

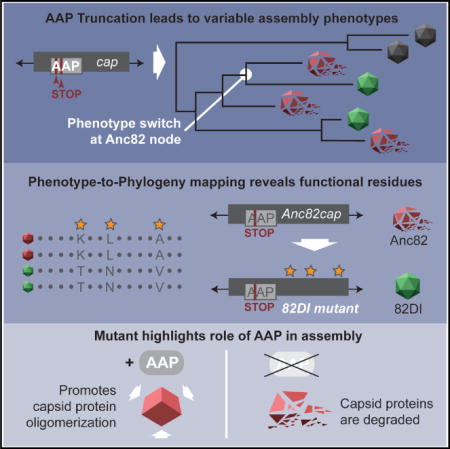
SUMMARY
The adeno-associated virus (AAV) vector is a preferred delivery platform for in vivo gene therapy. Natural and engineered variations of the AAV capsid affect a plurality of phenotypes relevant to gene therapy, including vector production and host tropism. Fundamental to these aspects is the mechanism of AAV capsid assembly. Here, the role of the viral co-factor assembly-activating protein (AAP) was evaluated in 12 naturally occurring AAVs and 9 putative ancestral capsid intermediates. The results demonstrate increased capsid protein stability and VP-VP interactions in the presence of AAP. The capsid’s dependence on AAP can be partly overcome by strengthening interactions between monomers within the assembly, as illustrated by the transfer of a minimal motif defined by a phenotype-to-phylogeny mapping method. These findings suggest that the emergence of AAP within the Dependovirus genus relaxes structural constraints on AAV assembly in favor of increasing the degrees of freedom for the capsid to evolve.
Graphical abstract
In Brief: Maurer et al. describe a phenotype-to-phylogeny mapping strategy correlating phenotypic variation in AAVs to a reconstructed phylogeny, revealing capsid structure-function relationships relevant to that phenotype. Dependence on the viral co-factor AAP for capsid assembly is examined, and capsid functional motifs, in addition to mechanistic roles of AAP, are elucidated.
INTRODUCTION
Gene therapy is gaining momentum as a viable treatment option for a broad spectrum of disease indications. Viral vectors are proving to be a highly effective modality for gene therapy, and the adeno-associated virus (AAV) is currently a preferred platform for many in vivo gene transfer applications. The AAV capsid serves as the interface between the therapeutic transgene and the host, interacting with host cell machinery and performing functions to accomplish a transduction event, including attachment to host cell glycans (Bell et al., 2012; Kern et al., 2003; Wu et al., 2000), binding to cell surface receptor(s) (e.g., AAVR) to trigger its endocytosis (Pillay et al., 2016), endosomal escape (Girod et al., 2002), trafficking to the nucleus (Grieger et al., 2006), entry through the nuclear pore complex (Nicolson and Samulski, 2014), and uncoating the single-stranded DNA (ssDNA) genome in the nucleoplasm (Thomas et al., 2004). While the capsid enacts these processes, it maintains the integrity of its genomic cargo, protecting it from both the host cellular milieu and the external environment. Assembling this highly functional protein shell is therefore the primary requirement for any virus or gene transfer vector, but AAV capsid assembly mechanisms remain poorly understood.
The AAV capsid is a non-enveloped, icosahedral 60-mer of three repeating monomers: VP1, VP2, and VP3. A single transcript expressed from the AAV cap gene containing nested open reading frames (ORFs) is alternately spliced, resulting in three distinct protein products that share C-terminal identity the length of VP3. A 1:1:10 stoichiometry of VP1:VP2:VP3 in the assembled capsid is thought to be a consequence of the relative abundance of each protein, which, in turn, is regulated by splice product abundance and a non-canonical ACG translation start codon for VP2. VP3-only particles can be assembled and packaged with the ssDNA genome; however, they are non-infectious because of the absence of a PLA2 domain encoded in the VP1-unique region of the cap ORF (Girod et al., 2002). The current model for capsid assembly suggests that, after translation, AAV2 VP proteins oligomerize into trimers or pentamers, potentially in the cytoplasm, and that further assembly into the icosahedron occurs in the nucleolus (Wistuba et al., 1995, 1997). The genome is thought to be inserted through the five-fold pore of the pre-formed capsid by the AAV Rep proteins to form the mature virion (Bleker et al., 2006).
The assembly-activating protein (AAP) is a non-structural protein expressed from a non-canonical CTG start codon of an overlapping reading frame embedded within the cap gene (Sonntag et al., 2010). AAP has so far been implicated as an absolute requirement for assembly of AAV virions composed of only VP3 units for several serotypes, but AAP’s mechanism of action in the assembly process is still unknown. AAP has been suggested to have a scaffolding function (Naumer et al., 2012), but evidence supporting this function is lacking. AAP of AAV2 (referred to here as AAP2) has been shown to shuttle VP3 proteins to the nucleolus (Earley et al., 2015), but recent findings demonstrate that AAV2 is unique in this respect, with no other AAV serotype demonstrating strict nucleolar assembly (Earley et al., 2017). To date, most studies of AAP’s requirement for particle assembly employ VP3-only particles, and studies of the full VP1, VP2, and VP3 capsid were only very recently published (Grosse et al., 2017).
During the infectious process, the AAV capsid is subject to various selective pressures (host immunity, tissue tropism, etc.) that drive evolution, which has led to the biodiversity of serotypes and naturally occurring variants the AAV field has sampled and studied extensively (Gao et al., 2005). Indeed, this structural variation correlates with phenotypic variation in aspects relevant to AAV’s use as a gene transfer vector; e.g., production yields (Vandenberghe et al., 2009), transduction efficiency (Gao et al., 2004; Thomas et al., 2004), and tissue and cell tropism (Foust et al., 2009; Landegger et al., 2017). Based on these observations, numerous efforts to further modulate AAV function by engineering its capsid are underway (Grimm and Zolotukhin, 2015; Kotterman and Schaffer, 2014; Vandenberghe et al., 2009). This process, both in nature and in the laboratory, is constrained substantially because it may affect a multitude of functionalities. One primary constraint is that of assembly because no mutation is allowable that prevents the expression or interferes with multimerization of the viral proteins. It is obvious that only if and when an assembly can be built from a given VP monomer, the selective advantage of a (set of) de novo mutation(s) can be evaluated, either during natural evolution or in a lab-based engineering context (Lochrie et al., 2006). Notwithstanding the extensive research on AAV capsid biology, AAV assembly, its cofactors, and structural requirements have not been extensively studied.
This basic premise has been the focus of our past studies as well as those presented here. Specifically, we previously demonstrated that, through ancestral sequence reconstruction of the AAV capsid, one could preserve the assembly function while modulating 8.6%–40.5% of its sequence. This work was built on the assumption that ancestral AAVs were assembly-competent and that our computational and statistical modeling effort was able to approximate the molecular state of these putative evolutionary intermediates. Indeed, we predicted the primary structure of 9 putative bifurcation points within the lineages of AAV serotypes 1–3 and 6–9, each of which, to varying degrees, demonstrated assembly, viral genome packaging, and in vitro infectivity (Zinn et al., 2015). These reagents now provide us with a toolset to interrogate viral and vector phenotypes across much shorter genetic distances than previously possible.
Here, we leveraged this toolkit of AAVs to interrogate AAP function, aiming to deepen our understanding of AAP’s role in the assembly process and delineate structural determinants within the capsid relevant to assembly and AAP function. These results will further inform engineering approaches to improve AAV technology relevant to gene therapy applications, guide optimization of AAV vector manufacturing, and provide an approach named phenotype-to-phylogeny mapping to elucidate structure-function relationships in complex assemblies such as icosahedral viral capsids.
