

Abstract
Single cancer cell–sequencing studies currently use randomly selected cells, limiting correlations among genomic aberrations, morphology, and spatial localization. We laser-captured microdissected single cells from morphologically distinct areas of primary breast cancer and corresponding lymph node metastasis and performed whole-exome or deep-target sequencing of more than 100 such cells. Two major subclones coexisted in different areas of the primary tumor, and the lymph node metastasis originated from a minor subclone in the invasive front of the primary tumor, with additional copy number changes, including chr8q gain, but no additional point mutations in driver genes. Lack of metastasis-specific driver events led us to assess whether other clonal and subclonal genomic aberrations preexisting in primary tumors contribute to lymph node metastasis. Gene mutations and copy number variations analyzed in 5 breast cancer tissue sample sets revealed that copy number variations in several genomic regions, including areas within chr1p, chr8q, chr9p, chr12q, and chr20q, harboring several metastasis-associated genes, were consistently associated with lymph node metastasis. Moreover, clonal expansion was observed in an area of morphologically normal breast epithelia, likely driven by a driver mutation and a subsequent amplification in chr1q. Our study illuminates the molecular evolution of breast cancer and genomic aberrations contributing to metastases.
Keywords: Genetics, Oncology
Keywords: Breast cancer, Genetic variation, Molecular genetics
Introduction
Increasing application of next-generation sequencing in cancer genome research is illuminating the genetics underlying the molecular mechanism of tumorigenesis and primary tumor growth. However, comprehensive understanding of the evolutionary progress and molecular mechanism of tumor metastasis, the primary cause of death, remains unclear (1, 2). Insight into the molecular mechanism of tumor spread and metastasis is crucial to clinical efforts to prevent metastasis. Several theories and hypotheses as to how and why tumors metastasize have been put forth. Some studies have shown that the probability of metastases correlates with primary tumor size (3), while other studies focused on functional features of the primary tumor, such as epithelial-mesenchymal transition (EMT) (4, 5), tumor microenvironment (6), and physical factors (7, 8). More recent genomic studies of paired primary tumors and metastasis have shown that internal factors and intrinsic mechanisms, especially genomic variation, likely trigger and promote tumor metastasis (9), but only a few genes have been identified in these comparisons (10). A shift in mutational rates of cancer genes in metastasis has been observed, but only TP53 showed a significantly increased mutational rate when analyzing pair samples of multiple cancer types, including breast cancer (11). Yates et al. recently reported that most distant metastases acquired additional mutations in a wider repertoire of cancer genes, while only a few additional mutated driver genes were found in lymph node metastasis of breast cancers (12). Moreover, other studies have shown that metastasis-related aberrations originate from the primary tumor or certain subclones thereof (13–15), suggesting that metastasis-related genes are altered in both primary tumors and metastasis, thus hindering identification by genomic comparison of paired primary tumor and metastasis.
Single-cell sequencing is one of the most promising and valuable techniques in cancer research and medical science (16, 17). Single-cell sequencing has successfully illuminated intratumoral heterogeneity as well as clonal and tumor evolution analysis (18, 19). However, these studies have not provided detailed insight of the morphologic spatial localization of specific cells within the tumor, nor revealed the relationship between clonal evolution and spatial location of intratumor cells. We used laser-capture microdissection (LCM) of single cells from H&E-stained tissue sections followed by single-cell sequencing and targeted duplex deep sequencing of pools of thousands of cells from different spatial locations of breast tumors to define the microheterogeneity and spatial distribution of intratumoral subclones and in primary tumor and synchronous lymph node metastasis (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI97449DS1). Furthermore, we explored the molecular mechanisms of lymph node metastasis in 4 sequencing data sets of primary breast tumors from patients with or without lymph node metastasis and copy number variation (CNV) analysis of selected genes in another 170 breast cancers.
Results
LCM isolation and sequencing of single cells from primary tumor and lymph node metastases of an ER+ breast cancer patient.
Tissues from a woman undergoing mastectomy for a 32 mm primary estrogen receptor–positive (ER+) breast cancer were obtained, including tissue blocks from the primary tumor and from adjacent morphologically normal ductal tissue (>20 mm from any pathological detectable tumor cell) as well as the sentinel axillary lymph node. Single cells and cell pools were isolated by LCM of H&E-stained sections from distinct morphological areas of the primary tumor, the periphery (MeP) and central (MeC) areas of the tumor-infiltrated lymph node, a morphologically normal area of the tumor-infiltrated lymph node (MeD), and the morphologically normal ductal tissue, and sequenced. Small macroscopically dissected pieces (2 × 2 mm) of the primary tumor (PT4), lymph node metastasis, and morphologically normal breast tissue (BN-T) (>20 mm from any pathological detectable tumor cell) were also sequenced. To determine the patient’s germline, we also sequenced pieces of normal skin (Skin-T) and normal lymphocytes from the axillary nodes (Ly-T).
A total of 97 single cells from 9 morphologically distinct areas were whole-genome amplified and whole-exome sequenced, including the invasive front (InvF, defined as malignant cells embedded in the border of solid tumor growth and fatty tissue) (Figure 1, A, F, I and J), 3 distinct areas within the solid invasive growth (PT1, PT2, and PT3) (Figure 1, A and F–H), an area of ductal carcinoma in situ (DCIS), and invasive cancer cells localized in close proximity to the selected DCIS (Figure 1, A and K–N), MeC and MeP, normal lymphocytes from the same lymph node (Ly) (Figure 1, A–C), and normal breast epithelial cells (BN) (Figure 1, A, D and E) using Illumina’s HiSeq 2000 and HiSeq 2500 (Supplemental Figure 2A and Supplemental Table 1). The mean depth of single-cell exome sequencing was 45× with a median 10× coverage up to 53%. For whole-exome sequencing (WES) of the small tissue pieces, the depths of the primary tumor (PT4), lymph node metastasis, BN-T, Ly-T, and Skin-T were 87.25×, 71.65×, 99.33×, 105.32×, and 123.77×, respectively.
Figure 1. Subclone distribution within different tissues of the analyzed breast cancer patient.

(A) Schematic representation of the tissues analyzed by WES of single cells and cell pools and the distribution of the different subclones within these areas. MT, metastasis. (B) Axillary lymph node containing large MT stained for estrogen receptor. (C) Magnification of an area of the lymph node exhibiting normal architecture and morphology, but also containing disseminated single cancer cells. (D) H&E-stained section of the BN-T including normal breast ducts. (E) Magnification of an area of D showing normal breast epithelia. (F) H&E-stained section of the primary tumor biopsy provides a spatial overview of the 3 solid growth tumor areas selected for LCM in addition to the InvF. (G and H) Solid growth area 2 (PT2). (I and J) Border of solid growth area 1 (PT1) including the InvF. H and J are stained for Ki-67, while G and I are stained for ER. Original magnification: ×12.5 (D, E, F); ×100 (B, C, K, L, M, N); ×50 (G, H, I, J). Section containing area of DCIS stained with H&E (K), and for ER (L), CK14 (M), and Ki-67 (N). (M) The intact myoepithelial layer is visualized by staining for CK14.
Typically, the allele dropout (ADO) (20) rate is used to assess whole-genome amplification (WGA) uniformity of single-cell–sequencing data. However, we defined and used the K value, which is less affected by sequencing depth of single cells, as a measure of single-cell data quality (see Methods for further details; Supplemental Figure 2, B–E). For mutation calling of single cells, we developed a pipeline optimized to avoid false positives and focused on mutations identified in at least 2 separate single cells (see Methods for further details). As a result, the predominant C→A signature of substitution in single cells, presumably caused by oxidized guanine bases (8-oxo-guanine) in the raw cellular DNA or in early cycles of WGA, was significantly reduced (Supplemental Figure 3, A and B). After filtering the putative germline mutations using normal skin tissue as a reference, a total of 218 single nucleotide variants (SNVs) and 4 indels from the 97 single cells remained. To validate the SNVs identified by single-cell exome sequencing as well as achieve a deeper sequencing depth and determine the mutational frequencies in the bulk tumor, we designed a targeted sequencing panel that covered the 218 identified SNVs together with the top 20 breast cancer genes and performed targeted deep sequencing on 78 selected single cells (median depth of 157×) (Supplemental Figure 2, A and C). In addition, 7 cell pools (see Methods for details), including 4 distinct areas within the invasive tumor (PT1, PT2, PT3, and PT4), InvF, lymph node metastasis, and BN (total depth of 4177× for cell pools), also underwent targeted duplex sequencing. The duplex method (21) was used to analyze the 7 cell pools, achieving a total duplex consensus sequence (DCS) depth of 1500× (see Methods for further details).
