

Abstract
Purpose of review
With the advent of the genome-wide association study (GWAS), our understanding of the genetics of addiction has made significant strides forward. Here, we summarize genetic loci containing variants identified at genome-wide statistical significance (P<5×10−8) and independently replicated, review evidence of functional or regulatory effects for GWAS-identified variants, and outline multi-omics approaches to enhance discovery and characterize addiction loci.
Recent findings
Replicable GWAS findings span 11 genetic loci for smoking, eight loci for alcohol, and two loci for illicit drugs combined and include missense functional variants and noncoding variants with regulatory effects in human brain tissues traditionally viewed as addiction-relevant (e.g., prefrontal cortex [PFC]) and, more recently, tissues often overlooked (e.g., cerebellum).
Summary
GWAS have discovered several novel, replicable variants contributing to addiction. Using larger samples sizes from harmonized datasets and new approaches to integrate GWAS with multiple ‘omics data across human brain tissues holds great promise to significantly advance our understanding of the biology underlying addiction.
Keywords: GWAS, omics, brain, nicotine/smoking, alcohol, drugs
Introduction
Addiction is a chronic, relapsing disease that alters the brain’s reward circuitry and consequently leads to compulsive drug seeking and other behavioral changes. The long-lasting biological effects of drug exposure cause a multitude of adverse effects throughout the body. Despite these well-known health consequences and widespread public health campaigns to curb use of addictive drugs, prevalence remains high. Among individuals aged 12 and older in the U.S. in 2015, an estimated 30.2 million (11.3%) smoked cigarettes daily in the past month; 15.7 million (5.9%) had an alcohol use disorder and 7.7 million (2.9%) had an illicit drug use disorder in the past year [1]. Individuals with addiction often have strong desires to quit, but rates of successful treatment and recovery are low. For example, among adult U.S. smokers during 2015, an estimated 68% wanted to quit, 55% had made a quit attempt in the past year, but only 7% had recently quit [2].
Addiction to drugs of abuse follows a recurring cycle, with each of the three stages being driven by a major neurobiological circuit: basal ganglia (including ventral tegmental area and nucleus accumbens) for the binge/intoxication stage; extended amygdala and habenula for the withdrawal/negative affect stage; and prefrontal cortex (PFC), insula, and allocortex for the preoccupation/anticipation (craving) stage [3]. This framework has expanded as knowledge of the complex neurocircuitry of addiction has evolved [3, 4].
Inter-individual differences in neurobiological circuits, due to genetic variation and their downstream effects, alter susceptibility to developing addiction. Although addiction is multifactorial, heritability estimates have indicated that around 40%–60% of the population variability in becoming addicted to nicotine, alcohol, or illicit drugs is attributable to genetic factors [3, 5]. Some genetic factors may influence an overarching susceptibility to developing addiction; thus, their effects are shared across different drugs of abuse. In contrast, other genetic factors may underlie susceptibility to developing specific drug addictions. To pinpoint the genetic factors underlying addiction, variants selected from biologically plausible candidate genes were long-studied in hypothesis-driven studies, but genetic variant associations were not firmly established until the advent of the agnostic, hypothesis-generating genome-wide association study (GWAS) approach. As seen across the field of complex human disease genetics, the potential of GWAS to make novel discoveries continues today with ever increasing sample sizes and statistical power, denser genomic coverage, and phenotype harmonization. This review outlines GWAS reports to date for nicotine, alcohol, or other drug addictions and focuses on common variants (mostly, single nucleotide polymorphisms [SNPs]) with robust evidence for association (i.e., identified at the standard genome-wide significance threshold [P<5×10−8] and replicated in an independent dataset).
GWAS for cigarette smoking
The earliest GWAS in the addiction field was conducted for nicotine dependence [6, 7], implicating nicotinic acetylcholine receptor genes on chromosomes 15q25 (CHRNA5-CHRNA3-CHRNB4) and 8p11 (CHRNB3-CHRNA6), which have since been firmly established in other studies of nicotine dependence and related smoking phenotypes (Table 1). To date, at least 26 GWAS analyses (largest N=74,035 [8]) have been conducted for self-reported phenotypes [6, 9–11, 8, 12–27], including ever vs. never smoking, former vs. current smoking, cigarettes per day (CPD), nicotine dependence defined by Fagerström Test for Nicotine Dependence (FTND) or Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV) [6, 9–11, 8, 12–27], nicotine withdrawal defined by DSM-IV, as well as smoking biomarkers [28–32]. The FTND, a validated, expert-recommended, low-burden questionnaire of six items used to assess severity of physiological nicotine dependence symptoms among cigarette smokers [33], is the most widely used measure of nicotine dependence.
Table 1.
Evidence supporting 11 genetic loci identified at genome-wide statistical significance (P<5×10−8) in GWAS of cigarette smoking phenotypes/biomarkers and independently replicated or extended to a related phenotype/biomarker. For each locus, replication (or extension) evidence is presented for GWAS-identified lead SNP/indel or proxy variants. P values shown in bold correspond to the first GWAS demonstrating genome-wide significant evidence for SNP/indel association. Loci are sorted by chromosome, and studies supporting each locus are presented chronologically.
| Genetic locus | Chr | Lead GWAS SNP/indela | SNP/indel annotation | Smoking phenotype or biomarker | N, by ancestry | P | |||
|---|---|---|---|---|---|---|---|---|---|
| European | African | Asian or Pacific Islander | Latin o/Hispanic | ||||||
| UGT2B10-UGT2A3b | 4q13 | rs115765562c | Upstream | Cotinine glucuronidation [30] | 437 | 364 | 985 | 453 | 1.6×10−155 |
| rs114612145c (r2=0.31, D′=0.94) | Downstream | Cotinine levels [32] | 4,548 | 0 | 0 | 0 | 5.9×10−10 | ||
| 755 | 0 | 0 | 0 | 0.020 | |||||
| PDE1C | 7p14 | rs215605 | Intronic | CPD [11] | 77,012 | 0 | 0 | 0 | 5.4×10−9 |
| Heavy vs. never [22] | 48,931 | 0 | 0 | 0 | 2.0×10−3 | ||||
| CHRNB3-CHRNA6 | 8p11 | rs6474412 | Upstream | CPD [11] | 84,956 | 0 | 0 | 0 | 1.4×10−8 |
| rs1451240 (r2=0.99, D′=1) | Upstream | Nicotine dependence [15] | 3,201 | 999 | 0 | 0 | 6.7×10−16 | ||
| DBH | 9q34 | rs3025343 | Intronic | Current vs. former [8] | 64,924 | 0 | 0 | 0 | 3.6×10−8 |
| Current vs. former [14] | 1,164 | 0 | 0 | 0 | 0.002 | ||||
| Current vs. former [16] | 0 | 11,644 | 0 | 0 | 0.03 | ||||
| FTND score [142] | 1,430 | 0 | 0 | 0 | 0.023 | ||||
| Heavy vs. never [22] | 48,931 | 0 | 0 | 0 | 1.2×10−5 | ||||
| Nicotine dependence [26] | 28,677 | 0 | 0 | 0 | 1.7×10−5 | ||||
| BDNF | 11p 14 | rs6265 | Missense | Ever vs. never [8] | 143,023 | 0 | 0 | 0 | 1.8×10−8 |
| Heavy vs. never [22] | 48,931 | 0 | 0 | 0 | 2.4×10−4 | ||||
| CHRNA5-CHRNA3-CHRNB4 | 15q 25 | rs16969968 | Missense | Nicotine dependence [6, 7] | 1,929 | 0 | 0 | 0 | 6.4×10−4 |
| CPD [11, 12, 8] | 73,853 | 0 | 0 | 0 | 5.6×10−72 | ||||
| CPD [14] | 3,440 | 0 | 0 | 0 | 1.5×10−4 | ||||
| CPD [16] | 0 | 11,480 | 0 | 0 | 0.027 | ||||
| CPD [143] | 28,772 | 0 | 0 | 0 | 3.2×10−25 | ||||
| Years of smoking [143] | 1.1×10−6 | ||||||||
| Pack-years [143] | 3.0×10−23 | ||||||||
| Current vs. former [143] | 6.9×10−10 | ||||||||
| CPD [144] | 12,364 | 0 | 0 | 0 | 5.2×10−6 | ||||
| CPD [145] | 1,9420 | 0 | 0 | 0 | 3×10−4 | ||||
| FTND score [146] | 815 | 0 | 0 | 0 | 0.0068 | ||||
| 1,121 | 0 | 0 | 0 | 0.0028 | |||||
| Heavy vs. light [25] | 0 | 0 | 0 | 5,085 | 2.2×10−7 | ||||
| Heavy vs. light [147] | 14,786 | 10,912 | 6,889 | 0 | 1.1×10−17 | ||||
| Heavy vs. light [148] | 140 | 0 | 0 | 0 | 0.007 | ||||
| Low vs. high dependence [148] | 0.01 | ||||||||
| Age of cessation [46] | 29,072 | 0 | 0 | 0 | 0.0042 | ||||
| Exhaled CO [28] | 1,521 | 247 | 0 | 0 | 1.7×10−8 | ||||
| Cotinine levels [32] | 4,548 | 0 | 0 | 0 | 6.9×10−17 | ||||
| Cotinine levels [144] | 12,364 | 0 | 0 | 0 | 2.7×10−11 | ||||
| rs1051730 (r2=0.87, D′=1) | Synonymous | CPD [9] | 15,771 | 0 | 0 | 0 | 6×10−20 | ||
| CPD [11, 12, 8] | 73,853 | 0 | 0 | 0 | 2.8×10−73 | ||||
| CPD [10] | 4,342 | 0 | 0 | 0 | 6.1×10−4 | ||||
| CPD [14] | 3,440 | 0 | 0 | 0 | 1.1×10−4 | ||||
| CPD [16] | 0 | 15,552 | 0 | 0 | 0.0079 | ||||
| CPD [149] | 788 | 0 | 0 | 0 | 0.004 | ||||
| Heavy vs. light [22] | 48,931 | 0 | 0 | 0 | 4.4×10−15 | ||||
| Heavy vs. light [150] | 1,264 | 0 | 0 | 0 | 0.0057 | ||||
| Adherence to NRT [151] | 633 | 0 | 0 | 0 | 0.044 | ||||
| NRT dose [151] | 0.026 | ||||||||
| rs2036527 (r2=0.67, D′=0.97) | Intergenic | CPD [16] | 0 | 15,554 | 0 | 0 | 1.8×10−8 | ||
| Nicotine dependence [152] | 1,428 | 0 | 0 | 0 | 9×10−6 | ||||
| Abstinence [153] | 0 | 1,295 | 0 | 0 | 0.004 | ||||
| rs34684276 (r2=0.88, D′=0.95) | Intronic | Nicotine dependence [21] | 17,074 | 0 | 0 | 0 | 3.5×10−17 | ||
| EGLN2c | 19q 13 | rs3733829 | Intronic | CPD [8] | 73,853 | 0 | 0 | 0 | 1.0×10−8 |
| CPD [154] | 1,395 | 0 | 0 | 0 | 0.026 | ||||
| exhaled CO [154] | 3.8×10−5 | ||||||||
| CYP2A6-CYP2B6d | 19q 13 | rs4105144 | Upstream | CPD [11] | 83,317 | 0 | 0 | 0 | 2.2×10−12 |
| CPD [14] | 3,440 | 0 | 0 | 0 | 3.9×10−5 | ||||
| Cotinine levels [155] | 845 | 0 | 0 | 0 | 1×10−4 | ||||
| rs8102683 (r2=0.21, D′=0.95) | Upstream | CPD [17] | 0 | 0 | 11,696 | 0 | 4.3×10−26 | ||
| 0 | 0 | 5,462 | 0 | 1.5×10−17 | |||||
| rs56113850 (r2=0.22, D′=0.53) | Intronic | NMR [29] | 1,518 | 0 | 0 | 0 | 5.8×10−86 | ||
| NMR [31] | 212 | 49 | 51 | 0 | 6.6×10−18 | ||||
| DNMT3Bd | 20q 11 | rs910083 | Intronic | Nicotine dependence [26] | 28,677 | 9,925 | 0 | 0 | 3.7×10−8 |
| Heavy vs. never [22, 26] | 48,931 | 0 | 0 | 0 | 3.6×10−4 | ||||
| NOL4L d | 20q 11 | rs57342388b | Intronic | Heavy vs. never [22] | 48,931 | 0 | 0 | 0 | 4.7×10−9 |
| Nicotine dependence [26] | 28,677 | 9,925 | 0 | 0 | 0.0017 | ||||
| CHRNA4 | 20q 13 | rs2273500 | Splice acceptor | Nicotine dependence [21] | 24,543 | 0 | 0 | 0 | 8.0×10−9 |
| Heavy vs. never [22, 26] | 48,931 | 0 | 0 | 0 | 1.4×10−6 | ||||
CPD, cigarettes per day; CO, carbon monoxide; FTND, Fagerström Test for Nicotine Dependence; NMR, nicotine metabolite ratio; NRT, nicotine replacement therapy
For loci with more than one GWAS lead or proxy SNP indicated, r2 and D′ values are shown in reference to all 1000 Genomes phase 3 panels as calculated using LDlink [117].
