

Abstract
It has become clear that the standard cartoon, in which macromolecular particles prepared for electron cryo-microscopy are shown to be surrounded completely by vitreous ice, often is not accurate. In particular, the standard picture does not include the fact that diffusion to the air-water interface, followed by adsorption and possibly denaturation, can occur on the time scale that normally is required to make thin specimens. The extensive literature on interaction of proteins with the air-water interface suggests that many proteins can bind to the interface, either directly or indirectly via a sacrificial layer of already-denatured protein. In the process, the particles of interest can, in some cases, become preferentially oriented, and in other cases they can be damaged and/or aggregated at the surface. Thus, although a number of methods and recipes have evolved for dealing with protein complexes that prove to be difficult, making good cryo-grids can still be a major challenge for each new type of specimen. Recognition that the air-water interface is a very dangerous place to be has inspired work on some novel approaches for preparing cryo-grids. At the moment, two of the most promising ones appear to be: (1) thin and vitrify the specimen much faster than is done currently or (2) immobilize the particles onto a structure-friendly support film so that they cannot diffuse to the air-water interface.
Keywords: Interfacial adsorption, preferential orientation, protein denaturation, affinity support films
Graphical Abstract
1. INTRODUCTION
The requirements are quite demanding for preparing thin specimens of randomly disbursed biological macromolecules that can be used for high-resolution electron microscopy [1, 2]. Ideally, these vitrified, aqueous specimens should be no more than 100 nm in thickness, and possibly as thin as 20 nm or 30 nm. Such thin specimens are required because, among other reasons, the mean free path for inelastic scattering [3] is estimated to be about 350 nm or less for high-energy (300 keV) electrons. In addition, the macromolecular particles must remain fully hydrated after being inserted into the vacuum of the electron microscope. The most practical way to maintain a well-hydrated state has proven to be to put a few μL of sample onto a thin, holey film, supported on a fine-mesh, 3 mm diameter metal grid, and then blot off excess sample with filter paper. This is usually done in an environment of controlled temperature and humidity, in order to minimize evaporation of the remaining water. The resulting, thin sample then is rapidly quenched to low temperature; for further detail see [4] and for historical background see [5].
A very simple picture has been used for decades to explain why blotting and subsequent quenching results in nearly ideal specimens, at least some of the time. As is illustrated in Figure 1, macromolecular particles are imagined to be embedded within a vitrified layer of buffer. According to this picture, the spatial distribution, orientation and structure of the macromolecules are expected to be identical to what they previously were in bulk solution, unperturbed by the process of making and freezing the thin film. If every grid were as shown in this picture, regardless of what protein complex was used, then all of them would give superb images. Many samples do, in fact, give superb results in electron microscopy, thus leading to the widely-held belief that the standard picture shown in Figure 1 is, indeed, correct.
Figure 1.

Cartoon showing the standard picture that is envisioned in order to explain why embedding macromolecular complexes within a thin film of vitrified buffer should preserve the structure in a near-native state. Macromolecular particles are randomly distributed in the sample when on a holey support film, just as they were in the test tube. When everything above the dotted line is blotted away, a thin film remains in the hole. This thin film is then vitrified by plunging into cryogen, leaving the particles embedded in amorphous ice.
Many macromolecules, however, prove to be difficult to prepare in the form of single-particle cryo-EM specimens (referred to here as cryo-grids), leading one to doubt whether the standard picture is always correct. As a result, an effort has begun to develop more sophisticated models, which take into account the fact that the required thin, aqueous films have a very high surface-to-volume ratio in the brief moment before vitrification. Such models take into account the fact that macromolecular particles can–indeed must–diffuse and collide with the air-water interface, where they may adsorb and even possibly denature before vitrification occurs. Other consequences include preferential orientation of particles, –or in some – cases the number of particles seen does not correspond to their concentration in bulk. Although numerous cautionary remarks about these hazards were, in fact, made in Section 6.6 of the review by Dubochet et al. [6], and more recently in a retrospective by Taylor and Glaeser [7], little has yet been done to address the issue in a systematic way.
The primary goal in this Opinion is to critically examine, on the basis of the known behavior of proteins at air-water interfaces, what might be wrong with what has been the standard picture of single-particle cryo-EM specimens, which is shown in Figure 1. The heart of this critique is presented in Section 4, preceded first by a limited review (Section 2) of some of the literature showing that many proteins do adsorb to air-water interfaces, followed by a similarly short review (Section 3) of work showing that many proteins do, in fact, denature shortly after they have first been adsorbed. Finally, the critique of what is wrong with the standard picture is followed, in Section 5, by a discussion of alternative approaches that are being taken for preparing cryo-EM specimens, all of which can be seen as ways to address unwanted adsorption of particles to the air-water interface.
2. MANY PROTEINS ARE KNOWN TO ADSORB TO AIR-WATER INTERFACES
2.1 Adsorption progresses in stages
Adsorption of proteins to the air-water interface has historically been pictured to progress through at least three distinct steps. A review published in 1950, for example, spoke of an initial adsorption of proteins at the interface “in the globular form”, followed by “unrolling of the peptide chains at the interface”, and subsequent “aggregation of the unrolled chains into a coagulum” [8]. Later, it seemed perhaps self-evident to include a step in which additional proteins bind to the layer of denatured-protein at the air-water interface, after which further denaturation and aggregation might follow [9]. A contrary view has long been presented in the literature, however, at least for some proteins [10]. For example, [11] concluded that preferential orientation and some structural deformation of bovine serum albumin may occur, but nevertheless there is no denaturation.
In either event, the first step involves structurally intact particles colliding with and sticking to the interface. Initial adhesion to a clean air-water interface presumably involves dewetting of individual hydrophobic side chains or even small hydrophobic patches, both of which normally exist on the surfaces of native proteins. This initial-adsorption step can be diffusion limited, i.e. the activation energy for binding to the (hydrophobic) interface may be very small, and thus the sticking coefficient (the number of times that particles stick, relative to the number of times that they impinge upon an interface) can be close to 1.0. If that is the case, the initial rate of adsorption is expected to be proportional to the bulk concentration of the particles.
