg kg−1) was necessary to significantly reduce the chronotropic response to photoactivation of SAN neurons (Fig. 11 A). This is consistent with the paradigm where neuronal activation generates high [NE]cleft within restricted cardiac synaptic volumes thereby overcoming the antagonist concentration necessary to inhibit the response of diffuse NE stimulation. The morphology of ECG waves remained unchanged during photostimulation, with the exception of the prolongation of p‐wave duration, as was more evident at the highest propranolol doses, both under basal conditions and during phostimulation (Table 3). The same pharmacological inhibition assay was performed in a subset of experiments with the β‐1 selective blocker, metoprolol, confirming that higher doses of the antagonist were required to ablate photostimulated chronotropic responses than that sufficient to minimize resting HR (Fig. 11 B).
Figure 11. Assessment of the efficiency of neurocardiac coupling in vivo .
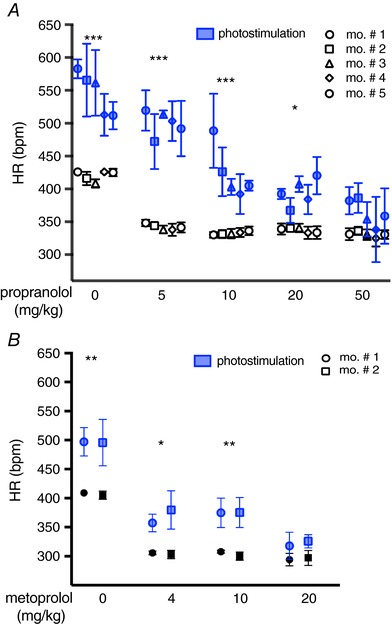
A, dose‐effect analysis of the treatment with the β‐AR antagonist, propranolol, on the chronotropic response to neuronal photostimulation. Blue symbols identify responses to the photostimulation train. Each point is the average of six to 10 repeats, and error bars show the 95% confidence interval (CI). Experiments were repeated in n = 5 different mice, as shown in the caption. Multiple comparison was performed with two‐way ANOVA followed by Tukey's test. Asterisks in each group represent comparison of resting vs. photostimulated HR values (*** P < 0.001; * P < 0.05). B, dose‐effect analysis of the treatment with the β1‐AR antagonist, metoprolol, on the chronotropic response to neuronal photostimulation. Blue symbols identify responses to the photostimulation train. Each point is the average of four to six repeats, and error bars show the 95% CI. Experiments were repeated in n = 2 different mice, as shown in the caption. Multiple comparison was performed with two‐way ANOVA followed by Tukey's test. Asterisks in each group represent comparison of resting vs. photostimulated HR values (** P < 0.01; * P < 0.05).
Table 3.
ECG parameters evaluated before and during 20 Hz photostimulation in the presence of different dosages of propranolol (10 vs. 50 mg kg−1)
| ECG parameter (ms) | Baseline | Illumination | Statistics |
|---|---|---|---|
| Propranolol 10 | |||
| PR | 55.31 ± 0,08 | 53.78 ± 4.33 | NS |
| P duration | 12.77 ± 0,07 | 18.93 ± 3.51 | ** |
| QRS | 12.60 ± 0.02 | 13.04 ± 0.10 | NS |
| Propranolol 50 | |||
| PR | 55.61 ± 0,04 | 59.58 ± 0.43 | NS |
| P duration | 22.54 ± 0,29 | 28.52 ± 1.31 | ** |
| QRS | 12.77 ± 0.02 | 13.45 ± 0.08 | NS |
The analysis was performed in five mice and tested with paired ANOVA.
** P < 0.01. NS, not significant.
In summary, our in vivo data, showing the spatial selectivity of SN activation of SAN responses, the functional evidence of elevated [NE]cleft, and the kinetics of neuroeffector coupling, are all in support of the working model where the establishment of a specialized extracellular domain enables sympathetic neurotransmission to the heart to occur in a quasi‐synaptic fashion (Zaglia & Mongillo, 2017).
