

Abstract
ONC201 is a first-in-class, orally active antitumor agent that upregulates cytotoxic TRAIL pathway signaling in cancer cells. ONC201 has demonstrated safety and preliminary efficacy in a first-in-human trial in which patients were dosed every 3 weeks. We hypothesized that dose intensification of ONC201 may impact antitumor efficacy. We discovered that ONC201 exerts dose- and schedule-dependent effects on tumor progression and cell death signaling in vivo. With dose intensification, we note a potent anti-metastasis effect and inhibition of cancer cell migration and invasion. Our preclinical results prompted a change in ONC201 dosing in all open clinical trials. We observed accumulation of activated NK+ and CD3+ cells within ONC201-treated tumors and that NK cell depletion inhibits ONC201 efficacy in vivo, including against TRAIL/ONC201-resistant Bax–/– tumors. Immunocompetent NCR1-GFP mice, in which NK cells express GFP, demonstrated GFP+ NK cell infiltration of syngeneic MC38 colorectal tumors. Activation of primary human NK cells and increased degranulation occurred in response to ONC201. Coculture experiments identified a role for TRAIL in human NK-mediated antitumor cytotoxicity. Preclinical results indicate the potential utility for ONC201 plus anti–PD-1 therapy. We observed an increase in activated TRAIL-secreting NK cells in the peripheral blood of patients after ONC201 treatment. The results offer what we believe to be a unique pathway of immune stimulation for cancer therapy.
Keywords: Oncology
Keywords: Apoptosis pathways, Cancer immunotherapy, NK cells
Introduction
We previously identified ONC201/TIC10 as a small molecule that upregulates endogenous TNF-related apoptosis-inducing ligand (TRAIL) in tumor and normal cells, and restores autocrine and paracrine antitumor activity within tumor cells (1). The search for TRAIL-inducing compounds (TICs) was specifically aimed at identifying compounds that do not rely on p53, leading to the discovery of ONC201/TIC10. The novel anticancer therapeutic ONC201 upregulates endogenous TRAIL expression through dual blockade of Akt and ERK, which releases Foxo3a to enter the nucleus and transcriptionally activate the TRAIL gene (1). Our prior work using shTRAIL and RIK-2 (a TRAIL-blocking antibody) demonstrated the relevance of TRAIL to the mechanism of action of ONC201 (1–3). As we investigated the kinetics of cell death, we discovered that at early time points ONC201 activates the integrated stress response, inducing eIF2-α–dependent ATF4 and CHOP and increasing TRAIL death receptor 5 (DR5) expression (4–6). We also recently demonstrated potent antitumor effects on colorectal cancers initiated by cancer stem/progenitor cells (CSCs) (7). In vivo, first-in-class small molecule ONC201 exhibits a broad spectrum of anticancer activity, a wide safety margin, robust stability, aqueous solubility, blood-brain barrier penetration, and favorable pharmacokinetics (1–5, 7–14).
Prior evidence has demonstrated that TRAIL can inhibit cancer metastasis (15–18). Inactivating mutations in the TRAIL-R1 and TRAIL-R2 genes have been observed in metastases in several tumor types, including mammary tumors and melanoma (19, 20). The TRAIL pathway is part of the innate host immune surveillance mechanism against cancer and involves activation of the extrinsic cell death pathway selectively in cancer cells. As part of the immune system, NK cells respond to cellular signals that can trigger their activation, releasing perforins and granzymes and inducing cellular lysis within the tumor. In addition, NK cells secrete TRAIL and produce cytokines including IFN-γ that promote apoptosis in tumor cells and recruit other immune-like cells (21–23).
The therapeutic promise of ONC201 in preclinical in vivo studies in solid tumors, hematological malignancies, and cancer stem cells prompted its ongoing clinical development. In phase I clinical testing with ONC201, patients, including those with prostate cancer, were treated every 3 weeks, and the drug showed safety and promising efficacy in multiple tumor types (13, 24). The recommended phase II dose (RP2D) of ONC201 was determined to be 625 mg given orally every 3 weeks to patients with advanced cancer (24).
To maximize the clinical benefits of ONC201 and further elucidate its mechanism of action, we investigate here the impact of dose intensification on ONC201’s antitumor efficacy, and unravel its anti-metastasis properties and ability to induce an immune response leading suppression of to tumor growth. Through the use of syngeneic mouse models, coculture of established human NK and tumor cells, and primary normal and cancer patient NK cell data, we have uncovered an unanticipated immune-stimulatory antitumor effect of ONC201 involving NK and T cells along with a potent anti-metastasis effect. We further explored the mechanism to determine that TRAIL plays a role in both ONC201 anti-metastasis and ONC201-induced NK cell cytotoxicity. Finally, we identified key chemokines and cytokines that are upregulated by ONC201 treatment. We explored the consequences of NK cell depletion in vivo and, in preliminary experiments, the prospects of combining ONC201 with anti–programmed cell death 1 (anti–PD-1) therapy. Our findings reveal aspects of the mechanism of action of the antitumor compound ONC201, including insights into its anti-metastasis and pro–immune response activity, and potentially more efficacious dosing regimens for the drug in clinical trials.
Results
ONC201 dose intensification negatively impacts tumor growth and metastasis.
Given that every 3-week ONC201 dosing was well tolerated in the clinic, we explored the potential to augment its antitumor efficacy through dose intensification in preclinical models. We found that dose intensification of ONC201 significantly increased the extent of tumor growth inhibition in colorectal HCT116 p53–/–, HT29, and breast MDA-MB-231 human tumor xenografts (Figure 1, A–C, and Supplemental Figure 1, A–C and F; supplemental material available online with this article; https://doi.org/10.1172/JCI96711DS1). Doses of 50 mg/kg and 100 mg/kg impacted primary tumor growth in a drug administration frequency–dependent manner in the aggressive HT29 xenograft (Supplemental Figure 1E). Importantly, some tumors were completely ablated after 100 mg/kg weekly dosage for a month in HCT116 p53–/– or MDA-MB-231 tumor–bearing mice (Supplemental Figure 1, A and C). In mice, we found no significant difference in efficacy over a range of ONC201 doses from 25 to 100 mg/kg via the oral compared with i.p. route every 2 weeks (Supplemental Figure 2A). Moreover, weekly oral dosing of ONC201 appeared as effective as daily treatment (Supplemental Figure 2, B and C). This suggests that the pharmacodynamic properties of ONC201 are maximal with weekly administration, and that weekly administration is more efficacious than less-frequent dosing in mice. Importantly, administering a cumulative dose of 600 mg/kg ONC201 given via 6 weekly 100 mg/kg doses did not cause toxicity, affect weight, or decrease survival of the mice (Supplemental Figure 3, A–C).
Figure 1. ONC201 dose intensification negatively impacts tumor growth and metastasis.

(A) Mouse cohorts received 25 mg/kg or 100 mg/kg weekly, every 2, 3, or 4 weeks. Shown is the percent tumor growth of both dose and frequency of ONC201 over time in athymic female nu/nu mice using HCT116 p53–/– colorectal cancer xenografts. (B) Final HT29-luciferase tumors harvested after 4 weeks from cohorts administered weekly vehicle or 100 mg/kg ONC201 treatment. Blue arrows indicate metastatic tumors. (C) Final HT29-luciferase bioluminescence of cohorts administered 100 mg/kg at different frequencies and harvested after 4 weeks, with corresponding RLU for the whole cohort. n = 6 HT29 and HCT116 p53–/–. Percent tumor calculated by diving each tumor volume by the average tumor volume in the vehicle-treated group and multiplying by 100. *P < 0.05, **P < 0.01 compared with vehicle using 2-sided Wilcoxon’s rank-sum test. Data represent mean ± SD.
ONC201 triggers dual Akt/ERK inactivation, integrated stress response signaling, and TRAIL upregulation.