The second step, at least when it does occur, involves partial or complete unfolding of the native-protein structure at the interface. In some cases the thickness of the resulting protein monolayer is estimated to be less than 2 nm [12, 13]. Unfolding of the native structure at an air-water interface is imagined to involve a rapid, step-by-step movement of hydrophobic residues from the interior of a protein to air, while still leaving the hydrophilic residues on the aqueous side of the interface. As is discussed in Section 3.1, the energy landscape of protein unfolding at interfaces is thus expected to trend monotonically downhill, interrupted only by small activation barriers as the reaction progresses, which is very different from what it is in bulk.
The third step envisioned in this review, as mentioned above, involves the adsorption of additional, structurally intact proteins, possibly via a mixture of hydrophilic and hydrophobic interactions with the pre-existing layer of denatured proteins. Since the binding of proteins to hydrophilic surfaces is often much weaker than it is to hydrophobic surfaces [14–16], the sticking coefficient may be much lower for a denatured-protein monolayer than it is for a pristine air-water interface.
The fourth step envisioned here involves a process of structural remodeling of the second “layer” of proteins that adsorbed in step 3, which is discussed in the previous paragraph. This reorganization can lead to significant changes in the viscosity and elasticity of material previously adsorbed to the interface, [17–19]. If thin, i.e. “two dimensional” aggregates of material are observed instead of randomly dispersed single particles, it may be that adsorption to and reorganization on a denatured monolayer is the reason. Historically, these changes in viscosity and elasticity have generally been reported to occur more slowly than the first three steps, however. As a result, this process may or may not happen on the time scale typically used to make cryo-grids, depending upon the specific protein in question.
2.2 Adsorption has predictable consequences for cryo-specimens
The potential consequences of the first two steps, when thinking about what might happen when cryo-grids are made, have been described previously in panels C through F of Figure 5 in [7]. In some cases, the particles of interest were imagined to remain intact, but it is also possible that intact protein complexes might be seen in only one or a few preferred orientations. In other cases, it was envisioned that individual domains might become structurally damaged when in contact with the air-water interface, and in still other cases no intact particles might be seen because a completely denatured-protein monolayer had been formed.
If formation of a denatured-protein monolayer occurs rapidly on the time scale of thinning and vitrification, as experiments to be described in Section 3 suggest, but structurally intact particles are nevertheless seen in cryo-EM images, it may be that these are mostly bound to a monolayer of denatured protein as opposed to the air-water interface. Experiments reported by [20] present a clear example of such behavior. Electron microscopy was used to demonstrate that a continuous membrane of denatured apoferritin was formed within one second of when the protein solution, at a concentration of 1 mg/mL, first touched the air-water interface. After waiting 1 minute, a few intact ferritin molecules became stuck to this membrane, and the number continued to grow with time, eventually becoming so numerous that 2-D crystalline arrays were formed. A similar, but less thoroughly documented behavior was also reported for 20S proteasome particles.
The use of cryo-EM tomography is one way to establish whether particles of interest are bound at the interface. Preferential orientation is another indication that particles are bound in some way to the interface rather than being freely suspended in solution. Adsorption to the interface is also implicated whenever the number of particles seen per unit area exceeds the number that is present in a thin slab of the initial sample. Calculated values for the number of particles expected in the projection of an 80 nm thick sample are given in Figure 12 of [21], for a range of sample concentrations and particle sizes. As an example, if the particle size is 1 MDa and the sample concentration is 0.5 mg/mL, the average spacing between particles should be 100 nm. When the particles are seen to be almost in close contact to one another, as is the case for two examples shown in the Supplementary Information of [22], the spacing between particles is clearly much less than it was in the initial sample. Further, if the number of particles seen increases with how long the sample is incubated on the grid, prior to blotting, the most likely explanation is that they bind to and accumulate at the air-water interface. The other alternative is that the bulk concentration increases due to evaporation, but this is itself quite worrisome.
It is not uncommon that some types of large, macromolecular complexes do not remain intact and/or they form aggregated material when confined to the thin layer of sample left after blotting. In these cases one must consider that major structural changes may have occurred after adsorption of intact particles, possibly even adsorption to a monolayer of already denatured proteins. Other alternatives are considered in Section 4, below, but remodeling of a ”second” layer of bound protein is consistent with the historical picture that binding and unfolding does not always stop with the first layer of denatured protein.
3. DENATURATION OF ADSORBED PROTEINS CAN BE VERY FAST
Returning in more detail to the issue of unfolding of proteins at the air-water interface, formation of a denatured-protein monolayer can be a very fast process, limited–as was historically appreciated [9, 23] – only by the rate at which proteins can diffuse to the interface. At a concentration of 1 mg/mL, for example, which is typical of the values used to make cryo-grids, there is enough protein within 1 or 2 μm of the air-water interface to form such a monolayer. It takes only a fraction of a second for protein molecules to diffuse that short a distance, as is explained in Section 4.1. Rapid formation of a denatured monolayer is thus likely to occur for protein concentrations that are commonly used to make cryo-grids.
A simple way to measure how rapidly proteins can form a denatured monolayer first emerged from a related effort to measure the thickness of such layers. The latter measurement required that a known amount of protein be applied to the surface of a Langmuir trough, and that all of the protein was transferred to the surface rather than some of it becoming dispersed into the sub phase solution. A method to achieve the desired, quantitative transfer was first developed by Trurnit, who arranged to have the protein flow down the surface of a glass road as a thin “curtain” before it reached the trough [24]. Under these conditions, he found that trypsin, human serum albumin, and human gamma globulin were all quantitatively (>99%) transferred to the air-water interface within a few seconds when the thickness of the curtain was only 10 μm. As expected, the time required was correlated with the protein’s diffusion constant. In addition, somewhat longer times, up to 10 s, were required for 99% transfer when the thickness of the curtain was increased to 14 μm.
In a more recent experiment, which used time-resolved X-ray reflectivity to observe protein unfolding at the air-water interface, [25] concluded that “… lysozyme molecules initially adsorbed at an air-water interface unfold within 1 s”, i.e. faster than the time-resolution of the experiment. In addition, molecular dynamics simulations of lysozyme molecules placed in contact with a hydrophobic surface, in this case graphite rather than air, suggest that unfolding to an ensemble of partially or even completely spread states may actually happen within one or a few nanoseconds [26].