Sequencing of single cells and cell pools delineated the structure of subclones in the breast tumors.
In total, 127 of the 218 SNVs were verified by DCSs of cell pools. In addition, we found additional SNVs that, although not supported by DCSs, were only present in normal breast epithelia samples, including 22 detected in at least 2 BN cells and at least 1 of the 3 whole-genome amplified normal breast epithelial cell pools (BNM) (see Methods for further details), which were included in further analysis (Supplemental Table 2). The 69 candidate mutations that failed validation (94% were C>T and C>A) showed trinucleotide mutation signatures that significantly correlated with amplified errors (R = 0.85), but not true mutations (R = 0.17), and thus were excluded from further analysis (Supplemental Figure 4). The 149 SNVs showed 3 distinct patterns of allele frequencies in 7 cell pools and the small tissue pieces (Supplemental Figure 3C and Supplemental Figure 5) and thus could be divided into 3 subsets: 78 SNVs were only present in the breast cancer samples (subset 1, Supplemental Table 3); 49 SNVs were only found in morphologically normal breast epithelial cells (subset 2, Supplemental Table 4); and a minor fraction (22 SNVs) were identified in some cells from several areas that were thus not restricted to a single specific area from this patient (subset 3; Supplemental Table 5). Subset 1 SNVs were found only in bulk tissue, cell pools, and single cells isolated from tumor tissues. Subset 2 SNVs were found only in bulk tissue, cell pools, and single cells isolated from the normal breast epithelial tissue. For subset 3 SNVs, the allele frequencies were low in almost all of the cancer cell pools and bulk tissues (1.47% by mean), while for some subset 3 SNVs, allele frequencies were relatively higher in Ly-T (Supplemental Table 5). Interestingly, a truncating mutation of TET2, a gene related to clonal expansion of lymphocytes usually present in the blood of normal elderly persons, was identified in subset 3 (22). Thus, the mutations in subset 3 are likely a result of age-related lymphocyte genetic aberrations in this 92-year-old patient.
Focusing on the 78 breast cancer cell–specific mutations of subset 1, we identified a number of clonal mutations and 4 distinct subgroups of mutations that differed from the clonal mutations by integrating their distribution in single cells (Figure 2C) and data from cluster analysis of their allele frequencies in 6 cancer cell pools (Figure 2A).
Figure 2. SNV analysis of breast cancer single cells and cell pools identified 2 dominant subclones and additional spatial location–specific subclones.

(A) AF of breast cancer–specific SNVs in 6 cell pools of distinct morphologically defined breast tumor areas (lymph node metastasis, InvF, and 4 distinct areas within the solid invasive growth [PT1, PT2, PT3, PT4]). Presumed clonal SNVs located in CNV regions were not shown for distorted VAF. Blue, clonal SNVs; red, subclone 1–specific SNVs; green, subclone 2–specific SNVs; purple, metastases-specific SNVs; black, DCIS-specific SNVs. (B) Fraction of the 2 dominant subclones in each cell pool estimated by least square fit (red, subclone 1; green, subclone 2). (C) Heatmap of breast cancer–specific SNVs identified in breast cancer single cells (dark blue, mutated; sky blue, WT; white, WT and sequencing depth of less than ×8). Clonal SNVs predicted not to change protein structure and clonal SNVs with median sequencing depths of less than ×50 in the 57 single cells are not shown, while all subclonal SNVs are shown. Colored boxes encapsulate the group-specific SNVs (the same color code for each group as in A is used), except for the orange box, which denotes clonal SNVs located in CNV regions. (D) PCA of SNVs based on allele frequencies in breast cancer cell pools (the same color code for each group as in A is used). Two dimensions were shown (d = 2). (E) The evolutionary tree of the single cell–sequenced breast cancer. Accumulation of chromosome gains and losses as well as somatic mutations are represented by red, blue, and purple, respectively.
One subgroup consisting of 3 SNVs (GATAD2A, MYH6, MAGEC3) was specific to lymph node metastasis single cells, consistent with their presentation of DCSs, which were only identified in the lymph node metastasis cell pool, except for GATAD2A, which was also present in DCSs of the InvF cell pool. Another subgroup consisting of 3 SNVs (ARHGEF28, UBR5, KIF3C) was specific to DCIS single cells and present at relatively low allele frequencies in DCSs of the cell pools PT3 and PT4, which were the only 2 cell pools containing cells from the DCIS areas. The third subgroup, consisting of 3 SNVs (OR10K2, CHIC1, PDE1C), was specifically present in 7 single cells isolated from the InvF; however, DCSs for these SNVs were not only identified in the InvF cell pool, but also in cell pools from other invasive tumor areas, including PT1 and PT2, suggesting a genetic relationship between InvF and PT1/PT2. Moreover, the fourth subgroup of 9 SNVs exhibited the opposite distribution compared with the third mutation subgroup, being present in single cells from all tumor areas except the InvF. This subgroup of mutations formed a separate class in the principal component analysis (PCA) of allele frequencies in the cancer cell pools (Figure 2D). The combined data indicated that single cells from the InvF belonged to a distinct subclone (defined as subclone 2) as opposed to single cancer cells in the other areas (defined as subclone 1). These 9 SNVs were also identified in cell pools of PT1 and PT2, with allele frequencies significantly lower than those of clonal SNVs (P < 1 × 10–8, 2-tailed t test), similar to the cell pool of InvF. However, the fourth group of SNVs did not exhibit allele frequencies (P > 0.05) lower than clonal SNVs in PT3, PT4, and metastasis cell pools, while the third group of SNVs was absent in both single cells and cell pools of these 4 areas. Our results indicated that the tumors in this patient comprised 2 dominant subclones, subclone 1 and subclone 2, that coexisted in InvF, PT1, and PT2 in different proportions. The other morphological areas, including PT3, PT4, DCIS, and metastasis, consisted only of subclone 1 cancer cells (Figure 2B).
CNVs in morphologically distinct areas.
CNV analysis was performed on the cell pools and the macroscopically dissected tissue pieces. Gains of chr1q, chr7p, chr7q, and chr16p and losses of chr16q and chr17p were identified in all the breast cancer areas, consistent with the subsequent FISH result (Supplemental Figures 6 and 7; Supplemental Table 6), suggesting that they were early clonal events. In addition, chr3q and chr8q gains and chr8p loss were detected in all the breast cancer areas with the exception of InvF and PT2, which mainly comprised subclone 2 cells, indicating that copy number changes of these 3 chromosome arms were specific to subclone 1. In contrast, chr22 loss was found to be specific to subclone 2, as it was mainly present in the InvF and PT2 and not in the metastasis, PT3, and PT4. PT1 showed a mixed pattern of CNVs corresponding to the 2 subclones, in agreement with the mixed SNV characteristics corresponding to 2 subclones observed in the SNV allele frequency (AF) analysis (Supplemental Figure 6). It is worth noting that the AF of some SNVs located on chr1q and chr3q exceeded 50%, including PIK3CA (E545K), implying that these SNVs were presented in the amplified allele and therefore occurred before gains of the 2 chromosomal regions.
Sequencing of single cells and cell pools identified the evolutionary tree of metastasis and micrometastasis within a sentinel axillary lymph node.