We previously established that a single dose of ONC201 can lead to blockade of Akt and ERK that lasts up to 96 hours in vivo. Here we determined that there was no detectable effect on Akt/ERK or the integrated stress response (ISR) after 30 days following a single ONC201 dose. However, we found that the dual inhibition of Akt/ERK by ONC201 can still occur in a dose- and frequency-dependent manner in vivo, demonstrating the importance of dose intensification on long-term ONC201 pharmacodynamics in the clinic (Figure 2A). The degree of ISR activation as monitored by CHOP or ATF4 mRNA induction was significantly increased with more frequent me of ONC201 (every 1–2 weeks versus every 3–4 weeks). On the other hand, increasing the ONC201 dose did not further amplify CHOP upregulation at 50 or 100 mg/kg relative to 25 mg/kg given weekly (Figure 2B and Supplemental Figure 4A). We observed a frequency- but not a dose-dependent effect on serum TRAIL levels. Maximum serum TRAIL levels were achieved (150 pg/ml) when ONC201 was administered weekly even at low doses of 25 mg/kg (Figure 2C and Supplemental Figure 4, B and C). Similar trends were observed with respect to overall TRAIL expression (Supplemental Figure 4D).
Figure 2. ONC201 triggers dual ERK/Akt inactivation, ISR signaling, and TRAIL upregulation in tumor cells.

(A) HT29 tumor xenograft protein lysates analyzed by Western blots treated with (left) 25 mg/kg at varying frequencies and (right) increasing dose up to 100 mg/kg and varying frequency of administration as indicated. (B) Induction of ATF4, CHOP, and TRAIL mRNA in HT-29 xenografted tumors following (left) increasing dose with weekly administration of ONC201 or (right) frequency of ONC201 dosing at 50 mg/kg. (C) Serum TRAIL levels measured by nonspecific ELISA compared following administration of ONC201 at a 25 mg/kg dose at different frequencies in MDA-MB-231 xenograft–bearing mice. Treatments are indicated by arrows. AUC values are shown above. For Western blots and qRT-PCR: n = 6, run twice in triplicate of each sample. For ELISA, n = 4 run in duplicate, samples were frozen until end of mouse experiment and then run through the assay. All samples were harvested 4 weeks after treatment began, unless otherwise indicated. *P < 0.05, **P < 0.01, ***P < 0.005, compared with vehicle unless indicated, 2-sided Wilcoxon’s rank sum test; for C, only the last time point was analyzed for statistical significance. Data represent mean ± SD.
ONC201 inhibits metastasis, and migration and invasion, via a TRAIL-dependent mechanism.
Given the potential anti-metastatic effects of TRAIL signaling, we hypothesized that ONC201 — as a compound that upregulates TRAIL and DR5 as part of its mechanism — would suppress metastatic tumor development. We observed that ONC201 reduced (in the case of the 25-mg/kg cohort) or inhibited metastases (in the case of higher-dosed cohorts) in these subcutaneous HT29 models (Figure 3A and Supplemental Figure 5, A–C) and a secondary model where the subcutaneous primary tumor was surgically removed and metastases were allowed to grow before ONC201 treatment (Figure 3B). Increasing the ONC201 dose and administration frequency reduced the number, size, and incidence of metastases (Supplemental Figure 5, A–C). In orthotopic MDA-MB-231 or subcutaneous HT29 xenograft–bearing mice, weekly MRI analysis and end-of-life bioluminescence imaging results confirmed that metastases develop independently of the primary tumor xenograft (Figure 3A and Supplemental Figure 5, D–F). We observed a similar anti-metastasis effect of ONC201 in the immunocompetent subcutaneous CT26 xenograft model in syngeneic BALB/c mice and in the athymic nude mice injected with HCT116-GFP that had their subcutaneous tumor removed when it reached a diameter of 10 mm, with only the vehicle-treated mice showing metastatic cell populations (Figure 3B and Supplemental Figure 5G). This anti-metastasis effect was further observed in metastatic tumors of mice — as documented through CT imaging — that had received HT29 or HCT116 p53–/– xenografts by tail vein injection and were treated with vehicle or ONC201. Overall, the size of the metastases on the lungs decreased in the ONC201-treated cohorts, while the vehicle-treated mice had growing tumors (Figure 3, C and D, and Supplemental Figure 5H). We also observed an impact on metastasis in mice treated with ONC201 or vehicle immediately after receiving tail vein injections (Supplemental Figure 5I).
Figure 3. ONC201 inhibits metastasis in vivo.

(A) Metastasis imaging by gross analysis (top panels) and histology of liver in HT29 xenograft–bearing mice treated with vehicle or 100 mg/kg ONC201 for 3 weeks (middle; original magnification, ×10). Bottom: MRI of lung of HT29 subcutaneous xenograft mouse treated with vehicle after 4 weeks following inoculation. The panels show imaging of 1 vehicle-treated mouse from different MRI viewpoints. The tumor is indicated by a blue arrow. (B) HCT116-GFP tumor lesions from mice with primary tumor surgically removed and tumor metastases allowed to grow for 1–3 weeks before treatment, as identified by fluorescence imaging for vehicle or 100 mg/kg ONC201. (C) Estimated size by CT imaging before and after treatment in ONC201- and vehicle-treated in HT29 xenograft cohorts. (D) Representative CT images of HT29 xenograft mice treated through tail vein injection. Yellow: tumor burden; green: lung tissue. Same mouse, day 0 and day 14. For mouse tumor studies, n = 6 mice in subcutaneous HT29 in A and tail vein HT29 in C and D; n = 4 in HCT116-GPF. In C, 2 vehicle-treated mice died before end of study. All samples were harvested 4 weeks after treatment began unless otherwise indicated. *P < 0.05 compared with the vehicle, 2-side Wilcoxon’s rank-sum test. Data represent mean ± SD.
ONC201 suppressed cell migration in vitro in HCT116 and HT29 cells as demonstrated by Boyden chamber and xCELLigence migration assays (Supplemental Figure 6, A–D). TRAIL inhibition by the TRAIL-depleting RIK-2 antibody or shTRAIL attenuated the effect of ONC201 on prevention of cell migration and invasion of MBA-MB-231 cells (Figure 4A and Supplemental Figure 6E). Invasion as assessed via a scratch assay was also impaired by ONC201, but was partially restored following treatment with RIK-2 (Supplemental Figure 6F). In in vivo experiments using the same tail vein method, wherein mice were treated with ONC201 or vehicle once tumors were noted, we observed a decrease in metastasis size and number in MDA-MB-231 WT tumor–bearing mice as compared with MDA-MB-231 shTRAIL tumor–bearing mice (Figure 4, B and C). This impact was also noted in mice treated immediately after tail vein injection of tumor cells (Supplemental Figure 5, G–I). Interestingly, there was a slight difference observed in cleaved caspase-3 levels between the tumor cohorts, but there was a statistically significant difference in the number of Ki-67+ cells between the vehicle- and ONC201-treated mice bearing the WT tumors, and no significant difference in the mice bearing the shTRAIL tumors (Figure 4, D–F). These results suggest that ONC201 inhibits metastasis at least in part by downregulating tumor cell migration and invasion in a TRAIL-dependent manner (17).
Figure 4. ONC201 inhibits migration/invasion and metastasis in a partially TRAIL-dependent manner.