Rapid denaturation of proteins, once they collide with the air-water interface, implies that the activation barrier for unfolding must be much smaller than what it is in bulk solution, where unfolding is normally a rare event. This was already recognized by [8], for example, who represented the reaction diagram for denaturation and aggregation at the air-water interface as a series of monotonically decreasing steps in free energy, separated by small activation barriers between each step. In a perhaps more modern view, [27] represented the hypothetical reaction pathway by a 1-dimensional, “rough” energy landscape in which individual, local energy barriers were similar to what they are for the unfolding pathway in bulk solution. These relatively small barriers were imagined, for example, to represent structural transitions of “foldons”, i.e. independent folding units much smaller than a domain. Unlike the case in bulk solution, however, the free-energy landscape at the air-water interface was imagined to decrease monotonically. This picture is similar to the results obtained in the molecular dynamics simulations of [26], cited above, except that these reflected just the internal energy component and did not include the entropy component.
Whether creation of a denatured-protein monolayer is universally a fast process cannot be said, however, since kinetic experiments on denaturation at the air-water interface have focused on a limited set of readily available proteins. Furthermore, few of these experiments have been concerned with the rate at which a denatured protein monolayer is formed at high protein concentration (e.g. 1 mg/mL).
4. CRITIQUE: WHAT IS WRONG WITH THE STANDARD PICTURE?
The standard picture shown in Figure 1 claims that biological macromolecules are preserved in a state that faithfully represents what the sample looked like in the test tube. Since, according to this picture, nothing harmful could happen to the particles when cryo-grids are made, it was assumed by some to be the biochemist’s fault if the sample on the grid was not usable for high-resolution structural studies. Even the observation of preferred orientation of particles was often thought to be due to particles having an asymmetric shape, and thus being forced to become oriented within the confined volume between two air-water interfaces. This “passive orientation” picture was not questioned, since there is little doubt that orientation in the confined space happens for filamentous particles such as Tobacco Mosaic Virus, microtubules, or actin filaments.
On the other hand, what if the sample in the test tube really is in very good condition, but yet it is found to be unusable when on the grid? In that case there would have to be something wrong with the standard picture. Once this possibility is admitted, it is obvious that interaction with the air-water interface should have been included as part of the picture. Indeed, biochemists would not intentionally bubble and foam their samples – i.e. create thin layers with an extremely high surface-to-volume ratio. Creating such a thin layer, however, is exactly what microscopists must do to the samples.
4.1 The standard picture ignores diffusion and collision with the air-water interface
The very first thing that can be wrong with the standard picture is that it does not reflect the fact that, prior to vitrification, macromolecules can diffuse and collide with any nearby air-water interface. In particular, as is shown in Figure 2, this must happen at the air-water interface that is created when the sample is first applied over the open holes on the grid, long before blotting and thinning begins. The cartoon applies only to cases when the aliquot of sample stays on one side of the grid, of course, which often is the case–but not always.
Figure 2.

Cartoon showing – not to scale – the two air-water interfaces that exist when an aliquot of sample is deposited onto a holey support film. The individual hole sizes in the thin film are typically about 1 μm, while the ~3 μL aliquot deposited onto the grid typically covers a diameter of 3 mm. The interface at the top of the drop is usually ignored because it presumably will be blotted away, along with excess sample. The second interface, on the bottom, i.e. within the holes, is seldom discussed, and it is more complicated to say whether this second interface will also be blotted away. Either the top interface or the bottom interface presumably remains, however, when preferential orientation is observed, and especially whenever the number of particles seen is greater than is expected, as is discussed in Section 2.2.
Secondly, the standard picture does not consider how quickly particles collide with the air-water interface, assuming that they start out only micrometers, or less, from such an interface. The time to reach the interface can easily be estimated using the equation
| Equation 1 |
where t is the time needed to diffuse a mean-squared distance, 〈χ2〉; q =2,4, or 6 depending upon whether diffusion occurs in 1, 2, or 3 dimensions, respectively; and D is the diffusion coefficient of the particle. As an example, if D =10μm2/ (a reasonable value for a mega Dalton sized particle), the time to diffuse a distance of 1 μm in three dimensions is less than 1 second, and the time to diffuse a distance of 100 nm, the thickness of useable areas left after blotting, is 100 times less than that.
4.2 The standard picture has ignored discrepancies in particle concentration relative to the initial sample
There is growing awareness that adsorption to the air-water interface may have occurred in cases when the observed number of particles is far greater than expected. As mentioned in Section 2.2, examples for the expected number of particles can be found in Figure 12 of [21]. Increasing the number of structurally intact particles by adsorption to the air-water interface can actually be beneficial, as is also true for adsorption to a continuous support film. This is because, in the absence of interfacial adsorption, the number of particles in an image may be far less than desired.
In addition, it now is well accepted that adsorption to the air-water interface is responsible for the unwanted, preferential orientation of particles mentioned in Section 2.2, especially when it is clear that this is not due to the shape of the particle and the small thickness of the vitrified ice in which it is embedded. Preferential orientation can prevent one from getting a high-resolution, 3-D reconstruction [28], unless it is possible to tilt the specimen to high angle and still obtain high-resolution images, as has been done by [29]. It thus is desirable to fully understand why preferential orientation happens. It is worth considering not just a model in which particles are adsorbed directly to the air-water interface, possibly with little structural damage, but also a model in which a denatured-protein monolayer is first formed, which then serves as a kind of support film, as is imagined in Figure 3.
Figure 3.

Cartoon showing healthy particles adsorbed to a sacrificial skin of denatured protein. It is hypothesized that the first particles to collide with the air-water interface form a denatured monolayer, perhaps 1 nm to 2 nm thick. Structurally intact particles (may) then stick to the monolayer, sometimes reaching a much higher concentration than in bulk. When everything above the dotted line is blotted away, the remaining thin film is quenched by plunging into cryogen, leaving the particles embedded in vitreous ice.
