

Abstract
Chronic obstructive pulmonary disease (COPD) is an incurable inflammatory lung disease that afflicts millions of people worldwide, and it is the fourth leading cause of death. Systemic comorbidities affecting the heart, skeletal muscle, bone, and metabolism are major contributors to morbidity and mortality. Given the surprising finding in large prospective clinical biomarker studies that peripheral white blood cell count is more closely associated with disease than inflammatory biomarkers, we probed the role of blood growth factors. Using the SHIP-1–deficient COPD mouse model, which manifests a syndrome of destructive lung disease and a complex of comorbid pathologies, we have identified a critical and unexpected role for granulocyte-CSF (G-CSF) in linking these conditions. Deletion of G-CSF greatly reduced airway inflammation and lung tissue destruction, and attenuated systemic inflammation, right heart hypertrophy, loss of fat reserves, and bone osteoporosis. In human clinical translational studies, bronchoalveolar lavage fluid of patients with COPD demonstrated elevated G-CSF levels. These studies suggest that G-CSF may play a central and unforeseen pathogenic role in COPD and its complex comorbidities, and identify G-CSF and its regulators as potential therapeutic targets.
Keywords: Inflammation, Pulmonology
Keywords: COPD
Introduction
Chronic obstructive pulmonary disease (COPD) is an umbrella term representing a group of closely related and incurable inflammatory lung diseases, comprising bronchitis, bronchiolitis, and emphysema (1). COPD afflicts around 300 million people worldwide, responds very poorly to current treatments, and has escalated to the world’s fourth leading cause of death. COPD patients exhibit lung inflammation, lung tissue destruction, and small airway narrowing, collapse, and obstruction. In the emphysematous disease presentation, this hyperinflates the lungs, making exhalation progressively more difficult, and in patients with very severe disease, causes marked and intractable breathlessness that, even on the mildest exertion, is extremely debilitating and distressing (2, 3). In addition to the disease burden attributable to declining lung function, systemic comorbidities, often designated the “COPD comorbidome,” are extremely common, and contribute significantly to reduction in quality of life and the economic cost of the disease. These systemic comorbidities, notably cardiovascular disease, bone loss, and osteoporosis, and in patients with very advanced disease, cachectic loss of fat and skeletal muscle reserves among other conditions, are very common and variable in COPD and contribute substantially to disease morbidity in diverse patient groups (4). Around half of all COPD patients die from cardiovascular comorbidities including myocardial infarction, right heart failure, and stroke. Furthermore, acquired defects in mucosal immunity lead to recurrent viral and bacterial infectious exacerbations and irreversible colonization of the lungs, with an aberrant metagenome in many patients (5). Indeed, there is evidence that COPD patients have increased intestinal permeability, and smokers have disruptions in their normal flora, which may present a link between the lung and gut microbiome influencing disease susceptibility (6). These infections and colonization are believed to further worsen comorbidities by driving systemic inflammation (7). The clustering of these disease traits has driven the search for unifying causative mechanisms.
It is surprising, given the important progress in identifying novel gene variants in genome-wide association studies (8, 9), that the trait most strongly associated with COPD in large prospective biomarker studies is a simple elevation in peripheral WBC count (10–12), which is directly linked to severity of lung destruction, propensity for exacerbations, and extent of comorbidities. Thus, we reasoned that probing the molecular basis of WBC regulation and studying the role of hematological CSFs might lead to novel insights into COPD and its comorbidome. Furthermore, myeloid lineage innate inflammatory cells, such as macrophages and neutrophils, are markedly elevated in the COPD lung and they are critically dependent on CSFs for their development and activation, thereby linking bone marrow myelopoiesis with tissue-damaging inflammation, and hence host defense (13). Granulocyte macrophage-CSF (GM-CSF), originally discovered as a blood cell growth factor released from endotoxin-inflamed lungs (14), is one such factor, and animal studies have shown that neutralization of GM-CSF ameliorates experimental COPD (15). Granulocyte-CSF (G-CSF) is another important myeloid growth factor; however, it has been largely ignored because of its traditional role as a neutrophil-specific factor. G-CSF is produced by stromal cells, endothelium, and macrophages, and the major stimuli for its production are bacterial products and proinflammatory cytokines (16). G-CSF stimulates the production of neutrophils from the bone marrow and promotes their survival, priming, and function. Recombinant human G-CSF (filgrastim) is used clinically in chemotherapy-induced neutropenia (17, 18) and for the mobilization of peripheral blood progenitor cells for transplantation (reviewed in ref. 19). G-CSF–deficient mice are neutropenic and also exhibit reduced numbers of bone marrow myeloid precursors (20), and while they are susceptible to acute bacterial infection (21), they handle chronic bacterial infection (22). More recent studies have shown that despite G-CSF’s role in innate immunity, G-CSF has pathogenic roles in inflammatory autoimmune diseases such as inflammatory arthritis (23), experimental allergic encephalomyelitis (24), and uveoretinitis (25). Furthermore, G-CSF is induced in airway inflammation and in response to cigarette smoke (26–29), but its role in COPD and its complex comorbidome has not been investigated.
To probe the role of G-CSF in COPD, we have used mice deficient in the regulatory lipid phosphatase SH2 domain–containing inositol 5′ phosphatase-1 (SHIP-1). SHIP-1, which is orthologous to INPP5D (inositol polyphosphate-5-phosphatase D) in humans, is found only in hematopoietic cells where it acts as a critical negative regulator of multiple inflammatory signaling pathways (30). We have previously described that SHIP-1–deficient mice spontaneously develop a marked inflammatory lung disease with features reminiscent of clinical COPD, including inflammation, emphysema, and small airway fibrosis (31–33). The lung inflammatory infiltrate comprises activated macrophages, neutrophils, lymphocytes, and eosinophils (32, 34). In addition to lung inflammation, SHIP-1–/– mice exhibit systemic inflammation and extramedullary hematopoiesis, which drives splenomegaly and the expansion of both erythroblasts and myeloid cells in the periphery (35). SHIP-1 also regulates hematopoiesis and SHIP-1–/– mice display elevated WBC count due to leukocytosis (36).
Here we demonstrate using genetic complementation that deletion of G-CSF not only leads to a dramatic improvement in lung health, but most surprisingly, ameliorates the associated disease comorbidome in SHIP-1–deficient mice. By targeting a single factor we have been able to ablate the diverse pathologies in multiple organ systems encompassed by COPD. We have furthermore demonstrated that G-CSF levels are elevated in the lungs of patients with COPD. Our studies are the first to our knowledge to strongly implicate G-CSF and its regulators as promising new therapeutic targets in the treatment of COPD and the systemically manifested complex COPD comorbidome.
Results
G-CSF levels are elevated in the lungs and periphery of SHIP-1–deficient mice.
We and others have previously reported elevated levels of G-CSF in SHIP-1–/– mice (31, 37), which suggested to us that it may be playing a pathogenic role by supporting the production of neutrophils and stimulating hematopoietic stem cell mobilization. To confirm and extend these studies, we measured levels of G-CSF in the lung and circulation, finding that it was significantly elevated in bronchoalveolar lavage fluid (BALF) and serum of a large cohort of SHIP-1–/– mice (Figure 1A).
Figure 1. G-CSF is elevated in SHIP-1–/– mice and contributes to lung fibrosis, consolidation of airspaces, and emphysema.

(A) G-CSF levels in the BALF and serum of 12-week-old control (C57 = C57BL/6) and SHIP-1–/– mice determined by ELISA; n ≥ 6 per group. (B) Masson’s trichrome–stained sections of lungs from the indicated 12-week-old mice. Scale bars: 100 μm. Images are representative of n = 3–14 per group. (C) Quantification of airspace size of the lung sections in (B) by mean linear intercept; pooled data from n = 3–14 per group. S–/– = SHIP-1–/–, G–/– = G-CSF–/–. (D) Levels of MMP-9 in BALF of the indicated 12-week-old mice determined by multiplex assay; pooled data from n = 4–14 per group. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 by Mann-Whitney U test (A) or ANOVA (C and D).
Deletion of G-CSF in SHIP-1–/– mice ameliorates lung pathology.
To determine the role of G-CSF in disease pathogenesis in SHIP-1–deficient mice, we generated SHIP-1–/– G-CSF–/– double-knockout (DKO) mice, with the striking finding that lung disease was dramatically reduced, evidenced by significantly less inflammation and fibrosis in the lung parenchyma and small airways of DKO mice (Figure 1B). Quantification of the size of airspaces in the lungs of SHIP-1–/– mice by determining mean linear intercept revealed significant emphysema compared with control lungs (Figure 1C), while removal of G-CSF from SHIP-1–/– mice significantly reduced this phenotype, restoring airspace sizes towards those of control mice (Figure 1C). MMPs are implicated in the pathophysiology of COPD, as they degrade the extracellular matrix and cause parenchymal tissue damage (1). To determine whether the reduced emphysema phenotype in DKO mice correlated with alterations in MMP production, we measured MMP-9 in BALF by Luminex assay. These studies revealed a significant elevation in MMP-9 in SHIP-1–/– lungs compared with control lungs, while levels in DKO lungs were significantly reduced (Figure 1D).