(A) xCelligence migration kinetics assay in MDA-MB-231 (top) and MDA-MB-231 (bottom) shTRAIL groups. (B) Size of MDA-MB-231 WT and shTRAIL tumors from tail vein–injected mice after treatment for 2 weeks and then resection. Mice were treated once tumor burden was noted (tumor burden tended to align with the spinal cord). (C) Representative image of MDA-MB-231 WT and shTRAIL tumors after resection. (D) Quantification of Ki-67+ cells in MDA-MB-231 WT and shTRAIL xenografts of ONC201- and vehicle-treated cohorts. n = 12 slides, 3 per tumor. (E) Quantification of cleaved caspase-3–positive (CC3-positive) cells in MDA-MB-231 WT and shTRAIL xenografts of ONC201- and vehicle-treated cohorts. n = 12 slides, 3 per tumor. (F) Representative images of both at ×20 magnification. For mouse tumor studies, n = 5 for MDA-MB-231 and MDA-MB-231 shTRAIL. For in vitro migration/invasion studies, n = 4 run 2 separate times. All samples were harvested 4 weeks after treatment began unless indicated. *P < 0.05 compared with vehicle unless indicated using 2-sided Wilcoxon’s rank-sum test. For IHC studies, when 3 IHC slides were analyzed per tumor, the mean of each tumor was compared using a 2-sided Wilcoxon’s rank-sum test. Data represent mean ± SD.
ONC201 induces CD3+ NK cell accumulation, which plays an important role in the antitumor effect.
Given the potential role of NK cells in preventing metastases through the secretion of TRAIL (25), we investigated the presence of NK cells and other immune cells in ONC201-treated mice within the leukocyte population in colorectal MC38 and CT26 mouse tumors (Supplemental Figures 7 and 8). ONC201 induced activation and accumulation of T and NK cells within tumors, blood, and spleen in 2 WT syngeneic mouse models and the NCR1-GFP mouse model (C57BL/6 × 129/SvJ [BL6/129] background) (Figure 5, A–C and Supplemental Figure 9, A–D). Importantly, upregulation of NK+ and CD3+ cells was observed in blood of non-tumor-bearing mice, indicating that the immune effect was not due to extrinsic signaling from ONC201-treated tumors alone (Supplemental Figure 9E). These ONC201-stimulated NK cells expressed granzyme at a higher MFI and expressed IFN-γ (Figure 5D and Supplemental Figure 9F). Importantly, ONC201 activated human primary NK cells from healthy donors by dramatically increasing IFN-γ expression and causing increased degranulation of NK cells by increasing LAMP1 expression in the presence of target cells (Figure 6 and Supplemental Figure 10). Recruitment of CD4+ and CD8+ cells to the tumor was also observed (Supplemental Figure 9G). The susceptibility of HCT116 Bax–/– and RKO-ONC201–resistant cells to ONC201 in vivo, despite their resistance to ONC201 in vitro, implicated involvement of an immune response in ONC201-mediated tumor suppression (Figure 7A and Supplemental Figure 11A). Importantly, depletion of NK cells with the anti–asialo-GM1 antibody significantly attenuated the observed ONC201 antitumor efficacy in CT26, MC38, and Bax–/– tumor models despite there still being an increased presence of CD3+ T cells in the syngeneic models (Figure 7, A and B, and Supplemental Figure 11, B and C). We found no significant impact from ONC201 compared with vehicle on perforin-induced cytotoxicity in MC38 tumors in either WT or perforin-KO (Prf–/–) C57BL/6 mice (Supplemental Figure 11, D and E). Interestingly, we saw no impact on T cell depletion using a CD8a inhibitor in combination with ONC201 in MC38 mice, suggesting that NK cells may have a greater impact on antitumor efficacy during ONC201 treatments than CD3+ cells (Supplemental Figure 11, F and G).
Figure 5. ONC201 induces NK accumulation and activation and CD4/CD8+CD3+ T cell accumulation.

(A) Analysis of NK and T cells in MC38 xenografts in NCR1-GFP mice. (B) Immunofluorescence analysis of GFP expression of MC38 xenograft tumors from BL6/129-NCR1-GFP mice and DAPI at ×20 and ×100 magnification. (C) Comparison of T cell and NK cell populations in CT26 tumors in BALB/c mice. (D) Analysis of NK cells and NK cell granzyme expression and MFI in CT26 tumors in BALB/c mice. (E) Relative CT26 tumor volume after 4 weeks in BALB/c mice treated with vehicle, 100 mg/kg ONC201, PD-1, or ONC201+PD-1. (F) Final CT26 tumor volume after 4 weeks in BALB/c mice treated with vehicle, 25 mg/kg ONC201, PD-1, or ONC201+PD-1. For mouse studies: NCR1-GFP mice, n = 4; CT26 PBMC experiment, n = 7; CT26 PD-1 100 mg/kg experiment in E, n = 7; CT26 PD-1 25 mg/kg experiment in F, n = 7. **P < 0.01, ***P < 0.001, relative to vehicle using 2-sided Wilcoxon’s rank-sum test. For immunofluorescence: green, GFP; blue, DAPI. Merge performed by ImageJ. Analysis was performed on 4 sections per tumor; quantitation is shown in Supplemental Figure 9B. Data represent the mean ± SD.
Figure 6. ONC201-induces primary NK cell activation and degranulation.

(A) Human primary NK cell IFN-γ titration with increasing doses of ONC201. (B) Analysis of 5 healthy donors using 1 μM ONC201 treatment. (C) LAMP1 expression in the absence/presence of target cells using vehicle or 1 μM ONC201 treatment. Cells were treated overnight in ONC201 or vehicle. For healthy human samples, n = 5. *P < 0.05 relative to vehicle using 2-sided Wilcoxon’s rank-sum test. Data represent mean ± SD.
Figure 7. ONC201-induced NK activation and accumulation play an important role in antitumor effect.

(A) Final tumor volumes of Bax–/– (top) and CT26 (bottom) mice treated with 100 mg/kg ONC201 weekly for 4 weeks and GM1 every 5 days as prescribed. (B) NK cell enrichment by ONC201 and depletion by GM1 are confirmed with flow cytometry in the CT26-bearing mice. (C) Fluorescence microscopy from coculture of HCT116 p53–/––GFP and NK cells or NK media treated as indicated for 48 hours. (D) Quantitation of fluorescence microscopy. Multiplex analysis of (E) IP-10 and (F) IFN-α2a levels within conditioned media from ONC201-treated HCT116 cells. More cytokines were analyzed as indicated in Supplemental Figure 15. 10 μM ONC201, 2 mg/ml RIK-2. Green: GFP, blue: calcein AM; red: PI. Merge was performed by using ImageJ. Non-GFP blue+ cells are NK cells. Flow cytometry was performed on PI+ CD19–CD45+ cells. For mice, n = 10 for CT26 and Bax–/– GM1 studies. For cocultures, n = 3 and samples were run separately twice. For multiplex, n = 3 run in duplicate (6 samples total). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared with vehicle using 2-sided Wilcoxon’s rank-sum test; for multiplex ELISA study, 3 separate conditioned media samples per cohort were harvested and run in duplicate. The mean of each duplicate was taken and compared using a 2-sided Wilcoxon’s rank-sum test. Data represent mean ± SD.
To investigate the impact of ONC201’s pro-immune response on metastasis, we analyzed metastases from tail vein–injected HCT116, HCT116 Bax–/–, and MC38 mice. We saw no increase in lymphocyte populations, including of NK cells or CD3+ cells, in any of these metastases (Supplemental Figure 12).
ONC201’s efficacy derives from direct tumor cell death and NK cell–related tumor cell death.
Using the TRAIL-resistant HCT116 Bax–/– xenografts compared with WT HCT116 xenografts, we determined that ONC201’s impact on Bax–/– tumor growth could be significantly attenuated when NK cells were depleted in vivo, whereas the NK cell depletion in the sensitive WT cell line only slightly impacted antitumor efficacy (Supplemental Figure 13). However, there was still an antitumor effect from ONC201 treatment on HCT116 Bax–/– cells in NSG mice carrying no NK cells, T cells, or myeloid cells (Supplemental Figure 13, C and D). We noted a slight decrease in the number of Ki-67+ cells when mice were treated with ONC201 and no change in cleaved caspase-3 (Supplemental Figure 13, E–G).
PD-1 inhibitors in combination with ONC201 may enhance antitumor efficacy.