Of the 11 loci, UGT2B10-UGT2A3 is the only one implicated for biomarkers but not extended to nicotine dependence or related smoking phenotypes.
rs115765562, as originally reported, has since merged into rs34100980. rs57342388 has the same alignment as rs143125561. rs114612145, as originally reported, has since merged into rs77107237.
Most GWAS analyses have been conducted in European ancestry populations [6, 9–11, 8, 12–14, 18, 19, 21, 22, 29, 32], although one focused on Hispanics [25], one focused on Japanese [17], eight included or focused exclusively on African Americans [15, 16, 20, 23, 24, 26, 28, 27], and two others included multiple ancestries [30, 31]. Two GWAS analyses examined copy number variants [17, 23], while all others focused on SNP and insertion/deletion (indel) variants.
Table 1 presents the 11 genetic loci with common SNPs/indels implicated at genome-wide significance (P<5×10−8) and replicated in one or more independent datasets for the same phenotype/biomarker or extended to a related smoking phenotype/biomarker. The UGT2B10-UGT2A3 locus was identified in cotinine biomarker GWAS, but this finding has not been extended to smoking phenotypes [32]; the 10 other GWAS-identified loci influence susceptibility of smoking phenotypes. Lead SNPs with the smallest P value from each distinct genetic locus are presented, although several loci include many significantly associated SNPs, often strongly correlated with the lead SNPs. For example, 360 SNPs in the CHRNA5-CHRNA3-CHRNB4 locus were significantly associated in GWAS meta-analysis of FTND-defined nicotine dependence [21], and 719 SNPs in the CYP2A6-CYP2B6 locus were significantly associated in GWAS meta-analysis of the nicotine metabolite ratio [29]. SNPs in some GWAS-identified loci also influence susceptibility of developing smoking-related health outcomes, including: CHRNA5 with lung cancer (most recently [34]), lung function [35], airflow obstruction [36], COPD (most recently [37]), and peripheral arterial disease [9]; the nicotine metabolizing gene CYP2A6 with lung cancer [11], lung function [35], and emphysema [38]; and CHRNB3-CHRNA6, CHRNA4, and the DNA methyltransferase gene DNMT3B with lung cancer [11, 21, 26]. Beyond common SNPs, the CHRNA5-CHRNA3-CHRNB4 [39–42] and CHRNA4 [40, 43, 44] loci also harbor smoking-associated rare variants, albeit some do not have replication evidence or occur in an isolated population. Genome-wide significant SNP associations in other loci have been reported but not yet independently replicated [8, 13, 20, 22, 24].
The GWAS-identified SNPs each explain a small percent of the variance in nicotine dependence or related smoking phenotypes, but their discoveries have revealed important neurobiology that impacts susceptibility to developing addiction and exerts clinically important effects downstream, as best illustrated by CHRNA5. Following the discovery of its association with nicotine dependence (Table 1), the CHRNA5 missense SNP rs16969968 was found to alter α5 receptor function [45] and later its high-risk rs16969968-AA genotype was associated with a 4-year median delay in quitting smoking and a 4-year earlier median age of lung cancer diagnosis, even when adjusted for CPD [46]. Moreover, the haplotype carrying the rs16969968-A risk allele interacts with cessation treatment, with smokers at highest nicotine dependence risk being less likely to quit smoking overall but responding most effectively to pharmacologic treatment [47], demonstrating the potential for personalized cessation treatment based on genetic risk variants for nicotine dependence. Independent of rs16969968, noncoding CHRNA5 SNPs have been found to tag cis-expression quantitative trait loci (cis-eQTL) and cis-methylation QTL (cis-meQTL) variants that regulate CHRNA5 RNA expression (rs588765 [48] and rs880395 [49]) and DNA methylation (rs11636753 [50]), respectively, in postmortem human PFC. Consistent with these findings, mouse models have indicated that genetically altered CHRNA5 RNA expression profoundly influences behavioral traits characteristic of nicotine dependence [51–53].
While the regulatory effects of CHRNA5 SNPs have mainly centered on PFC, widely recognized for its involvement in addiction [3, 54], noncoding SNPs nearby genes identified in more recent GWAS analyses have highlighted gene regulatory effects in unexpected brain tissues. The CHRNA4 splice site acceptor SNP rs2273500 (Table 1) was indicated as a cis-eQTL SNP for CHRNA4 in intralobular white matter [21]. The DNMT3B intronic SNP rs910083 (Table 1) [26] and CHRNA2 upstream SNP rs117804171, discovered for its association with lung cancer but then extended to smoking [34], were each indicated as cis-eQTL SNPs for their proximal gene in cerebellum, which has been often overlooked despite evidence for its involvement in the neurobiology of addiction [55–57]. Follow-up of these and other GWAS discoveries for functional or regulatory effects, in normal physiological vs. smoking-exposed states, is needed across the wide array of human brain tissues to expand our neurobiological understanding of initiating smoking, becoming a regular smoker, developing nicotine dependence, and ultimately improving cessation treatment strategies.
GWAS for alcohol
At least 34 GWAS analyses (largest N=112,117 [58]) have been reported for alcohol use disorder, related alcohol phenotypes, or alcohol biomarkers from studies focused on European [13, 18, 59–74, 58, 75, 76], European and African [77–83], or Asian [84–88] ancestries, or multiple ancestries combined [89]. Table 2 presents the genome-wide significant and replicable SNP associations for self-reported phenotypes in or near seven loci—serpin family C member 1 (SERPINC1), glucokinase regulator (GCKR,) shugoshin 1 (SGOL1), β-klotho (KLB), alcohol dehydrogenase (ADH) cluster, autism susceptibility candidate 2 (AUTS2), and aldehyde dehydrogenase 2 (ALDH2); an eighth locus—transferrin (TF)—was implicated for biomarkers of excessive alcohol intake but has not been associated with alcohol use disorder or related phenotypes. Emerging evidence suggests that the ADH gene cluster contains more than one independent association signal [58]. Other genome-wide significant loci have been reported, but independent replication has not been attained [13, 59, 60, 64, 68, 70, 73, 74, 58].
Table 2.
Evidence supporting eight genetic loci identified at genome-wide statistical significance (P<5×10−8) in GWAS of alcohol phenotypes/biomarkers and independently replicated or extended to a related phenotype/biomarker. For each locus, replication (or extension) evidence is presented for GWAS-identified lead SNP/indel or proxy variants. P values shown in bold correspond to the first GWAS demonstrating genome-wide significant evidence for SNP association. Loci are sorted by chromosome, and studies supporting each locus are presented chronologically.