4.3 Other mechanisms for causing particle damage have also been ignored
Other mechanisms have been mentioned for how particles might be damaged, and these, too, are not reflected in the standard picture. These include (1) shear forces might damage the particles as excess buffer is drawn from the grid during blotting; (2) evaporation of water after blotting might change the buffer composition enough to cause damage; or (3) something harmful might leach from filter paper [30]. Of these three, it seems unlikely that harmful material is released from the filter paper. The first two suggestions deserve further comment, however.
Shear forces cannot be avoided during the brief period during which excess sample is blotted from the grid, estimated to be as short as 100 ms [31]. Shearing forces often appear to be big enough to cause flow-induced orientation of filamentous macromolecular assemblies such as tobacco mosaic virus (TMV), microtubules and actin filaments. While orientation per se of such filamentous structures is not necessarily a problem, the forces can be big enough to also stretch and even break the filaments [32]. Unfortunately, it is not clear how to model the shearing forces in order to estimate their magnitude theoretically. In one attempt to do so, Zheng et al. [31] assumed that blotting is done through the holes of a support film. They suggested that the maximum gradient in flow velocity (referred to as the “shear rate”) would be between 104 and 106 s−1. This value is still well below the value of 107 s−1 that is expected to damage small, globular proteins [33, 34] or, by extension, individual protein domains. Less certain, however, is whether the shear force generated during blotting might strip off subunits from large complexes or otherwise damage flexible macromolecular complexes. In this regard it is important that optimizing buffer conditions in order to enhance the thermodynamic stability in bulk solution is expected to protect a particle from being damaged by shear [33]. The same should also be true for cross-linking with a bifunctional reagent.
While a small amount of evaporation of water can probably be tolerated by most samples, some will be more sensitive than others to the resulting increase in ionic strength. In the most extreme case, however, complete evaporation might occur in areas of a grid that were especially thin to begin with. This will necessarily remove the bulk water that normally surrounds the particles, and possibly begin to remove the more tightly bound “structural” water, even though the ambient humidity is kept high. With some experience, it may be possible for one to avoid areas that have dried out, but in other cases the situation may be too ambiguous to tell. In spite of these hazards, evaporation should not be a problem as long as the ambient humidity is kept as close as possible to 100 %, given the practical limitations of tools to measure and maintain high humidity; the grid and tweezers are at the same temperature as (or lower than) the ambient atmosphere; and the grid is plunged into cryogen as soon as possible after retracting the filter paper.
5. WHAT OPTIIONS ARE AVAILABLE WHEN SPECIMENS NEED TO BE IMPROVED?
This section is concerned only with those hypothetical cases in which interaction of macromolecular particles with the air-water interface causes preferential orientation or even damages their structure in some way. Structural damage sustained during isolation and purification, while always a concern, is assumed at the moment to not be in question. As was acknowledged at the end of Section 4.3, there can also be other reasons why specimens that are perfectly good in the test tube end up as not being usable on the grid. Once again, for the sake of discussion, it is assumed that these also are not a problem.
Table 1 identifies six different approaches that can be tried in order to protect particles from becoming damaged by interaction with the air-water interface. The underlying concepts can be grouped into the following: (1) stabilize the structure in solution such that the particle is less likely to unfold; (2) block the air-water interface with a surfactant, thus making it more difficult for the particle to adsorb to the interface; (3) apply, thin and then quench the sample rapidly enough to outrun the adsorption and/or denaturation process; and (4) immobilize the particles on a structure-friendly support film in order to prevent them from diffusing to and interacting with the air-water interface.
Table 1.
Approaches that have been identified as possible ways to improve the quality of cryo-grids for specimens that have proven to be “difficult”. In each case, examples of ways to implement a given approach are provided, and comments are made about caveats and known weak points of each.
| APPROACHES | EXAMPLES | COMMENTS |
|---|---|---|
| Stabilize the structure by optimizing the buffer conditions |
|
|
| Stabilize macromolecular complexes by chemical crosslinking |
|
|
| Minimize interaction with the air-water interface by adding a pre-emptive, structure-friendly surfactant |
|
|
| Minimize interaction by ultrafast thinning and quenching |
|
|
| Adsorption to carbon (or other) films to prevent diffusion to the air-water interface |
|
|
| Immobilization onto structure- friendly affinity grids |
|
|
5.1 Optimizing structural stability in bulk solution
It has been shown experimentally that the relative surface activity of various proteins correlates with their stability in solution [35]. Optimizing the buffer conditions in order to improve the chance of success in making cryo-EM specimens [36] thus seems well worth trying. Two examples of how the thermodynamic stability can be optimized are: (1) the addition of so-called “stabilizing cosolutes”, such as glycerol or trehalose or (2) optimization of pH, ionic strength, and ionic composition of the buffer for each type of particle. Stabilization of the structure in bulk solution is not certain to reduce binding to the air-water interface, of course, and thus preferred orientation–if present–may persist. Nor is it certain to reduce the danger of denaturing at the air-water interface if binding does occur, since the energy landscape for unfolding is expected to be completely different between bulk and the interface (see Section 2). Unfortunately, some buffer additives may be impractical to use for making cryo-EM samples, even though they may be optimal for the structure of the particle. An example is glycerol, which greatly slowed denaturation of apoferritin at the air-water interface [20], but which is generally avoided in cryo-EM because it causes increased bubbling and beam-induced motion.
Covalent cross-linking of macromolecular complexes can be an orthogonal way to stabilize the structure of a particle in bulk solution. It is well-established that cross-linking can make it possible to prepare cryo-EM grids of particles that otherwise were not usable [37–39]. Polymerase II pre-initiation complexes [40, 41], human 26S proteasomes [42], and pre-catalytic spliceosomes [43] are three examples in which cross-linking led to successful high-resolution structure determinations. Even though cross-linking can sometimes make it possible to prepare otherwise difficult particles, that may not always be the case. Among the issues to be aware of, cross-linking might not be expected to prevent binding to the interface in a preferred orientation, nor is it certain to prevent subsequent denaturation at the interface. In this regard, it is worth pointing to the example of lysozyme, a small protein with four internal disulfide crosslinks, which is rapidly denatured at the air-water interface [23]. Other issues to be aware of are: (1) cross-linking may permanently trap off-pathway conformations, even if they would otherwise be rare, and (2) reaction with a high mole-ratio of bifunctional cross-linker is likely to change the surface charge of a particle, since the reagent will react with all lysine residues, whether they are cross-linked or not.