We noted potent antitumor effects in vivo despite the presence of increased PD-1 expression in CD3+ cells within ONC201-treated tumors, and given that ONC201 is efficacious in athymic nu/nu mice, we sought to leverage the ONC201-induced T cell effect in immunocompetent mice. We therefore explored the combination of ONC201 with anti–PD-1 therapy and preliminarily found evidence for more potent in vivo tumor suppression with the combination versus anti–PD-1 therapy alone (Figure 5, E and F). This effect was not seen in CT26-bearing mice treated with low doses of ONC201 or in MC38-bearing mice (Supplemental Figure 14, A–G). Since we suspected that PD-1 may be present within T cells or NK cells (26, 27), we examined the impact of PD-1 on CD3+ cells in terms of ONC201 treatment using a combination of anti–PD-1 and the NK-depleting antibody anti-GM1. We saw only a slight but not significant difference in anti–PD-1–treated cohorts when NK cells were depleted (Supplemental Figure 14, F and G).
ONC201-induced NK activation/accumulation and antitumor effect are mediated through a pro-immune mechanism whereby several key factors, including TRAIL, IFN-α2a, and IP-10 (CXCL10), are upregulated.
We further investigated mechanisms by which NK cells contribute to ONC201 antitumor activity in vitro. To further explore the potential mechanism of ONC201-induced NK activation and accumulation, we performed a multiplex ELISA-based assay and identified immune activating and recruiting factors that are upregulated in conditioned media of tumor cells in response to ONC201 treatment. This included IFN-α2a, IL-12p70, and IFN-γ–induced protein 10 (IP-10, or CXCL10) (Figure 7, E and F, and Supplemental Figure 15). Cell culture experiments with NK92 cells confirmed in vivo results indicating that ONC201 induces NK cell activation and TRAIL secretion (Supplemental Figure 16, A and B). Complementing our in vivo data, coculture studies of NK cells or conditioned media with tumor cells, including the ONC201-resistant HCT116 Bax–/– cells, confirmed that the ONC201 antitumor effect occurs as a result of ONC201 activation of NK cells, since neither the inactive NK cells nor ONC201 treatment alone significantly induce tumor cytotoxicity in a model that is resistant to direct drug effects (Supplemental Figure 13 and Supplemental Figure 16C). Moreover, addition of the TRAIL-sequestering antibody RIK-2 reduced but did not eliminate the cytotoxic activity toward tumor cells. These results indicate that ONC201 efficacy is potentiated by ONC201 activation of NK cells to produce TRAIL (Figure 7, C and D, and Supplemental Figure 16, C and D). Cell viability was further confirmed by a CTG assay and showed that ONC201-activated NK cells induce cytotoxicity at levels similar to those observed with NK cells activated by IL-2 and IFN-γ (Supplemental Figure 16, E and F). Peripheral blood samples from patients who received ONC201 treatment showed an increase in the number of activated TRAIL-secreting NK cells after ONC201 treatment up to 3 days after treatment, further establishing that activated NK cells are upregulated by ONC201 treatment and express TRAIL (Table 1 and Figure 8, A–C).
Table 1. Characteristics of patients 1–5 treated with ONC201.
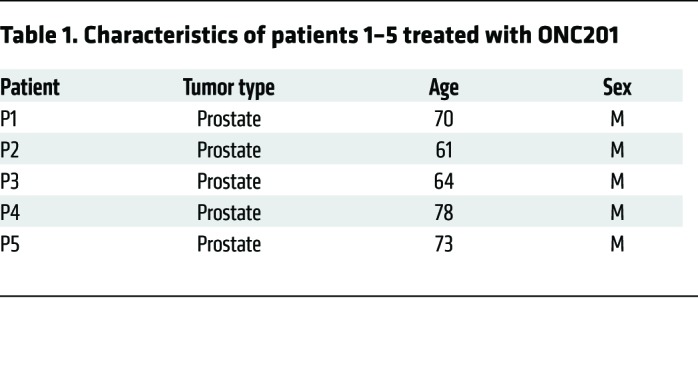
Figure 8. NK cells are increased in peripheral blood of patients following treatment with ONC201.

(A) Percentage of NK cells among all lymphocytes in peripheral blood of patients described in Table 1. (B) Percentage of NK cells expressing TRAIL. (C) Percentage of NK cells expressing granzyme B. P values are as indicated compared with day 0 using a paired 2-sided t test. Data represent mean ± SD.
Discussion
We demonstrate in this study that dose intensification of ONC201 increases antitumor drug efficacy, in part by amplifying ONC201’s ability to induce TRAIL and activate the ISR, without impacting toxicity. In vivo, dose intensification sustains a more potent growth and survival pathway inhibition. The impact of these results is of immediate relevance to clinical trials, where the frequency of administration is being increased to weekly dosing as a new RP2D. We uncover an aspect of the antitumor effect of ONC201: namely, powerful blockade of metastasis that may be at least in part due to ONC201’s ability to inhibit tumor cell migration and invasion. We unexpectedly discovered that ONC201 stimulates mobilization and activation of NK cell activity, including infiltration of tumors in tumor-bearing mice, and cytotoxic effects of NK cells toward ONC201-sensitive or -resistant tumor cells. Optimum pharmacodynamic effects of ONC201 were observed with weekly dosing even though the drug half-life is 10 hours (9). Dose intensification of ONC201 increased the extent of pro-survival kinase (Akt and ERK) inhibition, leading to increased TRAIL expression and signaling in vivo, causing a more efficacious tumor growth inhibition and anti-metastasis effects. Given the impact of ONC201 on the number and size of colorectal xenograft metastases in both the surgical and tail vein injection models, we can conclude that ONC201 suppresses metastases in vivo. While the role of TRAIL in ONC201’s anti-metastasis effect is apparent in the in vitro studies, there was not a significant impact on TRAIL inhibition within the tumor in vivo. This may be due to the impact of TRAIL induction from ONC201 globally, and further studies suppressing TRAIL within the entire mouse may help clarify the importance of TRAIL in vivo. Further, we cannot rule out other yet-to-be-discovered mechanisms that may play a role in the ability of the drug to inhibit migration and invasion. Further studies are warranted to investigate other pathways activated by ONC201 that may play a role in the anti-metastasis effect. ONC201’s ability to reduce migration is not dependent on its cytotoxic activity, and downregulation of the TRAIL pathway indicates that ONC201 exerts its anti-metastasis effects partially via a TRAIL-dependent mechanism (13). Also, given that the strongest impact on tumor growth within the MDA-MB-231 and shTRAIL tumors seemed to be from a decrease in Ki-67+ cells, ONC201 may reduce metastasis by impeding other key events in tumor metastasis unrelated to TRAIL.
The increased level, accumulation within tumors, and activation of NK cells both in vivo and in culture with human primary NK cells due to ONC201 treatment were unexpected and appeared to promote tumor cytotoxicity in part through NK cell TRAIL secretion. As expected, the TRAIL-sequestering antibody did not completely abrogate NK cell–mediated cytotoxicity in cell coculture, demonstrating that NK cells play a role in ONC201 efficacy that goes beyond TRAIL production. The significant cytotoxicity in the TRAIL-resistant Bax–/– cells in cocultures with both NK cells and conditioned media indicates that NK cells promote cytotoxicity through other tumor-suppression mechanisms, namely cytokine secretion and direct NK–tumor cell contact (28). However, there was still efficacy in vivo, including within the Bax–/– xenograft–carrying NSG mice. Given that there was a slight decrease in Ki-67+ cells and there was an impact on tumor growth during ONC201 treatment regardless of immune-related cytotoxicity, it is likely that ONC201 may require both its intrinsic-direct cytotoxic mechanisms and a possible immune response to suppress more ONC201-resistant tumors in vivo. Finally, although there appears to be an impact on degranulation in human primary NK cells, interestingly there was no impact of ONC201 treatment in Prf–/– compared with WT mice. Since MC38 tumors were still affected by ONC201 treatment, this may indicate that the role of direct NK cell killing is not necessary for ONC201’s pro-immune effects. Further studies of whether this could be mitigated in other tumors may clarify this further.