| Genetic locus | Chr | Lead GWAS SNPa | SNP annotation | Alcohol phenotype or biomarker | N, by ancestry | P | |||
|---|---|---|---|---|---|---|---|---|---|
| European | African | Asia n | Latino/His panic | ||||||
| SERPINC1 | 1q25 | rs1799876 | Intronic | Maximum drinks in 24-hour period [82] | 2,328 | 0 | 0 | 0 | 4.0×10−8 |
| Weekly alcohol intake [89] | 47,967 | 0 | 0 | 0 | 0.031 | ||||
| GCKR | 2p23 | rs780094 | Intronic | Daily alcohol intake [73] | 98,477 | 0 | 0 | 0 | 3.6×10−9 |
| rs4665985 (r2=0.41, D′=0.68) | Downstream | Drinker vs. non-drinker [89] | 71,071 | 2,475 | 6,034 | 7,047 | 5.0×10−4 | ||
| Weekly alcohol intake [89] | 47,967 | 1,310 | 2,746 | 4,374 | 2.3×10−8 | ||||
| SGOL1 | 3p24 | rs11128951 | Upstream | Maximum drinks in 24-hour period [69] | 2,687 | 0 | 0 | 0 | 4.3×10−8 |
| Drinker vs. non-drinker [89] | 71,071 | 0 | 0 | 0 | 0.019 | ||||
| TF-SRPRBb | 3q22 | rs1799899 | Missense | CDT% [64] | 5,181 | 0 | 0 | 0 | 3.4×10−35 |
| 1,509 | 0 | 0 | 0 | 9.7×10−6 | |||||
| rs3811647 (r2=0.02, D′=0.96) | Intronic | Total transferrin [64] | 5,181 | 0 | 0 | 0 | 1.3×10−10 | ||
| 1,509 | 0 | 0 | 0 | 5.3×10−19 | |||||
| 745 | 0 | 0 | 0 | 8.8×10−9 | |||||
| rs1534166 (r2=0.07, D′=0.86) | Intronic | CDT concentration [64] | 1,509 | 0 | 0 | 0 | 5.8×10−12 | ||
| 5,181 | 0 | 0 | 0 | 3.0×10−7 | |||||
| KLB | 4p14 | rs11940694 | Intronic | Daily alcohol intake [73] | 98,477 | 0 | 0 | 0 | 9.2×10−12 |
| Weekly alcohol intake [58] | 112,117 | 0 | 0 | 0 | 8.4×10−19 | ||||
| rs7686419 (r2=0.47, D′=0.78) | Upstream | Drinker vs. non-drinker [89] | 71,071 | 2,475 | 6,034 | 7,047 | 4.1×10−5 | ||
| Weekly alcohol intake [89] | 47,967 | 1,310 | 2,746 | 4,374 | 3.4×10−10 | ||||
| ADH1B-ADH1C | 4q23 | rs1789891 | Intronic | DSM-IV-defined cases vs. controls [65] | 3,501 | 0 | 0 | 0 | 1.3×10−8 |
| DSM-IV-defined cases vs. controls [156] | 2,103 | 0 | 0 | 0 | 7.2×10−5 | ||||
| rs1229984 (r2=0.02, D′=0.89) | Missense | Cases vs. controls with varied definitions [91] | 5,850 | 0 | 12,266 | 0 | 1.0×10−36 | ||
| Ever vs. never [85] | 0 | 0 | 6,720 | 0 | 3.6×10−4 | ||||
| DSM-IV-defined cases vs. controls [157] | 3,789 | 1,721 | 0 | 0 | 6.6×10−10 | ||||
| Maximum drinks in 24-hour period [157] | 3,777 | 1,698 | 0 | 0 | 3.2×10−13 | ||||
| DSM-IV-defined cases vs. controls [86] | 0 | 0 | 1,371 | 0 | 2.6×10−21 | ||||
| DSM-IV symptom count [81] | 7,001 | 0 | 0 | 0 | 2.9×10−18 | ||||
| Maximum drinks in a 24-hour period [82] | 2,587 | 0 | 0 | 0 | 6.0×10−15 | ||||
| DSM-IV-defined cases vs. controls [156] | 2,103 | 0 | 0 | 0 | 8.4×10−6 | ||||
| Drinker vs. non-drinker [89] | 71,071 | 2,475 | 6,034 | 7,047 | 8.0×10−4 | ||||
| Weekly alcohol intake [89] | 47,967 | 1,310 | 2,746 | 4,374 | 3.0×10−4 | ||||
| rs2066702 (r2=0.01, D′=0.96) | Missense | DSM-IV symptom count [81] | 0 | 4,638 | 0 | 0 | 2.2×10−13 | ||
| Maximum drinks in 24-hour period [82] | 0 | 5,064 | 0 | 0 | 2.5×10−10 | ||||
| rs145452708 (r2<0.01, D′=1) | Intronic | Weekly alcohol intake [58] | 112,117 | 0 | 0 | 0 | 1.2×10−30 | ||
| rs141973904 (r2<0.01, D′=1) | Intronic | Alcohol use disorder identification test (AUDIT) score [76] | 20,328 | 0 | 0 | 0 | 4.4×10−7 | ||
| AUTS2 | 7 | rs6943555 | Intronic | Average consumption (grams/day/kg body weight) [60] | 26,316 | 0 | 0 | 0 | 4.1×10−9 |
| Weekly alcohol intake [89] | 47,967 | 0 | 0 | 0 | 0.0050 | ||||
| ALDH2 | 12q24 | rs2074356 | Intronic | Average consumption (grams alcohol/d ay) [84] | 0 | 0 | 1,721 | 0 | 5.8×10−46 |
| 0 | 0 | 1,113 | 0 | 1.3×10−16 | |||||
| rs671 (r2=0.49, D′=0.83) | Missense | Ever vs. never [85] | 0 | 0 | 6,720 | 0 | 3.6×10−211 | ||
| DSM-IV-defined cases vs. controls [86] | 0 | 0 | 396 | 0 | 8.4×10−8 | ||||
| Past year drinkers vs. nondrinkers [87] | 0 | 0 | 23,199 | 0 | 5.2×10−215 | ||||
| Alcohol dependence [88] | 0 | 0 | 595 | 0 | 4.7×10−8 | ||||
| Maximum drinks in a 24-hour period [88] | 0 | 0 | 595 | 0 | 1.5×10−16 | ||||
| Flush response [88] | 0 | 0 | 595 | 0 | 4.8×10−26 | ||||
| Drinker vs. non-drinker [89] | 0 | 0 | 6,034 | 0 | 2.3×10−72 | ||||
| Weekly alcohol intake [89] | 0 | 0 | 2,746 | 0 | 5.4×10−4 | ||||
| rs11066280 (r2=0.44, D′=0.88) | 5′ untranslated | Past year drinkers vs. nondrinkers [87] | 0 | 0 | 23,199 | 0 | 3.3×10−215 | ||
| Daily alcohol intake [87] | 0 | 0 | 6,211 | 0 | 4.0×10−21 | ||||
CDT, carbohydrate-deficient transferrin (biomarker of excessive alcohol intake)
For loci with more than one GWAS lead SNP indicated, r2 and D′ values are shown in reference to all 1000 Genomes phase 3 panels as calculated using LDlink [117].
Of the 8 loci, TF is the only one implicated for biomarkers but not extended to alcohol use disorder or related alcohol phenotypes.
The GWAS-identified variants include missense functional SNPs that alter the alcohol metabolism pathway converting alcohol to acetaldehyde via ADH enzymes and then to acetate via ALDH enzymes. For example, the ADH1B SNP allele rs1229984-T is a missense variant that alters enzymatic activity resulting in up to 100-fold higher rate of alcohol to toxic acetaldehyde metabolism, causing the alcohol flush reaction, and reducing susceptibility to developing alcohol drinking problems [90]. Moreover, the rs671-A allele in ALDH2, another missense variant, alters the latter step of the alcohol metabolism pathway by greatly reducing ALDH2 activity and rendering high acetaldehyde concentrations upon alcohol exposure [85]. Both the ADH1B rs1229984-T and ALDH2 rs671-A alleles occur frequently in East Asian populations but infrequently in other world populations, and their highly significant associations with reduced susceptibility to drinking alcohol and developing an addiction were studied as candidate SNPs based on their functional consequences on enzymatic activity before they were highlighted via agnostic GWAS [85, 91]. Associations of these SNPs or their proxies on alcohol-related disease outcomes include coronary heart disease [85], various cancers [92, 93], renal function [94], blood pressure [95], and cirrhosis [96]. Beyond these functional missense SNPs, the intronic AUTS2 SNP rs6943555 was implicated as a cis-eQTL for AUTS2 in PFC [60], and the intronic KLB SNP rs11940694 was implicated as a cis-eQTL for the replication factor RCF1 gene in cerebellum [58]. Little else is known about the biological relevance of other noncoding SNPs that may exert independent effects on regulation of genes that underlie alcohol metabolism in liver or the neurobiology of developing an alcohol use disorder.
GWAS for other specific drugs
Relatively few GWAS have been reported for cannabis, stimulants, or opioids. Unlike GWAS of cigarette smoking and alcohol phenotypes, GWAS of these specific drugs have had limited success at identifying and replicating variant associations.
For cannabis, four of the five reported GWAS had no genome-wide significant SNP associations [97–100], most notably including a large GWAS meta-analysis (total N=32,330 of European ancestry) [99]. The most recent GWAS meta-analysis (total N=14,754 European Americans and African Americans) identified three loci that attained genome-wide significance, but these findings await independent replication [101].
For stimulants, four GWAS have been reported. Two focused on methamphetamine and found no genome-wide significant associations [102, 103]. The other two GWAS focused on amphetamine [104] and cocaine [105], and each reported a single genome-wide significant SNP association, but these findings also await replication.
For opioids, six GWAS have been reported. Three SNP-based GWAS were small (N<1,000), and no SNPs attained genome-wide significance [106–108]. The three larger GWAS, including two SNP-based [109, 110] and one copy number variant-based [111], each found genome-wide significant associations in different genetic loci, but these association signals all await independent replication. GWAS analyses have not yielded genome-wide significant evidence for any of the biologically plausible opioid receptor genes. However, such evidence emerged from a targeted study of cis-eQTL SNPs of the μ-opioid receptor gene (OPRM1); smallest P=4.3×10−8 for intronic SNP rs3778150 with total N=16,729 of European or African American ancestry [112]. Much attention has focused on OPRM1 SNP associations but with inconclusive findings from mostly studies of single cohorts. The OPRM1 SNP rs1799971, which results in an amino acid change in the μ-opioid receptor, has been the most extensively studied. In an accumulation of evidence for rs1799971, meta-analysis of >28,000 European ancestry participants from 25 studies supported a modest association with general substance dependence, rather than opioids or any substance specifically [113]. As we previously reported, the cis-eQTL SNP rs3778150 may explain the prior inconsistent associations, as the rs1799971-A allele conferred increased heroin addiction risk only in the presence of the rs3778150-C allele that lowers OPRM1 expression levels and plausibly disrupts the opioid system of a key compensatory response to exogenous opioid exposure [112]. The association pattern observed between heroin addiction and the haplotype carrying rs3778150-C and rs1799971-A has been extended to subjective alcohol response [114].
Relatedly for opioid use disorder phenotypes, two GWAS of clinical opioid dosing have been reported [115, 116]. Most recently, a genome-wide significant association of methadone dosing among 383 opioid-dependent African Americans was observed for the SNP rs73568641 (P=2.8×10−8), located upstream of OPRM1 [116]. The rs73568641 association was extended to morphine dosing for post-operative pain in an independent dataset of 241 African Americans (P=0.039) [116]. Rs73568641 is in weak linkage disequilibrium (r2<0.01 and D′=0.54 across 1000 Genomes African ancestry reference panels [117]) with the cis-eQTL SNP rs3778150, suggesting that variants marking more than one signal across the OPRM1 gene region may be driving different opioid phenotypes. However, association analyses of these SNPs conditioned on one another are needed to formally assess independence of the signals.
GWAS for illicit drugs combined
Given the high levels of co-morbidity across drugs of abuse and the plausible shared heritability underlying susceptibility to developing addiction in general, three GWAS have combined cases addicted to any illicit drug [18, 118, 119]. Two of these GWAS reported genome-wide significant and replicable associations. First, a GWAS of general substance dependence liability, based on factor analysis of DSM-IV symptoms, identified the intergenic SNP rs2952621 (P=1.8×10−8, N=2,322 European Americans) and replicated the finding in an independent dataset (P=0.02, N=2,647 European Americans) [118]. Rs2952561 is a SNP of unknown biological consequence that is located upstream of the uncharacterized gene LOC151121.
Next, GWAS of people who inject drugs vs. controls, revealed an African American-specific association for the intronic SNP rs9829896 in the lysine acetyltransferase gene (KAT2B; P=4.6×10−8, N=3,742), which replicated in an independent dataset (P=0.0016, N=755) [119]. Follow-up analyses in postmortem human PFC implicated rs9829896 as a cis-eQTL SNP, specifically in African Americans, for KAT2B and other genes in KAT2B-containing pathways, including OPRM1 and the CREB binding protein (CREBBP) genes [119] that have been highlighted in prior SNP- or pathway-based analyses of opioid phenotypes [109, 112, 116].
Potential for future GWAS to broaden the known genetic etiology unique to and shared among addiction phenotypes
Prior GWAS have utilized varied addiction phenotypes and biomarkers as well as study designs. A major distinction among the case-control studies involves the assessment of substance use and misuse for controls. Some studies have used population-based, unassessed controls when study controls were not available, for example [119], or when intending to increase sample size and statistical power with minimal offset from misclassification due to the relatively low prevalence of addiction, particularly for illicit drugs in healthy cohorts [112]. Other studies have focused on assessed controls with prior exposure or history of misuse but without symptoms of addiction, for example [110], aimed at detecting genetic variants associated with progression from misuse to later stages of addiction. Relatedly, studies with assessed controls have either adjusted for co-morbid exposure and/or addiction to other substances as covariates, under the premise of identifying genetic variants associated with specific substances, for example [105, 109]. Other studies have not adjusted for other substance co-morbidities [110], which may enhance detection of generalizable genetic variant associations. To our knowledge, there has been no formal testing to outline the best scenarios for future addiction studies to balance statistical power with the use of unassessed vs. assessed controls. Ultimately, study design decisions will depend on the primary research question and expected misclassification rate. For strategizing adjustment for co-morbid addictions, simulation testing of genetically correlated outcomes and covariates suggests that the impact of covariate adjustment depends on the direction and magnitude of correlation with the outcome [120].