We have previously shown that SHIP-1–/– mice have a markedly increased BAL cell count (31, 32); however, this phenotype was dramatically blunted in DKO mice (Figure 2A). Differential counts of the BAL revealed that not only were alveolar macrophages (AMΦs) reduced, but neutrophil, eosinophil, and lymphocyte populations were largely absent in DKO mice (Figure 2A). Cytology of the BAL cells revealed that the morphology of the AMΦs in SHIP-1–/– and DKO mice was strikingly different (Figure 2B). While SHIP-1–/– BAL macrophages were enlarged and strongly pigmented, as we have previously reported (31, 32), the macrophages observed in the BAL of DKO mice were small and predominantly quiescent in appearance (Figure 2B).
Figure 2. G-CSF deficiency abrogates inflammatory cell infiltration and corrects AMΦ phenotype in SHIP-1–/– mice, while SHIP-1 deficiency in AMΦs enhances responsiveness to G-CSF.

(A) Total BAL cells and differential BAL cell counts of the indicated 12-week-old mice; pooled data from n = 4–7 per group. C57 = C57BL/6, S–/– = SHIP-1–/–, G–/– = G-CSF–/–. (B) Images of cytospins from the indicated 12-week-old mice. Scale bar: 100 μm. Images are representative of n = 4–7 per group. (C) Flow cytometry of CD11b versus CD11c expression on AMΦs from the indicated 12-week-old mice to segregate AMΦ subpopulations into subset R or Ri and Rii in SHIP-1–/– mice; representative of n = 10–12 per genotype in a minimum of 3 experiments. (D) Levels of chemokines KC and MIP-2 in the BALF of the 12-week-old indicated groups determined by multiplex assay; pooled data from n = 9–10 per group. (E) qRT-PCR analysis of G-CSFR gene expression in AMΦs from C57BL/6 mice (C57), CD11b– and CD11b+ AMΦs from SHIP-1–/– mice, sorted neutrophils (PMN) and bone marrow cells (BM) from 12-week-old mice; n = 2–6 per group pooled from 2 experiments. Assessment of (F) ERK and (G) STAT-3 phosphorylation by flow cytometry in AMΦs from C57BL/6 mice (C57) or SHIP-1–/– mice (S–/–) treated with media or 10 ng/ml G-CSF for 15 minutes. Data are expressed as a ratio to the unstimulated control; pooled data from n = 3–9 per group from 2 experiments. *P < 0.05; **P < 0.01; ****P < 0.0001 by ANOVA (A and D) or Mann-Whitney U test (F and G).
Previous studies by our laboratory have shown that AMΦs do not ordinarily express the leukocyte integrin CD11b, but its expression is induced in settings of viral infection and LPS challenge, or under conditions of chronic lung inflammation such as in SHIP-1–/– mice (32). To determine whether increased CD11b was an innate feature of SHIP-1–/– AMΦs or whether it correlated with local inflammation and the heightened activation status of the cells, we compared CD11b expression on AMΦs from SHIP-1–/– and DKO mice. Flow cytometry analyses showed that SHIP-1–/– mice had both CD11b– and CD11b+ AMΦ subpopulations (populations Ri and Rii), while the AMΦs of DKO mice, like control mice, showed minimal CD11b expression (population R) (Figure 2C).
Various chemotactic factors orchestrate the recruitment of inflammatory cells to the inflamed lungs of COPD patients (1). The chemotaxis of neutrophils is induced by activation of the CXC chemokine receptor CXCR2, which binds several CXC chemokines including CXCL1, CXCL2, and CXCL8 (IL-8), which are elevated in the sputum of COPD patients (38, 39). To determine whether the reduction of total cell number as well as the loss of neutrophils were due to altered neutrophil chemoattractants, we measured levels of the murine CXCL1 and CXCL2 homologs keratinocyte-derived chemokine (KC) and macrophage inflammatory protein-2 (MIP-2). We found that the BALF from SHIP-1–/– mice had significantly elevated KC and MIP-2 levels compared with controls, and that these levels were significantly reduced in the BALF of DKO mice (Figure 2D).
SHIP-1–/– lung macrophages are hyperresponsive to G-CSF.
The G-CSF receptor (G-CSFR, CD114) is not only expressed in the granulocyte lineage but can be found on platelets and other hematopoietic cells including monocytes. However, the role of G-CSFR signaling in AMΦs remains uncertain, and thus we first examined G-CSFR expression in AMΦs purified from BAL using quantitative real-time PCR (qRT-PCR). These studies revealed that G-CSFR was expressed by C57BL/6 AMΦs and by the CD11b– and CD11b+ AMΦ subsets in SHIP-1–/– mice, albeit at lower levels than in bone marrow cells and neutrophils (Figure 2E).
SHIP-1 is known to be recruited to the G-CSFR as an inhibitory signaling mediator downstream of G-CSF signaling (40). Since AMΦs expressed G-CSFRs, we determined whether SHIP-1–/– AMΦs were hyperresponsive to G-CSF–induced signaling responses, which may provide an explanation for their activated phenotype (Figure 2, B and C), given the presence of elevated G-CSF levels in the lung (Figure 1A). In order to assess this, AMΦs were stimulated with G-CSF in vitro followed by detection of phosphorylated proteins by phospho-flow cytometry. Interestingly, AMΦs from SHIP-1–/– mice exhibited increased induction of phosphorylated Erk (p-Erk) upon exposure to G-CSF, compared with control AMΦs (Figure 2F). Induction of phosphorylated STAT-3 (p-STAT-3) was also significantly increased in SHIP-1–/– AMΦs compared with controls (Figure 2G). These results indicate that the MAPK and JAK/STAT pathways downstream of G-CSFR in SHIP-1–/– AMΦs are hyperresponsive to G-CSF stimulation.
Systemic inflammation is markedly reduced in SHIP-1–/– G-CSF–/– mice.
Since we observed a dramatic improvement in lung inflammation and pathology in DKO mice, we next sought to determine whether other indices of inflammation were moderated. An inflammatory trait consistently observed in SHIP-1–/– mice is splenomegaly (31, 41) and this was markedly reduced in DKO mice (Figure 3A), as was the coexistent splenic neutrophilia (Figure 3B) and splenic histiocytosis (Figure 3C). Splenomegaly can be a marker of systemic inflammation due to extramedullary hematopoiesis and this is a well-known trait of SHIP-1–/– mice (31, 35, 36). Extramedullary myelopoiesis was strikingly dampened in DKO mice, with numbers of myeloid progenitors reduced to the levels seen in control mice (Figure 3D). Erythrocytosis/polycythemia is another feature of SHIP-1–/– mice (35) and very interestingly, numbers of nucleated erythroid cells, which were dramatically increased in SHIP-1–/– mice, were markedly attenuated by G-CSF deficiency (Figure 3E).
Figure 3. G-CSF deficiency reduces systemic inflammation and extramedullary hematopoiesis and restores oxygen saturation and heart health in SHIP-1–/– mice.

(A) Spleen weight of the indicated 12-week-old mice; n = 3–12 per group pooled from 3 experiments. C57 = C57BL/6, S–/– = SHIP-1–/–, G–/– = G-CSF–/–. Absolute numbers of (B) splenic neutrophils and (C) splenic macrophages of the indicated 12-week-old mice, determined by spleen cell numbers and flow cytometry; n = 4–12 per group pooled from 3 experiments. (D) Quantification of myeloid progenitor cells in the spleen of 12-week-old mice that are responsive to the indicated myeloid growth factors, indicated as mean ± SEM. (E) Absolute numbers of splenic nucleated erythroid cells in the indicated 12-week-old mice, determined by spleen cell counts and flow cytometry; n = 4–12 per group pooled from 3 experiments. (F) Pulse oximetry measurements of arterial oxygen saturation in conscious 12-week-old mice; n = 4–7 per group pooled from 2 experiments. (G) Assessment of RVH in heart sections from the indicated 12-week-old mice, determined by calculating the ratio of the right ventricular wall area to left ventricular wall area plus septum area (RV/[LV + S]); pooled data from n = 5–7 per group. Data in A–C and E–G analyzed by ANOVA. *P < 0.05; **P < 0.01; ****P < 0.0001.
B cell abnormalities are still present in SHIP-1–/– G-CSF–/– mice and are not responsible for lung disease.