The immune effects of ONC201 play a role in its mechanism of action in vivo that is highly relevant in ONC201-resistant tumors and contributes to antitumor effects in models where ONC201 sensitivity is observed in the absence of NK cells. We did not observe an increase in lymphocytes or immune cells in the larger metastases treated with ONC201. This could be due to the limited size of the metastases, given that they were substantially smaller than our subcutaneous primary tumors. It is possible given limitations of the size of metastases in mice that the role of immune stimulation with regard to the impact of ONC201 on metastases may be better investigated in the context of the ONC201 clinical trials with human patients. Also, there could be an effect within the tumor microenvironment, as it is possible that ONC201’s pro-immune response may not bypass a stroma-heavy tumor, which may represent a difference from subcutaneously implanted tumors, which are not surrounded by epithelium from the lung as occurs in patients. Further, studies into the effect of ONC201 on the tumor microenvironment should be pursued to establish whether ONC201 can cause the tumor microenvironment to secrete antiinflammatory cytokines and protect the tumor. Nonetheless, ONC201 has a potent anti-metastasis effect that is relevant to its use as cancer therapy in patients.
Importantly, our multiplex assay indicated that in response to ONC201 treatment, colorectal tumor cells secrete immune-promoting factors. Of these, IFN-α2a, which induces sustained changes in NK cell responsiveness; IL-12p70, which enhances the cytotoxic activity of NK cells and induces differentiation and growth in T cells; and IP-10/CXCL10, a key chemoattractant for T and NK cell recruitment, are all of interest and further establish that ONC201 has an immune-enhancing role. We plan to pursue further the mechanism of action of these extrinsic signaling pathways on NK cell activation as a result of ONC201 treatment. The induced cytotoxicity in coculture is comparable to that resulting from the activation of NK cells by increased IL-2 and IFN-γ, without the added toxicity that these 2 cytokines provoke, making ONC201 a potentially attractive method of increasing the immune response (28, 29). The increase in CD3+ T cells shows a distinct promotion of the immune system, with CD4+ and CD8+ T cells being present in the tumor. However, CD8a inhibition did not attenuate ONC201’s efficacy, suggesting that the T cells play less of a role in ONC201’s pro-immune effect. Further, the combination of anti–PD-1 therapy with ONC201 showed some increased efficacy in comparison to the anti–PD-1 monotherapy in CT26 tumors treated with high doses of ONC201, indicating that alleviating T cells of PD-1 expression may enhance ONC201’s potency in vivo. However, we did not observe a clear advantageous impact of ONC201 and anti–PD-1 therapies in MC38 tumors or in CT26 tumors treated with lower ONC201 doses. This could be due to the inability of the TCR to recognize the tumors, given that the tumors are not mismatch repair (MMR) deficient and may not secrete enough clonal neoepitopes. It is also possible that with lower ONC201 doses, there may have been weaker signals for immune stimulation. Further studies of whether there could be an advantage to ONC201 and anti–PD-1 therapies in combination may clarify these issues. Overall, the impact of ONC201 on immune surveillance contributes to its efficacy in general and is particularly critical for promoting efficacy in some tumors that are resistant to ONC201 in vitro. There is probably sufficient preliminary evidence to support further testing of ONC201 plus anti–PD-1 in the clinic.
We demonstrate that higher or more frequent ONC201 dosing enhances the antitumor response, metastasis inhibition, and promotion of antitumor immunity. Dose intensification of ONC201 is now being explored in clinical trials to assess the proper regimen for different patient cohorts and is thus of immediate impact in the clinic. It is of clinical relevance that the pharmacodynamic effect of ONC201 extends for days to weeks beyond its serum half-life of 10 hours (9), and thus the most efficacious dosing in the clinic needs to be modeled by the pharmacodynamics characteristics. Additionally, we found no evidence that dosing based more on pharmacokinetics characteristics, i.e., daily dosing, provides any advantage over twice-weekly or weekly dosing. An important outcome of this study is the suggestion that, along with other biomarkers, monitoring of NK cell numbers, the state of NK cell activation in the blood, and NK infiltration in post-treatment biopsies of patient tumors has the potential to yield useful correlative clinical information with ONC201 efficacy. Initial patient data from a phase II clinical trial demonstrate that patients who received ONC201 treatments exhibit increased NK cell populations in their peripheral blood. These NK cells were activated and secreted TRAIL, similar to what was observed in mice. The presence of these NK cells in tumor sites and in larger ONC201-treated patient populations will be further assessed. The activation of primary human NK cells by ONC201 may be considered for combination therapy in clinical trials using adoptive transfer of therapeutic NK cells. We note that unlike the effects of phosphatase and tensin homolog (PTEN) (30) or the Src inhibitor dasatinib (31), ONC201 inhibition of Akt and ERK pathways with consequent NK cell activation leads to potent antitumor cytotoxic effects both in coculture experiments and in vivo. Moreover, unlike treatment with elotuzumab, which can stimulate NK cell antitumor activity against multiple myeloma (32), ONC201 administration is not associated with the well-known toxicities of IL-2 or TNF-α. The unique immune-stimulatory effect of ONC201 provides a rationale for combination therapy with complementary targeted therapeutics against cancer or checkpoint immunotherapy. With dose intensification, ONC201 has a greater likelihood of sustained TRAIL pathway activation in vivo, immune stimulation, and anti-metastasis effects.
Methods
Reagents and cell-based assays.
All cell lines were obtained from ATCC or discussed previously (1). NK92 cells were provided by Kerry Campbell’s laboratory at Fox Chase Cancer Center. CT26 and MC38 cells were provided by Scott Waldman’s laboratory at Thomas Jefferson University (Philadelphia, Pennsylvania, USA). ONC201 was obtained from Oncoceutics.
Western blot analysis and IHC.
Western blot analysis was conducted as previously described, with NuPAGE 4%–12% Bis-Tris gel, and visualized with ECL Prime Western Blotting Detection Reagent (Amersham) or SuperSignal West Femto (Thermo Scientific) and x-ray film and a CytoSMART Live Imaging System (Lonza). For all cell lysis buffers, fresh protease inhibitor (Roche) was added immediately. All antibodies were purchased from Cell Signaling Technology except anti-DR5 (Abcam ab1675) (catalog and clone numbers are listed in Supplemental Table 1). After fixation, the tumor samples were embedded in paraffin, and 8-μm sections were cut and mounted on slides. The sections were then processed and analyzed using IHC with TRAIL, Ki-67, cleaved caspase-3, CD3, and GFP antibodies, similar to the previously described method (1). CD31 and Ki-67 levels were calculated by independent blind-scoring and the use of the Vectra 3.0 Automated Quantitative Pathology Imaging system and Inform 2.0 software (PerkinElmer) courtesy of the Fox Chase Cancer Center Biosample Repository.
In vivo studies.
All animal experiments were conducted in accordance with the IACUC of Fox Chase Cancer Center. For subcutaneous xenografts, 6-week-old female athymic nu/nu mice (Taconic Biosciences) were inoculated with 1 × 106 cells of the HT29-luciferase, HCT116 p53−/−, HCT116 Bax–/–, RKO-ONC201–resistant, or HCT116-GFP cell line in each rear flank, or MDA-MB-231–luciferase in the lower mammary fat pads orthotopically, in a 150-μl suspension of 1:1 Matrigel (BD). For subcutaneous xenografts in syngeneic models, 1.0 × 106 CT26 cells were inoculated into 6-week-old female BALB/c mice (Taconic Biosciences), or 1.0 × 106 MC38 cells were inoculated into 6-week old female C57BL/6 mice (Taconic Biosciences), 6-week-old female BL6/129-NCR1-GFP mice (The Jackson Laboratory), or 6-week-old female C57BL/6-Prf1tm1Sdz/J mice (The Jackson Laboratory). All subcutaneous tumors were allowed to establish for 1–3 weeks after injection until they reached a volume of approximately 150–200 mm3 before treatment initiation. Mice were monitored every 3 days, and tumors volumes were measured using calipers. For the surgical method, mice were anesthetized using isoflurane after the tumors reached 1,000 mm3, and to remove the tumor, an incision to the surrounding skin was made and the tumor scraped out. All bleeding was controlled, and the wound closed using surgical wound clips. Mice were monitored every 6 hours for 2 days until they recovered. For tail vein injections, the same cell number as above for each tumor type was placed into 150 μl PBS and injected through the tail vein. Mice were monitored daily for the first 3 days.