We fully expect that future GWAS using larger sample sizes from expanded meta-analyses and large-scale biobanks with data on cigarette smoking, alcohol, and other drugs will identify and replicate additional loci with robust statistical evidence. These loci will likely include some variants that underlie specific addictions, while others are shared across different drugs of abuse and across psychiatric diseases that are highly co-morbid with addiction. This notion of shared genetic susceptibility is supported by schizophrenia-associated common genetic variation having significant correlations with smoking-associated genetic variation: for example, rg=0.14 for nicotine dependence, rg=0.12–0.14 for CPD, and rg=0.10 for ever/never smoking [121, 122]. Polygenic risk scores (weighted summations of top ranking subsets of genetic variant associations) have provided additional evidence for shared genetic etiologies. Polygenic risk scores based on GWAS-identified variants for schizophrenia or bipolar disorder have shown significant associations with smoking, alcohol, and substance use disorder phenotypes [123–125]. Early examples of genetic variants that demonstrate such pleiotropy, i.e., exert effects on more than one outcome, across addiction and other psychiatric diseases include: ZNF804A SNP rs1344706, the first genome-wide significant finding for schizophrenia [126] and later extended to heroin addiction [127, 128]; and the BDNF SNP rs6265 and CHRNA5 SNPs rs16969968 and rs1051730 implicated at genome-wide significance for smoking (Table 1) and extended to schizophrenia/bipolar disorder outcomes [129, 130]. As the sample sizes for addiction GWAS expand to sizes comparable with other psychiatric disease GWAS, polygenic risk scores and top loci that comprise these scores are likely to reveal even more extensive sharing. Once established, polygenic risk scores may have clinical utility for predicting individuals at highest risk for developing addiction or guiding treatment strategies.
Ongoing sequential integration of multiple ‘omics data
Establishing robust statistical evidence for a GWAS-identified variant marks the starting point in the path to elucidating its biological consequences as related to disease outcomes. Its annotation in the context of DNA, RNA, and protein sequences often provides the first clue. Coding SNPs that alter the protein’s amino acid sequence can be undoubtedly important for risk of developing complex human diseases, including addiction (e.g., CHRNA5 SNP rs16969968 for smoking [Table 1] and ADH1B SNP rs1229984 and ALDH2 SNP rs671 for alcohol [Table 2]). However, >95% of GWAS discoveries come from noncoding SNPs [131]. Noncoding SNPs can alter gene activity by influencing DNA methylation, RNA expression, protein expression, or metabolite levels and are thus well-recognized for their disease risk potential. Indeed, the GWAS catalog is heavily enriched for regulatory variants, such as eQTLs [131–133], and variants residing in sequences that are highly conserved, sensitive to cleavage by DNase I, or mark enhancer, promoter, or protein binding sites [133].
Integration of GWAS with other ‘omics data types (such as, DNA methylation or RNA expression) commonly happens in a sequential fashion to infer functional or regulatory effects of top GWAS findings. Each ‘omics type is analyzed separately, and comparisons are made across results. For example, rs910083 was identified in the largest GWAS meta-analysis of nicotine dependence, and its association was extended to heavy vs. never smoking in the UK Biobank (Table 1); rs910083 was then followed-up via a cis-eQTL analysis using RNA expression data measured across multiple postmortem human brain tissues in the Genotype-Tissue Expression (GTEx) and Brain eQTL Almanac projects, leading to the implication of rs910083 as a DNMT3B cis-eQTL in cerebellum [26]. This type of sequential integration in moving from GWAS to functional or regulatory characterization has also yielded other important discoveries in addiction [21, 48, 119].
A reversal in the direction of sequential integration is an alternative, but not as widely utilized, approach that first identifies functional or regulatory variants to narrow the search space, thereby reducing the multiple testing burden of GWAS to uncover disease associated variants that may have otherwise been missed. The OPRM1 cis-eQTL SNP rs3778150 finding for heroin addiction serves as an example of moving from gene regulation into GWAS [112].
In taking integration to the next level, PrediXcan [134] is a promising new gene-based method that utilizes genome-wide SNP genotypes and RNA-sequencing data in GTEx to build genetically regulated gene expression models in a diverse set of tissues and applying those models in GWAS with disease phenotypes of interest. The PrediXcan framework is applicable to other sources of ‘omics data (e.g., DNA methylation) in GTEx or elsewhere.
Importance of brain tissue specificity for multi ‘omics analyses
These sequential integration examples highlight a challenge for the field of addiction, and psychiatric disease more broadly, because functional and regulatory effects can be highly tissue-specific [135] and brain is the most relevant tissue for studying the neurobiology of addiction. GWAS genotypes, other ‘omics data in brain, and addiction phenotypes are seldom available in the same dataset. Other ‘omics data derived in blood or other peripheral tissues that are easily accessible in living participants may offer informative biomarkers of addiction but overall provide poor indicators of neurobiology. Poor correlations in RNA expression levels between blood and brain has been repeatedly shown [136, 137, 135]. Most recently, pilot GTEx analyses with RNA expression measured in 43 tissues showed that blood vs. brain had the most distinct expression profiles among the tissue comparisons [135]. There is also limited overlap of meQTL and eQTL [136, 137, 135] variants mapped in blood vs. brain. Thus, when analyzing gene regulatory potential to study the neurobiology of addiction, it is critical to use disease-relevant brain tissue. Reliance on blood-specific regulatory effects could even lead to erroneous conclusions, as illustrated by the cis-eQTL SNP rs880395 having opposing directions of association with CHRNA5 expression in lymphoblastoid cell lines, as compared to frontal cortex [49]. Moreover, gene regulatory effects can differ across the different brain tissues, so comprehensive functional and regulatory assessment will require the availability of ‘omics data across multiple brain tissues, including ones traditionally viewed as being addiction-relevant (e.g., PFC, nucleus accumbens) and others often overlooked (e.g., cerebellum). GTEx, Brain eQTL Almanac, and others provide an unprecedented opportunity to carry out comprehensive analyses in normal brains. Future studies are needed for more thorough and well-powered assessment of brain tissues in participants with addiction phenotype data.
Upcoming concurrent integration of multi ‘omics data
Where once a single ‘omics type was used, studies that capture multiple ‘omics data types in the same dataset are emerging. As these datasets become available, concurrent integration that jointly assess all data may unveil relationships not evident when each data type is analyzed separately by virtue of increased statistical power and their explicit biological relationships (Figure 1). It is expected that genetic variants with large effect sizes are identifiable in sequential analyses but that concurrent integration will enable the identification of genetic variants with moderate-sized, but multi-faceted, functional or regulatory effects. This approach can operate in a bidirectional fashion, with datasets rich in ‘omics informing follow-up in large-scale GWAS of addiction phenotypes/biomarkers and vice versa (Figure 1), and accelerate understanding of the biology underlying statistical associations with addiction phenotypes.
Figure 1.
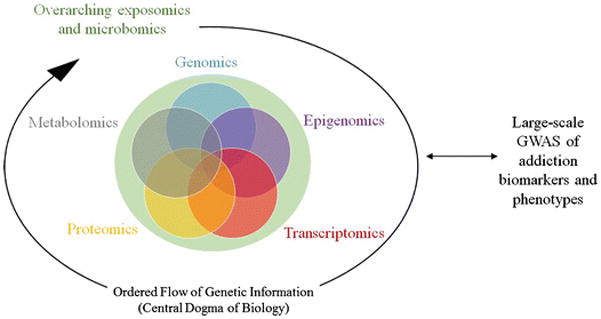
Concurrent integrative analysis approach with multiple types of ‘omics data used to inform discovery or characterize top findings from large-scale addiction GWAS. Genomics encompasses SNP/indel genotypes, rare variants, and structural variants including copy number variants; epigenomics includes DNA methylation and histone modification; and transcriptomics refers to expression of all RNA types. ‘Omics data may pertain to endogenous factors along the flow of information according to the Central Dogma of Biology or exogenous factors such as environmental exposures.
New methods are being developed that will allow researchers to analyze data concurrently, as applied in plant [138] and mouse model [139] studies. In an early adaptation of this approach in humans [140], the Two-Way Orthogonal Partial Least Squares (O2PLS) method to RNA expression and metabolomics data in 466 Finnish participants recapitulated signals detected in sequential analyses of the two data types and detected new signals by modeling the data types together. Colocalization (COLOC) [141] also enables concurrent integrative analyses. Comparisons of these and other methods will be needed to establish standard analytic practices.
Conclusions
GWAS analyses have identified several genetic loci with statistically robust and reproducible SNP/indel associations, cementing the polygenic nature of addiction. However, with only a fraction of the heritability explained (for example, 15% of the variance in nicotine dependence [121] and 13% of the variance in alcohol consumption [58] explained by common SNPs together) and limited knowledge of the neurobiological pathways leading to addiction, much remains to be discovered. We fully expect that GWAS analyses conducted with sample sizes into the hundreds of thousands and millions will replicate previously suggested, but currently unreplicated, genetic variants and will also unveil novel variants. As evidenced by the GWAS-identified variants identified to date (mainly SNPs), novel variants will likely exert small effect sizes on developing addiction but potentially uncover previously unrecognized neurobiological pathways. Additional heritability may also be explained by complex interactions of genetic variants with one another and with prominent environmental exposures, which will likely require very large sample sizes with harmonized exposures and addiction phenotypes across multiple datasets. In the future expansion of GWAS sample sizes, we emphasize the importance of including diverse ancestry groups that rival the current representation of European ancestry to address important health disparities, identifying variants that manifest under certain genetic, cultural, or environmental backgrounds and narrowing the resolution of association signals that arise from the varied linkage disequilibrium patterns across diverse populations.
New genetic discoveries are also likely to arise from leveraging novel analytic approaches such as integrating GWAS with multiple ‘omics data generated across addiction-relevant brain tissues, even at the resolution of individual cell types and single cells. With the ever-expanding postmortem human brain resources being made available to the scientific community, the field has begun to unravel the biology of top GWAS-identified variants, including a few functional missense SNPs that alter susceptibility to developing nicotine dependence or alcohol use disorder and several noncoding SNPs that perturb gene regulation and influence nicotine dependence and substance use disorders. The recent availability of ‘omics data across multiple brain tissues has offered unparalleled opportunities to study the neurobiological underpinnings of addiction and led to somewhat unexpected connections, such as smoking- and alcohol-associated cis-eQTL SNPs specific to cerebellum [26, 34, 58]. In this era of precision medicine, genetic variant discoveries hold great promise in tailoring prevention and treatment strategies for addiction. Discovery of more genetic loci and biological characterization of the associated variants is needed to enhance our understanding of the neurobiology underlying addiction and lessen its widespread health consequences on individuals and the public health burden.
Acknowledgments
This work was supported by NIDA grants R01s DA035825 and DA042090 (PI: Dana Hancock) and R01 DA036583 (PI: Laura Bierut).
Footnotes
Compliance with Ethics Guidelines
Conflict of Interest
Dana B. Hancock reports grants from National Institutes of Health.
Christina A. Markunas reports grants from National Institutes of Health.
Laura J. Bierut reports grants from National Institutes of Health. In addition, Dr. Bierut is listed as an inventor on U.S. Patent 8,080,371,”Markers for Addiction” covering the use of certain
SNPs in determining the diagnosis, prognosis, and treatment of addiction.