RESULTS
The Requirement for AAP Ranges Broadly across All AAV Clades
To test whether AAP is required to assemble capsids from the full complement of VP proteins (i.e., VP1, VP2, and VP3), AAP expression was abolished from rep-cap trans plasmids by an early stop codon in the AAP reading frame, a silent mutation in VP (Figure 1A; AAPstop60). AAPstop60s were generated for 12 serotypes, including at least one member of each AAV clade, with the aim of a comprehensive assessment of AAP requirements across mammalian AAV serotypes. Considering AAP’s non-canonical CTG start codon, AAPstop60 mutations were positioned so that they would be sufficiently downstream of potential alternate start codons but upstream of regions shown to be essential for AAP2 function (Naumer et al., 2012). To verify loss of AAP protein, a hemagglutinin tag was inserted in the C-terminal region of the AAP ORF in two representative serotypes (Figure 1A; AAP-hemagglutinin [HA]). Whole-cell lysates transfected with these constructs were analyzed by western blot (Figure 1B), confirming that AAPstop60 results in loss of full-length AAP or any shorter protein product translated from alternate starts. A double band in the AAP-HA lane supports the likelihood of additional downstream start codons and corroborates the late placement of the stop codons (Figure 1B).
Figure 1. The Requirement for AAP Ranges Broadly across All Clades.
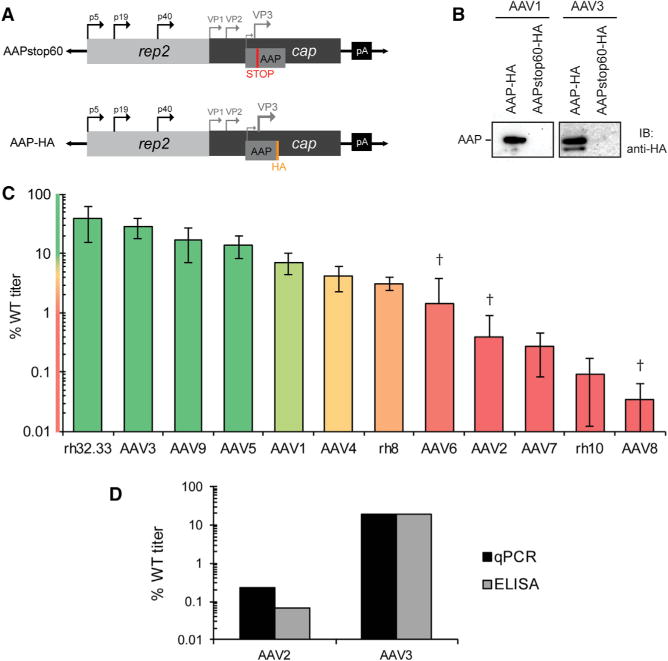
(A) AAPstop60 and AAP-HA construct maps. Black arrows, transcription start sites at p5, p19, and p40 viral promoters; gray arrows, cap gene product translation start codons. The early stop codon (red) was introduced by site-directed mutagenesis (°60 amino acids [aa]) into the AAP ORF. The HA tag (orange) was inserted at a conserved BsiWI site near the AAP C terminus.
(B) AAV1 and AAV3 AAP-HA constructs were generated in both the WT AAP and AAPstop60 context. Lysates from transfected HEK293 cells were harvested after 36 hr, clarified by centrifugation, and interrogated for AAP by western blot with anti-HA antibody.
(C) Vector produced from WT or AAPstop60 rep-cap constructs was titrated by qPCR to quantify DRPs. AAPstop60 titers are reported as a percentage of each WT serotype titer and represent the average of at least 3 independent experiments ± SEM. Bar color corresponds to heatmap color on the y axis. Dagger indicates AAPstop60 titer below background level for at least one trial (no cap gene control). See Table S2 for statistics.
(D) Vector produced from WT and AAPstop60 constructs of AAV2 and AAV3 were titrated by A20 capsid ELISA and reported as a percentage of WT titer (average of two experiments; see Table S1 for individual data).
See also Figure S4.
Recombinant AAV vectors were produced from AAPstop60 and wild-type (WT) AAP plasmids and titrated by qPCR quantifying DNase-resistant particles (DRPs). AAPstop60 vector titers reveal that, when all three VP proteins are present, AAP is not strictly required to assemble the virion in several serotypes (Figure 1C). Rather, the AAP requirement ranges broadly across serotypes, with AAPstop60 vectors producing as high as 39% of the WT titer for rh32.33 and as low as 0.035% of the WT titer for AAV8. This observation is in contrast with previous findings demonstrating AAP’s absolute requirement for assembling VP3-only capsids, in particular AAV9 and AAV1 (Sonntag et al., 2011).
The advantage of DRP titration is the ability to quantify virus of any capsid serotype with the same vector genome absent of differential bias in measurement. However, DRP measures the amount of assembled particles that also underwent viral genome packaging, a process that occurs downstream of capsid assembly. Moreover, DRP does not assess for non-packaged, empty AAV virions. To directly assay capsid assembly and rule out the possibility that serotypes with low AAPstop60 titers were due to a packaging defect, we performed an A20 capsid ELISA, which recognizes a conformational epitope only present in assembled AAV2 capsids. A20 cross-reacts with AAV3, allowing us to assay assembly directly for an AAV that requires AAP (AAV2) and one that accomplishes assembly in the AAPstop60 context (AAV3). A20 ELISA data for both serotypes corroborated the DRP indirect measure of assembly (Figure 1D). Given that there are no reagents available that allow comparative particle titration on a protein level across serotypes, based on the AAV2 versus AAV3 data, we assumed that AAPstop60 constructs do not influence viral genome packaging, and DRP measures were used from here on as a surrogate for assembly.
A Role for AAP in VP Protein Stability
To ensure that the observed range of AAP dependence for assembly was not due to variation in VP translation efficiencies imposed by alternate codon usage in the AAPstop60 mutants, we interrogated VP protein levels produced by WT and AAPstop60 constructs. The B1 monoclonal antibody detects a conserved linear epitope on VP proteins under denaturing conditions across all AAVs tested in this study, except AAV4 and rh32.33, allowing us to assay nearly all serotypes. No appreciable difference in VP2 or VP3 protein levels and a slight decrease in VP1 levels were observed for AAPstop60s that produce 10% or higher of their respective WT titers (Figure 2A), whereas AAPstop60s with titers below this threshold showed a dramatic decrease in VP protein levels (Figure 2B). To examine whether the observed decreases in VP levels were due to a potential translational defect, we co-expressed AAP2 in trans with AAPstop60 (rescue; Figure 2B). Appreciable restoration of VP protein was observed for all affected serotypes. Furthermore, no difference was observed between WT, AAPstop60, or rescue transcript levels in all three splice isoforms (Figure 2C), indicating that VP protein loss in the absence of AAP most likely occurs post- or co-translationally. To interrogate degradation as the mechanism for instability, we treated AAV8 AAPstop60-transfected cells with increasing concentrations of the proteasome inhibitor bortezomib, the E1 inhibitor MLN7243, or the vacuole-specific H+ ATPase inhibitor bafilomycin (Figure 2D). This allowed us to examine the earliest and latest steps of the ubiquitin-proteasome pathway, as well as late steps of lysosomal degradation or autophagy by inhibiting the required acidification. Inhibiting lysosomal acidification resulted in a mild but dose-dependent rescue of AAV8 VP3 protein. Proteasomal inhibition is accompanied by a robust rescue in AAV8 VP proteins in a dose-dependent manner, but this was not concomitant with rescue of assembled capsids (Figure 2E). E1 inhibition provided equally mild to moderate VP rescue independent of drug concentration. Collectively, these results suggest that instability of VP proteins in the absence of AAP can primarily be attributed to proteasomal degradation and that this may in part be ubiquitin-independent. Lysosomal or autophagosomal degradation may also degrade a proportion of VP proteins.
