

Abstract
Thiazolidinediones (TZDs) are the only antidiabetic drugs that reverse insulin resistance. They have been a valuable asset in the treatment of type 2 diabetes, but their side effects have curtailed widespread use in the clinic. In this issue of the JCI, Kraakman and colleagues provide evidence that deacetylation of the nuclear receptor PPARγ improves the therapeutic index of TZDs. These findings should revitalize the quest to employ insulin sensitization as a first-line approach to managing type 2 diabetes.
Targeting PPARγ provides hope for insulin sensitivity
Elevated insulin levels in the face of hyperglycemia, the sine qua non of insulin resistance and type 2 diabetes, have been recognized since Berson and Yalow first measured insulin in plasma of patients with maturity-onset diabetes (1). Hence, we have known for almost four decades that insulin therapy does not get at the root of the problem, yet the treatment of type 2 diabetes continues to rely largely on increasing insulin levels, for example, by stimulation of endogenous secretion with incretin therapy or by pharmacological dosing of insulin itself. Thus, the development of thiazolidinediones (TZDs), drugs that improve insulin sensitivity, was heralded as a breakthrough in the early 1990s (2). Twenty-five years later, TZDs are much less widely prescribed than in their heyday due to bona fide as well as perceived toxicities (3). In this issue of the JCI, Kraakman et al. provide hope that PPARγ, the main biological target of TZDs, can be targeted to sensitize patients to insulin while ameliorating toxicity of this class of drugs (4).
PPARγ is a member of the nuclear receptor superfamily of ligand-responsive transcription factors (5). The recognition that PPARγ is selectively expressed in fat cells (6, 7) and that TZDs stimulate adipocyte differentiation (8) converged in the discovery that TZDs function as high-affinity PPARγ ligands (9), establishing a link between PPARγ and insulin sensitivity. Genetic linkage of PPARγ to type 2 diabetes comes both from rare variants with powerful effects inherited in Mendelian fashion (10) and from a common single nucleotide polymorphism with small and variable effects (11). By the late 1990s, there was great optimism for conquering type 2 diabetes with TZD medications that bind to PPARγ, whose function as a nuclear receptor was reassuring, since many safe and effective drugs have related targets, including fibrates that act on PPARα (12).
Tarnished reputation of PPARγ
Unfortunately, a variety of factors drove PPAR from a promising therapeutic target of TZDs to one with a tarnished reputation. Some of the supposed side effects of TZDs were either overblown or idiosyncratic, most notably a putative increase in cardiovascular mortality attributed to the TZD drug rosiglitazone that has been dismissed by the FDA in recent years (13). Moreover, two large trials published 11 years apart demonstrate that pioglitazone significantly reduces cardiovascular mortality (14, 15). However, reproducible side effects. such as edema and bone loss, are more problematic, and prescriptions for TZDs have plummeted in recent years (16).
The fact remains that no other class of drugs improves insulin sensitivity as TZDs do. Can these benefits be salvaged? The salutary effects of TZDs have a strong genetic influence that is in part related to single nucleotide polymorphisms that affect PPARγ binding to the genome (17). Indeed, not all patients have metabolic improvement with TZDs, and only a minority of patients suffer from side effects, such as edema (3). Thus, a better understanding of the pharmacogenetics of TZD risks and benefits may allow a personalized approach to therapy.
Other nuclear receptors offer clues
Perhaps lessons can be learned from other prescription drugs that target nuclear receptors. In some cases, these medications are used to treat hormone deficiencies, such as hypothyroidism. Endocrinologists know that thyroid hormone must be carefully replaced, aiming for the normal physiological range, because the principle of “too much of a good thing” leads to hyperthyroid symptoms, including untoward effects on heart, bone, and volume status. Although the physiological ligand for PPARγ remains a mystery, perhaps applying this principle to TZD therapy would improve the therapeutic index of TZDs.
Another successful approach to targeting other nuclear receptors has been to develop pharmaceuticals that bind in unique ways, leading to context-dependent biological effects. This class includes selective estrogen receptor modulators (SERMs) that bind to different conformations of estrogen receptor (ER) and thereby recruit coregulatory molecules that mediate tissue-specific regulation of gene expression (18). For example, tamoxifen acts as an ER antagonist in breast cancer, where it has beneficial antitumor activity, but functions as an ER agonist in bone where a full antagonist would lead to bone loss (19). The success with SERMs has spurred attempts to develop analogous selective PPAR modulators (SPPARMs), which may or may not be chemically related to TZDs and function, depending on gene and cell type, as full agonists, partial agonists, or antagonists (20).