5.2 Blocking the air-water surface with a surfactant
Passivating the air-water interface with a monolayer of surfactant seems to be another good thing to do. Macromolecules are expected to have only weak interactions with polar head groups, except in specific cases where they contain a natural ligand for the particle. The rate at which proteins interact with the air-water interface can be slowed considerably by first applying a phospholipid monolayer to the air-water interface [9, 13]. The rate of protein adsorption nevertheless depends, as might be expected, on the surface pressure of the lipid monolayer [44]. In fact, added detergent is known to be effective in preventing preferential orientation of some types of particles [2, 45], and fluorinated Fos-choline-8 has been found to be a useful additive for a number of specimens [46], but added detergent does not always solve the problems that occur in preparing “difficult” samples. One shortcoming of adding detergents or other surfactants to samples may be that the surface pressure of the resulting monolayer may still not be high enough to prevent the particles of interest from pushing the surfactant molecules to one side, thereby penetrating the monolayer and binding to the interface.
As indicated both in Section 2 and in Section 4, a denatured-protein monolayer also acts as a surfactant. Such a sacrificial layer might then bind additional copies of the protein, which may or may not remain structurally intact. As long as a cryo-grid shows randomly oriented, structurally intact particles, it is of little practical importance to determine whether a denatured-protein monolayer is first formed, as is imagined in Figure 3. In other words, it is only important to consider that the standard picture, shown in Figure 1, is wrong when it fails to explain why there are only few well-preserved particles, or why the particles show preferential orientation.
5.3 Thin and quench the sample faster than adsorption can occur
A third way to prevent labile particles from becoming damaged is to thin and quench the sample very rapidly. The idea here is to outrun the process of interacting with the air-water interface, and thus to actually achieve the condition envisioned in the standard picture, i.e. Figure 1. The fastest method developed so far combines a novel, “self-blotting” type of grid [47] with the Spotiton technology [48] for delivering sample volumes as small as tens of pL. If this or other technology can be developed to the point where interaction with the air-water interface is out-run, it will be necessary to use high sample concentrations in order to have the desired number of particles per unit area in the EM images. This is because the number of particles in an image may be less than desired, even when using a concentration of 1 mg/mL, a point made previously in Section 4.
5.4 Immobilize particles on a structure-friendly support film
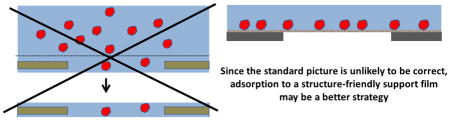
Another approach to improving how specimens are made is to avoid altogether the chance of there being unwanted interactions between particles and the air-water interface. This can be done by immobilizing particles on an appropriate, structure-friendly support film. Care must be taken, of course, to record images only in areas where the thickness of the remaining, vitrified buffer solution is greater than the diameter of the particles. This is because unwanted contact between immobilized particles and the air-water interface can still occur if the sample becomes too thin, as is schematically shown, in Figure 4, to almost be about to happen. If the binding affinity is high, adsorption to a support film has the additional advantage that the number of particles seen in images can be quite high, even when the solution concentration is as low as tens of nM. While this approach has potential for becoming a method that works for nearly every type of specimen, and to do so nearly 100% of the time, achieving that goal still requires further development. At present, three different types of support film are being used.
Figure 4.

Immobilized particles can still be contacted by the air-water interface if the remaining buffer is too thin. Although use of affinity support films may provide a path to reliably prepare cryo-grids for every type of specimen, a remaining problem is to find a way to keep the air-water interface from touching the immobilized particles. As the green-colored interface indicates, the situation currently is safe, but further thinning, as suggested by the arrows, may not be a good thing.
Continuous carbon support films
Evaporated carbon film, made hydrophilic by exposure to a glow discharge at low vacuum, is currently the standard support film used to make specimens. Evaporated carbon films can also be chemically functionalized in a better-characterized way than is provided by glow-discharge treatment [49]. While using evaporated carbon films has worked well for some particles, it still is not effective for others. Perhaps a sub-microscopic, patchy-mosaic of hydrophilic and hydrophobic areas remains on the surface after exposure to a glow-discharge plasma or other chemical modifications. In any event, the resulting surfaces are not satisfactory for all types of macromolecules. In addition, the structural noise of a thin-carbon support film is believed to become unacceptable for smaller particles.
Graphene-based support films
For these reasons attention has recently turned to using single-atom thick graphene oxide [50–52] or hydrogen-plasma treated graphene [53] as a support film. Graphene still has some worrisome unknowns, however. While graphene oxide flakes are fully hydrophilic, the chemical nature and distribution of oxygen adducts on the surface are still not well characterized. As is true for evaporated-carbon films exposed to a glow discharge, it also is not yet known whether graphene oxide surfaces consist of a submicroscopic, patchy-mosaic of hydrophilic and hydrophobic areas. The same concerns may also be an issue for plasma-treated graphene. It thus remains to be determined how general it is that either can be a useful support film for macromolecules that otherwise had been difficult to prepare for cryo-EM.
Affinity support films with known biochemical functionality
Various types of biochemical-affinity grids are currently being investigated, with the intent that they would serve as structure-friendly support films for immobilizing particles. At present there are at least three types. (1) Monolayers of Ni-NTA derivatized phospholipid picked up on graphene oxide [54] or on holey-carbon support films (optionally backed with evaporated carbon) [55, 56]. These are intended for use with his-tagged versions of macromolecules of interest. (2) Antibodies adsorbed to evaporated films of carbon [57, 58], which provide an alternative way to pull down specific macromolecules. The antibodies themselves are either adsorbed non-specifically or protein A is adsorbed nonspecifically and then antibodies are bound to the immobilized protein A. (3) Streptavidin monolayer-crystals, which were used by [59, 60] to pull down membrane proteins incorporated into biotinylated liposomes [61].