ONC201 injections were administered subcutaneously at indicated doses in 20:80 DMSO/PBS or orally in 10:70:20 DMSO/PBS/Cremophor EL as described previously (1). GM1 was delivered every 5 days (Wako, 986-10001). PD-1 was injected at 200 μg every 3 days (Bio X Cell, BE1046). CD8a inhibitor was injected at 400 μg twice a week (Bio X Cell, 0061). PD-1 inhibition and staining were confirmed by staining one ONC201 monotherapy–treated tumor with Bio X Cell PD-1; eBioscience PD-1–PE; or Bio X Cell PD-1 for 2 hours, followed by staining with PD-1–PE. The resulting staining indicated that Bio X Cell PD-1 is a true inhibitor of PD-1 (Supplemental Figure 14F). Tumor volumes were measured according to the formula (L*W2)/2.
In vivo pathology and toxicology.
Toxicity during the course of ONC201 treatment was adjudged by body weight decrease of greater than 10%, tumor growth of more than 10% of body weight, or a body condition scoring less than 2. Serum and plasma samples were collected through orbital bleeding and cardiac puncture before sacrifice, and were immediately stored at 4°C and processed by Antech Diagnostics for complete blood count and chemistry panels. Results were analyzed by board-certified toxicologists. Tumors were measured postmortem through caliper and water density examination. Organ and tumor samples were processed in 10% formalin and fixed in paraffin. Hematoxylin-stained samples were analyzed by a board-certified pathologist to determine whether tumor cells existed on any organs or necrosis occurred in tumors. Board-certified veterinary pathologists also indicated whether signs of toxicity were present.
In vivo mouse tumor imaging.
For luciferase cell lines, D-luciferin from Gold Biotechnology was administered weekly according to the manufacturer’s instructions (60 μl, 50 mg/ml stock), and imaging was performed on a Xenogen IVIS system. MRI imaging was performed in a vertical-bore 7-T magnet with a Bruker DRX300 spectrometer and ParaVision 3.0 software (Bruker) through the Fox Chase Imaging Core Facility. Custom-built transfer cassettes constructed to compress the mice to an equal thickness of 15.0 mm were used within the MRI scanner to keep the mouse still and administer isoflurane gas. GFP imaging was performed using the Maestro in vivo imaging system (Cambridge Research & Instrumentation) and the Nuance multispectral imaging system (Cambridge Research & Instrumentation). Image data sets were converted and tumor volumes calculated using PARAVISION. Final images were converted to ImageJ (NIH) format. For CT imaging, all mice were imaged in a Sofie Biosciences G8 scanner under isoflurane. CT data sets were acquired with an isotropic voxel size of 200 μm3. Mice were maintained under isoflurane anesthesia for the duration of the scan. For analysis, the thoracic cavity was segmented into compartments comprising the heart, lung air space, lung tissue, and discrete tumors on the CT data sets. The lung air space was segmented on the CT scans with a connected nearest-neighbor seed growing algorithm, using 1 voxel in the lung as a seed point, and manually setting the seed thresholds. The lung tissue present outside the air space, as well as any structures identified as tumors, was determined manually using a 3D region-of-interest tool. All image analyses were performed with VivoQuant software (Invicro Inc.).
ELISAs.
A total of 100–150 μl blood was collected through orbital blood draw as described above, and plasma was collected in EDTA tubes and serum collected in heparin separating tubes. Tubes were spun at 1,000 g for 15 minutes. Samples were analyzed using a Human TRAIL (also known as TNFSF10) Quantikine ELISA kit (R&D Systems). All analyses were performed according to the manufacturer’s directions.
For conditioned media studies, a mesoscale multiplex ELISA was used. Conditioned media was harvested from the cell lines at designated times and doses. Three separate conditioned media samples per cohort were harvested and run in duplicate. The mean of each duplicate was taken and compared using a 2-sided Wilcoxon’s rank-sum test.
In vitro tumor migration and invasion assays.
Boyden assays were performed using the QCM ECMatrix Cell Invasion Assay (Millipore, ECM550) and the Cultrex cell migration assay (R&D Systems, 3465-096-K). Cell migration and invasion were also assessed using the real-time xCelligence system (ACEA Biosciences Inc.). Invasion assays in the xCelligence system were conducted with Matrigel (33). Wound healing assays were performed with the CytoSelect Wound Healing Assay kit (Cell Biolabs Inc., CBA-120T). Cell viability was confirmed by Trypan blue or Cell TiterGlo; data were only included if cell viability was greater than 75%.
In vitro tumor and NK cell coculture.
NK92 cells were maintained in 4% IL-2 media and before experiments were maintained in low–IL-2–containing (1%) media. Tumor cell lines were plated at 30,000 cells/well on an 8-well chamber slide for 24 hours. NK92 cells were washed, suspended in fresh media, and then plated over the tumor cells at a concentration of 80,000 cells/ml. Alternatively, conditioned media (media as shown in the figures) from the NK cells cultured for 48 hours was added to the tumor cells. After 48 hours of coculturing, NK92 cells were removed, and both NK and tumor cells were stained with calcein blue and ethidium homodimer. Non-GFP-expressing tumor cells were labeled with anti–EPCAM-1 antibody (1:200), followed by Alexa Fluor 488 secondary antibody (1:200). NK92 cells were labeled with CD56-APC (BioLegend, 318309). Immunofluorescence was performed on the Nuance multispectral imaging system. As an alternative for measuring tumor cell viability in coculture, the CellTiter-Glo assay (Promega, as directed) was used directly after NK cells were removed. Plates were analyzed using the IVIS system described above in In vivo mouse tumor imaging. As controls, NK92 cells were incubated in media with a level of high IL-2 (4%) and 400 U/ml IFN-γ.
In vitro primary NK cell analysis.
Primary NK cells were acquired from peripheral blood of healthy donors per the IRB protocol. Lymphocytes were isolated through Lymphoprep (STEMCELL Technologies, 07801) centrifugation, and NK cells were isolated using the EasySep Human NK Cell Isolation Kit and magnet per the manufacturer’s instructions (STEMCELL Technologies, 17955). NK cells were then incubated overnight with 1% IL-2 with or without ONC201. IFN-γ assay was performed by incubating cells with IFN-γ and then with IFN-γ-PE and antibodies listed in Supplemental Figure 10A. LAMP1 assay was performed by incubating NK cells alone, with 721.221 target cells, or with target cells and 100 ng/ml rituximab. NK cells only degranulate in the presence of 721.221 target cells, allowing us to measure LAMP1 expression of activated NK cells, and rituximab, which increases degranulation, was used as the positive control. Cohorts were as shown in Figure 6C: peripheral blood lymphocytes (PBL), PBL ONC: NK cells alone or with ONC201; PBL+721.221, PBL+721 ONC: NK cells incubated with target cells with or without ONC201; PBL+721+Rxb, PBL+721.221+Rxb ONC: NK cells incubated with target cells and rituximab with or without ONC201. Cells were then rinsed and stained with LAMP1 and antibodies listed in Supplemental Figure 10B. Gating strategies are shown in Supplemental Figure 10.
Flow cytometry.