The improved therapeutic index of some SPPARMs may derive from submaximal agonism that results directly from relatively weak binding affinity for PPARγ (21). However, other SPPARMs with high affinity for PPARγ must work by other mechanisms. New light has been shed on this by recent advances in understanding posttranslational modifications (PTMs) of PPARγ. The first well-studied PPARγ PTM was its serine phosphorylation on residue 112 of fat-specific isoform PPARγ2 (S84 in the PPARγ1 isoform), which reduced PPARγ activity (22, 23). In addition, the binding of TZDs as well as non-TZD ligands has been shown to reduce phosphorylation of PPARγ on S273 (24). New chemical entities targeting this phosphorylation event activate a subset of genes associated with toxicity, while retaining insulin sensitization, and thus could be promising clinical candidates (25). Another therapeutic strategy would be to selectively target the activity of kinases modulating PPARγ phosphorylation. PPARγ activity is also regulated by other PTMs, including sumoylation and ubiquitinylation, which have been reviewed elsewhere (26).
Prevention of PPARγ acetylation mitigates TZD side effects
The focus of the paper by Kraakman et al. is on the regulation of PPARγ activity by acetylation (4). This group previously reported that PPARγ is acetylated on lysine residues 268 and 293 (K268/K293) and that sirtuin-mediated deacetylation of these sites was associated with selective induction of brown adipocyte genes and repression of visceral white adipose genes, leading to insulin sensitization (27). In the present work, the authors utilize the power of mouse genetics to generate an elegant knockin mouse model in which PPARγ residues K268 and K293 have been mutated to arginine, a related amino acid that is not subject to the reversible pathways that modulate lysine acetylation. These 2KR mice were found to respond to rosiglitazone with the same beneficial metabolic effects seen in wild-type mice, including improvement of glucose tolerance, insulin resistance, and hepatic steatosis. Importantly, however, the 2KR mice were protected from clinically problematic side effects associated with TZD treatment, including bone loss and fluid retention. One caveat of the study is that, in the absence of TZD treatment, the 2KR mice exhibited browning of their white adipose tissue and resistance to diet-induced obesity compared with control mice, which made the study of TZD treatment difficult to control for potential confounding effects of differences in body weight. The authors did their best to control for this by studying a cohort of 2KR mice that was weight matched with a control group and found that the mutant mice responded as well or better to TZD.
Concluding remarks
The ability of PPARγ acetylation to shift the balance between the beneficial and adverse effects of TZDs raises new hope for targeting PPARγ in the clinic (Figure 1). Sirtuin activators are being developed for clinical use (28) and could in principle be combined with TZDs or with non-TZD PPARγ ligands that have an improved safety profile on their own. Moreover, the mechanism by which PPARγ becomes acetylated remains to be explored. If this is enzymatic, then it could be possible to develop selective acetylation inhibitors that improve the therapeutic index of PPARγ ligands. It is exciting to imagine harnessing the insulin-sensitizing power of PPARγ ligands without the accursed side effects that currently limit their clinical use.
Figure 1. Shifting the balance of benefits and risks of TZD therapy.
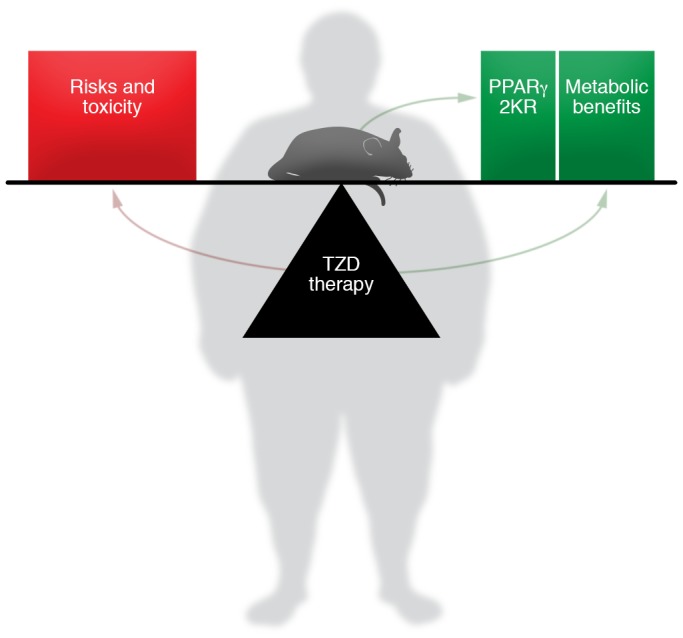
Kraakman et al. (4) show that mice with the PPARγ-2KR mutation that prevents acetylation maintain the benefit of TZD therapy with fewer risks. If this work translates to humans, it may lead to the resurgence of TZD therapy via drugs that deacetylate PPARγ. It also may be possible to develop SPPARMs that achieve this goal by stabilizing the conformation of deacetylated PPARγ.
Acknowledgments
Work on PPARγ in the author’s laboratory is supported by NIH grant R01DK49780 and the JPB Foundation.