We and others have previously observed B cell abnormalities and autoimmune disease in SHIP-1–/– mice (31, 42). To determine whether B cell defects were influenced by G-CSF–mediated inflammation, we examined the B cell compartment. Although splenic B cell frequency was partially restored in DKO mice (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI98224DS1), DKO B cells retained their activated phenotype, mirroring that observed in SHIP-1–/– mice (Supplemental Figure 1B), and DKO mice, like SHIP-1–/– mice, had elevated levels of serum immunoglobulins (Supplemental Figure 1C). Furthermore, DKO mice, like SHIP-1–/– mice, developed autoimmune disease exhibiting circulating autoantibodies (Supplemental Figure 1D) and glomerulonephritis (Supplemental Figure 1E). Collectively, these data indicate that G-CSF–mediated inflammation does not underlie B cell abnormalities in SHIP-1–/– mice. To confirm that B cells and B cell–mediated autoimmune disease do not play a role in the lung pathology manifested by SHIP-1–/– mice, we generated SHIP-1–/– mice that lacked B cells (SHIP-1–/– μMT–/– mice). These mice still exhibited severe lung pathology, in a manner akin to SHIP-1–/– mice (Supplemental Figure 1, F and G), indicating that lung disease in SHIP-1–/– mice is not a consequence of their underlying B cell–mediated autoimmune disease.
G-CSF deficiency rescues reduced arterial oxygen saturation and right ventricular hypertrophy in SHIP-1–/– mice.
Erythrocytosis in SHIP-1–/– mice is likely to be due to extramedullary erythropoiesis (35); however, additional potential mechanisms could involve those related to hypoxia/hypoxemia (low blood/tissue oxygen), a common lung disease comorbidity. To explore this possibility, we performed noninvasive arterial oxygen sensing on conscious animals using a pulse oximeter, finding that SHIP-1–/– mice had a modest, albeit significant reduction in arterial oxygen saturation, while DKO mice showed similar arterial oxygen saturation to control mice (Figure 3F).
Right ventricular hypertrophy (RVH) is a common comorbidity of COPD linked with pulmonary hypertension (43). To determine whether RVH was a feature of SHIP-1–/– mice, we analyzed hearts from 12-week-old mice. There was a significant increase in RVH in SHIP-1–/– mice, whereas RVH was absent in DKO mice, whose hearts were comparable to those from control and G-CSF–/– mice (Figure 3G). These data suggest that lung disease may contribute to RVH in SHIP-1–/– mice but could also point to other mechanisms inducing pressure overload, including pulmonary leukostasis, pulmonary embolism, or infarction.
Comorbidities in SHIP-1–/– mice are dependent on G-CSF–mediated inflammation.
SHIP-1–/– mice have reduced survival due to their attendant lung disease, and they also develop other phenotypes including systemic inflammation, polycythemia, and osteopenia (31, 36, 41, 44). Survival studies revealed that the median survival of DKO mice was 326 days, which was significantly improved compared with that of SHIP-1–/– mice, which was 168 days (P < 0.01). Initial characterization of DKO mice revealed that they were healthier in appearance than SHIP-1–/– mice, having improved body condition and increased body weight (Figure 4A). The proinflammatory cytokine IL-6 is elevated in the serum of COPD patients and is associated with systemic inflammation and poor prognosis (45). To determine whether G-CSF contributes to the systemic inflammatory phenotype in SHIP-1–/– mice, sera were tested for levels of IL-6 (Figure 4B). As found previously (46), SHIP-1–/– mice displayed elevated levels of IL-6 in serum, with a trend toward reduction in DKO mice (Figure 4B). Systemic inflammation arising due to spillover from the lungs can also induce acute-phase protein production in the liver and contribute to systemic disturbances (4). We therefore examined gene expression of C-reactive protein (CRP) and serum amyloid A (SAA) in the liver by qRT-PCR. Although no significant differences were observed in CRP expression between groups (Figure 4C), SAA expression was significantly elevated in SHIP-1–/– mice compared with controls, and this was reduced in the absence of G-CSF (Figure 4D). Interestingly, SAA has been reported to be a marker of acute exacerbations in COPD patients and to block resolution of inflammation (47, 48).
Figure 4. G-CSF deficiency improves body condition and restores fat and bone parameters in SHIP-1–/– mice.

(A) Body weights of the indicated 12-week-old mice; pooled data from n = 4–20 per group. C57 = C57BL/6, S–/– = SHIP-1–/–, G–/– = G-CSF–/–. (B) IL-6 levels in serum of the indicated 12-week-old mice measured by ELISA; pooled data from n= 4–16 per group. qRT-PCR analysis of (C) CRP and (D) SAA gene expression in liver from the indicated 12-week-old mice; pooled data from n = 3–12 per group. (E) Weights of distinct muscle groups in the indicated 12-week-old mice, expressed as a ratio to body weight; pooled data from n = 6–12 per group. (F) Measurement of distinct anatomical white and brown fat in the indicated 12-week-old mice; pooled data from n = 6–12 per group. (G) qRT-PCR analysis of ATGL in brown fat of 12-week-old mice; pooled data from n = 4–7 per group. (H) Micro-CT analysis of trabecular and cortical bone in 6-week-old male mice; pooled data from n = 3–6 per group. All data analyzed by ANOVA. *P < 0.05; **P < 0.01; ****P < 0.0001.
Low body weight and wasting syndrome are common comorbidities in a subset of COPD patients with advanced disease that correlate with poor prognosis (4). We therefore assessed indices of overall body condition in 12-week-old mice, including muscle mass and body fat mass. Assessment of muscle mass in distinct muscle groups of the hind limb of 12-week-old mice did not show any differences between the groups (Figure 4E), indicating that catabolism of muscle had not developed at this age. However, assessment of white and brown body fat isolated from 3 distinct anatomical locations revealed that SHIP-1–/– mice displayed a significant loss of fat mass, with a trend towards restoration in DKO mice (Figure 4F). To determine whether aberrant lipolysis could account for the differences in body fat mass in SHIP-1–/– mice, we assessed expression of adipose triglyceride lipase (ATGL) in the brown fat (Figure 4G). SHIP-1–/– mice displayed increased ATGL gene expression, potentially contributing to increased lipolysis, with a trend towards reduction in DKO mice.
Osteoporosis is a significant comorbidity in COPD patients (49). SHIP-1–/– mice also display a low bone mass phenotype due to elevated osteoclastogenesis, and formation of osteoclasts that are hyperresorptive (44). To determine whether removal of G-CSF could alter the development of osteoporosis in DKO mice, we performed high-resolution micro-CT of long bones. As previously observed, SHIP-1–/– mice displayed significantly lower trabecular bone volume and trabecular thickness, with a trend towards restoration in DKO mice (Figure 4H). Cortical bone area and cortical thickness were also significantly lower in SHIP-1–/– mice, with protection of cortical thickness observed in DKO mice (Figure 4H).
SHIP-1–/– mice lacking G-CSF exhibit no further impairment of mucosal immune responses.
Given the impaired immunity reported for G-CSF–deficient mice (20) and the susceptibility of SHIP-1–/– mice to LPS-induced toxicity (50), we next examined the response of DKO mice to respiratory challenge by transnasal treatment with LPS followed by quantification of the influx of neutrophils and monocytes into the lung after LPS treatment (Figure 5, A and B). Control mice mounted a strong response, reflected by increased neutrophil frequency and absolute number at day 1, which was reduced by day 3 and resolved at day 10, with monocytes and macrophages peaking at day 3 and resolving at day 10. G-CSF–/– mice displayed a robust neutrophil response to LPS that abated more quickly than that of control mice and interestingly, they exhibited an enhanced monocyte response. By comparison, SHIP-1–/– mice were impaired at all time points after LPS treatment, with reduced neutrophil and monocyte infiltration into the lung. In contrast, although DKO mice were impaired in the same manner as SHIP-1–/– mice in their ability to mount a neutrophil response to LPS treatment, they mounted an early compensatory monocyte response at day 1, which increased further at day 3 and was largely resolved by day 10. Cytology of the BALF at day 1 showed the clear presence of neutrophils in DKO mice, and interestingly, the presence of enlarged macrophages (Figure 5A). These results demonstrate that complete removal of G-CSF genetically can impair mucosal immunity but does not further impair immune responses in SHIP-1–/– mice.
Figure 5. G-CSF deficiency does not further impair the response of SHIP-1–/– mice to pulmonary challenge.

(A) Images of cytospins from the indicated 12-week-old mice at the indicated days after transnasal LPS challenge. Scale bar: 100 μm. Images are representative of n = 2–6 mice per group collected over 4 experiments. (B) Frequency of neutrophils (CD45intLy6G+CD11b+) and monocytes (CD45+Ly6G–CD11b+) in BALF at the indicated time points after LPS challenge measured by flow cytometry; pooled data from n = 2–6 per group collected over 4 experiments. Graphical data represent mean ± SEM for each group at each time point. C57 = C57BL/6, S–/– = SHIP-1–/–, G–/– = G-CSF–/–.
G-CSF is elevated in human COPD samples.