Eric O. Johnson reports grants from National Institutes of Health.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Recently published papers of particular emphasis have been highlighted as:
• Of importance
•• Of major importance
- 1.Center for Behavioral Health Statistics and Quality. Key substance use and mental health indicators in the United States: Results from the 2015 National Survey on Drug Use and Health. 2016. (HHS Publication No. SMA 16–4984, NSDUH Series H-51) [Google Scholar]
- 2.Babb S, Malarcher A, Schauer G, Asman K, Jamal A. Quitting Smoking Among Adults - United States, 2000–2015. MMWR Morb Mortal Wkly Rep. 2017;65(52):1457–64. doi: 10.15585/mmwr.mm6552a1. [DOI] [PubMed] [Google Scholar]
- 3••.Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3(8):760–73. doi: 10.1016/S2215-0366(16)00104-8. This article from the Directors of NIAAA and NIDA, respectively, provides a comprehensive review, updated from their 2010 review in Neuropsychpharmacology (see reference 4), of the key brain tissues involved at each addiction stage and their neurocircuit connections. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35(1):217–38. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agrawal A, Verweij KJ, Gillespie NA, Heath AC, Lessov-Schlaggar CN, Martin NG, et al. The genetics of addiction-a translational perspective. Transl Psychiatry. 2012;2:e140. doi: 10.1038/tp.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bierut LJ, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau OF, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet. 2007;16(1):24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PA, et al. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet. 2007;16(1):36–49. doi: 10.1093/hmg/ddl438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42(5):441–7. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452(7187):638–42. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caporaso N, Gu F, Chatterjee N, Sheng-Chih J, Yu K, Yeager M, et al. Genome-wide and candidate gene association study of cigarette smoking behaviors. PLoS One. 2009;4(2):e4653. doi: 10.1371/journal.pone.0004653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet. 2010;42(5):448–53. doi: 10.1038/ng.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu JZ, Tozzi F, Waterworth DM, Pillai SG, Muglia P, Middleton L, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet. 2010;42(5):436–40. doi: 10.1038/ng.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lind PA, Macgregor S, Vink JM, Pergadia ML, Hansell NK, de Moor MH, et al. A genomewide association study of nicotine and alcohol dependence in Australian and Dutch populations. Twin Res Hum Genet. 2010;13(1):10–29. doi: 10.1375/twin.13.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siedlinski M, Cho MH, Bakke P, Gulsvik A, Lomas DA, Anderson W, et al. Genome-wide association study of smoking behaviours in patients with COPD. Thorax. 2011;66(10):894–902. doi: 10.1136/thoraxjnl-2011-200154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rice JP, Hartz SM, Agrawal A, Almasy L, Bennett S, Breslau N, et al. CHRNB3 is more strongly associated with Fagerstrom Test for Cigarette Dependence-based nicotine dependence than cigarettes per day: phenotype definition changes genome-wide association studies results. Addiction. 2012;107(11):2019–28. doi: 10.1111/j.1360-0443.2012.03922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.David SP, Hamidovic A, Chen GK, Bergen AW, Wessel J, Kasberger JL, et al. Genome-wide meta-analyses of smoking behaviors in African Americans. Transl Psychiatry. 2012;2:e119. doi: 10.1038/tp.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumasaka N, Aoki M, Okada Y, Takahashi A, Ozaki K, Mushiroda T, et al. Haplotypes with copy number and single nucleotide polymorphisms in CYP2A6 locus are associated with smoking quantity in a Japanese population. PLoS One. 2012;7(9):e44507. doi: 10.1371/journal.pone.0044507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGue M, Zhang Y, Miller MB, Basu S, Vrieze S, Hicks B, et al. A genome-wide association study of behavioral disinhibition. Behav Genet. 2013;43(5):363–73. doi: 10.1007/s10519-013-9606-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loukola A, Wedenoja J, Keskitalo-Vuokko K, Broms U, Korhonen T, Ripatti S, et al. Genome-wide association study on detailed profiles of smoking behavior and nicotine dependence in a twin sample. Mol Psychiatry. 2014;19(5):615–24. doi: 10.1038/mp.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gelernter J, Kranzler HR, Sherva R, Almasy L, Herman AI, Koesterer R, et al. Genome-wide association study of nicotine dependence in American populations: identification of novel risk loci in both African-Americans and European-Americans. Biol Psychiatry. 2015;77(5):493–503. doi: 10.1016/j.biopsych.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21•.Hancock DB, Reginsson GW, Gaddis NC, Chen X, Saccone NL, Lutz SM, et al. Genome-wide meta-analysis reveals common splice site acceptor variant in CHRNA4 associated with nicotine dependence. Transl Psychiatry. 2015;5:e651. doi: 10.1038/tp.2015.149. This GWAS meta-analysis was the first to identify CHRNA4 as a genome-wide significant locus for nicotine dependence (total N=17,074 for discovery and 7,469 for replication). The top SNP rs2273500 was indicated as a splice site acceptor SNP and a cis-eQTL SNP for CHRNA4 in postmortem human intralobular white matter. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22•.Wain LV, Shrine N, Miller S, Jackson VE, Ntalla I, Artigas MS, et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir Med. 2015;3(10):769–81. doi: 10.1016/S2213-2600(15)00283-0. This UK Biobank study included a GWAS of heavy vs. never smoking (N=48,931) and identified NOL4L and 4 other novel genome-wide significant loci and extended associations of genome-wide significant loci—PDE1C, DBH, BDNF, and CHRNA4—from other studies with related smoking phenotypes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Begum F, Ruczinski I, Hokanson JE, Lutz SM, Parker MM, Cho MH, et al. Hemizygous Deletion on Chromosome 3p26.1 Is Associated with Heavy Smoking among African American Subjects in the COPDGene Study. PLoS One. 2016;11(10):e0164134. doi: 10.1371/journal.pone.0164134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin X, Bizon C, Tilson J, Lin Y, Gizer IR, Ehlers CL, et al. Genome-wide meta-analysis identifies a novel susceptibility signal at CACNA2D3 for nicotine dependence. Am J Med Genet B Neuropsychiatr Genet. 2017;174(5):557–567. doi: 10.1002/ajmg.b.32540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saccone NL, Emery LS, Sofer T, Gogarten SM, Becker DM, Bottinger EP, et al. Genome-wide association study of heavy smoking and daily/nondaily smoking in the Hispanic Community Health Study/Study of Latinos (HCHS/SOL) Nicotine Tob Res. doi: 10.1093/ntr/ntx107. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26•.Hancock DB, Guo Y, Reginsson GW, Gaddis NC, Lutz SM, Sherva R, et al. Genome-wide association study across European and African American ancestries identifies a SNP in DNMT3B contributing to nicotine dependence. Mol Psychiatry. doi: 10.1038/mp.2017.193. In press This largest GWAS meta-analysis of nicotine dependence (total N=38,602) identified DNMT3B at genome-wide significance and extended its association to heavy vs. never smoking (N=48,931 from the UK Biobank). The top SNP rs910083 was indiciated as a cis-meQTL SNP in fetal brain and a cis-eQTL SNP in adult cerebellum. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen KP, Smith AH, Herman AI, Farrer LA, Kranzler HR, Sofuoglu M, et al. A protocadherin gene cluster regulatory variant is associated with nicotine withdrawal and the urge to smoke. Mol Psychiatry. 2017;22(2):242–9. doi: 10.1038/mp.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bloom AJ, Hartz SM, Baker TB, Chen LS, Piper ME, Fox L, et al. Beyond cigarettes per day. A genome-wide association study of the biomarker carbon monoxide. Ann Am Thorac Soc. 2014;11(7):1003–10. doi: 10.1513/AnnalsATS.201401-010OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29•.Loukola A, Buchwald J, Gupta R, Palviainen T, Hallfors J, Tikkanen E, et al. A Genome-wide association study of a biomarker of nicotine metabolism. PLoS Genet. 2015;11(9):e1005498. doi: 10.1371/journal.pgen.1005498. This first GWAS of NMR was conducted using total N=1,518 and identified genome-wide significant associations of chromosome 9q13, which contained three independent signals all located in the vicinity of CYP2A6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30•.Patel YM, Stram DO, Wilkens LR, Park SS, Henderson BE, Le Marchand L, et al. The contribution of common genetic variation to nicotine and cotinine glucuronidation in multiple ethnic/racial populations. Cancer Epidemiol Biomarkers Prev. 2015;24(1):119–27. doi: 10.1158/1055-9965.EPI-14-0815. This multi-ancestry GWAS meta-analysis of cotinine and nicotine glucuronidation (total N=2,239) was the first to report genome-wide significant associations for the UGT2B10 locus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baurley JW, Edlund CK, Pardamean CI, Conti DV, Krasnow R, Javitz HS, et al. Genome-Wide Association of the Laboratory-Based Nicotine Metabolite Ratio in Three Ancestries. Nicotine Tob Res. 2016;18(9):1837–44. doi: 10.1093/ntr/ntw117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ware JJ, Chen X, Vink J, Loukola A, Minica C, Pool R, et al. Genome-Wide Meta-Analysis of Cotinine Levels in Cigarette Smokers Identifies Locus at 4q13.2. Sci Rep. 2016;6:20092. doi: 10.1038/srep20092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. [Accessed: October 23, 2017];PhenX Toolkit [database on the Internet] Available from: https://www.phenxtoolkit.org/index.php?pageLink=browse.protocoldetails&id=31001.