Many other ways to use streptavidin affinity grids exist, of course, including decoration of the monolayer crystals with biotinylated DNA, which then pulls down DNA-binding proteins [62]; use of genetic tags (e.g. streptavidin binding peptide or AviTag™); and random biotinylation of lysine residues on the surface of any purified macromolecule [63]. Streptavidin monolayer crystals offer a unique advantage because they are expected to be sensitive to dewetting. As a result, the resolution shown by the monolayer crystal can indicate whether the specimen is well hydrated, and thus the particles of interest are still likely to be well preserved [64]
Since affinity grids are based on well characterized and trusted biochemical methods, it is expected that immobilization onto such surfaces will carry few risks to the native structure of the particle. Nevertheless, current issues with affinity grids include (1) the possibility that preferred orientation may be a problem for tagged proteins or when using monoclonal antibodies, and (2) structural noise from the (affinity) support film may be greater than it is when particles are suspended in open holes, without any support film. On the other hand, if particles in open holes are adsorbed to a denatured monolayer of protein anyway, as proposed in Figure 3, this monolayer, too, will contribute structural noise, not unlike that of an ultrathin carbon support film.
CONCLUSIONS
Surface-induced denaturation, dissociation, and aggregation of biological macromolecules at the air-water interface – possibilities that have long been recognized in other contexts – may occur rapidly after a sample is deposited onto an EM grid and before it is vitrified. This possibility has not been adequately accounted for in the standard picture of what thin specimens look like. While the standard picture is consistent with results obtained for some specimens, it does not give any indication why preparing cryo-grids fails for other specimens. By adding the fact that particles may bind to the air-water interface, however, one can explain why chemical cross-linking or inclusion of surfactants sometimes makes it possible to prepare high-quality cryo-grids of otherwise “difficult” specimens. Recognition of the value of completely avoiding, rather than just mitigating, interactions with the air-water interface has led to the further development of novel approaches for preparing cryo-grids. One such approach is to rapidly apply, thin, and quench the sample, effectively outrunning unwanted interaction with the air-water interface. Another is to immobilize the sample on a structure-friendly support film, using, for example, binding interactions based on known biochemical functionality.
Highlights.
The standard picture of cryo-EM specimens is often inaccurate.
In many cases, particles of interest adopt a preferred orientation within the specimen.
Denaturation is also expected to play a significant role when making cryo-EM specimens.
Immobilization onto support films can avoid unwanted interactions with the air-water interface.
Care must still be taken to not thin the aqueous sample too much.
Acknowledgments
Many colleagues have contributed important information and suggestions before and during the period in which this review was written. In this regard I especially want to thank Prof. Bridget Carragher, Prof. Ed Egelman, Dr. Rafael Fernandez-Leiro, Dr. Bong-Yoon Han, Dr. Richard Henderson, Prof. Eva Nogales, and Prof. Holger Stark. This work was supported in part by NIH grant GM051487.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Passmore LA, Russo CJ. Specimen Preparation for High-Resolution Cryo-EM. In: Crowther RA, editor. Resolution Revolution: Recent Advances in Cryoem. 2016. pp. 51–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng Y, Grigorieff N, Penczek PA, Walz T. A Primer to Single-Particle Cryo-Electron Microscopy. Cell. 2015;161:438–49. doi: 10.1016/j.cell.2015.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grimm R, Typke D, Barmann M, Baumeister W. Determination of the inelastic mean free path in ice by examination of tilted vesicles and automated most probable loss imaging. Ultramicroscopy. 1996;63:169–79. doi: 10.1016/0304-3991(96)00035-6. [DOI] [PubMed] [Google Scholar]
- 4.Dobro MJ, Melanson LA, Jensen GJ, McDowall AW. Plunge freezing for electron cryomicroscopy. In: Jensen GJ, editor. Methods in Enzymology, Vol 481: Cryo-Em, Part a - Sample Preparation and Data Collection. 2010. pp. 63–82. [DOI] [PubMed] [Google Scholar]
- 5.Dubochet J. A Reminiscence about Early Times of Vitreous Water in Electron Cryomicroscopy. Biophysical Journal. 2016;110:756–7. doi: 10.1016/j.bpj.2015.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, et al. Cryo-Electron Microscopy of Vitrified Specimens. Quarterly Reviews of Biophysics. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- 7.Taylor KA, Glaeser RM. Retrospective on the early development of cryoelectron microscopy of macromolecules and a prospective on opportunities for the future. Journal of Structural Biology. 2008;163:214–23. doi: 10.1016/j.jsb.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cumper CWN, Alexander AE. The surface chemistry of proteins. Transactions of the Faraday Society. 1950;46:235–53. [Google Scholar]
- 9.James LK, Augenstein LG. Adsorption of enzymes at interfaces - film formation and effect on activity. Advances in Enzymology and Related Areas of Molecular Biology. 1966;28:1–40. doi: 10.1002/9780470122730.ch1. [DOI] [PubMed] [Google Scholar]
- 10.Wierenga PA, Egmond MR, Voragen AGJ, de Jongh HH. The adsorption and unfolding kinetics determines the folding state of proteins at the air-water interface and thereby the equation of state. Journal of Colloid and Interface Science. 2006;299:850–7. doi: 10.1016/j.jcis.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 11.Lu JR, Su TJ, Thomas RK. Structural conformation of bovine serum albumin layers at the air-water interface studied by neutron reflection. Journal of Colloid and Interface Science. 1999;213:426–37. doi: 10.1006/jcis.1999.6157. [DOI] [PubMed] [Google Scholar]