Version 1. 05/14/2018
Electronic publication
Version 2. 06/01/2018
Print issue publication
Footnotes
Conflict of interest: MAL serves as a member of scientific advisory boards for Pfizer and Lilly.
Reference information: J Clin Invest. 2018;128(6):2202–2204. https://doi.org/10.1172/JCI121392.
See the related article at PPARγ deacetylation dissociates thiazolidinedione’s metabolic benefits from its adverse effects.
References
- 1.Yalow RS, Berson SA. Plasma insulin concentrations in nondiabetic and early diabetic subjects. Determinations by a new sensitive immuno-assay technic. Diabetes. 1960;9:254–260. doi: 10.2337/diab.9.4.254. [DOI] [PubMed] [Google Scholar]
- 2.Hofmann CA, Colca JR. New oral thiazolidinedione antidiabetic agents act as insulin sensitizers. Diabetes Care. 1992;15(8):1075–1078. doi: 10.2337/diacare.15.8.1075. [DOI] [PubMed] [Google Scholar]
- 3.Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014;20(4):573–591. doi: 10.1016/j.cmet.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kraakman MJ, et al. PPARγ deacetylation dissociates thiazolidinedione’s metabolic benefits from its adverse effects. J Clin Invest. 2018;128(6):2600–2612. doi: 10.1172/JCI98709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lazar MA. Maturing of the nuclear receptor family. J Clin Invest. 2017;127(4):1123–1125. doi: 10.1172/JCI92949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chawla A, Schwarz EJ, Dimaculangan DD, Lazar MA. Peroxisome proliferator-activated receptor (PPAR)γ: adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology. 1994;135(2):798–800. doi: 10.1210/endo.135.2.8033830. [DOI] [PubMed] [Google Scholar]
- 7.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPARγ2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8(10):1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- 8.Kletzien RF, Clarke SD, Ulrich RG. Enhancement of adipocyte differentiation by an insulin-sensitizing agent. Mol Pharmacol. 1992;41(2):393–398. [PubMed] [Google Scholar]
- 9.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPARγ) J Biol Chem. 1995;270(22):12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 10.Barroso I, et al. Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402(6764):880–883. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- 11.Altshuler D, et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26(1):76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 12.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5(12):993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 13. FDA Drug Safety Communication: FDA eliminates the Risk Evaluation and Mitigation Strategy (REMS) for rosiglitazone-containing diabetes medicines. FDA website. https://www.fda.gov/Drugs/DrugSafety/ucm476466.htm Published December 16, 2015. Accessed April 21, 2018.
- 14.Dormandy JA, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366(9493):1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 15.Kernan WN, et al. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med. 2016;374(14):1321–1331. doi: 10.1056/NEJMoa1506930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma M, Nazareth I, Petersen I. Trends in incidence, prevalence and prescribing in type 2 diabetes mellitus between 2000 and 2013 in primary care: a retrospective cohort study. BMJ Open. 2016;6(1):e010210. doi: 10.1136/bmjopen-2015-010210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soccio RE, et al. Targeting PPARγ in the epigenome rescues genetic metabolic defects in mice. J Clin Invest. 2017;127(4):1451–1462. doi: 10.1172/JCI91211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295(5564):2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 19.Komm BS, Mirkin S. An overview of current and emerging SERMs. J Steroid Biochem Mol Biol. 2014;143:207–222. doi: 10.1016/j.jsbmb.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Balint BL, Nagy L. Selective modulators of PPAR activity as new therapeutic tools in metabolic diseases. Endocr Metab Immune Disord Drug Targets. 2006;6(1):33–43. doi: 10.2174/187153006776056620. [DOI] [PubMed] [Google Scholar]
- 21.Reginato MJ, et al. A potent antidiabetic thiazolidinedione with unique peroxisome proliferator-activated receptor gamma-activating properties. J Biol Chem. 1998;273(49):32679–32684. doi: 10.1074/jbc.273.49.32679. [DOI] [PubMed] [Google Scholar]
- 22.Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science. 1996;274(5295):2100–2103. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- 23.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem. 1997;272(8):5128–5132. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- 24.Choi JH, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by Cdk5. Nature. 2010;466(7305):451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi JH, et al. Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477(7365):477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Floyd ZE, Stephens JM. Controlling a master switch of adipocyte development and insulin sensitivity: covalent modifications of PPARγ. Biochim Biophys Acta. 2012;1822(7):1090–1095. doi: 10.1016/j.bbadis.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiang L, et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell. 2012;150(3):620–632. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinclair DA, Guarente L. Small-molecule allosteric activators of sirtuins. Annu Rev Pharmacol Toxicol. 2014;54:363–380. doi: 10.1146/annurev-pharmtox-010611-134657. [DOI] [PMC free article] [PubMed] [Google Scholar]