The SNV of GATAD2A (E110K) was present in most single cancer cells (12/16) from the lymph node metastasis (MeC, MeP, and MeD), but not from single cells in other areas (Figure 2C). The AF of this mutation in cell pools of MeC and MeP was similar to that of the clonal SNVs (P > 0.05, 2-tailed t test) (Figure 2A), indicating it is a clonal mutation in the lymph node metastasis. As mentioned above, this mutation was also found at low frequency in the cell pool of the InvF (AF of 4.9%) (Figure 2A and Supplemental Table 3), implying that the GATAD2A mutation was not exclusively restricted to metastases, but also existed in the subclone 1 lineage of the InvF, which indicates that the lymph node metastasis likely originated from subclone 1 of the InvF, even though subclone 1 constituted a small fraction thereof (Figure 1A). Furthermore, single cells from the lymph node metastasis obtained an additional SNV (R696H) in MYH6 compared with their ancestor in the primary tumor.
Eight single cells were isolated from a morphologically normal area of the tumor-infiltrated lymph node, as determined by H&E staining. Three were normal lymphocytes, and 5 were dispersed cancer cells according to analysis of the sequencing data. An additional SNV in MAGEC3 (P33Q) was identified to be common to 2 of the 5 single cancer cells and not present in other lymph node metastasis single cells (MeC and MeP), nor in any cell pools except for lymph node metastasis with a relatively low AF (2.4%) (Figure 2, A and C, and Supplemental Table 3). This suggests, based on the genomic evolution within these cells, that they likely disseminated from the lymph node metastasis, according to a linear model of metastasis (10). To further address the low frequency of cancer cells within the morphologically normal area of the tumor-infiltrated lymph node, we performed immunohistochemical analysis of the lymph node using a pan-cytokeratin antibody, which demonstrated the presence of single dispersed cancer cells within the apparently normal area that were not detected by H&E staining (Figure 1, B and C). Overall, these data suggest that the lymph node metastasis resulted from clonal expansion of a single cell derived from the InvF of the primary tumor and that single cancer cells may have dissociated from the lymph node metastasis, acquiring additional genetic aberrations and potentially giving rise to distant tumor spread. Moreover, single cells of DCIS were homologous with subclone 1, with several new SNVs developed in some of the single cells, indicating a separate branch of the phylogenetic tree. In summary, our data support the classical theory of breast cancer development: primary tumor develops into the InvF, which then metastasizes to the lymph node. But our data also suggest that DCIS may arise in parallel with the infiltrating carcinoma (Figure 1 A and Figure 2E).
Early events of carcinogenesis in normal breast cells.
Interestingly, 49 SNVs were exclusively identified in single cells and small, macroscopically dissected pieces of morphologically normal breast epithelial cells obtained well distant from the resected tumor border (>20 mm) (Figure 1, A, D and E), including another hot-spot mutation of PIK3CA (H1047R) located in the kinase domain (Figure 3A). This PIK3CA mutation differed from the hot-spot, helix domain PIK3CA (E545K) mutation identified in single cancer cells from the patient. The 49 mutations were not common to SNVs in single cancer cells or cancer cell pools, indicating that the adjacent morphologically normal breast epithelial cells exhibited genetic aberrations nonhomologous to breast cancer tissue.
Figure 3. Mutations and CNV of chr1q in normal breast epithelial single cells and cell pools.

(A) Heatmap depicting BN-T–specific SNVs identified by sequencing of 11 normal single breast epithelial cells and 3 normal cell pools (brown, mutated; pink, WT; white, WT and sequencing depth of less than 8×). (B) Two distinct haplotypes of chr1q in 4 samples, including Ly-T, lymph node metastasis tissue, BN-T, and a population of macroscopically dissected normal breast epithelial cells (BNM-3). The left panels show the distribution of SNP allele frequencies of haplotypes 1 (red) and 2 (blue) in the amplified genome region chr1q, while the horizontal axis shows VAF and the vertical axis the number of SNPs within the corresponding VAF. The right panels plot SNP allele frequencies of haplotypes 1 (red) and 2 (blue) across chr1q, while the horizontal axis shows coordinate of SNPs in chr1q and the vertical axis shows the AF. Haplotype 1 was amplified in MT, while haplotype 2 was amplified in BN-T and BNM. (C) LOH of chr1q in BN single cells. Variant allele frequencies of SNPs in 2 haplotypes of chr1q are shown as blue and red points (the same color code for each haplotype as in B). P values of LOH in each cell were calculated using Wilcoxon’s rank sum test. Error bars represent the values of median, upper, and lower quartiles and maximum and minimum.
Additionally, both normal breast epithelial tissue and cell pools exhibited amplification of chr1q, as confirmed by loss of heterozygosity (LOH) of chr1q in normal breast epithelial tissue (Figure 3B). However, the chr1q LOH was only identified in 2 (BN-3 and BN-11) of the 4 normal breast epithelial single cells (BN-1, BN-3, BN-11, and BN-12) that exhibited a PIK3CA mutation (P < 1 × 10–15, Wilcoxon’s rank sum test), and not in the other 2 (P > 0.05), implying that the chr1q amplification took place after the occurrence of some SNVs, including the PIK3CA mutation (Figure 3C). Interestingly, the duplicated haploid copy of chr1q in BN-T was not identical to that observed in the breast cancer samples (Figure 3B). Although another study observed that oncogene amplification was a major factor in clonal expansion of premalignant cells (23) in the breast, this seems not to be the trigger event of premalignant cells in our study, but suggests that point mutations might be drivers of clonal expansion of premalignant cells.
Somatic alterations associated with lymph node metastasis in a Chinese sample set of primary breast cancer.
Our single-cell analysis revealed that only 3 additional mutations were present in the lymph node metastasis compared with the cells they likely originated from within the primary tumor (subclone 1), and none seemed to be associated with breast cancer according to the Catalogue of Somatic Mutations in Cancer (COSMIC) database (https://cancer.sanger.ac.uk/cosmic) and previous large sample set studies (24). Several studies have found that the genomic aberrations that predispose tumor cells to metastasize may simultaneously be crucial for their selection advantage and may occur in the early stage of carcinogenesis, which makes it difficult to distinguish them from other driver genes (13, 14, 25). We therefore hypothesized that the lymph node metastasis in this patient may have been prompted by whole-tumor clonal events or subclonal events in subclone 1, which gave rise to lymph node metastasis. Among the 50 driver genes of breast cancer reported by Stephens et al. and The Cancer Genome Atlas Network (TCGA; http://www.cbioportal.org/study?id=brca_tcga_pub#summary) (24, 26), only 2 were mutated (PIK3CA, RUNX1) in the primary breast cancer, as determined by single-cell sequencing, and both were clonal mutations. In addition, several large fragment copy number aberrations were identified in our patient (chr1q, chr3q, chr7p, chr7q, chr8q, and chr16p gains and chr8p and chr17p losses) in which chr3q and chr8q gains and chr8p loss were specific to subclone 1. To assess which aberrations may contribute to breast cancer lymph node metastasis, we performed targeted sequencing on a Chinese sample set of 54 primary breast tumors, 28 from patients with and 26 without lymph node metastasis (Supplemental Table 7), using a target panel including 48 of the 50 breast cancer driver genes referred to above (Supplemental Tables 8, 9, and 10). We compared the mutation frequencies between samples with and without lymph node metastasis for each driver gene, and none were found to exhibit significance, with the exception of myeloid cell leukemia sequence 1 (MCL1), which was high-level amplified (≥5 copies) in 12 samples, 11 of which were from patients with lymph node metastasis (Figure 4, A and B, and Supplemental Figures 8 and 9). When only considering ER+ samples, a significant difference in the number of samples exhibiting highly amplified MCL1 between patients with and without lymph node metastasis remained (Supplemental Figure 10). The copy numbers of MCL1 were also significantly higher in patients with versus without lymph node metastasis (P = 0.01, Wilcoxon’s rank sum test), suggesting that MCL1 high-level amplifications, which were clonal events in our single-cell sample, may promote invasive and lymph node metastasis of breast cancer. High levels of MYC (chr8q) amplification were also more frequently found in patients with lymph node metastasis, but the difference did not reach statistical significance (Figure 4B and Supplemental Figure 11). We then performed whole-genome association analysis of CNV and identified 2 genome regions that exhibited significantly higher frequencies of amplification in the metastasis group, including chr1q and chr20q (Figure 5A).