Since we found that G-CSF is a key pathogenic factor in the SHIP-1–/– mouse model of COPD, we next determined G-CSF levels in a cohort of patients presenting to the respiratory clinic. Patients were admitted to the day clinic for endobronchial ultrasound for diagnosis of lung cancer based on chest abnormalities detected by X-ray. Patient diagnostic information was obtained after analysis and patients were grouped according to smoking and COPD status, with or without lung cancer diagnosis (Supplemental Table 1). These results showed that G-CSF levels were elevated in patients with COPD and/or smoker status compared with nonsmokers without COPD (Figure 6), indicating that G-CSF is induced in the human lung upon exposure to cigarette smoke, and correlates with COPD.
Figure 6. G-CSF levels are elevated in the BALF of smokers and COPD patients.
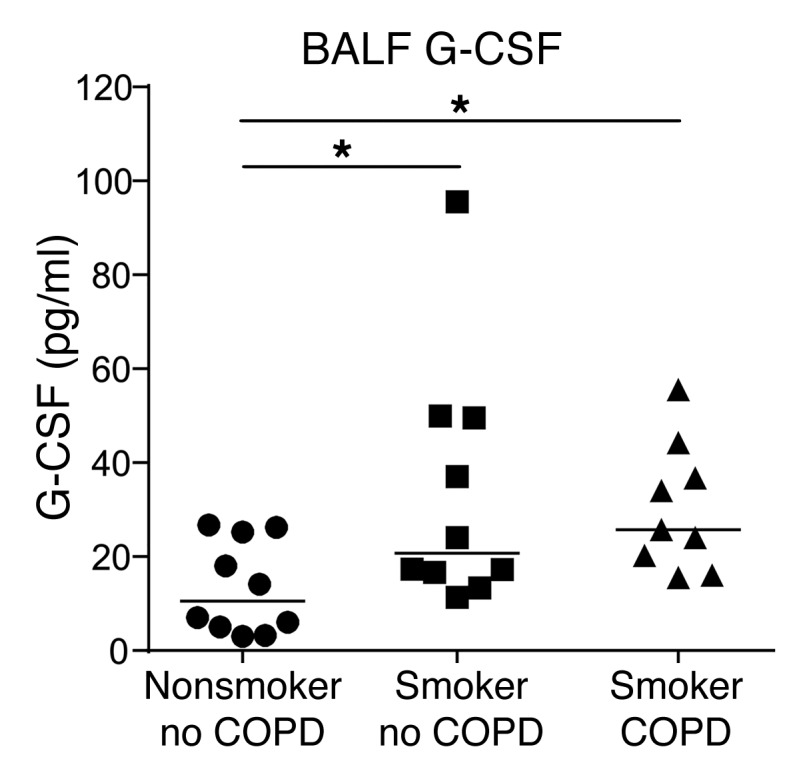
G-CSF levels in the BALF of the indicated patient groups measured by ELISA; pooled data from n = 9–10 per group. Data analyzed by Student’s unpaired t test. *P < 0.05.
Discussion
In this study, we have identified what we believe to be a novel therapeutic target, G-CSF, in a robust genetic model of COPD. Our study is the first to our knowledge to characterize the development of lung inflammation in conjunction with comorbid disease, and systematically evaluate the effect of the intervention on these pathologies. SHIP-1–/– mice develop a disease that resembles human COPD, characterized by progressive nonresolving lung inflammation, fibrosis, emphysema, and importantly, the development of comorbidities. We have found that removal of G-CSF from SHIP-1–/– mice was sufficient to reduce several COPD hallmarks, including immune cell infiltration and activation in the lung, fibrosis and emphysema, systemic inflammation and strikingly, the development of comorbid syndromes. Elevated levels of several inflammatory mediators that are present in the lung and systemic circulation of SHIP-1–/– mice are reduced in the absence of G-CSF, suggesting that it is an orchestrator of a cascade of inflammation in COPD, and the effect of its therapeutic targeting would not be limited to the lung but also to systemic disease. G-CSF may also be facilitating the chemotactic response of other cytokines, as illustrated by studies examining factors produced by lung epithelial cells following exposure to smoke extract (51).
While there have been no previous interventional studies investigating the role of G-CSF in COPD, there is some evidence to indicate that it can be a pathogenic factor in diverse respiratory conditions such as idiopathic pulmonary fibrosis (52), acute respiratory distress syndrome (53), acute chest syndrome (54), and sulfur-gas-induced fibrosis (55). With respect to COPD, it is well known that neutrophils are plentiful in the COPD lung (1) and their presence implicates G-CSF. Furthermore, there is strong evidence for G-CSF–dependent inflammation biomarkers in COPD such as proteinase 3 (56), neutrophil elastase and myeloperoxidase (57), and lipocalin (58). The clinical use of G-CSF for the recovery of neutrophil number and function in chemotherapy patients has also revealed critical toxicities, notably neutrophil-induced pulmonary toxicity, alveolar hemorrhage, acute respiratory distress syndrome, and hemoptysis (59, 60). Accordingly, recent research into G-CSF points to it as a therapeutic target in diseases where G-CSF and/or the activity of neutrophils is pathogenic (23–25). Given that clinical therapeutic antibodies can be conveniently dose-ranged, as has been shown in cultures of human bronchial epithelial cells and cigarette-smoke-induced mouse models (61, 62), we would advocate for doses that address excess G-CSF production aiming to restore G-CSF homeostasis rather than remove its activity completely, in order to preserve beneficial host defense. This is important, since COPD patients exhibit defects in mucosal immunity. We utilized LPS to determine the responsiveness of mice in a simplified sterile model of acute lung inflammation. We showed that although G-CSF–/– mice were somewhat impaired, they were able to mount a robust neutrophil response accompanied by an enhanced monocyte response. Interestingly, although SHIP-1–/– mice displayed diminished responses, removal of G-CSF did not lead to further impairment, and indeed DKO mice exhibited an enhanced monocyte response. Thus, productive neutrophil responses towards bacterial LPS can be attained in the absence of G-CSF, and would likely be even less impacted in therapeutic treatments that involve dose-ranging as outlined above. In future studies, the use of clinically relevant microorganisms in model systems such as Haemophilus influenzae, Streptococcus pneumonia, and Pseudomonas aeruginosa would provide further insights into the safety and effectiveness of targeting of G-CSF in the context of respiratory infection, acute exacerbations of COPD and, speculatively, potentially in other diseases such as cystic fibrosis and severe asthma where neutrophilic inflammation is prominent.
In COPD, neutrophils are thought to play a pathogenic role, as they promote a proinflammatory state and contribute to tissue destruction by the secretion of neutrophil elastase (63). AMΦs are also recognized as key mediators of pathological lung injury in COPD owing to their ability to produce inflammatory cytokines, neutrophil-attracting chemokines, and proteolytic enzymes that destroy lung parenchyma (1). Removal of G-CSF from SHIP-1–/– mice not only ablated neutrophils and neutrophil chemokines but resulted in significant reductions in AMΦ numbers and activation state, and a concomitant reduction in emphysema. Furthermore, systemic inflammation and induction of acute-phase reactants such as IL-6 and SAA, both of which are clinically validated human COPD biomarkers, were significantly reduced in the absence of G-CSF, a beneficial effect given the strong correlation between systemic inflammation and poor outcomes in COPD (45). Our studies also revealed that young SHIP-1–/– mice exhibited a modest decrease in arterial oxygen saturation to 95%, whereas DKO mice had normal blood oxygen. While this decrease was significant, it was small, and thus, it seems unlikely that this factor alone would lead to hypoxemia and compensatory erythrocytosis. It is of interest to examine aged mice as well as mice under exercise duress. Curiously, increased expression of the Ly-6 family member CD177 on neutrophils is a biomarker for polycythemia (64), a feature of SHIP-1–/– mice, and is increased upon exposure to G-CSF (65). This may present a feedback loop between G-CSF, neutrophils, and erythropoiesis that is deregulated in COPD patients. We have also found that young SHIP-1–/– mice developed RVH, which is a common COPD comorbidity, and this feature was ameliorated by deficiency of G-CSF. While it is entirely conceivable that lung disease has driven RVH, other factors may also contribute. Lung inflammation and the inflammatory milieu slows the pulmonary capillary transit time of leukocytes (66) and it is well known that SHIP-1 deficiency promotes leukocyte activation as well as platelet spreading (67), increasing the possibility of thrombus formation and pulmonary embolism. Since these are known to cause pulmonary arterial hypertension (68), it is possible that these factors might also contribute to RVH in SHIP-1–/– mice, and they are the subject of ongoing investigations.