- 12.Postel C, Abillon O, Desbat B. Structure and denaturation of adsorbed lysozyme at the air-water interface. Journal of Colloid and Interface Science. 2003;266:74–81. doi: 10.1016/s0021-9797(03)00571-x. [DOI] [PubMed] [Google Scholar]
- 13.Gidalevitz D, Huang ZQ, Rice SA. Protein folding at the air-water interface studied with x-ray reflectivity. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2608–11. doi: 10.1073/pnas.96.6.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramsden JJ. Experimental methods for investigating protein adsorption kinetics at surfaces. Quarterly Reviews of Biophysics. 1994;27:41–105. doi: 10.1017/s0033583500002900. [DOI] [PubMed] [Google Scholar]
- 15.Seigel RR, Harder P, Dahint R, Grunze M, Josse F, Mrksich M, et al. On-line detection of nonspecific protein adsorption at artificial surfaces. Analytical Chemistry. 1997;69:3321–8. doi: 10.1021/ac970047b. [DOI] [PubMed] [Google Scholar]
- 16.Gray JJ. The interaction of proteins with solid surfaces. Current Opinion in Structural Biology. 2004;14:110–5. doi: 10.1016/j.sbi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 17.Wierenga PA, Kosters H, Egmond MR, Voragen AGJ, de Jongh HHJ. Importance of physical vs. chemical interactions in surface shear rheology. Advances in Colloid and Interface Science. 2006;119:131–9. doi: 10.1016/j.cis.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Petkov JT, Gurkov TD, Campbell BE, Borwankar RP. Dilatational and shear elasticity of gel-like protein layers on air/water interface. Langmuir. 2000;16:3703–11. [Google Scholar]
- 19.Martin AH, Grolle K, Bos MA, Stuart MA, van Vliet T. Network forming properties of various proteins adsorbed at the air/water interface in relation to foam stability. Journal of Colloid and Interface Science. 2002;254:175–83. doi: 10.1006/jcis.2002.8592. [DOI] [PubMed] [Google Scholar]
- 20.Yoshimura H, Scheybani T, Baumeister W, Nagayama K. 2-dimensional protein array growth in thin layers of protein solution on aqueous subphases. Langmuir. 1994;10:3290–5. [Google Scholar]
- 21.Vinothkumar KR, Henderson R. Single particle electron cryomicroscopy: trends, issues and future perspective. Quarterly Reviews of Biophysics. 2016;49:1–25. doi: 10.1017/S0033583516000068. [DOI] [PubMed] [Google Scholar]
- 22.Herzik MA, Jr, Wu M, Lander GC. Achieving better-than-3-Å resolution by single-particle cryo-EM at 200 keV. Nature Methods. 2017;14:1075. doi: 10.1038/nmeth.4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graham DE, Phillips MC. Proteins at liquid interfaces .1. Kinetics of adsorption and surface denaturation. Journal of Colloid and Interface Science. 1979;70:403–14. [Google Scholar]
- 24.Trurnit HJ. A theory and method for the spreading of protein monolayers. Journal of Colloid Science. 1960;15:1–13. [Google Scholar]
- 25.Yano YF, Arakawa E, Voegeli W, Matsushita T. Real-time investigation of protein unfolding at an air-water interface at the 1 s time scale. Journal of Synchrotron Radiation. 2013;20:980–3. doi: 10.1107/S0909049513023741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raffaini G, Ganazzoli F. Protein adsorption on a hydrophobic surface: A molecular dynamics study of lysozyme on graphite. Langmuir. 2010;26:5679–89. doi: 10.1021/la903769c. [DOI] [PubMed] [Google Scholar]
- 27.Glaeser RM, Han B-G. Opinion: hazards faced by macromolecules when confined to thin aqueous films. Biophysics Reports. 2017;3:1–7. doi: 10.1007/s41048-016-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naydenova K, Russo CJ. Measuring the effects of particle orientation to improve the efficiency of electron cryomicroscopy. Nature Communications. 2017;8:629. doi: 10.1038/s41467-017-00782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan YZ, Baldwin PR, Davis JH, Williamson JR, Potter CS, Carragher B, et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nature Methods. 2017;14:793–6. doi: 10.1038/nmeth.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnold SA, Albiez S, Bieri A, Syntychaki A, Adaixo R, McLeod RA, et al. Blotting-free and lossless cryo-electron microscopy grid preparation from nanoliter-sized protein samples and single-cell extracts. Journal of Structural Biology. 2017;197:220–6. doi: 10.1016/j.jsb.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Zheng Y, Lin Z, Zakin JL, Talmon Y, Davis HT, Scriven LE. Cryo-TEM imaging the flow-induced transition from vesicles to threadlike micelles. Journal of Physical Chemistry B. 2000;104:5263–71. [Google Scholar]
- 32.Galkin VE, Orlova A, Vos MR, Schroder GF, Egelman EH. Near-Atomic Resolution for One State of F-Actin. Structure. 2015;23:173–82. doi: 10.1016/j.str.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaspe J, Hagen SJ. Do protein molecules unfold in a simple shear flow? Biophysical Journal. 2006;91:3415–24. doi: 10.1529/biophysj.106.089367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maa YF, Hsu CC. Protein denaturation by combined effect of shear and air-liquid interface. Biotechnology and Bioengineering. 1997;54:503–12. doi: 10.1002/(SICI)1097-0290(19970620)54:6<503::AID-BIT1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 35.Razumovsky L, Damodaran S. Surface activity-compressibility relationship of proteins at the air-water interface. Langmuir. 1999;15:1392–9. [Google Scholar]
- 36.Chari A, Haselbach D, Kirves J-M, Ohmer J, Paknia E, Fischer N, et al. ProteoPlex: stability optimization of macromolecular complexes by sparse-matrix screening of chemical space. Nat Meth. 2015;12:859–65. doi: 10.1038/nmeth.3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Southworth DR, Agard DA. Client-Loading Conformation of the Hsp90 Molecular Chaperone Revealed in the Cryo-EM Structure of the Human Hsp90:Hop Complex. Molecular Cell. 2011;42:771–81. doi: 10.1016/j.molcel.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kastner B, Fischer N, Golas MM, Sander B, Dube P, Boehringer D, et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nature Methods. 2008;5:53–5. doi: 10.1038/nmeth1139. [DOI] [PubMed] [Google Scholar]
- 39.Stark H. Grafix: Stabilization of fragile macromolecular complexes for single particle cryo-EM. In: Jensen GJ, editor. Methods in Enzymology, Vol 481: Cryo-Em, Part a - Sample Preparation and Data Collection. 2010. pp. 109–26. [DOI] [PubMed] [Google Scholar]