Figure 4. Comparison of mutations and CNVs in the Chinese sample set of 54 primary breast cancers between patients with and without lymph node metastasis revealed MCL1 is more frequently altered in primary cancers of patients with lymph node metastases.

(A) Heatmaps of CNVs of primary tumors from a Chinese sample set of 54 breast cancer patients with (upper panel) and without (lower panel) lymph node metastasis. Gain and loss are displayed by red and blue, respectively. (B) Mutational spectrum of 54 primary breast cancers with (left panel) and without (right panel) lymph node metastasis. The left panel (yellow bar plot) shows P values (Fisher’s exact test) for aberrant samples in the 2 subgroups for each gene. The middle panel (bar plot) shows the proportion of aberrant samples in each subgroup.
Figure 5. Genome regions associated with lymph node metastasis in breast cancers identified by whole-genome association analysis of frequencies of copy number gains and losses.

(A) Frequency of CNAs in the 54 Chinese primary breast cancer patients with (brown line) or without lymph node metastasis (green line) across the whole genome. chr1q (MCL1) and chr20 (BCL2L1, AURKA) gains were more frequent in primary tumors of patients with lymph node metastasis. The top panel shows the P values (Fisher’s exact test) of gain/loss frequency between the 2 subgroups. (B) Comparison of gain and loss frequencies between ER+ patients without lymph node metastasis (N0) and with high burden of lymph node metastasis (N2–N3) in the data sets of METABRIC (n = 403 vs. 78), TCGA (n = 265 vs. 106), and Nik-Zainal et al. (n = 131 vs. 63; ref. 28). The different genome regions are indicated by different colors.
Whole-genome association analysis of CNV frequency of genes reveals genome regions associated with lymph node metastasis.
To extend the findings from the Chinese breast cancer sample set and focus on ER+ breast cancer, the most frequent subtype, we interrogated the mutational status of each gene in the genome in ER+ breast tumors from 3 large data sets, including TCGA, Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) (http://www.cbioportal.org/study?id=brca_metabric#summary; ref. 27), and that published by Nik-Zainal et al. (28). For each data set, genome aberrations in tumors of patients with no lymph node metastasis (N0) were compared with those with high lymph node metastasis (N2 and N3 combined) according to the tumor-node metastasis (TNM) lymph node–staging system (described in Methods). Examining SNVs and indels surprisingly revealed no gene significantly associated with lymph node metastasis except TP53 in the METABRIC data set (Supplemental Figure 12 and Supplemental Figure 13, A and B). We then compared the frequencies of gain and loss for each gene in the whole genome between the 2 groups in each data set. For gains, 170 genes showed higher frequencies (P < 0.1, Fisher’s exact test) in N2–N3 versus N0 groups in all 3 data sets. These genes were located in chr8q (n = 2, including MYC), chr11q (n = 43, mostly around CCND1), chr12q (n = 112, mostly around CDK2/4), and chr20q (n = 13, mostly around AURKA; Supplemental Table 11). For losses, 631 genes showed higher frequencies (P < 0.1) in N2–N3 versus N0 groups in all 3 data sets, 431 located in chr1p and the others in chr3p (n = 20), chr9p (n = 109, mostly around CDKN2A), chr11q (n = 59), chr12p (n = 1), chr13q (n = 1), and chr18q (n = 2) (Figure 5B and Supplemental Table 12). The significance level of these genome regions remained largely unaffected after normalization for the prognostic factors of age, progesterone receptor (PR), and TP53 mutation status using a logistic regression model (Supplemental Table 13; Supplemental Figures 14 and 15). However, after being normalized for tumor size, which significantly correlated with lymph node metastasis status (P < 1 × 10–10 in TCGA sample set, Fisher’s exact test), genes in chr3p, and chr11q and CDKN2A at chr9p were no longer significant (P > 0.05), while genes in chr1p36.32-33 (the most significant region in chr1p), chr12q13.3 (including CDK2 and CDK4), parts of chr12q21.1-21.31 and chr20q13.3, and MYC at chr8q remained significant (P < 0.05) (Supplemental Figures 14 and 15), suggesting that CNVs of some genome regions contribute independently to lymph node metastasis, while others may contribute primarily by promoting increased tumor size.
Whole-genome association analysis of gene copy numbers confirms genome regions associated with lymph node metastasis.
For the TCGA samples with available detailed copy ratios, we subsequently compared logarithmic copy ratios (logR) for every gene between patients with N0 (265 samples) and N2–N3 (109 samples) for ER+ samples. While similar copy ratios (P ≥ 0.05, Wilcoxon’s rank sum test) were identified across most of the genome, several chromosome regions showed higher (including chr8q24.13-24.3, chr12q15, chr20q11.22, and chr20q13.2-13.33) or lower (including chr1p13-36, chr9p21, and chr18q12-23) copy ratios in the N2–N3 versus N0 groups (P < 0.02) (Figure 6A and Supplemental Table 14), including several known metastasis-related genes (Figure 6B), which significantly correlated with the gain/loss frequency analysis. After normalization for age, tumor size, PR, and TP53 mutation status, genes located in genome regions including chr1p, chr8q, and chr12q remained at significant correlation with lymph node stages (P < 0.05), indicating that these genome regions were mostly unaffected by the normalization, confirming that CNVs in chr1p, chr8q, and chr12q contributed to lymph node metastasis independently of tumor size (Supplemental Figure 16).
Figure 6. Genome regions associated with lymph node metastasis in breast cancers identified by whole-genome association analysis of detailed copy ratios.

(A) Differences in the average logR (outer circle) across the whole genome between primary ER+ breast cancers of patients with N0 (265 samples) and N2–N3 (106 samples) lymph node metastasis status (data from TCGA). P value of each gene (inner circle) was calculated for the logRs between the 2 groups (Wilcoxon’s rank sum test); red bars denote genes with P < 0.02. (B) Comparison of copy ratios of AURKA (chr20q), CDKN2A (chr9p), MYC (chr8q), MDM2 (chr12q), SMAD2 (chr18q), and SMC3 (chr10q) in the TCGA ER+ breast cancers grouped according to patient lymph node status (N0, N1, and N2–N3,). Significance of difference between N0 and N2–N3 groups was tested by Wilcoxon’s rank sum test. Each point represents the copy ratio of 1 sample. (C) Comparison of copy ratios of MCL1, MYC, and BCL2L1 in 170 Danish primary ER+ breast cancers grouped according to the number of positive lymph nodes of the patient (of n = 0, 0 < n < 3, and n ≥ 3) and recurrence status (recurrence [Recur] vs. without recurrence [Free]). Significance of difference between n = 0 and n ≥ 3 groups and between recurrence and without recurrence groups was tested by Wilcoxon’s rank sum test. Each point represents the copy ratio of 1 sample. Error bars in B and C represent the values of median and upper and lower quartiles.
Finally, to confirm the findings using the 3 data sets and the Chinese sample set, we performed CNV analysis of 3 genes by real-time quantitative PCR (qPCR), including MCL1 (located in chr1q), MYC (located in chr8q), and BCL2L1 (located in chr20q) (Figure 6C). A total of 170 Danish primary ER+ breast cancer samples (>60% tumor cells purity) were analyzed using, as the reference, TANC1, which is located on chr2q and is minimally affected by CNVs according to the TCGA and the Chinese sample set analysis (Supplemental Table 15). In patients with primary breast cancer samples exhibiting high MCL1 copy number (copy ratios ≥ 3), 80% (47/59) had at least 1 positive lymph node, while only 63% (69/110) of the remaining samples had lymph node metastasis (P = 0.037, Fisher’s exact test). We divided the samples into 3 groups based on the number of positive lymph nodes (n = 0, 3 > n > 0 and n ≥ 3). The median logRs for MYC were highest in the n ≥ 3 group (logR = 1.51) and lowest in the n = 0 group (logR = 1.28). A similar distribution was observed for BCL2L1 in 3 groups (logR = 1.08 and 1.16 for n = 0 and n ≥ 3, respectively). However, the difference in logR of both MYC (P = 0.10) and BCL2L1 (P = 0.13) between samples with n = 0 and n > 0 did not reach significance, likely due to the limited sample size. We then compared the logR for these 3 genes between samples with and without recurrence of disease and identified a higher logR of MYC (P = 0.028) and BCL2L1 (P = 0.0081) in the recurrence group.