Comorbidities are a significant health burden in the management of COPD patients, and in addition to the disease itself, often account for the majority of low quality of life and high healthcare costs associated with COPD (4). These syndromes include a diverse range of conditions including cardiovascular disease, osteoporosis, malnutrition, skeletal muscle loss, metabolic syndromes, and anemia (69). Removal of G-CSF from SHIP-1–/– mice not only alleviated lung disease in DKO mice, but significantly reduced comorbidities and led to increased body mass, improved body condition, and restored body fat mass. In COPD patients, muscle wasting manifested as sarcopenia and cachexia are most commonly noted in advanced disease, with reports predominantly focused on fat-free mass. It is interesting to note that in cancer patients, body fat mass depletion occurs concomitantly with cachexia driven by inflammatory cytokines such as IL-6 and TNF-α, which contribute to increased lipolysis as well as muscle catabolism (70). Poor nutritional status of COPD patients has been suggested as a potential cause of wasting phenotypes associated with poor survival; however, cachexia can occur in COPD patients despite adequate caloric intake (71). This suggests that body fat depletion may be an important metabolic consideration in COPD patient subgroups with advanced disease, especially with respect to the systemic increase in inflammatory cytokines, and indeed, higher body mass index in this patient subset is associated with better survival. Osteoporosis is a significant and debilitating comorbidity in COPD patients and develops in an analogous manner in SHIP-1–/– mice (44). SHIP-1 is known to negatively regulate osteoclast proliferation in the presence of excess cytokine (72), while macrophages from SHIP-1–/– mice have been reported to produce elevated levels of IL-1β (73, 74). The improved bone structure in SHIP-1–/– mice lacking G-CSF could reflect the loss of G-CSF’s actions to promote bone resorption, its action to suppress bone formation, or a combination of both. Pharmacologic administration of G-CSF has been reported to both stimulate osteoclast formation (75) and to rapidly block bone formation via a macrophage-mediated mechanism (76, 77) and by downregulation of osteoprotegerin (78). Inhibition of G-CSF may protect the skeleton through these different mechanisms.
This study shows clearly that G-CSF contributes to lung and systemic inflammation and importantly, a complex pattern of comorbid conditions in the SHIP-1–/– COPD mouse model. Although it is well known that neutrophils are important in COPD pathogenesis, and G-CSF is a key mediator of their survival, to the best of our knowledge this factor has not been previously explored in COPD. Our examination of human BAL samples revealed that G-CSF was elevated in patients with COPD and smoking status. Importantly, these results point to G-CSF or G-CSF–induced factors as biomarkers for lung inflammation in patients with diagnosed COPD or those predisposed to developing COPD.
In summary, our work is the first to our knowledge to show pathogenic involvement of G-CSF in COPD and its complex comorbidome, providing rationale for exploring G-CSF, as well as its regulation and signaling mechanisms, as therapeutic targets in clinical COPD.
Methods
Mice.
Mice were rederived and reared at the specific pathogen–free Monash Animal Research Platform, Clayton, Victoria, Australia upon relocation of the laboratory to Monash University. This change in facility and housing resulted in a change to the survival characteristics of the SHIP-1–/– mice compared with our previously published studies (31). SHIP-1–/– mice (JAX Inpp5dtm1Dmt) have been described previously (79) and C57BL/6 background mice were used (31). C57BL/6 background G-CSF–/– mice (JAX Csf3tm1Ard) (20) and μMT–/– mice (80) were also used. SHIP-1–/– mice and G-CSF–/– mice were intercrossed to generate double-knockout SHIP-1–/– G-CSF–/– (DKO) mice. The line was maintained by intercrossing female SHIP-1+/– G-CSF–/– mice with DKO males and genotyping progeny. Studies were performed on 6- to 12-week-old mice, except for autoimmune disease analyses, which were performed on 30-week-old mice. SHIP-1–/– mice and μMT–/– mice were intercrossed to generate B cell–deficient SHIP-1–/– μMT–/– mice; lung analysis studies were performed on 7- to 13-week-old mice.
Measurement of BALF and serum cytokines.
BALF and serum cytokine levels were measured using a Luminex multiplex assay (R&D Systems) according to the manufacturer’s instructions. Beads coated with capture antibodies against known cytokines, chemokines, and proteases were used simultaneously in the multiplex assay. Levels of mouse G-CSF (PeproTech), mouse IL-6 (BD Biosciences), and human G-CSF (R&D Systems) in BALF or serum were analyzed by ELISA kits according to the manufacturers’ protocols.
Lung fixation, histology, and airspace size assessment.
Lungs were inflation-fixed at exactly 25 cmH2O pressure in 10% neutral-buffered formalin and then paraffin embedded. Whole-lung sections (3 μm) were stained with H&E or Masson’s trichrome and analyzed using an Olympus BX51 microscope (Olympus Australia). Images were captured with an NP70 camera. Alveolar airspace size was determined by the mean linear intercept method (81). Evenly distributed lines (20 per genotype) were drawn over images of H&E–stained lung cross sections, and then number of intercepts with alveolar air walls counted. Size of airspace was determined by dividing the known length of the line by the average number of intercepts.
BAL and cytospins.
Lungs from lethally anesthetized mice were lavaged using 400 μl PBS, followed by 3 further lavages of 300 μl. Cells were counted, centrifuged onto glass slides using a Shandon CytoSpin 3 (Thermo Fisher Scientific), stained with Diff-Quik (Merck), and mounted. The BALF was centrifuged to pellet cells, and the supernatant was kept for cytokine measurement.
Cell preparation and flow cytometry.
Single-cell suspensions from BAL or spleen were prepared as described previously (82) and analyzed by flow cytometry on an LSRII flow cytometer (BD Biosciences) using the following monoclonal antibodies: FITC-anti-CD45 (rat IgG2b, κ, 1:500, catalog 553080; BD Biosciences), PE-Cy-7-anti-CD11c (Armenian hamster, IgG, 1:500, catalog 25-0114-82; eBioscience), APC-e780-anti-CD11b (rat IgG2b, κ, 1:500, catalog 47-0112-82; eBioscience), biotin-anti-Ly6C (rat IgM, κ, 1:500, catalog 557359; BD Biosciences), PE-anti-Ly6G (rat IgG2a, κ, 1:500, catalog 551461; BD Biosciences), biotin-anti–c-fms (rat IgG2a, κ, 1:500, catalog 13-1152-82; eBioscience), Ter119 (rat IgG2b, κ, 1:500, catalog 553673; BD Biosciences), FITC-anti-CD71 (rat IgG1, κ, 1:500, catalog 553266; BD Biosciences), APC-e780-B220 (rat IgG2a, κ, 1:500, catalog 47-0452-82; eBioscience), and eFluor 450 anti–MHC Class II (I-A/I-E) (rat IgG2b, κ, 1:500, catalog 48-5321-82; eBioscience). FlowJo software (Windows v10, FlowJo LLC) was used to analyze acquired data. Absolute number of cells was calculated from cell counts and proportions determined by flow cytometry.
Phospho-flow cytometry method.
Single-cell suspensions from BAL were prepared and stained as described above with a few modifications as follows. For phospho-flow cytometry, cells were incubated in serum-free cell culture media alone at 37°C for 15 minutes, and then stimulated with either 10 ng/ml of G-CSF (PeproTech) or media. Cells were placed on ice and the stimulation was stopped with ice-cold medium. Phospho-flow was performed with fixation and permeabilization buffers according to the manufacturer’s protocol (BD Biosciences). FlowJo software was used to analyze acquired data. Fluorescence-minus-one control samples were included for the phospho-specific antibodies and were used to normalize fluorescence values from the stained samples. Data are presented as the ratio of the normalized fluorescence intensity to the media-treated control cells.
Immunoglobulin ELISA.
Serum immunoglobulin titers were measured by ELISA as previously described (31). Briefly, 96-well immunoplates (Nunc) were coated with anti-mouse Ig overnight, before diluted sera and standards were added to wells prior to detection with isotype-specific secondary antibodies.
Autoantibody detection using slides of HEp-2 cells.
HEp-2 slides (Immuno Concepts) were stained according to the manufacturer’s instructions. Sera were diluted 1:40 in PBS and added to the slide wells and incubated for 30 minutes at room temperature. Slides were washed in PBS for 10 minutes, incubated in goat anti-mouse IgG(H+L) conjugated to Alexa 488 (Invitrogen), rewashed in PBS, and then coverslipped in aqueous mounting medium. Stained slides were imaged on the same day with an Olympus BX51 microscope and images were captured with an NP70 camera.
Intranasal LPS challenge.
Mice (10–12 weeks old) were lightly anesthetized with isoflurane and then challenged by intranasal administration of 30 μl of E. coli LPS (10 μg/mouse, serotype O114:B4; Sigma-Aldrich). Control mice received 30 μl of DMEM. Mice were euthanized at 1, 3, or 10 days after challenge. BAL cells were collected, counted, and analyzed by flow cytometry to quantify degree and type of inflammation, and by microscopy to examine cell morphology.
qRT-PCR.