Figure 2. VP Protein Levels in Natural Serotypes.
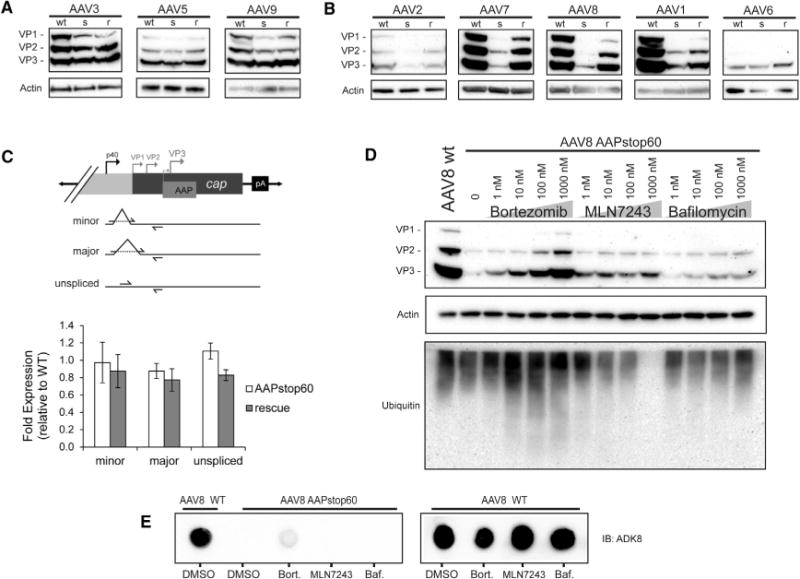
(A and B) HEK293 cells were transfected with helper and rep-cap plasmids as denoted above the lanes: WT, WT AAP; s, AAPstop60; r, AAPstop60 plus a CMV-driven AAP2. Whole-cell lysates were harvested after 36 hr and clarified by centrifugation, and VP levels were interrogated with B1 antibody (VP1/2/3). Actin was used as a loading control. Serotypes of rep-cap plasmids are indicated above each blot, with AAPstop60 titers ≥ 10% of the WT titer (A) and AAPstop60 titers < 10% of the WT titer (B).
(C) RNA quantified from AAV2 transfections as above, normalized to GAPDH, and reported relative to AAV2 WT. Minor and major splice isoforms as well as unspliced transcript levels were examined as denoted on the x axis and diagrammed on the right; primers are indicated by arrows. The graph represents the mean of three independent experiments ± SEM; there is no statistically significant difference between groups (see Table S2 for statistics).
(D) HEK293 cells transfected with helper and AAV8 WT or AAPstop60 rep-cap plasmids as indicated. At 24 hr, AAPstop60-transfected cells were treated with concentrations of bortezomib, MLN7243, or bafilomycin as indicated above the lanes and incubated for an additional 8 hr before whole-cell lysates were harvested as in (A) and (B). VP levels were interrogated by western blot with B1 antibody (top). The lot was stripped and reprobed for ubiquitin (bottom). Actin was used as a loading control (center).
(E) Lysates from (D) treated with DMSO or 1 μM bortezomib, MLN7243, or bafilomycin as listed below were assayed for the presence of assembled capsids by dot blot with the ADK8 antibody (which recognizes a conformational epitope only present in assembled AAV8 capsids). The experiment was repeated in the presence of AAP to control for any effects of the drugs on capsid assembly (right).
See also Figure S1.
In an attempt to examine the rate of AAV8 VP degradation, we blocked protein synthesis with cycloheximide (CHX) and harvested protein lysates at progressive time points (Figure S1A). As expected in AAPstop60 lysates, VP protein levels were too low to detect, even without CHX treatment and despite long exposure times with highly sensitive detection reagents. However, in the presence of AAP, VP protein levels remained consistent over all time points of CHX treatment. This is likely because capsids are assembled rapidly in the presence of AAP, and because assembled VP proteins are not susceptible to degradation, a VP band persists. Grosse et al. (2017) performed a similar experiment and found that unassembled AAV2 VP proteins have a half-life of 2–3 hr in the absence of AAP and that VP proteins are rapidly assembled into capsids when AAP is present. Importantly, when AAP is present in these experiments, CHX treatment also results in loss of AAP protein, so it is difficult to determine whether AAP directly affects monomer stability or whether stability is solely a result of assembly into the stable icosahedron. We were not able to recapitulate these experiments or results for AAV8, likely because of serotypical differences between AAV2 and AAV8 and/or experimental differences that could not be controlled for.
Given the spectrum of AAP phenotypes observed across the major clades, we similarly tested 9 putative evolutionary intermediates (AncAAVs) to the major AAV serotypes to gain insight into which elements of VP structure either impose the observed requirement for AAP or impart an ability for some VPs to perform these functions independently (Zinn et al., 2015). As with the nat ural serotypes, a broad range of requirements for AAP was observed for the AAPstop60 AncAAVs (Figure 3A).
Figure 3. The Requirement for AAP Exhibits Branch Specificity in the Context of a Putative AAV Phylogeny.
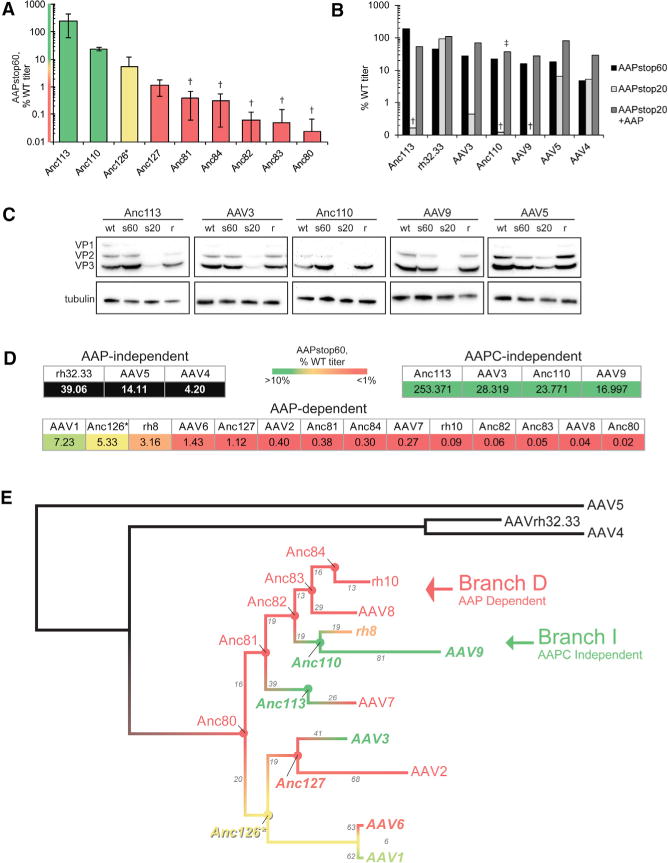
(A) AAPstop60s were generated for the nine putative ancestral AAVs. Vector produced from WT or AAP-stop60 rep-cap constructs was titrated by qPCR to quantify DRPs. AAPstop60 titers are reported as a percentage of each serotype’s WT titer and represent the mean of at least 3 independent experiments ± SEM. Bar color corresponds to heatmap color on the y axis and is also used in (D) and (E).