Discussion
Deciphering the molecular evolution of cancer is central to understanding cancer heterogeneity and its role in malignant progression. Although the evolution of multiregion breast tumors in patients has been described (25), the genetic architecture of morphologically distinct areas of breast tumors, so-called microheterogeneity, has not been detailed. We sequenced single cells and multi-cell pools isolated using LCM in multiple morphologically distinct regions of primary tumor and corresponding sentinel lymph node metastasis of a breast cancer patient and demonstrated that different subclones coexisted in multiregions in the primary tumor, including the InvF. This approach could be applied in the clinic to identify the malignant state of single cells at a distinct location, e.g., within an area of DCIS. Our data showed that the lymph node metastasis likely was derived from a minor subclone of the InvF. Our data also suggest that the sequenced DCIS cells arose from a subclone and were not a precursor or a separate entity, an observation in line with another recent study (29). Further, single cancer cells located in areas of morphologically normal lymph nodes distant from the lymph node metastasis had gained additional mutations compared with single cells in the solid lymph node metastasis, indicating a case of a linear model of metastasis. Interestingly, our analysis also showed that clonal expansion due to mutation of cancer driver genes was also present in adjacent BN-T and hematopoietic cells of this 92-year-old patient, supporting the view that clonal expansion due to mutated cancer-associated genes are prevalent in aged individuals and greatly increase the risk of cancer (30). Genome profiles of precancerous lesions could illuminate how cancer is initiated and help define the divergent events between “normal” and malignant, which are critical to early detection and treatment of cancer as well as possible prevention. Recent studies have revealed driver mutations in the normal skin and hematopoietic system by bulk sequencing (31, 32). However, genomic analysis, to our knowledge, has not been widely applied to studying of precancerous lesions of other tissues, as such areas are small and difficult to identify. Single-cell sequencing has the potential to overcome the problem of low cellularity and determine the heterogeneity and evolution of precancerous lesions.
We used single-cell sequencing to identify driver mutations and CNVs in the subclone giving rise to lymph node metastasis, and some similar features were also observed in additional primary breast tumors of patients with lymph node metastasis in 5 breast cancer sample sets. Importantly, high-level gain of MCL1 and amplification of chr8q were identified as candidates contributing to tumor spread to lymph nodes. In our single-cell–sequenced patient, high-level MCL1 (copy number ≥5) amplification was a clonal event, while chr8q amplification was present in a subclone of the primary tumor, giving rise to lymph node metastasis and indicating that CNVs, rarely considered in genomic analysis of metastasis, may be crucial drivers of lymph node metastasis. MCL1 is a prosurvival gene and a member of the BCL2 family, which governs the intrinsic apoptotic pathway (33) and has been shown to be highly expressed in some breast cancers, playing an important role in patient response to antitubulin chemotherapeutics (34). MCL1 expression has been associated with metastasis in colorectal (35), gastric (36, 37), and breast cancers (38–40). Our study also identified aberrations of several other genes or chromosome regions that were enriched in ER+ breast cancer patients with versus those without lymph node metastasis, including gains of chr12q and chr20q and losses of chr1p and chr9p, most of whose copy number alteration (CNA) have not previously been associated with breast cancer metastasis (Figure 6B). Several oncogenes on chr20q, such as BCL2L1 (41, 42) and AURKA (43), have been reported to have a function in breast cancer lymph node metastasis. CDKN2A, located in chr9p, has been proven to promote cancer metastasis (44) and has been identified as one of several metastasis-associated genes in a CRISPER mouse model (45). MYC overexpression has been reported to induce EMT (46), which was believed to be one of the mechanism of metastasis (5), though with some controversy (47, 48). In the 5 sample sets we studied, higher copy numbers of MYC were observed in lymph node–positive breast cancer patients, suggesting that MYC may contribute to, but is not a requirement for, metastases to the lymph nodes. Some presumably metastasis-related genes, including MYC, MCL1, AURKA, and TP53, regulated apoptosis and enhanced cell survival in new environments, which may be crucial for disease progression and metastasis. Although the significance level of the correlation between lymph node metastasis and some genes decreased when normalized for tumor size, there may still be an association either directly or indirectly, since these genes may promote tumor cell proliferation, invasion, and apoptosis resistance, which, in addition to influencing tumor size, may also stimulate metastatic spread to lymph nodes. Our results suggest that metastasis may partly result from molecular processes closely related to genetic aberrations, and we show, for what we believe is the first time, that the copy number status of genes is associated with breast cancer spread and lymph node metastasis.
Axillary lymph node status is the single most important prognostic indicator in the management of primary breast cancer patients. Of patients diagnosed without lymph node metastasis, 75% survive beyond 10 years, while only 40% of patients with lymph node metastasis survive after this period (49). Further, prognosis decreases as the number of tumor-positive lymph nodes increases. Metastasis in the axillary lymph nodes is not only a sign of later-stage breast cancer, but also a marker of an aggressive phenotype. Improving our understanding of the molecular mechanisms of the metastatic process, including the genetic basis, will improve clinical management of the disease.
In our study, the mutation burden (1.62 mutations per megabase) of our LCM sample was modest among breast cancers, and unique mutations of each distinct area were rare, suggesting that most of the mutations occurred before detectable neoplasia formation, consistent with another study on hepatocellular carcinoma (HCC) (50). Thus, WES has a limited power of detecting microheterogeneity in patients with a limited mutation burden, since only a few mutations could be used as identifiers of different clones. It is also worth noting that a high rate of false positive C>T mutations was identified in single cells, likely due to damage of the cellular DNA during tissue preparation, H&E staining, and WGA. Furthermore, we were unable to identify specific driver genes in genomic regions that seemed to increase the ability of cancer cells to metastasize to lymph nodes, since most of the genes showing enrichment of CNV in tumors with lymph node metastasis should be passenger genes, such as for chr1p and chr11q loss and chr12q gain. The size of the sample data sets used in our analysis (54–481 samples) is insufficient to precisely locate the true driver genes, which may be identified at the “peak” of significance in a data set with thousands of samples, as in the GWAS studies. Although larger sample sets are needed to address the molecular profile of metastasis, our results provide an initial basis from which to further examine metastasis-associated genetic aberrations.
Methods
Patient material and clinical information
Tumor tissue blocks of approximately 4 × 4 × 10 mm obtained from a 92-year-old female mastectomized for primary breast cancer, including removal of axillary lymph nodes (3/18 were tumor infiltrated), and having received no preoperative treatment, were embedded in OCT (Sakura Finetek), snap-frozen in isopentane, and stored at –80 °C. The tissue blocks were histologically verified using H&E-stained sections by a highly experienced breast cancer pathologist to contain normal mammary ductal tissue, primary breast tumor (invasive ductal carcinoma [IDC]), DCIS, lymph node metastasis, normal skin, and normal lymph nodes, respectively. The primary tumor was 32 mm IDC, grade 2. The following routine markers were investigated by immunohistochemistry: ER = 100%, Ki-67 = 5%, and HER2 = 2+. The patient received adjuvant Letrozol that was discontinued due to side effects and age-related morbidity after 6 months, and she is recurrence-free 5 years after primary surgery (April 2017). DNA from a Danish sample set of 170 ER+ primary tumors from postmenopausal breast cancer patients was analyzed for qPCR CNV. Primary breast cancers of a sample set of 54 Chinese patients (31 ER+, 22 ER–, 1 unknown) were analyzed by targeted sequencing. Mutation and CNV data from a sample set of 572 primary ER+ breast cancers from patients with known lymph node status were obtained from TCGA. Mutation and CNV data from another sample set of 255 primary ER+ breast cancers along with lymph node status was published by Nik-Zainal et al. (28). CNV data from the fifth sample set of 1,194 primary ER+ breast cancers from patients with known lymph node status were obtained from METABRIC. Since information on lymph node stage was not directly available for the METABRIC data set, tumor stages of 1 and 3 to 4 were used instead, as 97% of samples with stage 1 were also N0, and 96% of samples with stage 3 to 4 were with N ≥ 1 according to clinical information from the TCGA data set.