RNA was isolated from AMΦs and CD11b+Ly-6G+ neutrophils sorted from BAL and bone marrow cells using the RNeasy Kit (QIAGEN). RNA was extracted from the liver and brown adipose tissue in the same manner with a protocol adapted for tissue. After determination of quality and concentration (Nanodrop), up to 1 μg was converted to cDNA using a High-Capacity RNA-to-cDNA Kit (Life Technologies) and diluted in RNase-free water. Genes of interest were screened using prevalidated Taqman assays (Life Technologies) and the reaction mix contained 2 μl cDNA, 0.5 μl target primer, and 3.5 μl Taqman Universal PCR Mastermix diluted with RNase-free water. qRT-PCR samples were run for 40 cycles under single-plex conditions (QuantStudio 7, Life Technologies), and eukaryotic 18S rRNA (Applied Biosystems) was used as an endogenous control for all samples. For each target gene, a no-RT negative control was used to verify negligible nonspecific binding to genomic DNA. Cycle threshold (Ct) values were determined using automatic threshold analysis (QuantStudio Software) and taken as an average of triplicates. The relative expression of each target gene was calculated using the 2–ΔΔCt method.
Myeloid progenitor assay.
Hematopoietic progenitors were quantified in semisolid 0.3% agar cultures comprising 100,000 spleen cells as described previously (83). Colony formation was stimulated by M-CSF (10 ng/ml), IL-3 (10 ng/ml), GM-CSF (10 ng/ml), SCF (50 ng/ml), or G-CSF (10 ng/ml) (PeproTech). The number of colonies (>50 cells) was determined after 5–7 days in culture.
Pulse oximetry.
Cardiopulmonary parameters were measured on conscious mice using the MouseOx system with small collar clip (Starr Life Sciences). Recordings were taken every 3 seconds over 2 four-minute readings, following a 2-minute baseline measurement and averaged over the observation period.
Assessment of RVH.
The degree of RVH was assessed on H&E–stained heart sections from 12-week-old mice by measuring the ratio of the right ventricular wall area to left ventricular wall area plus septum area, or RV/(LV + S), as described previously (84).
Muscle and fat analyses.
Skeletal muscles (soleus, tibialis anterior, and gastrocnemius) from 12-week-old mice were dissected and weighed. Brown adipose tissue was collected from the anterior subcutaneous depot and gonadal white fat was dissected and weighed. Gene expression for ATGL was performed by qRT-PCR.
Micro-CT of bone.
Micro-CT of bone was performed on paraformaldehyde-fixed femora stored in 70% ethanol, from 6-week-old male mice using the SkyScan 1076 micro-CT system (Bruker) with the following settings: 9 μm voxel resolution; 0.5 mm aluminium filter; 50 kV; 220 μA; 2,300 ms exposure time; rotation 0.7°; and frame-averaging = 1. The images were reconstructed and analyzed using SkyScan Software programs NRecon (1.6.10.2), DataViewer (1.4.4), and CT Analyser (v 1.14.4.1). Femoral lengths were measured on the scanned images. The femoral trabecular and cortical regions of interest (ROIs) commenced at a distance equal to 15% or 30%, respectively, of the total femur length proximal to the distal end of the femur; for both, an ROI of 15% of the total femur length was analyzed. The lower adaptive threshold limits for trabecular and cortical analysis were equivalent to 0.35 g/mm3 and 0.73 g/mm3 calcium hydroxyapatite, respectively.
Patient samples.
Patient BALF was obtained as described previously (85). Briefly, the bronchoscope was wedged into a segmental bronchus distant from any lung lesion(s) and 20 ml of saline was instilled, retrieved, and discarded to clear the bronchoscope of bronchial secretions. A further 60–80 ml was instilled in 20-ml aliquots and retrieved via hand aspiration of the syringe. Samples were stored on ice immediately upon receipt. Cell-free BALF was obtained via centrifugation of the samples. Cells were counted, used for cytopins, and stored at –70°C for further analysis.
Statistics.
Data were analyzed by 2-tailed unpaired Student’s t test or nonparametric Mann-Whitney U test as appropriate according to normality of data when comparing 2 groups. Data involving 3 or more groups was tested by 1-way ANOVA or nonparametric ANOVA using GraphPad Prism 7 software. Multiple comparison tests were performed by Tukey’s or Dunn’s post hoc tests. A P value less than 0.05 was considered significant.
Study approval.
All animal experiments were approved by the Alfred Medical Research and Educational Precinct Animal Ethics Committee and were performed in accordance with National Health and Medical Research Council Australia guidelines for animal experimentation. Patient BAL was obtained by written informed consent and performed in accordance with the Human Research Ethics Committee of The Royal Melbourne Hospital and the National Statement on Ethical Conduct in Research Involving Humans.
Author contributions
ET, ML, CMNC, MJM, SLP, and NEM conducted experiments, acquired data, and analyzed data. DPS and LBI guided the clinical work and provided patient samples. ET, MJH, NAS, GPA, and MLH guided experiments, analyzed data, and edited the manuscript. ET, MJM, NAS, GPA, and MLH revised the final version of the manuscript. MLH conceived the project. ET and MLH wrote the complete manuscript.
Supplementary Material
Acknowledgments
The authors would like to thank Lovisa Dousha for technical assistance, Jelena Kezic for histology expertise, and members of the Alfred Medical Research and Education Precinct Flow Cytometry Core Facility, Monash Histology Platform, and Monash MicroImaging Platform for support. We are extremely grateful to Casey McLeod for providing invaluable assistance with patient records. We are also grateful for technical assistance provided by staff at the Monash Animal Research Platform. This research was supported by grants from the National Health and Medical Research Council of Australia (Project Grant APP1080274 to M.L. Hibbs and G.P. Anderson, and Research Fellowship APP0603124 to M.L. Hibbs) and a near-miss grant from the Central Clinical School, Monash University.
Version 1. 04/30/2018
Electronic publication
Version 2. 06/01/2018
Print issue publication
Footnotes
MJM’s present address is: CSL Limited, Parkville, Victoria, Australia.
SLP’s present address is: RMIT University, Bundoora, Victoria, Australia.
Conflict of interest: GPA has received advisory board and speaker honoraria from AstraZeneca/MedImmune (Gaithersburg, Maryland, USA), Boehringer Ingelheim (Ingelheim, Germany), GlaxoSmithKline (Brentford, United Kingdom [UK]), Janssen (Beerse, Belgium), Menarini (Chatswood, Australia), Mundipharma (Cambridge, UK), Novartis (Basel, Switzerland), and Pieris (Boston, Massachusetts, USA). In 2016, he undertook a sabbatical as Chief Scientist in the AstraZeneca RIA iMED drug discovery division in Cambridge, UK, funded by AstraZeneca.
Reference information: J Clin Invest. 2018;128(6):2406–2418.https://doi.org/10.1172/JCI98224.
Contributor Information
Evelyn Tsantikos, Email: Evelyn.Tsantikos@monash.edu.
Maverick Lau, Email: Maverick.Lau@monash.edu.