Discussion
Cardiac SNs have a fundamental role in the control of cardiac physiology, and their dysfunction is a common finding in several types of heart diseases (Kaye & Esler, 2005; El‐Armouche & Eschenhagen, 2009; Florea & Cohn, 2014; Franzoso et al. 2016). Although decades of investigations have clarified the molecular mechanisms underlying neurocardiac regulation and have described in great detail the intracellular signalling pathways dependent upon adrenergic input, fundamental aspects of the neurocardiac communication remain elusive. Here, we have presented new insights into the SN‐CM intercellular signalling dynamics, as revealed in vitro by live imaging of cAMP in co‐cultures and in vivo using SN optogenetics.
Our in vitro data indicate that sympathetic modulation of CM signalling requires direct cell–cell contact, which brings together the neuronal NE‐releasing and the CM NE‐sensing membranes. Such an interaction defines a low‐volume extracellular space, estimated in the order of 0.1 μm3, in which high [NE] is quickly achieved with only a few thousand molecules (i.e. few vesicles), as estimated by numerical simulation obtained in neuroeffector junctions with comparable geometry (Scepanovic, 2011). Such restricted signalling domains promote the activation of only a fraction of β‐ARs within the contacted CM membrane, as supported by the stringent selectivity of neuronal target activation, and allow high efficiency enabling CM responses to minimal neuronal activation. These results fit well with the observation that signalling complexes of the cAMP/PKA pathway, including β1‐ARs, as well as PKA anchoring proteins and targets, cluster within the CM membrane at the neuronal interaction site (Shcherbakova et al. 2007). Our imaging data describe the consequences of such arrangement, showing that cAMP synthesis initiates, upon neuronal stimulation, at the innervated CM site, and identify the complex formed by the neuronal varicosity and post‐synaptic CM membrane as the functional signalling unit in the neurogenic regulation of the heart. Our in vitro results were corroborated by our in vivo findings using optogenetics, which was exploited to achieve non‐invasive control of cardiac SNs, avoiding direct effects on heart electrophysiology. This property, together with the high spatial resolution in ChR2 activation, provided a flexible tool for probing the function of restricted groups of cardiac neurons in vivo, and address neuroeffector signalling kinetics. Although there are differences between the cellular environment in vitro and the intact innervated heart, our findings, in line with current literature (Landis, 1976; Shcherbakova et al. 2007; Oh et al. 2016), indicate that the neurocardiac contacts in two experimental settings share key ultrastructural and morphological features. The results of the in vivo experiments support the working model where neurogenic regulation of SAN automaticity is underpinned by high‐efficiency metabotropic synapses, with elevated speed and single beat precision. This is further sustained by previous numerical modelling of agonist kinetics in a neuroeffector junction of comparable morphology, showing a rise and decay time for [NE]cleft of only few ms (Bennett et al. 1995). Our results showing that high antagonist doses are required to ablate neuron‐dependent HR acceleration substantiate this model. Of note, these data are in line with clinical evidence showing that patients under β‐blocker treatment retain a certain degree of chronotropic sympathetic reflex, indicating the capacity of neurocardiac stimulation to release NE at a concentration competing with the binding of the antagonist to β‐ARs.