- 34.McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49(7):1126–1132. doi: 10.1038/ng.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lutz SM, Cho MH, Young K, Hersh CP, Castaldi PJ, McDonald ML, et al. A genome-wide association study identifies risk loci for spirometric measures among smokers of European and African ancestry. BMC Genet. 2015;16:138. doi: 10.1186/s12863-015-0299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilk JB, Shrine NR, Loehr LR, Zhao JH, Manichaikul A, Lopez LM, et al. Genome Wide Association Studies Identify CHRNA5/3 and HTR4 in the Development of Airflow Obstruction. Am J Respir Crit Care Med. 2012;186:622–32. doi: 10.1164/rccm.201202-0366OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hobbs BD, de Jong K, Lamontagne M, Bosse Y, Shrine N, Artigas MS, et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet. 2017;49(3):426–32. doi: 10.1038/ng.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castaldi PJ, Cho MH, San Jose Estepar R, McDonald ML, Laird N, Beaty TH, et al. Genome-wide association identifies regulatory Loci associated with distinct local histogram emphysema patterns. Am J Respir Crit Care Med. 2014;190(4):399–409. doi: 10.1164/rccm.201403-0569OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olfson E, Saccone NL, Johnson EO, Chen LS, Culverhouse R, Doheny K, et al. Rare, low frequency and common coding variants in CHRNA5 and their contribution to nicotine dependence in European and African Americans. Mol Psychiatry. 2016;21(5):601–7. doi: 10.1038/mp.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wessel J, McDonald SM, Hinds DA, Stokowski RP, Javitz HS, Kennemer M, et al. Resequencing of nicotinic acetylcholine receptor genes and association of common and rare variants with the Fagerstrom test for nicotine dependence. Neuropsychopharmacology. 2010;35(12):2392–402. doi: 10.1038/npp.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haller G, Druley T, Vallania FL, Mitra RD, Li P, Akk G, et al. Rare missense variants in CHRNB4 are associated with reduced risk of nicotine dependence. Hum Mol Genet. 2012;21(3):647–55. doi: 10.1093/hmg/ddr498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doyle GA, Chou AD, Saung WT, Lai AT, Lohoff FW, Berrettini WH. Identification of CHRNA5 rare variants in African-American heavy smokers. Psychiatr Genet. 2014;24(3):102–9. doi: 10.1097/YPG.0000000000000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thorgeirsson TE, Steinberg S, Reginsson GW, Bjornsdottir G, Rafnar T, Jonsdottir I, et al. A rare missense mutation in CHRNA4 associates with smoking behavior and its consequences. Mol Psychiatry. 2016;21(5):594–600. doi: 10.1038/mp.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McClure-Begley TD, Papke RL, Stone KL, Stokes C, Levy AD, Gelernter J, et al. Rare human nicotinic acetylcholine receptor alpha4 subunit (CHRNA4) variants affect expression and function of high-affinity nicotinic acetylcholine receptors. J Pharmacol Exp Ther. 2014;348(3):410–20. doi: 10.1124/jpet.113.209767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, et al. Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry. 2008;165(9):1163–71. doi: 10.1176/appi.ajp.2008.07111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen LS, Hung RJ, Baker T, Horton A, Culverhouse R, Saccone N, et al. CHRNA5 risk variant predicts delayed smoking cessation and earlier lung cancer diagnosis-a meta-analysis. J Natl Cancer Inst. 2015;107(5) doi: 10.1093/jnci/djv100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen LS, Baker TB, Piper ME, Breslau N, Cannon DS, Doheny KF, et al. Interplay of genetic risk factors (CHRNA5-CHRNA3-CHRNB4) and cessation treatments in smoking cessation success. Am J Psychiatry. 2012;169(7):735–42. doi: 10.1176/appi.ajp.2012.11101545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang JC, Cruchaga C, Saccone NL, Bertelsen S, Liu P, Budde JP, et al. Risk for nicotine dependence and lung cancer is conferred by mRNA expression levels and amino acid change in CHRNA5. Hum Mol Genet. 2009;18(16):3125–35. doi: 10.1093/hmg/ddp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang JC, Spiegel N, Bertelsen S, Le N, McKenna N, Budde JP, et al. Cis-Regulatory Variants Affect CHRNA5 mRNA Expression in Populations of African and European Ancestry. PLoS One. 2013;8(11):e80204. doi: 10.1371/journal.pone.0080204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hancock DB, Wang JC, Gaddis NC, Levy JL, Saccone NL, Stitzel JA, et al. A multiancestry study identifies novel genetic associations with CHRNA5 methylation in human brain and risk of nicotine dependence. Hum Mol Genet. 2015;24(20):5940–4. doi: 10.1093/hmg/ddv303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gallego X, Molas S, Amador-Arjona A, Marks MJ, Robles N, Murtra P, et al. Overexpression of the CHRNA5/A3/B4 genomic cluster in mice increases the sensitivity to nicotine and modifies its reinforcing effects. Amino Acids. 2012;43(2):897–909. doi: 10.1007/s00726-011-1149-y. [DOI] [PubMed] [Google Scholar]
- 52.Picciotto MR, Kenny PJ. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harb Perspect Med. 2013;3(1):a012112. doi: 10.1101/cshperspect.a012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilking JA, Stitzel JA. Natural genetic variability of the neuronal nicotinic acetylcholine receptor subunit genes in mice: Consequences and confounds. Neuropharmacology. 2015;96(Pt B):205–12. doi: 10.1016/j.neuropharm.2014.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. 2011;12(11):652–69. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miquel M, Vazquez-Sanroman D, Carbo-Gas M, Gil-Miravet I, Sanchis-Segura C, Carulli D, et al. Have we been ignoring the elephant in the room? Seven arguments for considering the cerebellum as part of addiction circuitry. Neurosci Biobehav Rev. 2016;60:1–11. doi: 10.1016/j.neubiorev.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 56.Moulton EA, Elman I, Becerra LR, Goldstein RZ, Borsook D. The cerebellum and addiction: insights gained from neuroimaging research. Addict Biol. 2014;19(3):317–31. doi: 10.1111/adb.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Strick PL, Dum RP, Fiez JA. Cerebellum and nonmotor function. Annu Rev Neurosci. 2009;32:413–34. doi: 10.1146/annurev.neuro.31.060407.125606. [DOI] [PubMed] [Google Scholar]
- 58•.Clarke TK, Adams MJ, Davies G, Howard DM, Hall LS, Padmanabhan S, et al. Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK Biobank (N=112 117) Mol Psychiatry. 2017;22(10):1376–84. doi: 10.1038/mp.2017.153. This largest ever GWAS for any alcohol phenotype reported genome-wide significant loci at more than one independent association signal at two previously implicated loci, ADH1B-ADH1C-ADH5 and KLB, and at three novel loci, GCKR, CADM2, and FAM69C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Treutlein J, Cichon S, Ridinger M, Wodarz N, Soyka M, Zill P, et al. Genome-wide association study of alcohol dependence. Arch Gen Psychiatry. 2009;66(7):773–84. doi: 10.1001/archgenpsychiatry.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schumann G, Coin LJ, Lourdusamy A, Charoen P, Berger KH, Stacey D, et al. Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc Natl Acad Sci U S A. 2011;108(17):7119–24. doi: 10.1073/pnas.1017288108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang KS, Liu X, Zhang Q, Pan Y, Aragam N, Zeng M. A meta-analysis of two genome-wide association studies identifies 3 new loci for alcohol dependence. J Psychiatr Res. 2011;45(11):1419–25. doi: 10.1016/j.jpsychires.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 62.Heath AC, Whitfield JB, Martin NG, Pergadia ML, Goate AM, Lind PA, et al. A quantitative-trait genome-wide association study of alcoholism risk in the community: findings and implications. Biol Psychiatry. 2011;70(6):513–8. doi: 10.1016/j.biopsych.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lydall GJ, Bass NJ, McQuillin A, Lawrence J, Anjorin A, Kandaswamy R, et al. Confirmation of prior evidence of genetic susceptibility to alcoholism in a genome-wide association study of comorbid alcoholism and bipolar disorder. Psychiatr Genet. 2011;21(6):294–306. doi: 10.1097/YPG.0b013e32834915c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kutalik Z, Benyamin B, Bergmann S, Mooser V, Waeber G, Montgomery GW, et al. Genome-wide association study identifies two loci strongly affecting transferrin glycosylation. Hum Mol Genet. 2011;20(18):3710–7. doi: 10.1093/hmg/ddr272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frank J, Cichon S, Treutlein J, Ridinger M, Mattheisen M, Hoffmann P, et al. Genome-wide significant association between alcohol dependence and a variant in the ADH gene cluster. Addict Biol. 2012;17(1):171–80. doi: 10.1111/j.1369-1600.2011.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang KS, Liu X, Zhang Q, Wu LY, Zeng M. Genome-wide association study identifies 5q21 and 9p24.1 (KDM4C) loci associated with alcohol withdrawal symptoms. J Neural Transm (Vienna) 2012;119(4):425–33. doi: 10.1007/s00702-011-0729-z. [DOI] [PubMed] [Google Scholar]
- 67.Edwards AC, Aliev F, Bierut LJ, Bucholz KK, Edenberg H, Hesselbrock V, et al. Genome-wide association study of comorbid depressive syndrome and alcohol dependence. Psychiatr Genet. 2012;22(1):31–41. doi: 10.1097/YPG.0b013e32834acd07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zuo L, Wang K, Zhang XY, Krystal JH, Li CS, Zhang F, et al. NKAIN1-SERINC2 is a functional, replicable and genome-wide significant risk gene region specific for alcohol dependence in subjects of European descent. Drug Alcohol Depend. 2013;129(3):254–64. doi: 10.1016/j.drugalcdep.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pan Y, Luo X, Liu X, Wu LY, Zhang Q, Wang L, et al. Genome-wide association studies of maximum number of drinks. J Psychiatr Res. 2013;47(11):1717–24. doi: 10.1016/j.jpsychires.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wetherill L, Kapoor M, Agrawal A, Bucholz K, Koller D, Bertelsen SE, et al. Family-based association analysis of alcohol dependence criteria and severity. Alcohol Clin Exp Res. 2014;38(2):354–66. doi: 10.1111/acer.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mbarek H, Milaneschi Y, Fedko IO, Hottenga JJ, de Moor MH, Jansen R, et al. The genetics of alcohol dependence: Twin and SNP-based heritability, and genome-wide association study based on AUDIT scores. Am J Med Genet B Neuropsychiatr Genet. 2015;168(8):739–48. doi: 10.1002/ajmg.b.32379. [DOI] [PubMed] [Google Scholar]
- 72.Adkins DE, Clark SL, Copeland WE, Kennedy M, Conway K, Angold A, et al. Genome-Wide Meta-Analysis of Longitudinal Alcohol Consumption Across Youth and Early Adulthood. Twin Res Hum Genet. 2015;18(4):335–47. doi: 10.1017/thg.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73•.Schumann G, Liu C, O’Reilly P, Gao H, Song P, Xu B, et al. KLB is associated with alcohol drinking, and its gene product beta-Klotho is necessary for FGF21 regulation of alcohol preference. Proc Natl Acad Sci U S A. 2016;113(50):14372–7. doi: 10.1073/pnas.1611243113. This GWAS meta-analysis was the first to report KLB as a genome-wide significant locus for daily alcohol intake (total N=98,477) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lu S, Zhao LJ, Chen XD, Papasian CJ, Wu KH, Tan LJ, et al. Bivariate genome-wide association analyses identified genetic pleiotropic effects for bone mineral density and alcohol drinking in Caucasians. J Bone Miner Metab. 2016 doi: 10.1007/s00774-016-0802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen XD, Xiong DH, Yang TL, Pei YF, Guo YF, Li J, et al. ANKRD7 and CYTL1 are novel risk genes for alcohol drinking behavior. Chin Med J (Engl) 2012;125(6):1127–34. [PMC free article] [PubMed] [Google Scholar]