Dagger indicates AAPstop60 titer below background (no cap gene control) for at least one trial, see Table S2 for statistics; asterisk indicates Anc126 produces at consistently low titers (below 1e9 genome counts (GC)/mL) for the WT and AAPstop60.
(B) AAPstop20s were generated for AAV4 and all AAV variants with AAPstop60 titers ≥ 10% by introducing early stop codons at ~20 amino acidsa into the AAP ORF. Vector was produced and titrated as in (A), adding the AAPstop20 condition (light gray bars) and AAPstop20 plus a CMV-driven construct expressing a homologous AAP (dark gray bars) (mean of two experiments; see Table S3 for individual data). Dagger indicates titer below background (no cap gene control) for at least one trial; Double dagger indicates rescue performed with AAP2.
(C) HEK293 cells were transfected with helper and rep-cap plasmids as denoted above the lanes: WT, WT AAP; s60, AAPstop60; s20, AAPstop20; r, AAPstop20 plus CMV-driven homologous AAP. Whole-cell lysates were harvested after 36 hr, and VP levels were interrogated by western blot. Tubulin was used as a loading control.
(D) Categorization of AAP phenotypes. Boxes below each serotype indicate the AAPstop60 percentage of the WT titer. Black boxes indicate AAP-independent (AAPstop20 titer is ≫1%). Serotypes with AAPstop60 titers > 10% (green) indicate assembly in the absence of the C-terminal two-thirds of AAP (AAPC-independent).
(E) Reconstructed AAV phylogeny; branches are colored as in (D). Gray numbers on branches indicate the number of divergent amino acids between the two serotypes flanking the branch segment.
See also Figures S1–S3 and S5.
Although the AAPstop60 early stop codon is placed upstream of domains shown to be required for AAP2 function, it is downstream of a highly conserved region (residues 52–57) in AAP (Figure S2, conserved core). This domain was shown by deletion analyses to be important for AAP2 function but not sufficient to assemble VP3-only capsids without more C-terminal portions of AAP present (Naumer et al., 2012). Because AAPs in other serotypes have not yet been tested by deletion analyses, and because the algorithms that generated the An-cAAVs were applied only to the VP ORF and may have unpredictable consequences on AAP (Zinn et al., 2015), we wanted to examine whether a partially functional N-terminal AAP (AAPN) was expressed from some AAPstop60 constructs, contributing to the observed varying requirement for AAP across the 21 AAVs we examined. For the 6 AAVs whose AAP-stop60 produces at least 10% of the WT titer and for AAV4, recently demonstrated to assemble VP3-only capsids without AAP (Earley et al., 2017), further upstream stop constructs (AAPstop20) were generated, placing the early stop codon at residue ~23 in the AAP ORF (silent mutations in VP). Of these, AAV5, rh32.33, and AAV4 AAPstop20 produce virus whereas AAV3, AAV9, Anc110, and Anc113 do not (Figure 3B). Although the B1 antibody does not detect AAV4 and rh32.33, levels of VP protein produced from the remaining AAPstop20 constructs mirror the titer (Figure 3C). Taken together with Figure 2, these results demonstrate that stability is a serotype-specific property of VP proteins that fall into one of three categories: independently stable, requiring only AAPN for stability, or requiring full-length AAP. Our results clearly illustrate a role for AAP in VP stability and provide an explanation for the broad range of requirement for AAP. The discrepancy between AAP-stop60 and WT titers, particularly for serotypes requiring only AAPN, points to additional shortcomings in some serotypes’ VPs for which AAP compensates and potentially discrete functions contained primarily in AAPN versus the C-terminal two-thirds of AAP (AAPC). Taking into consideration VP stability, AAPstop60 titers, and AAPstop20 titers, for clarity, we categorized the AAP phenotypes as AAP-independent, AAPC-independent, and AAP-dependent (Figure 3D). Additionally, we show that the AAPC-independent phenotype is a property of the VP proteins and not a result of a fully functional AAPN by demonstrating that AAPN of an AAPC-independent serotype cannot rescue viral production of an AAP-dependent serotype (Figure S3).
To examine whether serotypes with different AAP phenotypes’ VPs are subject to the same mechanisms of degradation, we blocked degradation of AAV3 proteins in the same manner as previously performed for AAV8 (Figure S1B). Proteasome inhibition with bortezomib provided dose-responsive robust rescue as with AAV8, and E1 inhibition with MLN7243 rescued VP at the highest dose. Unlike AAV8, inhibiting lysosomal acidification with bafilomycin robustly rescued AAV3 VP levels in a dose-dependent fashion, indicating that AAPC-independent serotypes (or at least AAV3) may be more susceptible to lysosomal degradation or autophagy. These results could also suggest that AAPN somehow promotes the proteasome as the primary means of degradation, whether by blocking lysosomal degradation or by other means, because AAPN is present in the AAV8 AAPstop60 lysates but absent in the AAV3 AAPstop20 lysates.
AAPC Does Not Affect Virion Morphology, Infectivity, or Stability
Although AAPN alone facilitates the production of appreciable quantities of virus for many serotypes, we next addressed whether AAPN-assembled particles retain the proper morphology as well as infectivity functions. Transmission electron microscopy (TEM) imaging of AAV3 WT and AAV3AAP-stop60 vectors indicates identical gross particle morphologies (Figure S4A). To examine whether AAPC loss affected infection capabilities, AAV9, AAV3, and their AAPstop60 vectors were tested on HEK293 cells in culture (Figures S4B and S4C), demonstrating that viruses assembled without AAPC retain infection capabilities. Additionally, we tested the melting temperature of these particles, and no appreciable difference was observed (Figure S4D).
The Requirement for AAP Exhibits Branch Specificity in the Context of a Putative AAV Phylogeny
As a next step toward identifying the VP structure responsible for assembly functions, we sought an overview of how AAP phenotypes diverge across the wide genetic range of AAV capsids tested, aiming to identify phenotypic differences across small genetic distances. AAP phenotypes of the 12 natural serotypes and the nine ancestral variants were correlated to the reconstructed phylogeny (Figure 3E). This revealed branch-specific AAP dependence profiles, with phylogenetic nodes illustrating a clear divergence in AAP phenotype. Among other apparent trends, Anc80, Anc81, Anc82, Anc83, and Anc84 comprise a fully dependent lineage that terminates in AAV8 and rh.10 and will thus be referred to as branch D (Figure 3E, red arrow). At the Anc82 node, a phenotypic switch from AAP-dependent to AAPC-independent is observed in its successor Anc110. The serotypes that diverge from Anc110, rh8 and AAV9, are also AAPC-independent; this branch was named branch I (Figure 3E, green arrow).
Phenotype-to-Phylogeny Mapping Analysis Reveals a Set of Residues Functioning in AAPC-Independent Assembly
Our observation that AAP phenotypes have branch-specific trends within the phylogeny presented the opportunity for a facile method to identify elements of the VP structure critical for assembly functions. Given that the VP sequence diverges by small increments along each of these branches, we hypothesized that a set of residues homologous only within the members of their respective branch were likely to functionally contribute to capsid assembly. To this end, a multiple sequence alignment was generated with the branch D and branch I members (Figure S5). Within the alignment, a total of 149 positions varied; however, at only twelve positions is the residue conserved within branch I, with a different but shared identity on branch D. Of these twelve, eight individual residues and two pairs of adjacent residues comprise 10 sites on VP. At some of these sites, residue identity diverges within branch I members; however, they share a chemical property that contrasts with branch D. For example, site 1 is a basic lysine in branch D serotypes compared with, in branch I, a threonine for Anc110 and rh8 and a serine in AAV9, both hydroxylic residues. The approach to identify a phenotypic switch along a reconstructed phylogeny and then interrogate the conserved differences across the two diverging lineages for the phenotype of interest to map the structural determinant(s) responsible was named phenotype-to-phylogeny mapping.
