


Abstract
A Co(0)-catalyzed intramolecular alkyne/benzocyclobutenone coupling through C–C cleavage of benzocyclobutenones is described. Co2(CO)8/P[3, 5-(CF3)2C6H3]3 was discovered to be an effective metal/ligand combination, which exhibits complementary catalytic activity to the previously established rhodium catalyst. In particular, the C8-substituted substrates failed in the Rh system, but succeeded with the Co catalysis. Experimental and computational studies show that the initially formed tetrahedral dicobalt-alkyne complex undergoes C1–C2 activation via oxidative addition with Co(0), followed by migratory insertion and reductive elimination to give the β-naphthol products.
Keywords: C–C activation, benzocyclobutenones, cobalt catalysis, cyclization, β-naphthol
Graphical abstract
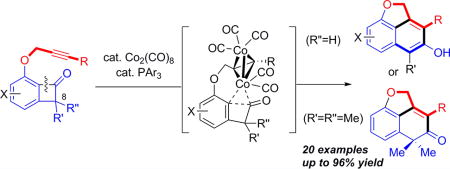
Catalytic carbon–carbon bond (C–C) activation has emerged as a useful tool for quickly building molecules with high complexity.1 Numerous elegant C–C activation transformations have been developed to date; however, the majority of them require the use of noble transition metals (TMs), such as Rh, Ru, Pd, and Ir. Clearly, it would be highly desirable if the same transformations can be catalyzed by Earth-abundant first-row TMs, which would offer a more costeffective option. In addition, complementary reactivity could be expected due to distinct properties of first-row TMs from noble metals. Despite the recent advance of Ni catalysis in various C–C activations,1r–u the use of other first-row TMs as catalysts has been rare.2 Our laboratory has been focusing on developing catalytic methods based on activation of ketone α-C–C bonds, and our prior works exclusively relied on using Rh catalysts.3 Herein, we disclose the first Co(0)-catalyzed C–C activation of ketones,4 in which an intramolecular coupling between benzocyclobutenones and alkynes was realized with an inexpensive cobalt precatalyst (Scheme 1). Preliminary mechanistic study suggests that the reaction is promoted by a tetrahedral dicobalt-mediated C–C cleavage.
Scheme 1.

“Cut and Sew” Reaction between Alkynes and Benzocyclobutenones
We have been engaged in the systematic development of “cut and sew” transformations1p for building bridged and fused rings, which involves oxidative addition of a TM into a C–C bond, followed by 2π insertion and reductive elimination. A suite of methods has been demonstrated for intramolecular “cut and sew” reactions with benzocyclobutenones,3a,b,d,m where insertion occurred selectively at the C1–C2 position. For example, the coupling with alkynes generates fused β-naphthols (see Scheme 1A).3d The DFT study by Liu and coworkers suggests that the reaction first cleaves the C1–C8 bond, and then through a decarbonylation/reinsertion sequence delivers Rh to the C2 position.5 Hence, one major limitation with the Rh system is that substitution at the C8 position is not well-tolerated, because of the steric constrain in the initial C–C cleavage step, which became a motivation to discover alternative catalytic systems. During our total synthesis of cycloinumakiol, a trace amount of the desired “cut and sew” product was detected when stoichiometric Co2(CO)8 was used.6 While the reaction was not catalytic, it inspired us to explore the use of Co2(CO)8 as the precatalyst for the intramolecular alkyne/benzocyclobutenone coupling (Scheme 1B).