For flow cytometry analyses, antibodies were purchased from eBioscience unless otherwise indicated (Supplemental Table 1). Analysis was performed by incubating cells for 30 minutes on ice with 20 μg/ml purified Fab antibody. Cells were stained with 1% FBS, 0.09% sodium azide in PBS, and red blood cells were lysed in red blood lysis buffer (155 mM NH4Cl, 12 mM NaHCO3, 0.1 mM EDTA in PBS). Cells were then washed and incubated in 2 μg/ml propidium iodide (PI) in staining buffer. Analyses were performed as shown in Supplemental Figures 7 and 8. Cellular concentrations of select antibodies were determined with LSRII (Beckton Dickinson) and analyzed with FlowJo software (Tree Star).
Patient analysis.
Cells from patients treated with ONC201 (ClinicalTrials.gov, NCT02250781) were obtained by peripheral blood draw. Peripheral blood mononuclear cells (PBMCs) were isolated from blood on the day of the blood draw by Ficoll-Hypaque density centrifugation and frozen prior to analysis in CryoStor solution (BioLife Solutions). Thawed cells were washed in PBS and incubated with fluorescent-labeled antibodies at 4°C for 20 minutes for extracellular staining, permeabilized, and incubated with fluorescent-labeled antibodies at 4°C for 30 minutes for intracellular staining. Analysis of live, non-debris, singlet leukocytes was performed as previously described (34) using an LSRII flow cytometer and FlowJo software.
Steps to maintain optimum viability of cryopreserved cells for future analyses were taken. Processing of patient blood samples occurred within 1 hour of blood draw, and slow freezing of cells was performed immediately after processing to increase viability. Cryopreserving these cells in this way and then thawing them all at the same time (and antibody staining and flowing them at the same time) ensures similar testing and analyses on all samples and increases the accuracy of the results. Further, all cells were cryopreserved in Cryostor CS10, a solution that mitigates temperature-induced molecular cell stress responses during freezing and thawing. As described by the manufacturer (and observed in our laboratory compared with 90% FBS/10%DMSO), this solution has been proven to be much more effective in reducing post-preservation necrosis and apoptosis compared with commercial and homemade formulations. Citations of cell preservation capabilities and comparisons with other solutions are available from the manufacturer (BioLife Solutions). Importantly, after thawing, viability analysis demonstrated greater than 80% live CD45+ cells across all samples, demonstrating the efficacy of our cryopreservation process and ensuring that analysis produced accurate results. As expected, the immune cell populations among PBMCs varied across the 5 patients and across the 3 time points at which samples were collected from each patient (as shown in Figure 8, where the NK cells varied across patients and over the 3 time points). However, overall, populations across the sum of samples analyzed were found to be at proportions that were still within the normal variation and range for patient samples, including T cells (53% [expected 45%–70% of PBMCs]), B cells (5% [expected 3%–15%]), NK cells (8% [expected 5%–20%]), monocytes (26% [expected [15%–35%]). In patients where the CD3–CD19– (non-T/non-B) population was increased (as in patient 1), the majority population was monocytes. This would not be an expected artifact of the cryopreservation process, as the myeloid lineage cells are more likely to be lost in cryopreservation than lymphocytes, yet here the proportions of monocytes were increased at a few individual time points. Cell proportions normalized with ONC201 treatment, and as in mouse model studies, NK cell proportions and function were augmented.
Statistics.
Data are presented as mean ± SD. To assess the statistical significance of the differences between group comparisons (vehicle versus ONC201, ONC201 versus ONC201+PD-1), of tumor volume before and after treatment in the tail vein, xenograft, RLU, blood serum, gene expression, and protein expression experiments, a 2-side Wilcoxon rank-sum test was used. For in vitro experiments, measurements from 3 biological replicates per treatment group were compared unless noted in the figure legends. When there were more than 2 observations made in the xenograft study, the means per mouse (for example, 3 IHC slides were analyzed per tumor, the mean of each tumor) were compared using a 2-sided Wilcoxon’s rank-sum test. For Supplemental Figure 6G, metastases were grouped as “<2” and “>2” for the comparison between WT and shTRAIL ONC201 groups using 2-sided Fisher’s exact test. For patient samples in Figure 8, statistical significance was determined by a paired 2-sided t test. For all tests, P < 0.05 was considered statistically significant. Unless otherwise noted in the figure legends, comparisons were made against the vehicle control.
Study approval.
For animal studies, all animals were housed and handled in accordance with the Institutional Animal Care and Use Committee of Fox Chase Cancer Center. All studies were performed in accordance with national animal protection laws. For patient studies at Rutgers, the IRB protocol used to obtain patient samples was #Pro20140000405, titled “A phase I single-agent open-label dose-escalation study of oral ONC201 in patients with advanced solid tumors.” Approval of the protocol was obtained from Health Sciences IRB — New Brunswick/Piscataway and was initially approved October 3, 2014. The latest version of this protocol was approved December 20, 2017, with an expiration date of June 5, 2018. All patients signed informed consent for trial participation. For healthy human NK study the IRB protocol was FCCC 99-802, and was last renewed on May 18, 2017, expiring May 17, 2018.
Author contributions
JW and WSED designed all the experiments. JW conducted the experiments and wrote the manuscript. CLK assisted in all animal experiments and qRT-PCR work. LZ assisted in all imaging, including IHC and coculture experiments. KSC and AWM assisted in the design of and experimental procedures for human primary NK cell work. KQC assisted with all pathological analysis and IHC analysis. HHH assisted with mouse CT imaging. CBC and JHN conducted NK cell studies utilizing patient-derived samples from the ONC201 clinical trial, and AZ analyzed the patient data and wrote the related manuscript sections. AJO contributed to the conceptualization and rationale of the dose intensification experiments. EAR contributed to the appropriate analytical methods. MDR assisted with mice for the HT29 dose intensification study when needed. JB and MS assisted with human clinical patient studies. WSED conceptualized and supervised the experiments and contributed as senior author, including editing of the manuscript and responsibility for oversight of conduct of the research.
Supplementary Material
Acknowledgments
This work was presented in part at the American Association for Cancer Research (AACR) meeting, April 16–20, 2016, in New Orleans, Louisiana, USA; the American Society for Clinical Oncology (ASCO) meeting, June 6, 2016, in Chicago, Illinois, USA; and the AACR meeting, April 2–5, 2017, in Washington, DC, USA; JW received the 2016 AACR-Triple Negative Breast Cancer Foundation training award for this work. We thank the Fox Chase Cancer Center Animal Facility and Small Animal Imaging Component of the Biological Imaging Facility at the Fox Chase Cancer Center, and acknowledge support from the National Cancer Institute (grant CA-006927) for assistance in animal imaging. We thank the Animal Histopatholgy Facility and Biosample Repository at the Fox Chase Cancer Center for assistance with pathology and IHC analysis. We also thank Anthony Lerro and the Fox Chase Cancer Center Animal Facility for assistance with the surgeries and tail vein injections. WSED is an American Cancer Society Research Professor. This work was supported by grants from the NIH (CA173453 to WSED), the American Cancer Society (to WSED), and the William Wikoff Smith Endowed Professorship in Cancer Research (to WSED).
Version 1. 03/13/2018
In-Press Preview
Version 2. 04/30/2018
Electronic publication
Version 3. 06/01/2018
Print issue publication
Footnotes
Conflict of interest: WSED is a cofounder of Oncoceutics Inc. and is fully compliant with institutional and NIH disclosure guidelines.
Reference information: J Clin Invest. 2018;128(6):2325–2338.https://doi.org/10.1172/JCI96711.
Contributor Information
Jessica Wagner, Email: Jessica.Wagner@fccc.edu.
C. Leah Kline, Email: lkline2cor5.15@gmail.com.
Lanlan Zhou, Email: lanlan.zhou@fccc.edu.
Kerry S. Campbell, Email: Kerry.Campbell@fccc.edu.
Alexander W. MacFarlane, Email: Alexander.MacFarlaneIV2@fccc.edu.
Anthony J. Olszanski, Email: Anthony.Olszanski@fccc.edu.