A Functional Motif Conferring AAPC-Independent Assembly and VP Protein Stability Is Transferable to a Heterologous Capsid
To test whether the 10 sites constitute a motif that functions in capsid assembly, we aimed to engraft the branch I identities onto a member of branch D and test whether the resulting hybrid gains AAPC-independent assembly function. Anc82, the node from which branch I diverges, was chosen as the background for these mutations; as the closest relative to the branch I serotypes, it is more likely to tolerate several targeted mutations and retain functionality than a more distant relative. All ten sites in Anc82 were mutated to branch I identities en masse by site-directed mutagenesis, creating a variant we named 82DI (Figure 4A). As predicted, 82DIAAPstop60 gained AAPC-independent assembly function (Figure 4B). To determine the minimal motif required to confer this phenotype, each site was individually reverted back to its branch D identity. All revertants are AAP-dependent (Figure 4C), corroborating that the 10 sites identified using phenotype-to-phylogeny mapping not only constitute a functional motif critical for capsid assembly but also comprise the minimal motif required for AAPC-independent assembly in this subset of serotypes.
Figure 4. Characterization of 82DI, an AAPC-Independent Gain-of-Function Mutant.
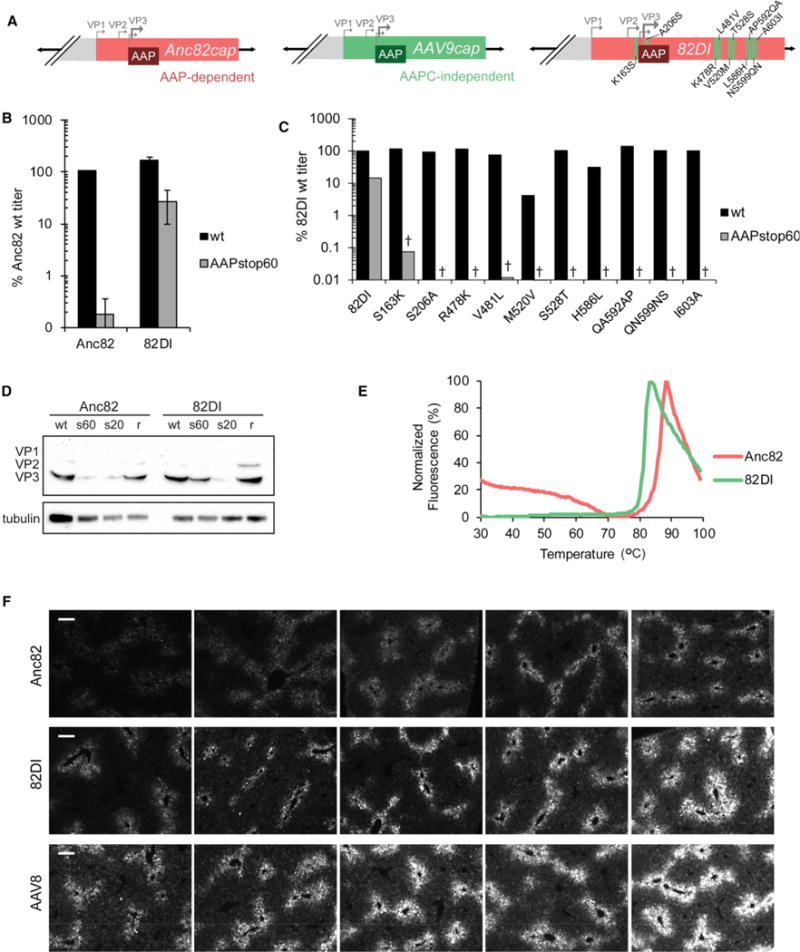
(A)82DI was generatedby introducing branch I residueidentities into an Anc82rep-cap plasmidby site-directedmutagenesis. A206Sisa silent mutation intheAAPORF.
(B) Vector produced from Anc82, 82DI, and their AAPstop60s was titrated by quantifying DRPs and is reported here as a percentage of the Anc82 WT titer. The graph represents the mean of four independent experiments ± SEM. See Table S2 for statistics.
(C) Each site in 82DI and 82DIAAPstop60 was reverted to its Anc82 identity individually by site-directed mutagenesis. Vector titers quantifying DRPs are reported here as a percentage of the 82DI WT titer and represent the mean of 2 trials (see Table S4 for individual data).
(D) HEK293 cells were transfected with helper and rep-cap plasmids as denoted above the lanes: WT, WT AAP; s60, AAPstop60; s20, AAPstop20; r, AAPstop20 plus CMV-driven AAP2. Whole-cell lysates were harvested after 36 hr, and VP levels interrogated by western blot. Tubulin was used as a loading control.
(E) Normalized SYPRO Orange fluorescence signals obtained for Anc82 and 82DI.
(F) GFP fluorescence detected in murine livers 30 days after systemic injection with 1 × 1011 VG/mouse of Anc82, 82DI, or AAV8. Each image is representative of an individual animal. Scale bars, 100 μm.
See also Figures S5 and S6.
We next assessed whether this dependent to independent (DI) motif affects VP protein stability. Consistent with other AAP-dependent serotypes (Figure 2B), Anc82 exhibits a dramatic decrease in VP levels under the AAPstop60 condition whereas 82DIAAPstop60 does not (Figure 4D). To properly categorize 82DI’s AAP phenotype, we generated AAPstop20 for Anc82 and DI and observed loss of protein in 82DIstop20, indicating that 82DI is AAPC-independent (Figure 4D).
To assess the broader effect of AAPC-independent assembly on the capsid as a whole, we further characterized 82DI’s biophysical properties and transduction capabilities compared with its parental strain Anc82. The melting temperature (Tm) of 82DI is 5 C lower than that of Anc82 (Figure 4E), a primary indication of a biophysically distinct entity (Pacouret et al., 2017). Considering the marked changes in Anc82 versus 82DI’s Tm and AAP phenotypes, we were compelled to test the infectivity of both variants. 82DI retains infectivity, and transduction may be increased moderately compared with Anc82 both in vitro (Figure S6A) and in vivo (Figures 4F and S6B).
Candidate Residues Contributing to AAP-Independent Assembly Lie at the VP Trimer Interface
To examine how this motif influences particle assembly, we mapped where these residues lie within the three-dimensional fold of VP and within an assembled capsid. Although crystal structures of AncAAVs are not available, the terminal branch D (AAV8) and branch I serotypes (AAV9) have been solved (DiMattia et al., 2012; Nam et al., 2007) and were used as surrogates to map the DI motif. Two of the 10 sites lie in the unstructured region of the VP N terminus, but only site 1 is outside of VP3. Of the eight sites within the structured region of VP, seven of them map to the three-fold interface of a VP trimer and contact a neighboring monomer (Figures 5A–5C). Comparing an AAV8 (AAP-dependent) trimer with an AAV9 (AAPC-independent) trimer at the atomic level, most of these sites exhibit compelling evidence for stronger inter-monomeric interactions within an AAPC-independent trimer compared with the AAP-dependent trimer (Figures 5D–5F). For example, a conserved glutamic acid forms a salt bridge with an adjacent monomer’s histidine at site 7 in the AAV9 trimer that cannot form with the leucine at site 7 in AAV8 (Figure 5D). On AAV9, a hydrogen bond forms between a conserved asparagine and a glutamine at site 8 of an adjacent VP monomer. On AAV8, this bond is unable to materialize because of a Gln-to-Ala substitution (Figure 5E). Site 10 lies at the three-fold axis, and beneath a conserved phenylalanine, the valine residues in AAV9 create a much larger network of hydrophobic interactions than the alanines in AAV8 (Figure 5F). Moreover, the site 10 interaction exists between all three monomers of the trimer simultaneously. These observations suggest that, in AAPC-independent serotypes, this motif aids in trimer stabilization and possibly nucleates capsid assembly in the absence of a full-length AAP.