References
- 1.Barnes PJ. Cellular and molecular mechanisms of asthma and COPD. Clin Sci. 2017;131(13):1541–1558. doi: 10.1042/CS20160487. [DOI] [PubMed] [Google Scholar]
- 2.Anderson GP. Advances in understanding COPD. F1000Res. 2016;5(F1000 Faculty Rev):2392. doi: 10.12688/f1000research.7018.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Donnell DE. Hyperinflation, dyspnea, and exercise intolerance in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3(2):180–184. doi: 10.1513/pats.200508-093DO. [DOI] [PubMed] [Google Scholar]
- 4.Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J. 2009;33(5):1165–1185. doi: 10.1183/09031936.00128008. [DOI] [PubMed] [Google Scholar]
- 5.Sze MA, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185(10):1073–1080. doi: 10.1164/rccm.201111-2075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Budden KF, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15(1):55–63. doi: 10.1038/nrmicro.2016.142. [DOI] [PubMed] [Google Scholar]
- 7.Leung JM, et al. The role of acute and chronic respiratory colonization and infections in the pathogenesis of COPD. Respirology. 2017;22(4):634–650. doi: 10.1111/resp.13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castaldi PJ, et al. The COPD genetic association compendium: a comprehensive online database of COPD genetic associations. Hum Mol Genet. 2010;19(3):526–534. doi: 10.1093/hmg/ddp519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wain LV, et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet. 2017;49(3):416–425. doi: 10.1038/ng.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müllerova H, et al. Hospitalized exacerbations of COPD: risk factors and outcomes in the ECLIPSE cohort. Chest. 2015;147(4):999–1007. doi: 10.1378/chest.14-0655. [DOI] [PubMed] [Google Scholar]
- 11.Lewis SA, Pavord ID, Stringer JR, Knox AJ, Weiss ST, Britton JR. The relation between peripheral blood leukocyte counts and respiratory symptoms, atopy, lung function, and airway responsiveness in adults. Chest. 2001;119(1):105–114. doi: 10.1378/chest.119.1.105. [DOI] [PubMed] [Google Scholar]
- 12.Koo HK, Kang HK, Song P, Park HK, Lee SS, Jung H. Systemic white blood cell count as a biomarker associated with severity of chronic obstructive lung disease. Tuberc Respir Dis (Seoul) 2017;80(3):304–310. doi: 10.4046/trd.2017.80.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8(7):533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 14.Burgess AW, Camakaris J, Metcalf D. Purification and properties of colony-stimulating factor from mouse lung-conditioned medium. J Biol Chem. 1977;252(6):1998–2003. [PubMed] [Google Scholar]
- 15.Vlahos R, et al. Neutralizing granulocyte/macrophage colony-stimulating factor inhibits cigarette smoke-induced lung inflammation. Am J Respir Crit Care Med. 2010;182(1):34–40. doi: 10.1164/rccm.200912-1794OC. [DOI] [PubMed] [Google Scholar]
- 16.Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine. 2008;42(3):277–288. doi: 10.1016/j.cyto.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bronchud MH, et al. Phase I/II study of recombinant human granulocyte colony-stimulating factor in patients receiving intensive chemotherapy for small cell lung cancer. Br J Cancer. 1987;56(6):809–813. doi: 10.1038/bjc.1987.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dührsen U, Villeval JL, Boyd J, Kannourakis G, Morstyn G, Metcalf D. Effects of recombinant human granulocyte colony-stimulating factor on hematopoietic progenitor cells in cancer patients. Blood. 1988;72(6):2074–2081. [PubMed] [Google Scholar]
- 19.Bendall LJ, Bradstock KF. G-CSF: From granulopoietic stimulant to bone marrow stem cell mobilizing agent. Cytokine Growth Factor Rev. 2014;25(4):355–367. doi: 10.1016/j.cytogfr.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Lieschke GJ, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84(6):1737–1746. [PubMed] [Google Scholar]
- 21.Seymour JF, Lieschke GJ, Grail D, Quilici C, Hodgson G, Dunn AR. Mice lacking both granulocyte colony-stimulating factor (CSF) and granulocyte-macrophage CSF have impaired reproductive capacity, perturbed neonatal granulopoiesis, lung disease, amyloidosis, and reduced long-term survival. Blood. 1997;90(8):3037–3049. [PubMed] [Google Scholar]
- 22.Mannering SI, Zhan Y, Gilbertson B, Lieschke GJ, Cheers C. T lymphocytes from granulocyte colony-stimulating factor-/- mice produce large quantities of interferon-gamma in a chronic infection model. Immunology. 2000;101(1):132–139. doi: 10.1046/j.1365-2567.2000.00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawlor KE, et al. Critical role for granulocyte colony-stimulating factor in inflammatory arthritis. Proc Natl Acad Sci U S A. 2004;101(31):11398–11403. doi: 10.1073/pnas.0404328101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rumble JM, et al. Neutrophil-related factors as biomarkers in EAE and MS. J Exp Med. 2015;212(1):23–35. doi: 10.1084/jem.20141015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldberg GL, et al. G-CSF and neutrophils are nonredundant mediators of murine experimental autoimmune uveoretinitis. Am J Pathol. 2016;186(1):172–184. doi: 10.1016/j.ajpath.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 26.Botelho FM, et al. Innate immune processes are sufficient for driving cigarette smoke-induced inflammation in mice. Am J Respir Cell Mol Biol. 2010;42(4):394–403. doi: 10.1165/rcmb.2008-0301OC. [DOI] [PubMed] [Google Scholar]
- 27.Didon L, et al. Lung epithelial CCAAT/enhancer-binding protein-β is necessary for the integrity of inflammatory responses to cigarette smoke. Am J Respir Crit Care Med. 2011;184(2):233–242. doi: 10.1164/rccm.201007-1113OC. [DOI] [PubMed] [Google Scholar]
- 28.Hansen MJ, et al. IL-17A and serum amyloid A are elevated in a cigarette smoke cessation model associated with the persistence of pigmented macrophages, neutrophils and activated NK cells. PLoS One. 2014;9(11):e113180. doi: 10.1371/journal.pone.0113180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mo Y, Chen J, Humphrey DM, Fodah RA, Warawa JM, Hoyle GW. Abnormal epithelial structure and chronic lung inflammation after repair of chlorine-induced airway injury. Am J Physiol Lung Cell Mol Physiol. 2015;308(2):L168–L178. doi: 10.1152/ajplung.00226.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pauls SD, Marshall AJ. Regulation of immune cell signaling by SHIP1: A phosphatase, scaffold protein, and potential therapeutic target. Eur J Immunol. 2017;47(6):932–945. doi: 10.1002/eji.201646795. [DOI] [PubMed] [Google Scholar]
- 31.Maxwell MJ, Duan M, Armes JE, Anderson GP, Tarlinton DM, Hibbs ML. Genetic segregation of inflammatory lung disease and autoimmune disease severity in SHIP-1-/- mice. J Immunol. 2011;186(12):7164–7175. doi: 10.4049/jimmunol.1004185. [DOI] [PubMed] [Google Scholar]
- 32.Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP, Hibbs ML. Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. J Immunol. 2012;189(2):946–955. doi: 10.4049/jimmunol.1200660. [DOI] [PubMed] [Google Scholar]
- 33.Duan M, et al. CD11b immunophenotyping identifies inflammatory profiles in the mouse and human lungs. Mucosal Immunol. 2016;9(2):550–563. doi: 10.1038/mi.2015.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oh SY, et al. Src homology 2 domain-containing inositol 5-phosphatase 1 deficiency leads to a spontaneous allergic inflammation in the murine lung. J Allergy Clin Immunol. 2007;119(1):123–131. doi: 10.1016/j.jaci.2006.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harder KW, et al. Perturbed myelo/erythropoiesis in Lyn-deficient mice is similar to that in mice lacking the inhibitory phosphatases SHP-1 and SHIP-1. Blood. 2004;104(13):3901–3910. doi: 10.1182/blood-2003-12-4396. [DOI] [PubMed] [Google Scholar]
- 36.Helgason CD, et al. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12(11):1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hazen AL, Smith MJ, Desponts C, Winter O, Moser K, Kerr WG. SHIP is required for a functional hematopoietic stem cell niche. Blood. 2009;113(13):2924–2933. doi: 10.1182/blood-2008-02-138008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Traves SL, Culpitt SV, Russell RE, Barnes PJ, Donnelly LE. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax. 2002;57(7):590–595. doi: 10.1136/thorax.57.7.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto C, et al. Airway inflammation in COPD assessed by sputum levels of interleukin-8. Chest. 1997;112(2):505–510. doi: 10.1378/chest.112.2.505. [DOI] [PubMed] [Google Scholar]
- 40.Hunter MG, Avalos BR. Phosphatidylinositol 3′-kinase and SH2-containing inositol phosphatase (SHIP) are recruited by distinct positive and negative growth-regulatory domains in the granulocyte colony-stimulating factor receptor. J Immunol. 1998;160(10):4979–4987. [PubMed] [Google Scholar]
- 41.Liu Q, et al. SHIP is a negative regulator of growth factor receptor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 1999;13(7):786–791. doi: 10.1101/gad.13.7.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Neill SK, et al. Monophosphorylation of CD79a and CD79b ITAM motifs initiates a SHIP-1 phosphatase-mediated inhibitory signaling cascade required for B cell anergy. Immunity. 2011;35(5):746–756. doi: 10.1016/j.immuni.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elwing J, Panos RJ. Pulmonary hypertension associated with COPD. Int J Chron Obstruct Pulmon Dis. 2008;3(1):55–70. doi: 10.2147/copd.s1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeshita S, et al. SHIP-deficient mice are severely osteoporotic due to increased numbers of hyper-resorptive osteoclasts. Nat Med. 2002;8(9):943–949. doi: 10.1038/nm752. [DOI] [PubMed] [Google Scholar]