Although our optogenetic investigations have been focussed on the sympathetic regulation of HR, and thus are limited to the analysis of SAN function, our in vitro (Fig. 3 A and B) and ex vivo (Fig. 2 A–C) data, in both experimental and human hearts, suggest that tight neurocardiac coupling also takes place in the ventricles. It is thus tempting to speculate that acute control of cardiac inotropism, and long‐term modulation of CM structure by SNs, may follow the same behaviour, and this will be the subject of future investigations. One may question whether the activation of a limited CM membrane (and receptor) portion by a minute varicosity could provide sufficient adrenergic input to sustain cardiac stimulation during fight‐or‐flight reactions. However, in both the SAN and ventricular myocytes, each cell forms several junctional sites with the same neuronal process, and may therefore receive NE simultaneously from multiple point sources, thus increasing the potency of adrenergic stimulation. In addition, because each cell is innervated by different processes, it is tempting to speculate that neuronal recruitment may be involved in modulating the degree of CM responses across the wide latitude of physiological regulation, spanning from the fine tuning of heart rhythm to drastic increase in cardiac stimulation of fight‐or‐flight responses. Our working model has several implications for cardiac pathology, including heart failure (HF), the first cause of cardiovascular mortality in Europe (47% of deaths). HF is commonly associated with altered function of cardiac autonomic control, although the underlying mechanisms are not entirely understood. The common tenet is that failing hearts have decreased responsiveness to β‐AR agonists, reduced NE content in sympathetic endings, increased venous spillover of neuronal NE and interstitial NE accumulation (Kaye & Esler, 2005; El‐Armouche & Eschenhagen, 2009; Florea & Cohn, 2014). All of these features would be explained by effects of the structural remodelling of failing hearts on NCJ organization, which may lead to the disruption of the junctional cleft, with subsequent interstitial NE diffusion, reduced efficiency of cardiac β‐adrenergic responses and decreased efficacy of pre‐synaptic NE re‐uptake. In further support of this, numerical modelling in similar neuroeffector junctions shows that the increase, by a few microns, of the neuron‐to‐target intermembrane distance, causes neurotransmitter diffusion and a dramatic reduction (<15%) in the number of activated post‐synaptic receptors (Bennett et al. 1995). In addition, our unpublished data show that prolonged interference with the mechanisms of neurocardiac coupling causes per se SN degeneration, thus reducing the number of functional neurocardiac contacts, and subsequently the cumulative neuronal input to target cells.
In conclusion, by inspecting a seemingly overlooked system, under the new light of fluorescence imaging and optogenetics, we propose a model of cellular interaction to explain neurocardiac regulation, which has implications in the understanding of heart physiology and pathology.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
VP, FDB, MF and APP performed the in vitro experiments, as well as analysed and critically discussed data. VP and FDB also contributed to the in vivo experiments. NP contributed to the in vivo optogenetics and analysed the ECG data. MF performed ultrastructural analysis on mouse heart slices. CB provided autopic human heart samples. MWK analysed and discussed data and contributed to the writing of the manuscript. TZ set up the experimental models, performed confocal IF analyses, in vivo optogenetics, analysed and critically discussed data, and wrote the manuscript. MM designed the study, analysed data and wrote the manuscript. All authors approved the final version of the manuscript submitted for publication and agree to be accountable for all aspects of the work, in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved, and that all persons designated as authors qualify for authorship, and have been listed.
Funding
This work was supported by the French Muscular Dystrophy Association (AFM‐Téléthon; grant no. 19996) to Tania Zaglia and Telethon‐Italy (GGP11224) to MM.
Translational perspective.
We studied, both in vitro and in vivo, the dynamics of intercellular signalling between myocardial sympathetic neurons and cardiomyocytes. Our findings indicate that specific interaction between the neuronal catecholamine‐releasing sites and cardiomyocyte membrane encloses a ‘synapse‐like’ domain, allowing fine and efficient regulation of heart activity under basal and stress conditions. Our results thus refine the understanding of basic cardiac physiology and suggest that alterations in the neurocardiac interaction may participate in the pathogenesis of cardiac diseases as common as heart failure. Our results extend the understanding of the mechanisms allowing the heart to adapt its performance to the blood demand of peripheral organs under basal conditions and during stress. Moreover, the present study may contribute to explaining the disease mechanisms causing an altered autonomic control of heart function in heart failure.
Acknowledgements
We are grateful to Drs Tullio Pozzan and Giorgio Carmignoto for critical discussion; Drs Martin Lohse and Jin Zhang for sharing EPAC1‐cAMPs and AKAR3 encoding plasmids; Dr Kees Jalink for sharing H187; and Anna Di Bona for technical assistance. We acknowledge the generous support of Mr Gilberto Benetton and Ms Margherita Zorzut.
Biographies
Valentina Prando is a neurobiologist who completed her PhD at the University of Padova. She has studied communication between cardiac sympathetic neurons and effectors in healthy samples and diseased models.

Francesca Da Broi received her PhD from the University of Padova. Her studies have focussed on in vitro imaging of second messenger dynamics and optogenetics.