Figure 5. Sites of Interest Map to the Trimer Interface, Suggesting Stronger Inter-monomeric Interactions in AAPC-Independent Serotypes.
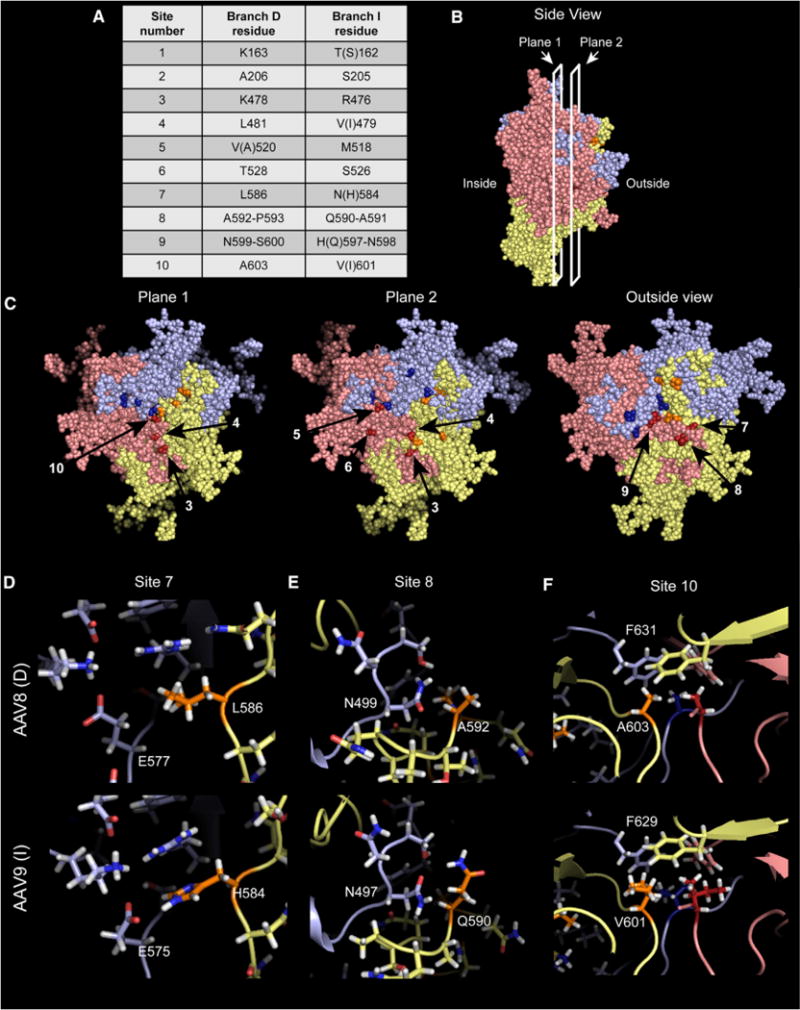
(A) Summary of the ten sites (twelve residues) identified by branch D/branch I multiple sequence alignment, numbered from the VP1 start codon. Branch D residues include Anc80, Anc81, Anc82, Anc83, Anc84, AAV8, and rh10; branch I residues include Anc110, rh8, and AAV9. Variations in identity for AAV8 and AAV9 are indicated in parentheses and are exclusive to these members of their respective branches. (B and C) Side view of an AAV9 trimer (B), showing planes of view for (C). Each monomer is represented as one color and each site of interest in a darker shade of that color. Numbered arrows indicate each site within the red monomer.
(D–F) Atomic-level view of sites 7 (D), 8 (E), and 10 (F) in AAV8 and AAV9 trimers.
AAPC-Independent Capsomere Nucleation
Next we hypothesized that VPs of AAPC-independent AAVs are able to associate into oligomers in the absence of AAPC, whereas AAP-dependent serotypes’ VPs do not strongly associate unless a full-length AAP is present. To test this, we evaluated VP-VP interaction of AAP-dependent and AAPC-independent AAVs by co-immunoprecipitation of VP3 with HA-tagged VP1 as bait (Figures 6A and 6B). The AAPC-independent VP1s tested, AAV3 and 82DI, were able to co-precipitate VP3 in the absence of a full-length AAP despite low VP3 levels in the input. Conversely, neither AAP-dependent AAV2 nor Anc82 VP1 co-precipitated significant VP3 despite appreciable input levels (Figure 6B). Addition of AAP2 allowed VP3 co-precipitation in AAP-dependent serotypes and co-precipitated an unknown VP species between VP2 and VP3’s predicted molecular weights in the AAPC-independent serotypes (marked with asterisk, Figure 6B). These may be VP2-like proteins translated from an alternate start codon or VP1 N-terminal cleavage/degradation products stabilized by AAP. Despite the appreciable increase in Anc82 and AAV3 VP3 input levels under the +AAP condition, these data support the above hypothesis.
Figure 6. AAP Promotes VP-VP Interactions.
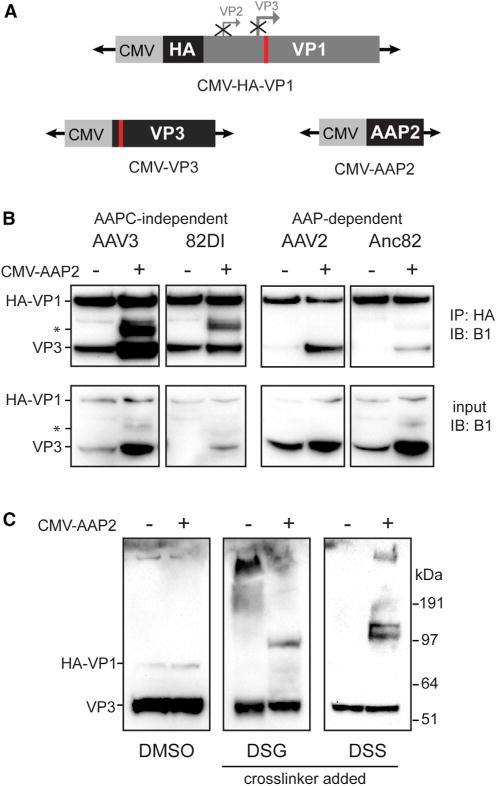
(A) Schematic of expression constructs for AAP2 and VP1 and VP3 of AAV2, AAV3, Anc82, and 82DI. In CMV-HA-VP1, the VP2 and VP3 start codons were modified to silence their expression, and the AAPstop60 mutation (red rectangle) was included.
(B) HEK293 cells were transfected with CMV-HA-VP1 and CMV-VP3 of the serotype indicated above each lane, with or without CMV-AAP2, and lysates were harvested after 48 hr. IP was performed using anti-HA antibody. VPs were detected by western blot using the B1 antibody.
(C) Lysates from CMV-HA-VP1 and CMV-VP3, with or without CMV-AAP2-transfected (all AAV2 proteins) HEK293 cells, were treated with DMSO, 5 mM disuccinimidyl glutarate (DSG), or 5 mM disuccinimidyl suberate (DSS), as indicated above the columns. VPs were detected by western blot with B1 antibody. Approximate molecular weights are shown at the right of each row.
See also Figure S7.