- 76.Sanchez-Roige S, Fontanillas P, Elson SL, Gray JC, de Wit H, et al. 23andMe Research Team. Genome-wide association study of alcohol use disorder identification test (AUDIT) scores in 20 328 research participants of European ancestry. Addict Biol. doi: 10.1111/adb.12574. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bierut LJ, Agrawal A, Bucholz KK, Doheny KF, Laurie C, Pugh E, et al. A genome-wide association study of alcohol dependence. Proc Natl Acad Sci U S A. 2010;107(11):5082–7. doi: 10.1073/pnas.0911109107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Edenberg HJ, Koller DL, Xuei X, Wetherill L, McClintick JN, Almasy L, et al. Genome-wide association study of alcohol dependence implicates a region on chromosome 11. Alcohol Clin Exp Res. 2010;34(5):840–52. doi: 10.1111/j.1530-0277.2010.01156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kendler KS, Kalsi G, Holmans PA, Sanders AR, Aggen SH, Dick DM, et al. Genomewide association analysis of symptoms of alcohol dependence in the molecular genetics of schizophrenia (MGS2) control sample. Alcohol Clin Exp Res. 2011;35(5):963–75. doi: 10.1111/j.1530-0277.2010.01427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zuo L, Gelernter J, Zhang CK, Zhao H, Lu L, Kranzler HR, et al. Genome-wide association study of alcohol dependence implicates KIAA0040 on chromosome 1q. Neuropsychopharmacology. 2012;37(2):557–66. doi: 10.1038/npp.2011.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gelernter J, Kranzler HR, Sherva R, Almasy L, Koesterer R, Smith AH, et al. Genome-wide association study of alcohol dependence:significant findings in African- and European-Americans including novel risk loci. Mol Psychiatry. 2014;19(1):41–9. doi: 10.1038/mp.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu K, Kranzler HR, Sherva R, Sartor CE, Almasy L, Koesterer R, et al. Genomewide Association Study for Maximum Number of Alcoholic Drinks in European Americans and African Americans. Alcohol Clin Exp Res. 2015;39(7):1137–47. doi: 10.1111/acer.12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen G, Zhang F, Xue W, Wu R, Xu H, Wang K, et al. An association study revealed substantial effects of dominance, epistasis and substance dependence co-morbidity on alcohol dependence symptom count. Addict Biol. 2016 doi: 10.1111/adb.12402. [DOI] [PubMed] [Google Scholar]
- 84.Baik I, Cho NH, Kim SH, Han BG, Shin C. Genome-wide association studies identify genetic loci related to alcohol consumption in Korean men. Am J Clin Nutr. 2011;93(4):809–16. doi: 10.3945/ajcn.110.001776. [DOI] [PubMed] [Google Scholar]
- 85.Takeuchi F, Isono M, Nabika T, Katsuya T, Sugiyama T, Yamaguchi S, et al. Confirmation of ALDH2 as a Major locus of drinking behavior and of its variants regulating multiple metabolic phenotypes in a Japanese population. Circ J. 2011;75(4):911–8. doi: 10.1253/circj.cj-10-0774. [DOI] [PubMed] [Google Scholar]
- 86.Park BL, Kim JW, Cheong HS, Kim LH, Lee BC, Seo CH, et al. Extended genetic effects of ADH cluster genes on the risk of alcohol dependence: from GWAS to replication. Hum Genet. 2013;132(6):657–68. doi: 10.1007/s00439-013-1281-8. [DOI] [PubMed] [Google Scholar]
- 87.Yang X, Lu X, Wang L, Chen S, Li J, Cao J, et al. Common variants at 12q24 are associated with drinking behavior in Han Chinese. Am J Clin Nutr. 2013;97(3):545–51. doi: 10.3945/ajcn.112.046482. [DOI] [PubMed] [Google Scholar]
- 88.Quillen EE, Chen XD, Almasy L, Yang F, He H, Li X, et al. ALDH2 is associated to alcohol dependence and is the major genetic determinant of “daily maximum drinks” in a GWAS study of an isolated rural Chinese sample. Am J Med Genet B Neuropsychiatr Genet. 2014;165B(2):103–10. doi: 10.1002/ajmg.b.32213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89•.Jorgenson E, Thai KK, Hoffmann TJ, Sakoda LC, Kvale MN, Banda Y, et al. Genetic contributors to variation in alcohol consumption vary by race/ethnicity in a large multi-ethnic genome-wide association study. Mol Psychiatry. 2017;22(9):1359–67. doi: 10.1038/mp.2017.101. This GWAS of alcohol consumption in the trans-ethnic Genetic Epidemiology Research in Adult Health and Aging cohort (N = 86,627 non-Hispanic whites, Hispanic/Latinos, East Asians and African Americans) provided genome-wide significant evidence in known loci ALDH2 and ADH1B and replicable evidence for the prior GWAS-identified KLB and GCKR loci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peng Y, Shi H, Qi XB, Xiao CJ, Zhong H, Ma RL, et al. The ADH1B Arg47His polymorphism in east Asian populations and expansion of rice domestication in history. BMC Evol Biol. 2010;10:15. doi: 10.1186/1471-2148-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li D, Zhao H, Gelernter J. Strong association of the alcohol dehydrogenase 1B gene (ADH1B) with alcohol dependence and alcohol-induced medical diseases. Biol Psychiatry. 2011;70(6):504–12. doi: 10.1016/j.biopsych.2011.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li R, Zhao Z, Sun M, Luo J, Xiao Y. ALDH2 gene polymorphism in different types of cancers and its clinical significance. Life Sci. 2016;147:59–66. doi: 10.1016/j.lfs.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 93.Zhao T, Wang C, Shen L, Gu D, Xu Z, Zhang X, et al. Clinical significance of ALDH2 rs671 polymorphism in esophageal cancer: evidence from 31 case-control studies. Onco Targets Ther. 2015;8:649–59. doi: 10.2147/OTT.S76526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Okada Y, Sim X, Go MJ, Wu JY, Gu D, Takeuchi F, et al. Meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat Genet. 2012;44(8):904–9. doi: 10.1038/ng.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet. 2015;47(11):1282–93. doi: 10.1038/ng.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cordell HJ, Han Y, Mells GF, Li Y, Hirschfield GM, Greene CS, et al. International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat Commun. 2015;6:8019. doi: 10.1038/ncomms9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Agrawal A, Lynskey MT, Bucholz KK, Kapoor M, Almasy L, Dick DM, et al. DSM-5 cannabis use disorder: a phenotypic and genomic perspective. Drug Alcohol Depend. 2014;134:362–9. doi: 10.1016/j.drugalcdep.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Verweij KJ, Vinkhuyzen AA, Benyamin B, Lynskey MT, Quaye L, Agrawal A, et al. The genetic aetiology of cannabis use initiation: a meta-analysis of genome-wide association studies and a SNP-based heritability estimation. Addict Biol. 2013;18(5):846–50. doi: 10.1111/j.1369-1600.2012.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99•.Stringer S, Minica CC, Verweij KJ, Mbarek H, Bernard M, Derringer J, et al. Genome-wide association study of lifetime cannabis use based on a large meta-analytic sample of 32 330 subjects from the International Cannabis Consortium. Transl Psychiatry. 2016;6:e769. doi: 10.1038/tp.2016.36. This meta-analysis was the largest GWAS conducted for any cannabis phenotype (total N=32,330). No genome-wide significant SNP associations were identified. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Minica CC, Dolan CV, Hottenga JJ, Pool R, Fedko IO, et al. Genome of the Netherlands C. Heritability, SNP- and Gene-Based Analyses of Cannabis Use Initiation and Age at Onset. Behav Genet. 2015;45(5):503–13. doi: 10.1007/s10519-015-9723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sherva R, Wang Q, Kranzler H, Zhao H, Koesterer R, Herman A, et al. Genome-wide Association Study of Cannabis Dependence Severity, Novel Risk Variants, and Shared Genetic Risks. JAMA Psychiatry. 2016;73(5):472–80. doi: 10.1001/jamapsychiatry.2016.0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Uhl GR, Drgon T, Liu QR, Johnson C, Walther D, Komiyama T, et al. Genome-wide association for methamphetamine dependence: convergent results from 2 samples. Arch Gen Psychiatry. 2008;65(3):345–55. doi: 10.1001/archpsyc.65.3.345. [DOI] [PubMed] [Google Scholar]
- 103.Ikeda M, Okahisa Y, Aleksic B, Won M, Kondo N, Naruse N, et al. Evidence for shared genetic risk between methamphetamine-induced psychosis and schizophrenia. Neuropsychopharmacology. 2013;38(10):1864–70. doi: 10.1038/npp.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hart AB, Engelhardt BE, Wardle MC, Sokoloff G, Stephens M, de Wit H, et al. Genome-wide association study of d-amphetamine response in healthy volunteers identifies putative associations, including cadherin 13 (CDH13) PLoS One. 2012;7(8):e42646. doi: 10.1371/journal.pone.0042646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gelernter J, Sherva R, Koesterer R, Almasy L, Zhao H, Kranzler HR, et al. Genome-wide association study of cocaine dependence and related traits: FAM53B identified as a risk gene. Mol Psychiatry. 2014;19(6):717–23. doi: 10.1038/mp.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nielsen DA, Ji F, Yuferov V, Ho A, Chen A, Levran O, et al. Genotype patterns that contribute to increased risk for or protection from developing heroin addiction. Mol Psychiatry. 2008;13(4):417–28. doi: 10.1038/sj.mp.4002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nielsen DA, Ji F, Yuferov V, Ho A, He C, Ott J, et al. Genome-wide association study identifies genes that may contribute to risk for developing heroin addiction. Psychiatr Genet. 2010;20(5):207–14. doi: 10.1097/YPG.0b013e32833a2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kalsi G, Euesden J, Coleman JR, Ducci F, Aliev F, Newhouse SJ, et al. Genome-Wide Association of Heroin Dependence in Han Chinese. PLoS One. 2016;11(12):e0167388. doi: 10.1371/journal.pone.0167388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gelernter J, Kranzler HR, Sherva R, Koesterer R, Almasy L, Zhao H, et al. Genome-wide association study of opioid dependence: multiple associations mapped to calcium and potassium pathways. Biol Psychiatry. 2014;76(1):66–74. doi: 10.1016/j.biopsych.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nelson EC, Agrawal A, Heath AC, Bogdan R, Sherva R, Zhang B, et al. Evidence of CNIH3 involvement in opioid dependence. Mol Psychiatry. 2016;21(5):608–14. doi: 10.1038/mp.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li D, Zhao H, Kranzler HR, Li MD, Jensen KP, Zayats T, et al. Genome-Wide Association Study of Copy Number Variations (CNVs) with Opioid Dependence. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112•.Hancock DB, Levy JL, Gaddis NC, Glasheen C, Saccone NL, Page GP, et al. Cis-Expression Quantitative Trait Loci Mapping Reveals Replicable Associations with Heroin Addiction in OPRM1. Biol Psychiatry. 2015;78(7):474–84. doi: 10.1016/j.biopsych.2015.01.003. This study mapped prefrontal cortex cis-eQTL SNPs for the long-studied candidate gene OPRM1, and in finding an associatoin between the cis-eQTL SNP rs3778150 and heroin addiciton, it was the first to report genome-wide significant associtaion evidence for any OPRM1 SNP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113•.Schwantes-An TH, Zhang J, Chen LS, Hartz SM, Culverhouse RC, Chen X, et al. Association of the OPRM1 Variant rs1799971 (A118G) with Non-Specific Liability to Substance Dependence in a Collaborative de novo Meta-Analysis of European-Ancestry Cohorts. Behav Genet. 2016;46(2):151–69. doi: 10.1007/s10519-015-9737-3. Using the largest sample size (N=28,689) ever to investigate the association between the long-studied missense OPRM1 SNP rs1799971 and substance dependence, no significant association with observed with heroin specifically, but a modest association was found with general substance dependence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Otto JM, Gizer IR, Deak JD, Fleming KA, Bartholow BD. A cis-eQTL in OPRM1 is Associated with Subjective Response to Alcohol and Alcohol Use. Alcohol Clin Exp Res. 2017;41(5):929–38. doi: 10.1111/acer.13369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nishizawa D, Fukuda K, Kasai S, Hasegawa J, Aoki Y, Nishi A, et al. Genome-wide association study identifies a potent locus associated with human opioid sensitivity. Mol Psychiatry. 2014;19(1):55–62. doi: 10.1038/mp.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116•.Smith AH, Jensen KP, Li J, Nunez Y, Farrer LA, Hakonarson H, et al. Genome-wide association study of therapeutic opioid dosing identifies a novel locus upstream of OPRM1. Mol Psychiatry. 