Benzocyclobutenone 1a with an alkyne moiety was employed as the model substrate. After a careful survey of the reaction parameters (see Table S1 in the Supporting Information), the desired β-naphthol product (2a) was afforded in 88% yield using Co2(CO)8/P[3, 5-(CF3)2C6H3]3 as the metal/ligand combination and pyridine N-oxide as the additive. Control experiments were carried out to understand the role of each reactant (see Table 1). In the absence of Co2(CO)8, no desired product was observed (entry 2 in Table 1). The reaction without pyridine N-oxide or the ligand still afforded the desired product, albeit in a lower yield (entries 3 and 4 in Table 1). Their roles are proposed to create open coordination sites7 and assist catalyst dissociation. Co(II) complexes, such as CoBr2 and salen Co complex, are not reactive (entries 5 and 6 in Table 1). On the other hand, cobalt nanoparticles, obtained from the decomposition of Co2(CO)8, showed no catalytic activity (entry 7 in Table 1), suggesting that the reaction is likely catalyzed by homogeneous Co species. The use of other monodentate phosphine ligands, such as PPh3 or SPhos, slightly lowered the yield (entries 9 and 10 in Table 1); in contrast, bidentate ligands, such as dppb, significantly inhibited the reaction, probably through blocking reactive sites on Co (entry 8 in Table 1). Lowering the reaction temperature to 90 °C slightly decreased the yield (entry 11 in Table 1). Surprisingly, raising the reaction temperature to 130 °C gave a much lower yield (entry 12 in Table 1), likely due to the accelerated decomposition of the Co catalyst or competitive alkyne trimerization reaction.8 In the previous Rh system, adding ZnCl2 was found to be beneficial to the reactivity;3d however, it was detrimental to the Co catalyst, although the exact reason remains unclear (entry 13 in Table 1).
Table 1.
Control Experiments

| ||
|---|---|---|
| entry | change from “standard condition” | yielda (%) |
| 1 | none | 88 |
| 2 | without Co2(CO)8 | 0 |
| 3 | without pyridine N-oxide | 46 |
| 4 | without P(3,5-C6H3(CF3)2)3 | 52 |
| 5 | CoBr2 instead of Co2(CO)8 | 0 |
| 6 | salenCo(II) instead of Co2(CO)8 | 0 |
| 7 | Co nanoparticlesb instead of Co2(CO)8 | 0 |
| 8 | dppbc instead of P(3,5-C6H3(CF3)2)3 | trace |
| 9 | PPh3 instead of P(3,5-C6H3(CF3)2)3 | 75 |
| 10 | SPhos instead of P(3,5-C6H3(CF3)2)3 | 75 |
| 11 | 90 °C instead of 110 °C | 76 |
| 12 | 130 °C instead of 110 °C | 55d |
| 13 | with 1.2 equiv ZnCI2 | 33 |
Unless otherwise noted, yields were determined by 1H NMR.
Obtained by heating Co2(CO)8 alone in 1,4-dioxane at 130 °C for 6 h.
18 mol % of dppb was used.
Isolated yield.
With the optimized conditions in hand, the scope of this reaction was examined (Table 2). Similar to the [Rh(cod)Cl]2-dppp system,3d substrates containing alkyl, aryl, or alkenyl alkynes were converted into the corresponding β-naphthols in good to excellent yields. Numerous functional groups, such as aryl chloride (2k), ethers, esters (2o), olefins (2f), benzylprotected alcohols (2d), and triisopropylsilyl (TIPS) ethers (2e) were well-tolerated. Introducing a methyl group at the propargylic position decreased the yield, which is possibly caused by the steric hindrance around the alkyne (2g). Generally, less bulky and more electron-rich alkyne moieties gave higher yields. Both electron-donating (2t) and withdrawing (2s) substituents on the benzocyclobutenone rings were well-tolerated. Unfortunately, substrates with a longer tether (1u) did not undergo the “cut and sew” transformation, instead only giving alkyne trimerization products.8 Unsurprisingly, the less-reactive olefin-tethered substrate (1v) gave no reaction under the catalytic conditions.3a
Table 2.
Substrate Scopea

|
All yields are isolated yields. The yields with Rh are from ref 3d. Others are all new substrates.