To begin examining whether AAP is promoting oligomerization into species of defined geometry such as trimers or pentamers or whether the increase in VP-VP interactions observed by co-immunoprecipitation (coIP) are more randomized associations, crosslinking agents were added to transfected cell lysates, and VP species were interrogated by western blot (Figure 6C). In the presence of AAP, a single supershifted VP band appears around 97 kDa when DSG (7-Å crosslinking arm) is added, and a slightly larger supershifted doublet appears when DSS (11-Å crosslinking arm) is added. Although it is difficult to determine the molecular weight of crosslinked species because of unpredictable migration patterns, the discrete banding suggests that AAP promotes VP-VP interactions of defined geometry or number of monomers but may also be indicative of increased association of VP with a host protein(s) involved in capsid assembly, an association promoted by AAP. To further ensure that we were interrogating the VP oligomerization process separately from its assembly into full capsids, the immunoprecipitation (IP) experiment was repeated with AAV2 VPs, adding rep, aap2, helper, and inverted terminal repeat (ITR)-flanked reporter genome plasmids in trans to allow quantification of assembled DRPs (Figure S7A). Appreciable quantities of genomes were detected only in the input and supernatant fractions but absent from the IP fraction. An ELISA to quantify fully assembled capsids mirrored these results (Figure S7B), indicating that only oligomerized VPs were precipitated. Taken together, these data support that, in addition to a role for AAP in VP protein stability, AAP also promotes oligomerization of VP proteins to nucleate assembly of the icosahedron, which could potentially increase the efficiency of the capsid assembly process.
DISCUSSION
The icosahedral capsid is an efficient protein superstructure employed by numerous viral taxa to protect, transport, and deliver nucleic acid and protein cargo to the host cell. AAV, with its non-enveloped T = 1 icosahedral capsid, is among the simplest mammalian viruses and can thus be considered a viral model organism to study the fundamentals of icosahedral protein structures and their functions. Capsid assembly is a primary requirement for any virus, but AAV’s mechanism of assembly is largely unknown.
The interest to study AAV assembly is not solely fundamental; AAV is a leading platform in therapeutic gene transfer, primarily for in vivo gene therapy approaches. Although preclinical and clinical studies continue to demonstrate AAV’s potential as a reagent for safe and efficient gene delivery to alleviate diseases with an unmet need, a bottleneck to its broader application is the production of sufficient vector quantities to treat these patient populations. A more profound understanding of assembly, a main determinant of viral vector yield, may allow improved processes for vector manufacturing. Furthermore, therapeutic programs using AAVs to date have utilized vector systems derived from naturally occurring serotypes. There is an increasing desire to modulate clinically relevant phenotypes, such as pre-existing immunity, tissue tropism, and specificity, by capsid engineering. However, many of these capsid modifications inadvertently affect viral assembly by affecting interactions with viral or cellular assembly co-factors (such as AAP) and/or disrupting the highly ordered icosahedral virion architecture, without which assembly cannot take place.
Here we aimed to study the role of AAP in the AAV assembly process, delineate features relevant to AAP function, and leverage this mechanistic information to improve control over preserving and improving particle formation. Several challenges hamper our ability to study assembly and AAP’s role in it. AAP is expressed from within the coding sequence of the very proteins it helps to assemble, complicating mutational studies of VP or AAP. Additionally, mutations in cap often affect yield; their role (e.g., AAP binding) is difficult to isolate because these same mutations could have a role in the secondary, tertiary, or quaternary structure. In an attempt to overcome these experimental challenges, we leveraged a toolset previously generated in our lab: a set of 9 assembly- and infectivity-competent intermediates to several of the known AAV serotypes. These AncAAVs are predicted structures at bifurcation points within the AAV lineage and provide a set of reagents to interrogate AAP function across a wide structural differential (e.g., AAV8 versus AAV9). This permitted a phenotype switch in how AAP interacted with Cap at the Anc82 branch point, which allowed for the identification of a minimal motif determining AAPC dependency. This phenotype-to-phylogeny mapping method allowed efficient elucidation of VP structural elements that relate to assembly functions and directly led to mechanistic insight into the function of AAP. This method holds other possible applications for mapping and mechanistic studies in AAV or complex assemblies in general.
The present study is a comprehensive look across the major AAV clades at assembly of the complete VP1/2/3 particle and in the natural transcriptional context, expanding greatly upon previous assembly and AAP studies using VP3-only capsids. The importance of VP1/2 in other viral functions has been demonstrated previously; VP1 harbors a phospholipase domain required for infectivity (Girod et al., 2002) in addition to basic regions that may function in trafficking the virion to the nucleus (Grieger et al., 2006; Popa-Wagner et al., 2012). Given that VP3-only capsids require AAP in all AAPC-independent serotypes tested by our group and others, and that the DI motif requires a VP1-unique site to confer AAPC independence, VP1/2-unique regions may aide capsid assembly. AAP may be partially redundant for such assembly functions lost from VP in AAP-dependent serotypes during evolution. We postulate that AAP arose to allow AAV more degrees of freedom to mutate its capsid toward fitness in its selective environment (e.g., immune escape) while maintaining assembly function. That the requirement for AAP is lessened at some branches of the modeled natural history may illustrate a selective advantage toward self-assembly, and our data of the D versus I branch illustrate that this can be accomplished by epistatic changes at the VP interface that minimize the necessity for AAP.
By varying the placement of early stop codons in the AAP reading frame, we were able to assay assembly both in the absence of any functional AAP protein and with what appears to be only the N-terminal ~60 amino acids expressed. Our results reveal that this portion alone can promote VP stability and allows for capsid assembly in roughly half of the 21 AAVs we examined. We demonstrate that, although AAP is required to reach the full production capability of AAV, the ability to assemble into capsids is fundamentally a property of VP. We identify 82DI, an AAPC-independent gain-of-function mutant differing from its parental strain Anc82 by only 12 amino acids on VP which requires only AAPN for VP protein stability and can bind other VPs in the absence of AAPC. Importantly, there are no differences in Anc82’s and 82DI’s AAP proteins. These observations suggest that AAPN may harbor chaperone activity and AAPC a scaffolding function; whether AAP chaperones by assisting proper folding or through less traditional means is unclear. The degree to which the scaffolding and chaperone functions are discrete is also unclear and likely varies between serotypes. We hypothesize that trimerization itself plays a role in VP stability, in addition to its obvious requirement for capsid assembly.
Stability of the assembled capsid has been examined in the post-entry stages of AAV infection but has not been thoroughly examined in the production/assembly-associated phases of the AAV life cycle. Ubiquitination or phosphorylation of specific surface-exposed residues has been implicated in the targeted degradation of the capsid post-entry (Gabriel et al., 2013; Sen et al., 2013; Yan et al., 2002; Zhong et al., 2008). Although it is possible that the DI mutations disrupt a residue or motif that targets VPs for degradation, it is equally possible that oligomerization shields such residues and that serotypes that can self-oligomerize or oligomerize using only AAPN lessen their dependence on AAP. Understanding how AAV protects itself from host defenses such as targeting VPs for degradation is a first step toward engineering capsids that can be produced more efficiently. The modest increase in 82DI titers over Anc82 (~2-fold) indicates that modulating AAP dependence could prove to be a useful strategy for increasing production titers.