Kathy Q. Cai, Email: Qi.Cai@fccc.edu.
Harvey H. Hensley, Email: harvey.hensley@fccc.edu.
Eric A. Ross, Email: eric.ross@fccc.edu.
Marie D. Ralff, Email: tuf01898@temple.edu.
Andrew Zloza, Email: az255@cinj.rutgers.edu.
Charles B. Chesson, Email: cbc96@cinj.rutgers.edu.
Jenna H. Newman, Email: jhn33@gsbs.rutgers.edu.
Howard Kaufman, Email: howard.kaufman@replimune.com.
Wafik S. El-Deiry, Email: wafik.eldeiry@gmail.com.
References
- 1.Allen JE, et al. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med. 2013;5(171):171ra17. doi: 10.1126/scitranslmed.3004828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen JE, et al. Identification of TRAIL-inducing compounds highlights small molecule ONC201/TIC10 as a unique anti-cancer agent that activates the TRAIL pathway. Mol Cancer. 2015;14(1):99. doi: 10.1186/s12943-015-0346-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen JE, et al. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget. 2016;7(45):74380–74392. doi: 10.18632/oncotarget.11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishizawa J, et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal. 2016;9(415):ra17. doi: 10.1126/scisignal.aac4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kline CL, Van den Heuvel AP, Allen JE, Prabhu VV, Dicker DT, El-Deiry WS. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2α kinases. Sci Signal. 2016;9(415):ra18. doi: 10.1126/scisignal.aac4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen JE, Crowder RN, Crowder R, El-Deiry WS. First-in-class small molecule ONC201 induces DR5 and cell death in tumor but not normal cells to provide a wide therapeutic index as an anti-cancer agent. PLoS One. 2015;10(11):e0143082. doi: 10.1371/journal.pone.0143082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prabhu VV, Allen JE, Dicker DT, El-Deiry WS. Small-molecule ONC201/TIC10 targets chemotherapy-resistant colorectal cancer stem-like cells in an Akt/Foxo3a/TRAIL-dependent manner. Cancer Res. 2015;75(7):1423–1432. doi: 10.1158/0008-5472.CAN-13-3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen JE, et al. Genetic and pharmacological screens converge in identifying FLIP, BCL2, and IAP proteins as key regulators of sensitivity to the trail-inducing anticancer agent ONC201/TIC10. Cancer Res. 2015;75(8):1668–1674. doi: 10.1158/0008-5472.CAN-14-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen JE, et al. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget. 2016;7(45):74380–74392. doi: 10.18632/oncotarget.11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner J, et al. The angular structure of ONC201, a TRAIL pathway-inducing compound, determines its potent anti-cancer activity. Oncotarget. 2014;5(24):12728–12737. doi: 10.18632/oncotarget.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin ZZ, Wang W, Fang DL, Jin YJ. mTOR inhibition sensitizes ONC201-induced anti-colorectal cancer cell activity. Biochem Biophys Res Commun. 2016;478(4):1515–1520. doi: 10.1016/j.bbrc.2016.08.126. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, Wang H, Ran L, Zhang Z, Jiang R. The preclinical evaluation of TIC10/ONC201 as an anti-pancreatic cancer agent. Biochem Biophys Res Commun. 2016;476(4):260–266. doi: 10.1016/j.bbrc.2016.05.106. [DOI] [PubMed] [Google Scholar]
- 13.Stein MN, et al. First-in-human clinical trial of oral onc201 in patients with refractory solid tumors. Clin Cancer Res. 2017;23(15):4163–4169. doi: 10.1158/1078-0432.CCR-16-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner J, Kline CLB, El-Deiry WS. Dose-intensified ONC201 to exert anti-metastatic efficacy and to promote intra-tumoral recruitment of NK-cells in mice [abstract] J Clin Oncol. 2016;34(15 suppl):11550 [Google Scholar]
- 15.Elrod HA, et al. Analysis of death receptor 5 and caspase-8 expression in primary and metastatic head and neck squamous cell carcinoma and their prognostic impact. PLoS One. 2010;5(8):e12178. doi: 10.1371/journal.pone.0012178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grosse-Wilde A, et al. TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J Clin Invest. 2008;118(1):100–110. doi: 10.1172/JCI33061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeda K, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med. 2001;7(1):94–100. doi: 10.1038/83416. [DOI] [PubMed] [Google Scholar]
- 18.Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol. 2002;168(3):1356–1361. doi: 10.4049/jimmunol.168.3.1356. [DOI] [PubMed] [Google Scholar]
- 19.Shin MS, et al. Mutations of tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1) and receptor 2 (TRAIL-R2) genes in metastatic breast cancers. Cancer Res. 2001;61(13):4942–4946. [PubMed] [Google Scholar]
- 20.Zhuang L, et al. Progression in melanoma is associated with decreased expression of death receptors for tumor necrosis factor-related apoptosis-inducing ligand. Hum Pathol. 2006;37(10):1286–1294. doi: 10.1016/j.humpath.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 21.Levy EM, Roberti MP, Mordoh J. Natural killer cells in human cancer: from biological functions to clinical applications. J Biomed Biotechnol. 2011;2011:676198. doi: 10.1155/2011/676198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gross E, Sunwoo JB, Bui JD. Cancer immunosurveillance and immunoediting by natural killer cells. Cancer J. 2013;19(6):483–489. doi: 10.1097/PPO.0000000000000005. [DOI] [PubMed] [Google Scholar]
- 23.Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8+ T cells. Nat Rev Immunol. 2011;11(10):645–657. doi: 10.1038/nri3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stein M, et al. First-in-human trial of ONC201 in patients with refractory solid tumors. J Clin Oncol. 2016;34(suppl):abstr 2514 [Google Scholar]
- 25.Smyth MJ, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J Exp Med. 2001;193(6):661–670. doi: 10.1084/jem.193.6.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Concha-Benavente F, Srivastava R, Kansv B, Ferris R. PD-1 is a marker of activation on tumor infiltrating NK cells in head and neck cancer. J Immunother Cancer. 2015;3(Suppl 2):P398. doi: 10.1186/2051-1426-3-S2-P398. [DOI] [Google Scholar]
- 27.Wei F, et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci U S A. 2013;110(27):E2480–E2489. doi: 10.1073/pnas.1305394110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zamai L, et al. NK cells and cancer. J Immunol. 2007;178(7):4011–4016. doi: 10.4049/jimmunol.178.7.4011. [DOI] [PubMed] [Google Scholar]
- 29.Jonasch E, Haluska FG. Interferon in oncological practice: review of interferon biology, clinical applications, and toxicities. Oncologist. 2001;6(1):34–55. doi: 10.1634/theoncologist.6-1-34. [DOI] [PubMed] [Google Scholar]
- 30.Briercheck EL, et al. PTEN is a negative regulator of NK cell cytolytic function. J Immunol. 2015;194(4):1832–1840. doi: 10.4049/jimmunol.1401224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hassold N, et al. Enhancement of natural killer cell effector functions against selected lymphoma and leukemia cell lines by dasatinib. Int J Cancer. 2012;131(6):E916–E927. doi: 10.1002/ijc.27537. [DOI] [PubMed] [Google Scholar]
- 32.Balasa B, et al. Elotuzumab enhances natural killer cell activation and myeloma cell killing through interleukin-2 and TNF-α pathways. Cancer Immunol Immunother. 2015;64(1):61–73. doi: 10.1007/s00262-014-1610-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisenberg MC, Kim Y, Li R, Ackerman WE, Kniss DA, Friedman A. Mechanistic modeling of the effects of myoferlin on tumor cell invasion. Proc Natl Acad Sci U S A. 2011;108(50):20078–20083. doi: 10.1073/pnas.1116327108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zloza A, et al. NKG2D signaling on CD8+ T cells represses T-bet and rescues CD4-unhelped CD8+ T cell memory recall but not effector responses. Nat Med. 2012;18(3):422–428. doi: 10.1038/nm.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.