Mauro Franzoso received his PhD from the University of Padova. He is a biotechnologist, whose main studies have focused on the neurotrophin‐mediated intercellular signalling between heart and neurons.
Anna Pia Plazzo received her PhD from Humboldt‐Universität, Berlin (Germany). Her studies focus on cell signalling and electrophysiology.
This research stems from the joint contribution of multiple scientists who participated with equal roles in the project.
Edited by: Don Bers & Bjorn Knollmann
Linked articles This article is highlighted by a Perspective by Sasse. To read this Perspective, visit https://doi.org/10.1113/JP276109.
Contributor Information
Tania Zaglia, Email: tania.zaglia@unipd.it.
Marco Mongillo, Email: marco.mongillo@unipd.it.
References
- Acker L, Pino E, Zorzo A, Henninger M & Boydes ES (2012). In vivo measurements of light propagation and brain heating: implications for optogenetics. Soc Neurosci 12, 525–534. [Google Scholar]
- Allen MD & Zhang J (2006). Subcellular dynamics of protein kinase A activity visualized by FRET‐based reporters. Biochem Biophys Res Commun 348, 716–721. [DOI] [PubMed] [Google Scholar]
- Anderson MJ & Cohen MW (1977). Nerve‐induced and spontaneous redistribution of acetylcholine receptors on cultured muscle cells. J Physiol 268, 757–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrenberg AB, Stainier DY, Baier H & Huisken J (2010). Optogenetic control of cardiac function. J Physiol 330, 971–974. [DOI] [PubMed] [Google Scholar]
- Bennett MR, Gibson WG & Robinson J (1995). Probabilistic secretion of quanta: spontaneous release at active zones of varicosities, boutons, and endplates. Biophys J 69, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2008). Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70, 23–49. [DOI] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G & Deisseroth K (2005). Millisecond‐timescale, genetically targeted optical control of neural activity. Nat Neurosci 8, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Boyle PM, Williams JC, Ambrosi CM, Entcheva E & Trayanova NA (2013). A comprehensive multiscale framework for simulating optogenetics in the heart. Nat Commun 4, 2370, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruegmann T, Malan D, Hesse M, Beiert T, Fuegemann CJ, Fleischmann BK & Sasse P (2010). Optogenetic control of heart muscle in vitro and in vivo. Nat Methods 7, 897–900. [DOI] [PubMed] [Google Scholar]
- Castaldi A, Zaglia T, Di Mauro V, Carullo P, Viggiani G, Borile G, Di Stefano B, Schiattarella GG, Gualazzi MG, Elia L, Stirparo GG, Colorito ML, Pironti G, Kunderfranco P, Esposito G, Bang ML, Mongillo M, Condorelli G & Catalucci D (2014). MicroRNA‐133 modulates the beta1‐adrenergic receptor transduction cascade. Circ Res 115, 273–283. [DOI] [PubMed] [Google Scholar]
- Choate JK, Klemm M & Hirst GD (1993). Sympathetic and parasympathetic neuromuscular junctions in the guinea‐pig sino‐atrial node. J Auton Nerv Syst 44, 1–15. [DOI] [PubMed] [Google Scholar]
- Chow LT, Chow SS, Anderson RH & Gosling JA (2001). Autonomic innervation of the human cardiac conduction system: changes from infancy to senility – an immunohistochemical and histochemical analysis. Anat Rec 264, 169–182. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D ( 2010). The role of the funny current in pacemaker activity. Circ Res 106, 434–446. [DOI] [PubMed] [Google Scholar]
- Dobrzynski H, Anderson RH, Atkinson A, Borbas Z, D'Souza A, Fraser JF, Inada S, Logantha SJ, Monfredi O, Morris GM, Moorman AF, Nikolaidou T, Schneider H, Szuts V, Temple IP, Yanni J & Boyett MR (2013). Structure, function and clinical relevance of the cardiac conduction system, including the atrioventricular ring and outflow tract tissues. Pharmacol Ther 139, 260–288. [DOI] [PubMed] [Google Scholar]