Compared to the Rh system, the Co catalysis gave comparable yields, but at a lower reaction temperature (110 °C vs 130 °C). However, one very significant difference is observed in the case of C8-substituted benzocyclobutenones. Using [Rh(cod)Cl2]2-dppp, 8-methyl-substituted substrate 1q exhibited low reactivity and only afforded 23% yield; in contrast, the Co system gave a more-than-doubled yield of product 2q. In addition, 8,8-dimethyl-substituted substrate 1r was completely unreactive with the Rh system, because of the bulkiness of the C1–C8 bond; however, the Co catalysis successfully provided the corresponding benzocyclohexenone product (2r) in 52% yield (Scheme 2). The enhanced reactivity of the Co catalyst with 8-substituted benzocyclobutenones suggests that the reaction with Co is unlikely initiated by cleavage of the C1–C8 bond; therefore, it may operate through a different mechanistic pathway from the Rh system.
Scheme 2.

Unique Reactivity with the 8,8-Dimethyl-Substituted Substrat
The unique reactivity of the Co system motivated us to explore the reaction mechanism. Consequently, several experiments were carried out. Analogous to the Pauson–Khand reaction,9 mixing of Co2(CO)8 and the substrate (1a) at room temperature rapidly generated a tetrahedral dicobalt-alkyne adduct (S2) (see Scheme 3a). This species was found to be catalytically active for the intramolecular benzocyclobutenone alkyne coupling, giving comparable yields as those with Co2(CO)8 (Scheme 3b). Considering the established high stability of the dicobalt-alkyne adduct,10 it is reasonable to assume that this intermediate may be involved in the catalytic cycle. On the other hand, running the reaction under a 13CO atmosphere yielded the desired product without much 13C incorporation. Given that the exchange between free CO and the coordinated CO on Co2(CO)8 is known to be facile,11 if the reaction proceeded through cleavage of the C1–C8 bond first, the subsequent CO desertion and reinsertion would have introduced substantial 13C into the product. The absence of significant 13C incorporation disfavored the pathway involving cleavage of the C1–C8 bond.
Scheme 3.

Preliminary Mechanistic Study
Having ruled out the pathway involving C1–C8 bond cleavage, a plausible catalytic cycle is proposed for the alkyne insertion (Figure 1). The reaction begins with decarbonylation of dicobalt-alkyne complex A, which is accelerated by pyridine N-oxide,7 and followed by coordination of the ketone carbonyl to form complex B. At this stage, two pathways are possible. Path a involves cyclometalation between the alkyne moiety and the carbonyl to form intermediate C that can undergo subsequent β-carbon elimination, reductive elimination and tautomerization to give the β-naphthol product and regenerates the catalyst. Path b involves direct oxidative addition into the benzocyclobutenone C1–C2 bond with one cobalt center. The resulting cobaltacycle D then undergoes migratory insertion into the alkyne to afford intermediate E, which, upon reductive elimination and tautomerization, forms the β-naphthol product and regenerates the catalyst.
Figure 1.

Plausible catalytic cycle.
To further explore the reaction mechanism, preliminary DFT study was conducted (without considering pyridine N-oxide and the phosphine ligand).12 The computation started from the active tetrahedral dicobalt complex, and the energy diagram is depicted in Figure 2 with 3D structures of key transition states TS1 and TS2 included. The computational study shows that oxidative addition into the distal C1–C8 bond exhibits extremely high activation barrier (TS1′, 50.5 kcal/mol), because of a highly strained transition state. The cyclometalation pathway (path a) also shows a high activation energy (TS1″, 56.7 kcal/mol), which is attributed to a noncoplanar structure of Co–C–C–O four-membered ring and, consequently, less-efficient orbital overlap. In contrast, the direct cleavage of the C1–C2 bond has a much lower energy barrier (TS1, 29.9 kcal/mol), supporting path b to be a more favorable pathway.13 The migratory insertion step was found to exhibit the highest overall activation energy (TS2, 36.6 kcal/mol) to give Int3. The following reductive elimination readily occurs to form the second C–C bond with a lower activation barrier. The two cobalt centers in the resulting intermediate (Int4, −28.7 kcal/mol) are stabilized by both C═C and C═O π-bond to form a dicobalt/enone complex via a μ2-carbonyl bridge.14 As a potential competitive pathway, decarbonylation from Int3 would lead to a six-membered cobalt cycle, the activation energy of which (TS3′, 8.2 kcal/mol) is 11.5 kcal/mol higher than that of the reductive elimination step; in addition, the free energy of the subsequent intermediate (Int4′, −1.6 kcal/mol) is 27.1 kcal/mol higher than Int4. Thus, the CO deinsertion pathway is neither thermodynamically favorable nor kinetically favorable. This observation is consistent with the result of the 13C-labeling study (vide supra), in which CO exchange was not significant. Computational methods and detailed information regarding each intermediates and transition states are available in the Supporting Information.