2017;22(3):346–52. doi: 10.1038/mp.2016.257. This GWAS identified a novel genome-wide signficant SNP association in OPRM1 with therapeutical methadone dose in 383 African Americans with opioid dependence. The association was extended to morphine dosage for treating surgical pain in an independent study sample of 241 African American children. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31(21):3555–7. doi: 10.1093/bioinformatics/btv402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wetherill L, Agrawal A, Kapoor M, Bertelsen S, Bierut LJ, Brooks A, et al. Association of substance dependence phenotypes in the COGA sample. Addict Biol. 2015;20(3):617–27. doi: 10.1111/adb.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119•.Johnson EO, Hancock DB, Levy JL, Gaddis NC, Page GP, Glasheen C, et al. KAT2B polymorphism identified for drug abuse in African Americans with regulatory links to drug abuse pathways in human prefrontal cortex. Addict Biol. 2016;21(6):1217–32. doi: 10.1111/adb.12286. This GWAS compared people who inject drugs with population controls and found a genome-wide significant and replicable association for the KAT2B SNP rs9829896 among African Americans (N=3,742 for discovery and 755 for replication). Rs9829896 was also associated with KAT2B RNA expression in postmortem prefrontal cortex of African Americans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aschard H, Vilhjalmsson BJ, Joshi AD, Price AL, Kraft P. Adjusting for heritable covariates can bias effect estimates in genome-wide association studies. Am J Hum Genet. 2015;96(2):329–39. doi: 10.1016/j.ajhg.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hartz SM, Horton AC, Hancock DB, Baker TB, Caporaso NE, Chen LS, et al. Genetic correlation between smoking behaviors and schizophrenia. Schizophr Res. 2017 doi: 10.1016/j.schres.2017.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–41. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Power RA, Verweij KJ, Zuhair M, Montgomery GW, Henders AK, Heath AC, et al. Genetic predisposition to schizophrenia associated with increased use of cannabis. Mol Psychiatry. 2014;19(11):1201–4. doi: 10.1038/mp.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Reginsson GW, Ingason A, Euesden J, Bjornsdottir G, Olafsson S, Sigurdsson E, et al. Polygenic risk scores for schizophrenia and bipolar disorder associate with addiction. Addict Biol. 2017 doi: 10.1111/adb.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hartz SM, Horton AC, Oehlert M, Carey CE, Agrawal A, Bogdan R, et al. Association Between Substance Use Disorder and Polygenic Liability to Schizophrenia. Biol Psychiatry. 2017 doi: 10.1016/j.biopsych.2017.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.O’Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40(9):1053–5. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- 127.Hancock DB, Levy JL, Gaddis NC, Glasheen C, Saccone NL, Page GP, et al. Replication of ZNF804A gene variant associations with risk of heroin addiction. Genes Brain Behav. 2015;14(8):635–40. doi: 10.1111/gbb.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sun Y, Zhao LY, Wang GB, Yue WH, He Y, Shu N, et al. ZNF804A variants confer risk for heroin addiction and affect decision making and gray matter volume in heroin abusers. Addict Biol. 2016;21(3):657–66. doi: 10.1111/adb.12233. [DOI] [PubMed] [Google Scholar]
- 129.Jackson KJ, Fanous AH, Chen J, Kendler KS, Chen X. Variants in the 15q25 gene cluster are associated with risk for schizophrenia and bipolar disorder. Psychiatr Genet. 2013;23(1):20–8. doi: 10.1097/YPG.0b013e32835bd5f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Notaras M, Hill R, van den Buuse M. A role for the BDNF gene Val66Met polymorphism in schizophrenia? A comprehensive review. Neurosci Biobehav Rev. 2015;51:15–30. doi: 10.1016/j.neubiorev.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 131.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6(4):e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133•.Markunas CA, Johnson EO, Hancock DB. Comprehensive evaluation of disease- and trait-specific enrichment for eight functional elements among GWAS-identified variants. Hum Genet. 2017;136(7):911–9. doi: 10.1007/s00439-017-1815-6. Prior studies have shown that top GWAS findings are enriched for specific functional elements (e.g., eQTLs). This study assessed a broad set of 8 functional elements and found significant enrichment for DNase sites, eQTLs, sequence conservation, enhancers, promoters, missense variants, and protein binding sites. Enrichment differences in blood vs. brain tissues were dependent on disease/trait and functional element. [DOI] [PubMed] [Google Scholar]
- 134••.Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47(9):1091–8. doi: 10.1038/ng.3367. This article presents the gene-based association method PrediXcan, which integrates genome-wide SNP genotypes and RNA-sequencing (or other ‘omics) data to build genetically regulated gene expression models and applies those models to GWAS datasets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135••.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–60. doi: 10.1126/science.1262110. The GTEx project has provided the scientific community with freely available RNA-sequencing data measured across multiple tissues from healthy individuals ( https://www.gtexportal.org/home/). Pilot GTEx analyses showed that blood vs. brain had the most distinct expression profiles among the tissue comparisons, emphasizing the importance of studying gene regulation specifically in brain to better understand the neurobiology of addiction and other psychiatric diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hernandez DG, Nalls MA, Moore M, Chong S, Dillman A, Trabzuni D, et al. Integration of GWAS SNPs and tissue specific expression profiling reveal discrete eQTLs for human traits in blood and brain. Neurobiol Dis. 2012;47(1):20–8. doi: 10.1016/j.nbd.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McKenzie M, Henders AK, Caracella A, Wray NR, Powell JE. Overlap of expression quantitative trait loci (eQTL) in human brain and blood. BMC Med Genomics. 2014;7:31. doi: 10.1186/1755-8794-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Srivastava V, Obudulu O, Bygdell J, Lofstedt T, Ryden P, Nilsson R, et al. OnPLS integration of transcriptomic, proteomic and metabolomic data shows multi-level oxidative stress responses in the cambium of transgenic hipI- superoxide dismutase Populus plants. BMC Genomics. 2013;14:893. doi: 10.1186/1471-2164-14-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kirwan GM, Johansson E, Kleemann R, Verheij ER, Wheelock AM, Goto S, et al. Building multivariate systems biology models. Anal Chem. 2012;84(16):7064–71. doi: 10.1021/ac301269r. [DOI] [PubMed] [Google Scholar]
- 140.Bouhaddani SE, Houwing-Duistermaat J, Salo P, Perola M, Jongbloed G, Uh HW. Evaluation of O2PLS in Omics data integration. BMC Bioinformatics. 2016;17(Suppl 2):11. doi: 10.1186/s12859-015-0854-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yang J, Wang S, Yang Z, Hodgkinson CA, Iarikova P, Ma JZ, et al. The contribution of rare and common variants in 30 genes to risk nicotine dependence. Mol Psychiatry. 2015;20(11):1467–78. doi: 10.1038/mp.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gabrielsen ME, Romundstad P, Langhammer A, Krokan HE, Skorpen F. Association between a 15q25 gene variant, nicotine-related habits, lung cancer and COPD among 56,307 individuals from the HUNT study in Norway. Eur J Hum Genet. 2013;21(11):1293–9. doi: 10.1038/ejhg.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Munafo MR, Timofeeva MN, Morris RW, Prieto-Merino D, Sattar N, Brennan P, et al. Association between genetic variants on chromosome 15q25 locus and objective measures of tobacco exposure. J Natl Cancer Inst. 2012;104(10):740–8. doi: 10.1093/jnci/djs191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Richmond-Rakerd LS, Otto JM, Slutske WS, Ehlers CL, Wilhelmsen KC, Gizer IR. A Novel Tobacco Use Phenotype Suggests the 15q25 and 19q13 Loci May be Differentially Associated With Cigarettes per Day and Tobacco-Related Problems. Nicotine Tob Res. 2017;19(4):426–34. doi: 10.1093/ntr/ntw260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen X, Chen J, Williamson VS, An SS, Hettema JM, Aggen SH, et al. Variants in nicotinic acetylcholine receptors alpha5 and alpha3 increase risks to nicotine dependence. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(7):926–33. doi: 10.1002/ajmg.b.30919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen LS, Saccone NL, Culverhouse RC, Bracci PM, Chen CH, Dueker N, et al. Smoking and genetic risk variation across populations of European, Asian, and African American ancestry--a meta-analysis of chromosome 15q25. Genet Epidemiol. 2012;36(4):340–51. doi: 10.1002/gepi.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Buczkowski K, Sieminska A, Linkowska K, Czachowski S, Przybylski G, Jassem E, et al. Association between genetic variants on chromosome 15q25 locus and several nicotine dependence traits in Polish population: a case-control study. Biomed Res Int. 2015;2015:350348. doi: 10.1155/2015/350348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kita-Milczarska K, Sieminska A, Jassem E. Association Between CHRNA3 and CHRNA5 Nicotine Receptor Subunit Gene Variants and Nicotine Dependence in an Isolated Population of Kashubians in Poland. Med Sci Monit. 2016;22:1442–50. doi: 10.12659/MSM.895907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sorice R, Bione S, Sansanelli S, Ulivi S, Athanasakis E, Lanzara C, et al. Association of a variant in the CHRNA5-A3-B4 gene cluster region to heavy smoking in the Italian population. Eur J Hum Genet. 2011;19(5):593–6. doi: 10.1038/ejhg.2010.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ware JJ, Aveyard P, Broderick P, Houlston RS, Eisen T, Munafo MR. The association of rs1051730 genotype on adherence to and consumption of prescribed nicotine replacement therapy dose during a smoking cessation attempt. Drug Alcohol Depend. 2015;151:236–40. doi: 10.1016/j.drugalcdep.2015.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Broms U, Wedenoja J, Largeau MR, Korhonen T, Pitkaniemi J, Keskitalo-Vuokko K, et al. Analysis of detailed phenotype profiles reveals CHRNA5-CHRNA3-CHRNB4 gene cluster association with several nicotine dependence traits. Nicotine Tob Res. 2012;14(6):720–33. doi: 10.1093/ntr/ntr283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhu AZ, Zhou Q, Cox LS, David SP, Ahluwalia JS, Benowitz NL, et al. Association of CHRNA5-A3-B4 SNP rs2036527 with smoking cessation therapy response in African-American smokers. Clin Pharmacol Ther. 2014;96(2):256–65. doi: 10.1038/clpt.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bloom AJ, Baker TB, Chen LS, Breslau N, Hatsukami D, Bierut LJ, et al. Variants in two adjacent genes, EGLN2 and CYP2A6, influence smoking behavior related to disease risk via different mechanisms. Hum Mol Genet. 2014;23(2):555–61. doi: 10.1093/hmg/ddt432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Timofeeva MN, McKay JD, Smith GD, Johansson M, Byrnes GB, Chabrier A, et al. Genetic polymorphisms in 15q25 and 19q13 loci, cotinine levels, and risk of lung cancer in EPIC. Cancer Epidemiol Biomarkers Prev. 2011;20(10):2250–61. doi: 10.1158/1055-9965.EPI-11-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Way M, McQuillin A, Saini J, Ruparelia K, Lydall GJ, Guerrini I, et al. Genetic variants in or near ADH1B and ADH1C affect susceptibility to alcohol dependence in a British and Irish population. Addict Biol. 2015;20(3):594–604. doi: 10.1111/adb.12141. [DOI] [PubMed] [Google Scholar]
- 157.Bierut LJ, Goate AM, Breslau N, Johnson EO, Bertelsen S, Fox L, et al. ADH1B is associated with alcohol dependence and alcohol consumption in populations of European and African ancestry. Mol Psychiatry. 2012;17(4):445–50. doi: 10.1038/mp.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]