LCM of single cells and cell pools
LCM of single cells was performed using the Arcturus PixCell IIe microscope (Life Technologies). Fresh-cut frozen tissue sections (7 μm) were placed on uncharged glass slides (Sigma-Aldrich) and briefly stained with H&E. Each section was thawed for 15 seconds and fixed in 70% EtOH for 30 seconds. After fixation, sections were dipped in DEPC-water for 15 seconds, followed by staining with hematoxylin for 30 seconds. Stained sections were dipped in DEPC-water until surplus dye was washed off and then put into autoclaved water for 30 seconds, DEPC-water for 30 seconds, 70% EtOH for 30 seconds, and 96% EtOH for 30 seconds. A droplet of eosin was added to each section by pipette and incubated for 5 seconds followed by 96% EtOH for 30 seconds, 100% EtOH for 30 seconds,100% EtOH for 30 seconds, and finally xylene for 4 minutes. Finally, the section was dried in a fume hood for 5 minutes. None of the reagents were reused during the protocol. The reagents used were as follows: DEPC-water (Sigma-Aldrich), EtOH (Sigma-Aldrich), hematoxylin (Sigma-Aldrich), and eosin (Sigma-Aldrich). Tissue sections were used for LCM for a maximum of 30 minutes to ensure good quality DNA, and multiple sequential sections were used per category of the various morphologically distinct areas. Single-cell isolation settings for the LCM, spot size 7.5 μm, were as follows: power, 25 mW; duration, 0.8; and target, 196 mV. For isolation of cell pools, the settings were as follows: power, 65–88 mW; duration, 2.3; and target, 259 mV. CapSure Macro LCM Caps (Applied Biosystems) were used, and immediately after capture, the cell or cells were transferred to 3 μl lysis buffer. The isolation was verified by viewing the tissue section after LCM and the cap. Following addition of precooled lysis buffer, the cells were immediately placed on dry ice and stored at –80 °C until DNA was amplified. A physiological saline blank was included as a negative control.
Multiple displacement amplification
WGA of single cells and normal breast epithelial cell pools was achieved using the REPLI-g Single Cell Kit according to the manufacturer’s manual (QIAGEN GmbH). Reactions in a total volume of 50 μl were performed at 30°C for 8 hours and terminated at 65°C for 3 minutes. Amplified DNA products were stored at –20°C.
Concentration measurement, amplification coverage estimation, and sequencing
The Qubit Quantitation Platform (Life Technologies) was used to measure the concentration of multiple displacement amplification (MDA) products to verify that the MDA was successful. Subsequently, all amplified DNA products yielding more than 30 ng/μl were examined using a panel of 10 housekeeping genes for PCR to estimate amplification coverage. The MDA products with successful amplification of at least 8 housekeeping genes were selected for further WES using the SureSelect Human All Exon 50Mb Kit (Agilent Technologies). All libraries were sequenced on either an Illumina HiSeq 2000 or HiSeq 2500. For target sequencing of single cells and cell pools, an Agilent custom sequence capture probe was used according to the manufacturer’s instructions. For the additional sample set of 54 primary breast cancer samples, DNA extracted from tumor tissue, lymph nodes, and peripheral blood was used for library construction, followed by enrichment using a custom sequence capture probe (Nimblegen, Roche) targeting 508 cancer-related genes (Supplemental Table 8).
qPCR of Danish sample set
TaqMan Copy Number Assays (Applied Biosystems) were used for all reactions as described by the manufacturer using 20 μl reactions run for 2 minutes at 50°C and 10 minutes at 95°C, followed by 50 cycles of 15 seconds at 95°C and 1 minute at 60°C. All samples were run in quadruple on the ABI QuantStudioTM 12K Flex System using software v1.2 (Applied Biosystems). The individual copy number assays investigated were MYC (Hs02758348_cn), BCL2L1 (Hs07178628_cn), MCL1 (Hs02097917_cn), and TANC1 (Hs00436935_cn), all labeled with 5′-FAM and 3′-MGB. These were duplexed with a company-provided reference assay targeting RNase P, labeled with 5′-VIC and 3′-TAMRA. For each real-time plate, we included a nontemplate control (water) and a technical control consisting of DNA from a human cancer cell line, CL16, with known altered copy numbers for the investigated genes to ensure contamination-free assays and interplate consistency, respectively. The raw data were analyzed using CopyCaller v2.0 (Applied Biosystems), which calculated a predicted copy number based on RNase P as an internal reference and a calibrator sample containing 2 copies per gene. Finally, the predicted copy numbers were recalculated to TANC1 as a reference gene and correlated with lymph node status using Wilcoxon’s rank sum test.
Bioinformatic analysis
Mutation calling of single-cell WES data.
All sequencing data were aligned to hg19 (UCSC) by Burrows-Wheeler Aligner (BWA) (http://bio-bwa.sourceforge.net/), followed by removal of duplication using PICARD (https://broadinstitute.github.io/picard/). Genome Analysis Toolkit (GATK) (https://software.broadinstitute.org/gatk/) was employed for local realignment around indels. For WES data, we used GATK UnifiedGenotyper to detect SNVs in each of the 97 single-cell samples with a quality threshold of 20. To avoid false-positive calling due to MDA and sequence errors, only mutations identified in at least 2 samples were selected as candidate mutations. Data suggesting a mutation of low quality in more than 50% of the samples were discarded. A mutation was considered low quality under the following conditions: (i) the average position of the mutation in the reads was less than 10 bp away from one of the ends; (ii) more than 30% of mutation-supporting reads had a map quality of less than 20; (iii) the difference of average mapping quality between reference-supporting reads and variant-supporting reads was 30 or more; (iv) the average mismatch number of mutation-supporting reads was 7 or more or the difference of average mismatch numbers between reference-supporting reads and variant-supporting reads was more than 3; (v) more than 30% of mutation-supporting reads were soft clipped; (vi) the difference of average indel length between reference-supporting reads and variant-supporting reads was more than 1; and (vii) more than 30% of mutation-supporting reads had multi-alignment. The SNVs with more than 3 supporting reads in Ly-T and Skin-T were considered to be germline mutations and filtered out.
K value and QC of single cell.
As ADO is affected by sequence depth and the criterion for mutation identification, we defined and utilized the K value (the sample excess kurtosis of allele frequencies of SNPs) to quantify amplification uniformity, and samples with K values of more than 0.2 were retained for analysis.
For example, in cases in which n equals the number of SNPs and xi equals the AF of ith SNP, the K value was defined as follows:
(Equation 1)
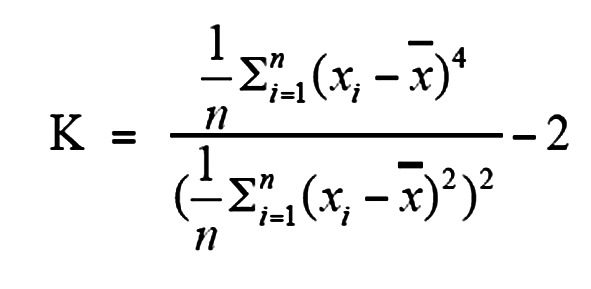
x ¯ is the mean value of xi. We also calculated ADO of each sample and found significant negative correlation between K values and ADO (Supplemental Figure 2B). For 97 WES-sequenced single cells, we selected 67 single cells accompanied by another 11 new cells for deep-target sequencing based on the K values of each (Supplemental Figure 2, A, C, D, and E). The cutoff of K values was set at 0.2. However, if the WES sequencing of 1 cell supported the presumed rare subclonal mutations, it was also included in further analysis, although the cell did not pass the cutoff.
Target deep sequencing of single cells and regenotyping of mutations.