Figure 2.

Density functional theory (DFT) calculated pathways for the intramolecular alkyne insertion.
In conclusion, we have discovered a simple Co system that can catalyze β-naphthol synthesis via an intramolecular alkyne/benzocyclobutenone coupling. To the best of our knowledge, this reaction represents the first example of Co-catalyzed C–C activation of ketones. Compared to the Rh system, the Co-catalyzed reaction not only operates at a lower temperature with similar yields, but also effectively tolerates C8-substituted substrates. Its unique mechanistic feature disclosed here should have broad implications for developing more efficient and economical C–C activation methods in the future.
Supplementary Material
Acknowledgments
This project was supported by NIGMS (No. R01GM109054). USTC is acknowledged for fellowship to Z.Z. and X.L. THU is acknowledged for fellowship to S.C. We thank Prof. Peng Liu from the University of Pittsburgh for DFT advice.
Footnotes
ASSOCIATED CONTENT
- Detailed experimental procedures, characterization of products, and computational discussion and data (PDF)
The authors declare no competing financial interest.
References
- 1.For selective reviews of C–C activation see: Rybtchinski B, Milstein D. Angew. Chem. Int. Ed. 1999;38:870–883. doi: 10.1002/(SICI)1521-3773(19990401)38:7<870::AID-ANIE870>3.0.CO;2-3.Murakami M, Ito Y. Top. Organomet. Chem. 1999;3:97–129.van der Boom ME, Milstein D. Chem. Rev. 2003;103:1759–1792. doi: 10.1021/cr960118r.Jun C-H. Chem. Soc. Rev. 2004;33:610–618. doi: 10.1039/b308864m.Satoh T, Miura M. Top. Organomet. Chem. 2005;14:1–20.Jun C-H, Park J-W. Top. Organomet. Chem. 2007;24:117–143.Necas D, Kotora M. Curr. Org. Chem. 2007;11:1566–1591.Korotvicka A, Necas D, Kotora M. Curr. Org. Chem. 2012;16:1170–1214.Jones WD. Nature. 1993;364:676–677.Seiser T, Saget T, Tran DN, Cramer N. Angew. Chem. Int. Ed. 2011;50:7740–7752. doi: 10.1002/anie.201101053.Murakami M, Matsuda T. Chem. Commun. 2011;47:1100–1105. doi: 10.1039/c0cc02566f.Seiser T, Cramer N. Org. Biomol. Chem. 2009;7:2835–2840. doi: 10.1039/b904405a.Cramer N, Seiser T. Synlett 2011. 2011:449–460.Aïssa C. Synthesis 2011. 2011:3389–3407.Murakami M, Ishida N. J. Am. Chem. Soc. 2016;138:13759–13769. doi: 10.1021/jacs.6b01656.Chen P-H, Billett BA, Tsukamoto T, Dong G. ACS Catal. 2017;7:1340–1360. doi: 10.1021/acscatal.6b03210.Dermenci A, Coe JW, Dong G. Org. Chem. Front. 2014;1:567–581. doi: 10.1039/c4qo00053f.Souillart L, Cramer N. Chem. Rev. 2015;115:9410–9464. doi: 10.1021/acs.chemrev.5b00138.C–C Bond Activation Dong G, editor. Topics in Current Chemistry. Vol. 346. Springer; Berlin: 2014. Murakami M, Chatani N, editors. Cleavage of Carbon–Carbon Single Bonds by Transition Metals. Wiley–VCH; Weinheim, Germany: 2016. Fumagalli G, Stanton S, Bower JF. Chem. Rev. 2017;117:9404–9432. doi: 10.1021/acs.chemrev.6b00599.