- El‐Armouche A & Eschenhagen T (2009). Beta‐adrenergic stimulation and myocardial function in the failing heart. Heart Fail Rev 14, 225–241. [DOI] [PubMed] [Google Scholar]
- Florea VG & Cohn JN (2014). The autonomic nervous system and heart failure. Circ Res 114, 1815–1826. [DOI] [PubMed] [Google Scholar]
- Franzoso M, Zaglia T & Mongillo M (2016). Putting together the clues of the everlasting neuro‐cardiac liaison. Biochim Biophys Acta 1863, 1904–15. [DOI] [PubMed] [Google Scholar]
- Gardner RT, Ripplinger CM, Myles RC & Habecker BA (2016). Molecular mechanisms of sympathetic remodeling and arrhythmias. Circ Arrhythm Electrophysiol 9, e001359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm M, Brown JH, Gardner RT, Ripplinger CM, Myles RC, Habecker BA, Anderson ME, Birren SJ, Fukuda K, Herring N, Hoover DB, Kanazawa H, Paterson DJ, Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P & Bamberg E (2010). Beta‐adrenergic receptor signaling in the heart: role of CaMKII molecular mechanisms of sympathetic remodeling and arrhythmias molecular and cellular neurocardiology: development, and cellular and molecular adaptations to heart disease Channelrhodopsin‐2, a directly light‐gated cation‐selective membrane channel. J Mol Cell Cardiol 48, 322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habecker BA, Anderson ME, Birren SJ, Fukuda K, Herring N, Hoover DB, Kanazawa H, Paterson DJ, Ripplinger CM, Gardner RT & Myles RC (2016). Molecular and cellular neurocardiology: development, and cellular and molecular adaptations to heart disease. J Physiol 594, 3853–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst GD, Choate JK, Cousins HM, Edwards FR & Klemm MF (1996). Transmission by post‐ganglionic axons of the autonomic nervous system: the importance of the specialized neuroeffector junction. Neuroscience 73, 7–23. [DOI] [PubMed] [Google Scholar]
- Jansen AS, Nguyen XV, Karpitskiy V, Mettenleiter TC & Loewy AD (1995). Central command neurons of the sympathetic nervous system: basis of the fight‐or‐flight response. Science (New York, NY) 270, 644–646. [DOI] [PubMed] [Google Scholar]
- Kaye D & Esler M (2005). Sympathetic neuronal regulation of the heart in aging and heart failure. Cardiovasc Res 66, 256–264. [DOI] [PubMed] [Google Scholar]
- Klarenbeek J, Goedhart J, van Batenburg A, Groenewald D & Jalink K (2015). Fourth‐generation epac‐based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS ONE 10, e0122513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis SC (1976). Rat sympathetic neurons and cardiac myocytes developing in microcultures: correlation of the fine structure of endings with neurotransmitter function in single neurons. Proc Natl Acad Sci USA 73, 4220–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy MN & Martin PJ (1989). Autonomic neural control of cardiac function In Physiology and Pathophysiology of the Heart, ed. Sperelakis N, pp. 361–379. Springer, Boston, MA. [Google Scholar]
- Liu J, Dobrzynski H, Yanni J, Boyett MR & Lei M (2007). Organisation of the mouse sinoatrial node: structure and expression of HCN channels. Cardiovasc Res 73, 729–738. [DOI] [PubMed] [Google Scholar]
- Lombardi F, Malliani A, Pagani M & Cerutti S (1996). Heart rate variability and its sympatho‐vagal modulation. Cardiovasc Res 32, 208–216. [DOI] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P & Bamberg E (2003). Channelrhodopsin‐2, a directly light‐gated cation‐selective membrane channel. Proc Natl Acad Sci USA 100, 13940–13945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev VO, Bunemann M, Hein L, Hannawacker A & Lohse MJ (2004). Novel single chain cAMP sensors for receptor‐induced signal propagation. J Biol Chem 279, 37215–37218. [DOI] [PubMed] [Google Scholar]
- Nussinovitch U, Shinnawi R & Gepstein L (2014). Modulation of cardiac tissue electrophysiological properties with light‐sensitive proteins. Cardiovasc Res 102, 176–187. [DOI] [PubMed] [Google Scholar]