A target panel was designed to cover the candidate mutations and top 20 breast cancer genes, including PIK3CA, TP53, MED12, CDH1, GATA3, MLL2, MLL3, PTEN, RB1, ARID1A, NF1, NCOR1, AKT1, ATM, FLT3, CREBBP, MAP2K4, BRCA1, RUNX1, and KIT. For the target deep sequencing of single cells, loci of candidate mutations in the target sequencing data were regenotyped by a likelihood ratio test to reveal mutational spectrums and reduce mutation loss rate. A sequencing error rate was estimated for each mismatch type. A test based on Poisson distribution was used to judge whether the supporting reads of an SNV were significantly higher than those generated by random sequencing errors. To avoid crossover contamination, only mutations with allele frequencies of no less than 1% were considered positive.
Identification of multinucleated cells and normal cells
Data analysis suggested that a few laser-captured single-cell samples contained both normal and cancerous nuclear material based on the fact that the allele frequencies of the validated tumorous SNVs were generally low in these samples. Data from such single-cell samples as might be contaminated by normal nuclei may distort the SNV spectrum because normal DNA simultaneously amplified with cancerous DNA dilutes the allelic frequencies of the tumor SNVs and possibly results in a high missing rate of SNVs despite these cells still being considered “good quality” according to K value or ADO with high genome coverage. Therefore, the single-cell samples with median AF of SNVs of less than 0.1 were eliminated from the downstream analysis.
Duplex analysis
For the duplex-sequencing data with a certain amount of DNA templates, paired reads were clustered by the “endogenous molecular barcodes” consisting of the first 12 bp in each end of the DNA templates. Initially, all the sequenced reads were sorted by alphabetical order of the barcode, and thus reads belonging to the same duplication clusters were ranked at adjacent positions. Reads with identical barcodes and similar sequences (with consistency >80%) were considered duplication clusters of 1 template, and reads in each duplication cluster were compared with each other to correct errors and then degenerated into a DCS. A valid DCS required at least 3 reads in the duplicated cluster in which reads from both sense and antisense strands were necessary. The different order of 2 paired end reads was used to identify the sense and antisense strands of the template. For each duplicated cluster, the reads were compared with each other to obtain the DCS based on the following criteria: in each position of the template, a base x (x = A/T/C/G) present in no less than 80% reads in both sense and antisense strand reads denoted that the base of this position in the DCS was x. If no x satisfied this condition, the base of this position in the DCS was denoted as N. The error rate of DCS was estimated to be 10–7 for each type of substitution according to our analysis.
CNV analysis of laser-capture microdissected cell pools and the Chinese sample set
Whole-genome shotgun (WGS) sequence data of metastasis, PT4, and BN-T and targeted sequencing data of cell pools were used for somatic CNV calling. The whole genome was segmented into 50 kb bins, each calculated with a read-depth copy ratio of case and control. For CNV calling of target sequencing, reads on or near the targeted region (within less than 500 bp) in both case and control data were filtered out and only the off-target reads were used to calculate the copy ratio. Correlation between GC content and copy ratio was fitted and normalized by the generalized least squares (GLS) model and the fluctuation was generally reduced (Supplemental Figure 17, A–D) after GC normalization. Subsequently, CLimAT (51) was used for segmentation and CNV detection using copy ratio as input. CNV analysis of metastasis was performed both by WGS and target sequencing based on the method described above, and the results showed very high correlation. Similar CNV analysis was used for the Chinese primary breast cancer sample set and the cell pools.
Tumor purity and copy number assessment of the Chinese sample set
As homozygous deletion of greater than 10 M base in cancer genomes is unlikely, we estimated the fraction of tumor cells based on the copy ratio of large deleted segments (>10 M). Note that a is the lowest copy ratio of all the large deleted segments, and consequently, the fraction of tumor cells was estimated to be (1 – a) × 2. For a few samples that did not contain large-segment deletions, the peaks in copy ratio distribution were used to calculate tumor purity. For each gene, the copy ratio was adjusted by tumor purity to obtain the copy number. Copy numbers of genes were not assigned to integers because mixtures of different subclones were prevalent, making definition of integral copy numbers difficult.
Mutation calling in primary breast cancers of patients in the Chinese sample set
A MuTect algorithm (52) with default parameters was employed to generate candidate somatic base substitution in 54 primary breast cancer samples. The candidate SNVs were further selected based on a sequence depth of more than 30 in both cancers and controls. dbSNP137 variations with AF records were filtered out. For short indels, we used Varscan2 to generate candidates (53), and a local alignment filter was used to filter out the suspected artificial indels by the following criteria: (i) more than 80% of indel supporting reads with map quality of less than 30; (ii) indel adjacent to any homopolymer (5 bp) or short tandem duplications (≥3 copies); and (iii) more than 30% of reads that covered the query indel supported other indels (no further than 40 bp away from the query indel). Somatic SNVs and indels were annotated by ANNOVAR (http://annovar.openbioinformatics.org/en/latest/), and only mutations that changed protein were retained for further analysis.
Whole-genome association analysis of CNV frequency of each gene
For each gene in the whole genome, we calculated the frequency of amplification in patients with low or high lymph node stages and compared the frequencies between the 2 subgroups using Fisher’s exact test. The same method was applied for the frequency of deletions.
Whole-genome association analysis of gene copy numbers in TCGA data
For each gene, we compared the copy ratios between tumors of patients with N0 (265 samples) and N2–N3 (106 samples) lymph node stages using Wilcoxon’s rank sum test. Genome regions consisting of more than 5 genes exhibiting P values of less than 0.02 were reported in Supplemental Table 14.
Statistics
Associations between the CNV and lymph node status of each gene were assessed using Fisher’s exact test, and P values of less than 0.05 were considered statistically significant. A logistic regression model (assigned 0 for N0/stage 1, and 1 for N2–N3/stages 3–4) was used to adjust for age, tumor size, PR, HER2, and TP53 mutation status, and the significance of coefficients was tested separately (t test, 2 tailed) for each gene in 2 relatively large data sets (METABRIC and TCGA) using a glm function in R. P values of less than 0.05 were considered statistically significant. The correlation between logR of each gene and lymph node status in the TCGA data set was assessed using Wilcoxon’s rank sum test, and P values of less than 0.05 were considered statistically significant. Results in Figure 3C and Figure 6, B and C, were reported by box plot, indicating the values of median and upper and lower quartiles. Exact numbers of samples in each data set are indicated in the corresponding figure legend. A 2-tailed t test was also used to compare mutation variant allele frequency (VAF) in cell pools. P < 0.05 was considered statistically significant, as indicated in Results.
Study approval
For the Danish sample set of 171 ER+ primary tumors from postmenopausal breast cancer patients, approval from the Ethical Committee of Southern Denmark (Odense, Denmark) and the Danish Data Protection Agency (Copenhagen, Denmark) was granted. For the primary breast cancers of a sample set of 54 Chinese patients, approval from the Ethical Committee of Xijing Hospital was granted. All tissue samples were collected in compliance with informed consent policy.
Data availability
Single-cell sequencing data and the sequencing data of the Chinese cohort are deposited in the NCBI Sequence Read Archive (SRA) under Bioproject (SRP103895; https://www.ncbi.nlm.nih.gov/bioproject/). The CNV data of the Danish cohort are provided in Supplemental Table 16.
Author contributions
HJD and LB managed the project. HJD, LB, and ZQ designed the analyses. HJD, LW, and TW provided clinical samples and clinical information. LB, ZQ, MBL, and YY performed experiments and sequencing and data analysis. HJD, LB, ZQ, and MBL wrote and edited the manuscript. HJD, JW, NB, HY, XZ, and LW obtained funding and supervised the project. All authors read and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
This study was supported by the Sino-Danish Breast Cancer Research Centre, financed by the National Natural Science Foundation of China (grant numbers 30890032 and 31161130357), the Danish National Research Foundation (Grundforskningsfonden), the National High Technology Research and Development Program of China (863 program, grant number 2015AA020408), the National Key Technology R&D Program (grant number 2014BAI09B04), the National Natural Science Foundation of China (grant numbers 81672593 and 81272899), and the Discipline Booster Plan of the Xijing Hospital (XJZT12Z07). Further support was obtained from the Danish Cancer Society, the Danish Research Council, the Academy of Geriatric Cancer Research (AgeCare), the Danish Center for Translational Breast Cancer Research (DCTB), A Race Against Breast Cancer, the National Experimental Therapy Partnership (NEXT), and Innovation Fund Denmark. The authors thank pathologist Anne Marie Bak Jylling (Department of Pathology, Odense University Hospital) for morphological interpretation of tissue sections, Lisbet Mortensen and Ole Nielsen (Department of Pathology, Odense University Hospital) for excellent technical assistance with the immunohistochemical stainings and FISH, and M. Kat Occhipinti for editorial assistance.