- 2.(a) Nakazawa H, Itazaki M, Kamata K, Ueda K. Chem. - Asian J. 2007;2:882–888. doi: 10.1002/asia.200700076. [DOI] [PubMed] [Google Scholar]; (b) Sherry BD, Fürstner A. Chem. Commun. 2009:7116–7118. doi: 10.1039/b918818e. [DOI] [PubMed] [Google Scholar]; (c) Dieskau AP, Holzwarth MS, Plietker B. J. Am. Chem. Soc. 2012;134:5048–5051. doi: 10.1021/ja300294a. [DOI] [PubMed] [Google Scholar]; (d) Perthuisot C, Edelbach BL, Zubris DL, Jones WD. Organometallics. 1997;16:2016–2023. [Google Scholar]; (e) Kurahashi T, de Meijere A. Synlett. 2005;17:2619–2622. [Google Scholar]; (f) Kurahashi T, de Meijere A. Angew. Chem. Int. Ed. 2005;44:7881–7884. doi: 10.1002/anie.200502596. [DOI] [PubMed] [Google Scholar]; (g) Zell D, Bu Q, Feldt M, Ackermann L. Angew. Chem. Int. Ed. 2016;55:7408–7412. doi: 10.1002/anie.201601778. [DOI] [PubMed] [Google Scholar]
- 3.(a) Xu T, Dong G. Angew. Chem. Int. Ed. 2012;51:7567–7571. doi: 10.1002/anie.201202771. [DOI] [PubMed] [Google Scholar]; (b) Xu T, Ko H-M, Savage N-A, Dong G. J. Am. Chem. Soc. 2012;134:20005–20008. doi: 10.1021/ja309978c. [DOI] [PubMed] [Google Scholar]; (c) Dermenci A, Whittaker RE, Dong G. Org. Lett. 2013;15:2242–2245. doi: 10.1021/ol400815y. [DOI] [PubMed] [Google Scholar]; (d) Chen P-H, Xu T, Dong G. Angew. Chem. Int. Ed. 2014;53:1674–1678. doi: 10.1002/anie.201310100. [DOI] [PubMed] [Google Scholar]; (e) Ko HM, Dong G. Nat. Chem. 2014;6:739–744. doi: 10.1038/nchem.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chen P-H, Sieber J, Senanayake C-H, Dong G. Chem. Sci. 2015;6:5440–5445. doi: 10.1039/c5sc01875g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhou X, Dong G. J. Am. Chem. Soc. 2015;137:13715–13721. doi: 10.1021/jacs.5b09799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zeng R, Dong G. J. Am. Chem. Soc. 2015;137:1408–1411. doi: 10.1021/ja512306a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Whittaker RE, Dong G. Org. Lett. 2015;17:5504–5507. doi: 10.1021/acs.orglett.5b02911. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Dermenci A, Whittaker RE, Gao Y, Cruz F, Yu Z, Dong G. Chem. Sci. 2015;6:3201–3210. doi: 10.1039/c5sc00584a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Zeng R, Chen P-H, Dong G. ACS Catal. 2016;6:969–973. doi: 10.1021/acscatal.5b02532. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Zhou X, Ko HM, Dong G. Angew. Chem. Int. Ed. 2016;55:13867–13871. doi: 10.1002/anie.201608158. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Deng L, Xu T, Li H, Dong G. J. Am. Chem. Soc. 2016;138:369–374. doi: 10.1021/jacs.5b11120. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Xia Y, Lu G, Liu P, Dong G. Nature. 2016;539:546–550. doi: 10.1038/nature19849. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Xia Y, Wang J, Dong G. Angew. Chem. Int. Ed. 2017;56:2376–2380. doi: 10.1002/anie.201611642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For stoichiometric indenylCo (I)-mediated cleavage of cyclobutenones see: Huffman MA, Liebeskind LS. J. Am. Chem. Soc. 1990;112:8617–8618.Huffman MA, Liebeskind LS, Pennington WT. Organometallics. 1992;11:255–266.For a Co(PPh3)2Cl-mediated cleavage of benzocyclobutenediones, see: Liebeskind LS, Baysdon SL, Goedken V, Chidambaram R. Organometallics. 1986;5:1086–1092.