- 45.Agustí A, et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS ONE. 2012;7(5):e37483. doi: 10.1371/journal.pone.0037483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maeda K, Mehta H, Drevets DA, Coggeshall KM. IL-6 increases B-cell IgG production in a feed-forward proinflammatory mechanism to skew hematopoiesis and elevate myeloid production. Blood. 2010;115(23):4699–4706. doi: 10.1182/blood-2009-07-230631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bozinovski S, et al. Serum amyloid a is a biomarker of acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(3):269–278. doi: 10.1164/rccm.200705-678OC. [DOI] [PubMed] [Google Scholar]
- 48.Bozinovski S, et al. Serum amyloid A opposes lipoxin A4 to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2012;109(3):935–940. doi: 10.1073/pnas.1109382109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarkar M, Bhardwaj R, Madabhavi I, Khatana J. Osteoporosis in chronic obstructive pulmonary disease. Clin Med Insights Circ Respir Pulm Med. 2015;9:5–21. doi: 10.4137/CCRPM.S22803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sly LM, Rauh MJ, Kalesnikoff J, Song CH, Krystal G. LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity. 2004;21(2):227–239. doi: 10.1016/j.immuni.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 51.Masubuchi T, et al. Smoke extract stimulates lung epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Pathol. 1998;153(6):1903–1912. doi: 10.1016/S0002-9440(10)65704-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashitani J, et al. Granulocyte-colony stimulating factor levels in bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis. Thorax. 1999;54(11):1015–1020. doi: 10.1136/thx.54.11.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aggarwal A, Baker CS, Evans TW, Haslam PL. G-CSF and IL-8 but not GM-CSF correlate with severity of pulmonary neutrophilia in acute respiratory distress syndrome. Eur Respir J. 2000;15(5):895–901. doi: 10.1034/j.1399-3003.2000.15e14.x. [DOI] [PubMed] [Google Scholar]
- 54.Abboud MR, et al. Elevated serum and bronchoalveolar lavage fluid levels of interleukin 8 and granulocyte colony-stimulating factor associated with the acute chest syndrome in patients with sickle cell disease. Br J Haematol. 2000;111(2):482–490. doi: 10.1046/j.1365-2141.2000.02358.x. [DOI] [PubMed] [Google Scholar]
- 55.Emad A, Emad Y. Increased granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) levels in BAL fluid from patients with sulfur mustard gas-induced pulmonary fibrosis. J Aerosol Med. 2007;20(3):352–360. doi: 10.1089/jam.2007.0590. [DOI] [PubMed] [Google Scholar]
- 56.Sinden NJ, Stockley RA. Proteinase 3 activity in sputum from subjects with alpha-1-antitrypsin deficiency and COPD. Eur Respir J. 2013;41(5):1042–1050. doi: 10.1183/09031936.00089712. [DOI] [PubMed] [Google Scholar]
- 57.Cockayne DA, et al. Systemic biomarkers of neutrophilic inflammation, tissue injury and repair in COPD patients with differing levels of disease severity. PLoS ONE. 2012;7(6):e38629. doi: 10.1371/journal.pone.0038629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eagan TM, et al. Neutrophil gelatinase-associated lipocalin: a biomarker in COPD. Chest. 2010;138(4):888–895. doi: 10.1378/chest.09-2718. [DOI] [PubMed] [Google Scholar]
- 59.Azoulay E, Attalah H, Harf A, Schlemmer B, Delclaux C. Granulocyte colony-stimulating factor or neutrophil-induced pulmonary toxicity: myth or reality? Systematic review of clinical case reports and experimental data. Chest. 2001;120(5):1695–1701. doi: 10.1378/chest.120.5.1695. [DOI] [PubMed] [Google Scholar]
- 60.D’Souza A, Jaiyesimi I, Trainor L, Venuturumili P. Granulocyte colony-stimulating factor administration: adverse events. Transfus Med Rev. 2008;22(4):280–290. doi: 10.1016/j.tmrv.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 61.Hsu AC, et al. Targeting PI3K-p110α suppresses influenza virus infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;191(9):1012–1023. doi: 10.1164/rccm.201501-0188OC. [DOI] [PubMed] [Google Scholar]
- 62.Liu G, et al. Fibulin-1 regulates the pathogenesis of tissue remodeling in respiratory diseases. JCI Insight. 2016;1(9):e86380. doi: 10.1172/jci.insight.86380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2013;48(5):531–539. doi: 10.1165/rcmb.2012-0492TR. [DOI] [PubMed] [Google Scholar]
- 64.Stroncek DF, Njoroge JM, Procter JL, Childs RW, Miller J. A preliminary comparison of flow cytometry and tube agglutination assays in detecting red blood cell-associated C3d. Transfus Med. 2003;13(1):35–41. doi: 10.1046/j.1365-3148.2003.00415.x. [DOI] [PubMed] [Google Scholar]
- 65.Wolff J, Brendel C, Fink L, Bohle RM, Kissel K, Bux J. Lack of NB1 GP (CD177/HNA-2a) gene transcription in NB1 GP- neutrophils from NB1 GP-expressing individuals and association of low expression with NB1 gene polymorphisms. Blood. 2003;102(2):731–733. doi: 10.1182/blood-2002-09-2831. [DOI] [PubMed] [Google Scholar]
- 66.Hogg JC, Doerschuk CM. Leukocyte traffic in the lung. Annu Rev Physiol. 1995;57:97–114. doi: 10.1146/annurev.ph.57.030195.000525. [DOI] [PubMed] [Google Scholar]
- 67.Maxwell MJ, Yuan Y, Anderson KE, Hibbs ML, Salem HH, Jackson SP. SHIP1 and Lyn kinase negatively regulate integrin alpha IIb beta 3 signaling in platelets. J Biol Chem. 2004;279(31):32196–32204. doi: 10.1074/jbc.M400746200. [DOI] [PubMed] [Google Scholar]
- 68.Rizkallah J, Man SFP, Sin DD. Prevalence of pulmonary embolism in acute exacerbations of COPD: a systematic review and metaanalysis. Chest. 2009;135(3):786–793. doi: 10.1378/chest.08-1516. [DOI] [PubMed] [Google Scholar]
- 69.Cavaillès A, et al. Comorbidities of COPD. Eur Respir Rev. 2013;22(130):454–475. doi: 10.1183/09059180.00008612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer. 2002;2(11):862–871. doi: 10.1038/nrc927. [DOI] [PubMed] [Google Scholar]
- 71.King DA, Cordova F, Scharf SM. Nutritional aspects of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2008;5(4):519–523. doi: 10.1513/pats.200707-092ET. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou P, Kitaura H, Teitelbaum SL, Krystal G, Ross FP, Takeshita S. SHIP1 negatively regulates proliferation of osteoclast precursors via Akt-dependent alterations in D-type cyclins and p27. J Immunol. 2006;177(12):8777–8784. doi: 10.4049/jimmunol.177.12.8777. [DOI] [PubMed] [Google Scholar]
- 73.Ganesan LP, et al. FcgammaR-induced production of superoxide and inflammatory cytokines is differentially regulated by SHIP through its influence on PI3K and/or Ras/Erk pathways. Blood. 2006;108(2):718–725. doi: 10.1182/blood-2005-09-3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ngoh EN, et al. Activity of SHIP, which prevents expression of interleukin 1β, is reduced in patients with Crohn’s disease. Gastroenterology. 2016;150(2):465–476. doi: 10.1053/j.gastro.2015.09.049. [DOI] [PubMed] [Google Scholar]
- 75.Takamatsu Y, Simmons PJ, Moore RJ, Morris HA, To LB, Lévesque JP. Osteoclast-mediated bone resorption is stimulated during short-term administration of granulocyte colony-stimulating factor but is not responsible for hematopoietic progenitor cell mobilization. Blood. 1998;92(9):3465–3473. [PubMed] [Google Scholar]
- 76.Winkler IG, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116(23):4815–4828. doi: 10.1182/blood-2009-11-253534. [DOI] [PubMed] [Google Scholar]
- 77.Winkler IG, et al. Hematopoietic stem cell mobilizing agents G-CSF, cyclophosphamide or AMD3100 have distinct mechanisms of action on bone marrow HSC niches and bone formation. Leukemia. 2012;26(7):1594–1601. doi: 10.1038/leu.2012.17. [DOI] [PubMed] [Google Scholar]
- 78.Christopher MJ, Link DC. Granulocyte colony-stimulating factor induces osteoblast apoptosis and inhibits osteoblast differentiation. J Bone Miner Res. 2008;23(11):1765–1774. doi: 10.1359/jbmr.080612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Q, et al. The inositol polyphosphate 5-phosphatase ship is a crucial negative regulator of B cell antigen receptor signaling. J Exp Med. 1998;188(7):1333–1342. doi: 10.1084/jem.188.7.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kitamura D, Rajewsky K. Targeted disruption of mu chain membrane exon causes loss of heavy-chain allelic exclusion. Nature. 1992;356(6365):154–156. doi: 10.1038/356154a0. [DOI] [PubMed] [Google Scholar]
- 81.Knudsen L, Weibel ER, Gundersen HJ, Weinstein FV, Ochs M. Assessment of air space size characteristics by intercept (chord) measurement: an accurate and efficient stereological approach. J Appl Physiol. 2010;108(2):412–421. doi: 10.1152/japplphysiol.01100.2009. [DOI] [PubMed] [Google Scholar]
- 82.Maxwell MJ, et al. SHIP-1 deficiency in the myeloid compartment is insufficient to induce myeloid expansion or chronic inflammation. Genes Immun. 2014;15(4):233–240. doi: 10.1038/gene.2014.9. [DOI] [PubMed] [Google Scholar]
- 83.Hibbs ML, et al. Mice lacking three myeloid colony-stimulating factors (G-CSF, GM-CSF, and M-CSF) still produce macrophages and granulocytes and mount an inflammatory response in a sterile model of peritonitis. J Immunol. 2007;178(10):6435–6443. doi: 10.4049/jimmunol.178.10.6435. [DOI] [PubMed] [Google Scholar]
- 84.Paddenberg R, et al. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir Res. 2007;8:15. doi: 10.1186/1465-9921-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Radhakrishna N, Farmer M, Steinfort DP, King P. A comparison of techniques for optimal performance of bronchoalveolar lavage. J Bronchology Interv Pulmonol. 2015;22(4):300–305. doi: 10.1097/LBR.0000000000000201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.