Version 1. 03/15/2018
In-Press Preview
Version 2. 04/23/2018
Electronic publication
Version 3. 06/01/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(6):2310–2324.https://doi.org/10.1172/JCI97449.
Contributor Information
Li Bao, Email: chengdu1125@hotmail.com.
Zhaoyang Qian, Email: qianzy@mail.ustc.edu.cn.
Maria B. Lyng, Email: maria.lyng@rsyd.dk.
Ling Wang, Email: sbn518057@hotmail.com.
Yuan Yu, Email: yuyuan@genomics.cn.
Ting Wang, Email: 715246184@qq.com.
Xiuqing Zhang, Email: zhangxq@genomics.org.cn.
Huanming Yang, Email: yanghm@genomics.org.cn.
Nils Brünner, Email: nbr@sund.ku.dk.
Jun Wang, Email: wangjun30@gmail.com.
Henrik J. Ditzel, Email: hditzel@health.sdu.dk.
References
- 1.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheung KJ, Ewald AJ. A collective route to metastasis: Seeding by tumor cell clusters. Science. 2016;352(6282):167–169. doi: 10.1126/science.aaf6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Minn AJ, et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc Natl Acad Sci U S A. 2007;104(16):6740–6745. doi: 10.1073/pnas.0701138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Budhu A, Wang XW. Transforming the microenvironment: a trick of the metastatic cancer cell. Cancer Cell. 2012;22(3):279–280. doi: 10.1016/j.ccr.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 5.Yu M, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339(6119):580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoshino A, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329–335. doi: 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marinari E, Mehonic A, Curran S, Gale J, Duke T, Baum B. Live-cell delamination counterbalances epithelial growth to limit tissue overcrowding. Nature. 2012;484(7395):542–545. doi: 10.1038/nature10984. [DOI] [PubMed] [Google Scholar]
- 8.Eisenhoffer GT, et al. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature. 2012;484(7395):546–549. doi: 10.1038/nature10999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu K, et al. Frequent alterations in cytoskeleton remodelling genes in primary and metastatic lung adenocarcinomas. Nat Commun. 2015;6:10131. doi: 10.1038/ncomms10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352(6282):169–175. doi: 10.1126/science.aaf2784. [DOI] [PubMed] [Google Scholar]
- 11.Robinson DR, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548(7667):297–303. doi: 10.1038/nature23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yates LR, et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. 2017;32(2):169–184.e7. doi: 10.1016/j.ccell.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gundem G, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547):353–357. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCreery MQ, et al. Evolution of metastasis revealed by mutational landscapes of chemically induced skin cancers. Nat Med. 2015;21(12):1514–1520. doi: 10.1038/nm.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yates LR, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21(7):751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Navin NE. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015;25(10):1499–1507. doi: 10.1101/gr.191098.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17(3):175–188. doi: 10.1038/nrg.2015.16. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512(7513):155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148(5):886–895. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wells D, Sherlock JK, Handyside AH, Delhanty JD. Detailed chromosomal and molecular genetic analysis of single cells by whole genome amplification and comparative genomic hybridisation. Nucleic Acids Res. 1999;27(4):1214–1218. doi: 10.1093/nar/27.4.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A. 2012;109(36):14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie M, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forsberg LA, et al. Signatures of post-zygotic structural genetic aberrations in the cells of histologically normal breast tissue that can predispose to sporadic breast cancer. Genome Res. 2015;25(10):1521–1535. doi: 10.1101/gr.187823.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yates LR, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21(7):751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nik-Zainal S, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534(7605):47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casasent AK, et al. Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell. 2018;172(1-2):205–217.e12. doi: 10.1016/j.cell.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martincorena I, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348(6237):880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. doi: 10.1038/ncomms12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kotschy A, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477–482. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 34.Wertz IE, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471(7336):110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 35.Radhakrishnan P, et al. Prolyl hydroxylase 3 attenuates MCL-1-mediated ATP production to suppress the metastatic potential of colorectal cancer cells. Cancer Res. 2016;76(8):2219–2230. doi: 10.1158/0008-5472.CAN-15-1474. [DOI] [PubMed] [Google Scholar]
- 36.Wu S, et al. miR-125b Suppresses Proliferation and Invasion by Targeting MCL1 in Gastric Cancer. Biomed Res Int. 2015;2015:365273. doi: 10.1155/2015/365273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee WS, et al. Myeloid cell leukemia-1 regulates the cell growth and predicts prognosis in gastric cancer. Int J Oncol. 2015;46(5):2154–2162. doi: 10.3892/ijo.2015.2890. [DOI] [PubMed] [Google Scholar]
- 38.Yang L, et al. Wnt modulates MCL1 to control cell survival in triple negative breast cancer. BMC Cancer. 2014;14:124. doi: 10.1186/1471-2407-14-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wee ZN, et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat Commun. 2015;6:8746. doi: 10.1038/ncomms9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young AI, et al. MCL-1 inhibition provides a new way to suppress breast cancer metastasis and increase sensitivity to dasatinib. Breast Cancer Res. 2016;18(1):125. doi: 10.1186/s13058-016-0781-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.España L, Fernández Y, Rubio N, Torregrosa A, Blanco J, Sierra A. Overexpression of Bcl-xL in human breast cancer cells enhances organ-selective lymph node metastasis. Breast Cancer Res Treat. 2004;87(1):33–44. doi: 10.1023/B:BREA.0000041579.51902.89. [DOI] [PubMed] [Google Scholar]
- 42.Méndez O, Fernández Y, Peinado MA, Moreno V, Sierra A. Anti-apoptotic proteins induce non-random genetic alterations that result in selecting breast cancer metastatic cells. Clin Exp Metastasis. 2005;22(4):297–307. doi: 10.1007/s10585-005-8751-x. [DOI] [PubMed] [Google Scholar]
- 43.Siggelkow W, et al. Expression of aurora kinase A is associated with metastasis-free survival in node-negative breast cancer patients. BMC Cancer. 2012;12:562. doi: 10.1186/1471-2407-12-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cui C, et al. P16-specific DNA methylation by engineered zinc finger methyltransferase inactivates gene transcription and promotes cancer metastasis. Genome Biol. 2015;16:252. doi: 10.1186/s13059-015-0819-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen S, et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160(6):1246–1260. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cowling VH, Cole MD. E-cadherin repression contributes to c-Myc-induced epithelial cell transformation. Oncogene. 2007;26(24):3582–3586. doi: 10.1038/sj.onc.1210132. [DOI] [PubMed] [Google Scholar]
- 47.Zheng X, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fischer KR, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472–476. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldhirsch A, et al. Thresholds for therapies: highlights of the St Gallen International Expert Consensus on the primary therapy of early breast cancer 2009. Ann Oncol. 2009;20(8):1319–1329. doi: 10.1093/annonc/mdp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ling S, et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc Natl Acad Sci U S A. 2015;112(47):E6496–E6505. doi: 10.1073/pnas.1519556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu Z, Liu Y, Shen Y, Wang M, Li A. CLImAT: accurate detection of copy number alteration and loss of heterozygosity in impure and aneuploid tumor samples using whole-genome sequencing data. Bioinformatics. 2014;30(18):2576–2583. doi: 10.1093/bioinformatics/btu346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koboldt DC, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell sequencing data and the sequencing data of the Chinese cohort are deposited in the NCBI Sequence Read Archive (SRA) under Bioproject (SRP103895; https://www.ncbi.nlm.nih.gov/bioproject/). The CNV data of the Danish cohort are provided in Supplemental Table 16.