- 5.Lu G, Fang C, Xu T, Dong G, Liu P. J. Am. Chem. Soc. 2015;137:8274–8283. doi: 10.1021/jacs.5b04691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu T, Savage NA, Dong G. Angew. Chem. Int. Ed. 2014;53:1891–1895. doi: 10.1002/anie.201310149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For selective examples of amine-N-oxide-promoted decarbonylation effect of dicobalt-alkyne adducts, see: Shambayani S, Crowe WE, Schreiber SL. Tetrahedron Lett. 1990;31:5289–5292.Jeong N, Chung Y-K, Lee B-Y, Lee S-H, Yoo S-E. Synlett. 1991;1991:204–206.
- 8.Vollhardt KPC. Angew. Chem. Int. Ed. Engl. 1984;23:539–556.For an undesired alkyne trimerization mediated by Co2(CO)8, see: Schore NE, Croudace MC. J. Org. Chem. 1981;46:5436–5438.
- 9.For selective reviews of the Pauson–Khand Reaction, see: Blanco-Urgoiti J, Añorbe L, Pérez-Serrano L, Domínguez G, Pérez-Castells J. Chem. Soc. Rev. 2004;33:32–42. doi: 10.1039/b300976a.Gibson SE, Mainolfi N. Angew. Chem. Int. Ed. 2005;44:3022–3037. doi: 10.1002/anie.200462235.Shibata T. Adv. Synth. Catal. 2006;348:2328–2336.
- 10.(a) Nicholas KM, Pettit R. Tetrahedron Lett. 1971;12:3475–3478. [Google Scholar]; (b) Melikyan GG, Nicholas KM. In: Modern Acetylene Chemistry. Stang PJ, Diederich F, editors. VCH; Weinheim, Germany: 1995. pp. 99–138. Chapter 4. [Google Scholar]; (c) Went MJ. In: Advances in Organometallic Chemistry. Stone FGA, West R, editors. Vol. 41. Academic Press; San Diego, CA: 1997. pp. 69–125. [Google Scholar]; (d) Omae I. Appl. Organomet. Chem. 2007;21:318–344. [Google Scholar]
- 11.Bor G. Inorg. Chim. Acta. 1969;3:196–200. [Google Scholar]
- 12.For a related DFT calculation of the traditional Pauson–Khand reaction, see: Yamanaka M, Nakamura E. J. Am. Chem. Soc. 2001;123:1703–1708. doi: 10.1021/ja005565+.
- 13.For a recent DFT calculation that shows selective insertion of Ni into benzocyclobutanone C1–C2 bond, see: Zou H, Wang Z-L, Huang G. Chem.—Eur. J. 2017;23:12593–12603. doi: 10.1002/chem.201702316.
- 14.For similar electronic structures of alkyne-bridged dicobalt complexes, see: Platts JA, Evans GJS, Coogan MP, Overgaard J. Inorg. Chem. 2007;46:6291–6298. doi: 10.1021/ic070278t.Overgaard J, Clausen HF, Platts JA, Iversen BB. J. Am. Chem. Soc. 2008;130:3834–3843. doi: 10.1021/ja076152c.de Bruin TJM, Michel C, Vekey K, Greene AE, Gimbert Y, Milet A. J. Organomet. Chem. 2006;691:4281–4288.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.