- 40.He Y, Yan C, Fang J, Inouye C, Tjian R, Ivanov I, et al. Near-atomic resolution visualization of human transcription promoter opening. Nature. 2016;533:359–65. doi: 10.1038/nature17970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plaschka C, Hantsche M, Dienemann C, Burzinski C, Plitzko J, Cramer P. Transcription initiation complex structures elucidate DNA opening. Nature. 2016;533:353–8. doi: 10.1038/nature17990. [DOI] [PubMed] [Google Scholar]
- 42.Haselbach D, Schrader J, Lambrecht F, Henneberg F, Chari A, Stark H. Long-range allosteric regulation of the human 26S proteasome by 20S proteasome-targeting cancer drugs. Nature Communications. 2017:8. doi: 10.1038/ncomms15578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plaschka C, Lin P-C, Nagai K. Structure of a pre-catalytic spliceosome. Nature. 2017;546:617–21. doi: 10.1038/nature22799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Macritchie F, Alexander AE. Kinetics of adsorption of proteins at interfaces .2. Role of pressure barriers in adsorption. Journal of Colloid Science. 1963;18:453–7. [Google Scholar]
- 45.Fernandez-Leiro R, Conrad J, Yang JC, Freund SMV, Scheres SHW, Lamers MH. Self-correcting mismatches during high-fidelity DNA replication. Nature Structural & Molecular Biology. 2017;24:140–3. doi: 10.1038/nsmb.3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson ZL, Chen J. Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell. 2017;168:1075–85. e9. doi: 10.1016/j.cell.2017.01.041. [DOI] [PubMed] [Google Scholar]
- 47.Razinkov I, Dandey VP, Wei H, Zhang Z, Melnekoff D, Rice WJ, et al. A new method for vitrifying samples for cryoEM. Journal of Structural Biology. 2016;195:190–8. doi: 10.1016/j.jsb.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jain T, Sheehan P, Crum J, Carragher B, Potter CS. Spotiton: A prototype for an integrated inkjet dispense and vitrification system for cryo-TEM. Journal of Structural Biology. 2012;179:68–75. doi: 10.1016/j.jsb.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Llaguno MC, Xu H, Shi L, Huang N, Zhang H, Liu QH, et al. Chemically functionalized carbon films for single molecule imaging. Journal of Structural Biology. 2014;185:405–17. doi: 10.1016/j.jsb.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bokori-Brown M, Martin TG, Naylor CE, Basak AK, Titball RW, Savva CG. Cryo-EM structure of lysenin pore elucidates membrane insertion by an aerolysin family protein. Nature Communications. 2016;7:11293. doi: 10.1038/ncomms11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pantelic RS, Meyer JC, Kaiser U, Baumeister W, Plitzko JM. Graphene oxide: A substrate for optimizing preparations of frozen-hydrated samples. Journal of Structural Biology. 2010;170:152–6. doi: 10.1016/j.jsb.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 52.Boland A, Martin TG, Zhang ZG, Yang J, Bai XC, Chang LF, et al. Cryo-EM structure of a metazoan separase-securin complex at near-atomic resolution. Nature Structural & Molecular Biology. 2017;24:414–8. doi: 10.1038/nsmb.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Russo CJ, Passmore LA. Controlling protein adsorption on graphene for cryo-EM using low-energy hydrogen plasmas. Nature Methods. 2014;11:649–52. doi: 10.1038/nmeth.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benjamin CJ, Wright KJ, Bolton SC, Hyun SH, Krynski K, Grover M, et al. Selective Capture of Histidine-tagged Proteins from Cell Lysates Using TEM grids Modified with NTA-Graphene Oxide. Scientific Reports. 2016:6. doi: 10.1038/srep32500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelly DF, Dukovski D, Walz T. Monolayer purification: A rapid method for isolating protein complexes for single-particle electron microscopy. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4703–8. doi: 10.1073/pnas.0800867105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelly DF, Dukovski D, Walz T. A practical guide to the use of monolayer purification and affinity grids. Methods in Enzymology, Vol 481: Cryo-Em, Part a - Sample Preparation and Data Collection. 2010:83–107. doi: 10.1016/S0076-6879(10)81004-3. [DOI] [PubMed] [Google Scholar]
- 57.Yu GM, Vago F, Zhang DS, Snyder JE, Yan R, Zhang C, et al. Single-step antibody-based affinity cryo-electron microscopy for imaging and structural analysis of macromolecular assemblies. Journal of Structural Biology. 2014;187:1–9. doi: 10.1016/j.jsb.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu G, Li K, Jiang W. Antibody-based affinity cryo-EM grid. Methods. 2016;100:16–24. doi: 10.1016/j.ymeth.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang LG, Ounjai P, Sigworth FJ. Streptavidin crystals as nanostructured supports and image-calibration references for cryo-EM data collection. Journal of Structural Biology. 2008;164:190–8. doi: 10.1016/j.jsb.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang LG, Sigworth FJ. Liposomes on a streptavidin crystal: a system to study membrane proteins by cryo-EM. In: Jensen GJ, editor. Methods in Enzymology, Vol 481: Cryo-Em, Part a - Sample Preparation and Data Collection. 2010. pp. 147–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang LG, Sigworth FJ. Structure of the BK potassium channel in a lipid membrane from electron cryomicroscopy. Nature. 2009;461:292–5. doi: 10.1038/nature08291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crucifix C, Uhring M, Schultz P. Immobilization of biotinylated DNA on 2-D streptavidin crystals. Journal of Structural Biology. 2004;146:441–51. doi: 10.1016/j.jsb.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 63.Han BG, Walton RW, Song A, Hwu P, Stubbs MT, Yannone SM, et al. Electron microscopy of biotinylated protein complexes bound to streptavidin monolayer crystals. Journal of Structural Biology. 2012;180:249–53. doi: 10.1016/j.jsb.2012.04.025. [DOI] [PubMed] [Google Scholar]
- 64.Han B-G, Watson Z, Cate JHD, Glaeser RM. Monolayer-crystal streptavidin support films provide an internal standard of cryo-EM image quality. Journal of Structural Biology. 2017;200:307–13. doi: 10.1016/j.jsb.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]