- Oh Y, Cho GS, Li Z, Hong I, Zhu R, Kim MJ, Kim YJ, Tampakakis E, Tung L, Huganir R, Dong X, Kwon C & Lee G (2016). Functional coupling with cardiac muscle promotes maturation of hPSC‐derived sympathetic neurons. Cell Stem Cell 19, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Kim HD, Miwa A, Kuriu T & Okado H (1999). Continual remodeling of postsynaptic density and its regulation by synaptic activity. Nat Neurosci 2, 804–811. [DOI] [PubMed] [Google Scholar]
- Rochais F, Vandecasteele G, Lefebvre F, Lugnier C, Lum H, Mazet JL, Cooper DM & Fischmeister R (2004). Negative feedback exerted by cAMP‐dependent protein kinase and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes: an in vivo study using adenovirus‐mediated expression of CNG channels. J Biol Chem 279, 52095–52105. [DOI] [PubMed] [Google Scholar]
- Saggin L, Gorza L, Ausoni S & Schiaffino S (1989). Troponin I switching in the developing heart. J Biol Chem 264, 16299–16302. [PubMed] [Google Scholar]
- Scepanovic D (2011). In Division of Health Sciences and Technology. Harvard‐MIT, Cambridge, MA. [Google Scholar]
- Sharma N, Deppmann CD, Harrington AW, St Hillaire C, Chen ZY, Lee FS & Ginty DD (2010). Long‐distance control of synapse assembly by target‐derived NGF. Neuron 67, 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova OG, Hurt CM, Xiang Y, Dell'Acqua ML, Zhang Q, Tsien RW & Kobilka BK (2007). Organization of beta‐adrenoceptor signaling compartments by sympathetic innervation of cardiac myocytes. J Cell Biol 176, 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater CR ( 2003). Structural determinants of the reliability of synaptic transmission at the vertebrate neuromuscular junction. J Neurocytol 32, 505–522. [DOI] [PubMed] [Google Scholar]
- Stieber J, Herrmann S, Feil S, Loster J, Feil R, Biel M, Hofmann F & Ludwig A (2003). The hyperpolarization‐activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. PNAS 100, 15235–15240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengrowski AM, Wang X, Tapa S, Posnack NG, Mendelowitz D & Kay MW (2015). Optogenetic release of norepinephrine from cardiac sympathetic neurons alters mechanical and electrical function. Cardiovasc Res 105, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M & Pozzan T (2002). Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science (New York, NY) 295, 1711–1715. [DOI] [PubMed] [Google Scholar]
- Zaglia T, Di Bona A, Chioato T, Basso C, Ausoni S & Mongillo M (2016). Optimized protocol for immunostaining of experimental GFP‐expressing and human hearts. Histochem Cell Biol 146, 407–419. [DOI] [PubMed] [Google Scholar]
- Zaglia T, Milan G, Franzoso M, Bertaggia E, Pianca N, Piasentini E, Voltarelli VA, Chiavegato D, Brum PC, Glass DJ, Schiaffino S, Sandri M & Mongillo M (2013). Cardiac sympathetic neurons provide trophic signal to the heart via beta2‐adrenoceptor‐dependent regulation of proteolysis. Cardiovasc Res 97, 240–250. [DOI] [PubMed] [Google Scholar]
- Zaglia T & Mongillo M (2017). Cardiac sympathetic innervation, from a different point of (re)view. J Physiol 595, 3919–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaglia T, Pianca N, Borile G, Da Broi F, Richter C, Campione M, Lehnart SE, Luther S, Corrado D, Miquerol L & Mongillo M (2015). Optogenetic determination of the myocardial requirements for extrasystoles by cell type‐specific targeting of ChannelRhodopsin‐2. Proc Natl Acad Sci USA 112, E4495–E4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zareen N & Greene LA (2009). Protocol for culturing sympathetic neurons from rat superior cervical ganglia (SCG). J Vis Exp 23, 988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A & Deisseroth K (2007). Multimodal fast optical interrogation of neural circuitry. Nature 446, 633–639. [DOI] [PubMed] [Google Scholar]