Our results suggest a model for the early steps of capsid assembly where, within the pool of VP proteins, equilibrium exists between monomers and oligomers (Figure 7). In the case of an AAPC-independent serotype, monomers are able to self-oligomerize, and the equilibrium favors oligomers. AAP-dependent serotypes’ unstable VPs are depleted, pushing the equilibrium to the left. At this stage of assembly, AAP is required for VP protein stability (Figure 2B) and oligomerization (Figure 6). It is unclear whether these are discrete functions—AAP blocks degradation, and subsequently acts as a scaffolding protein to assist oligomerization—or whether oligomerization itself stabilizes VP proteins. In the case of AAP-dependent AAV8, AAP is required for capsid assembly despite rescue of VP protein from proteasomal degradation (Figure 2E), suggesting that oligomerization itself contributes to VP stability. Conversely, an explanation that may be especially applicable to AAPC-independent serotypes is that, by stabilizing VP monomers and increasing the monomer pool, the equilibrium is able to move toward oligomers. It is difficult to dissect the effect of AAP on the stability of monomeric VP proteins, considering how rapidly VPs are assembled into the stable icosahedron in its presence. Experiments assaying AAP’s effect on monomer turnover rates necessitates blocking translation of new VP proteins (Grosse et al., 2017), which, unfortunately, also blocks translation of AAP protein, resulting in AAP loss over time and an inability to draw conclusions regarding the effect of AAP on monomer stability. It is unclear whether AAP interacts directly or indirectly with VP proteins; VP proteins purified from Sf9 cells by chemical extraction methods showed increased oligomerization when HeLa cell extract was added, suggesting that host factors are required or at least increase the efficiency of oligomerization (Steinbach et al., 1997). Whether AAP was present in these experiments is unknown because they were conducted well before its discovery.
Figure 7. Model for Early Steps of Capsid Assembly Across the AAP Phenotypes.
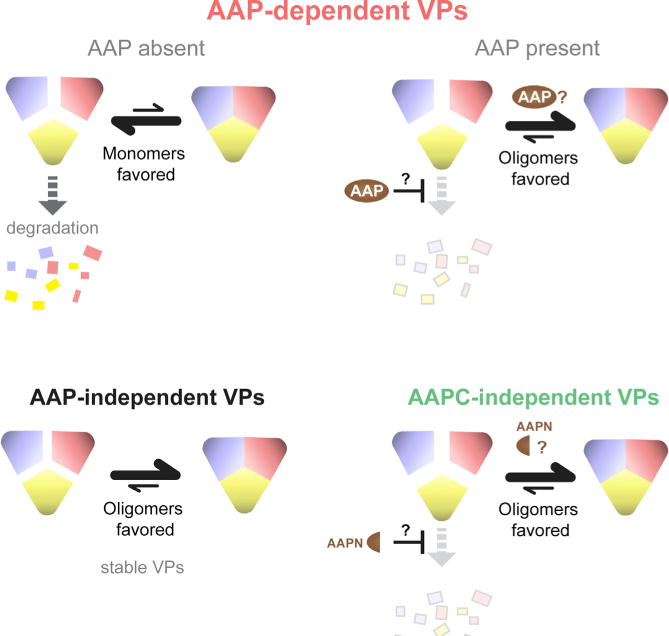
Whether a serotype is AAP-dependent, AAP-independent, or AAPC-independent, nucleating capsid assembly is likely dependent on both the stability and oligomerization of VP proteins. Our findings demonstrate that AAP is active in both functions. Whether these functions are discrete is unclear and indicated by question marks in the model.
Additional work is required, but our study provides evidence of a role for AAP in VP protein stability across multiple serotypes in addition to a potential role in VP oligomerization. We also postulate an evolutionary role for AAP: to lessen restrictions on capsid evolutionary mutations imposed by the requirement for the protein monomers to be assembled into a 60-mer icosahedral superstructure. Importantly, for this functionality to lead to an evolutionary benefit, it has to offset the additional constraints it imposes; namely, its coding out of frame within the cap ORF and the presumed need for AAP to bind VPs through a conserved motif to exert its effect. Because capsid assembly is the primary requirement for any virus or vector, our findings are relevant to the field of AAV gene therapy, which seeks to improve assembly to obtain higher yields in recombinant AAVs.
EXPERIMENTAL PROCEDURES
Vectors and Sequences
Adeno-associated viral vectors were pseudotyped with either extant or ancestral viral capsids. Extant capsids include AAV1 (GenBank [GB]: AAD27757.1), AAV2 (GB: AAC03780.1), AAV3 (GB: U48704.1), AAV4 (GB: U89790.1), AAV5 (GB: AAD13756.1), AAV6 (GB: AF028704.1), AAV7 (NC_006260.1) Rh.10 (GB: AAO88201.1), AAV8 (GB: AAN03857.1), AAV9 (GB: AAS99264.1), and Rh32.33 (GB: EU368926). Ancestral AAV capsids include Anc80L65, Anc81, Anc82, Anc83, Anc84, Anc110, Anc113, Anc126, and Anc127 (KT235804–KT235812). In this study, Anc83 has the following mutation in the presumed AAP ORF: Q1L (83AAP-KI).
Site-Directed Mutagenesis
AAPstop60, AAPstop20, and 82DI single revertant mutations were generated using the QuikChange II Site-Directed Mutagenesis Kit according to the manufacturer’s instructions. To generate 82DI, the QuikChange Lightning Multi Site-Directed Mutagenesis Kit was used according to the manufacturer’s instructions in two phases: first, five sites were mutated on an Anc82 backbone, and then the remaining five mutations were introduced into this quintuple mutant backbone. See Table S5 for primer sequences.
Crude Virus Preparations/Titration
Virus preparations to assay production in all serotypes and mutants were prepared as follows. Polyethyleneimine transfections of AAV cis ITR-CMV-EGFP-T2A-Luc-ITR (2 μg), AAV trans rep-cap (2 μg), and adenovirus helper plasmid (4 μg) were performed on HEK293 cells at 90% confluency in 6-well dishes. The polyetheleneimine (PEI) Max (Polysciences)/DNA ratio was maintained at 1.375:1 (w/w) in serum-free medium. Virus was harvested after 72 hr by three freeze/thaw cycles, followed by centrifugation at 15,000 × g.
For DRP titers, crude preparations were DNaseI-treated, and resistant (packaged) vector genome copies were used to titrate preparations by TaqMan qPCR amplification (Applied Biosystems 7500, Life Technologies) with primers and probes detecting cytomegalovirus (CMV) promoter regions of the transgene cassette.
Thermostability Assay
Thermostability of purified vector was assayed by AAV-ID (Pacouret et al., 2017). Briefly, a 500-μL sample of SYPRO Orange 50× was prepared using PBS2+ (21-030-CV, Corning Life Sciences, Corning, NY) as a solvent. 96-well plates were loaded with 45-μL samples supplemented with 5-μL SYPRO Orange 50×. PBS2+ and 0.25 mg/mL lysozyme (L6876, Sigma-Aldrich, St. Louis, MO, USA) solutions were used as negative and positive controls, respectively. Plates were sealed and centrifuged at 3,000 rpm for 2 min and subsequently loaded into a 7500 real-time PCR system (Thermo Fisher Scientific). Samples were incubated at 25°C for 2 min prior to undergoing a temperature gradient (25°C to 99°C, ~2°C/10 min, step and hold mode with 0.4°C temperature increments) while monitoring the fluorescence of the SYPRO Orange dye using the 6-carboxyl-X-rhodamine (ROX) filter cube available on both qPCR systems. Fluorescence signals (F) were normalized between 0% and 100%, and melting temperatures were defined as the temperature for which the numerical derivative dF/dT reached its maximum.
ELISAs
A20 capsid ELISAs were performed on crude virus preparations with the PROGEN AAV 2 titration ELISA kit (ref# PRATV) according to the manufacturer’s instructions.
hA1AT ELISAs were performed on 1:1,250–1:10,000 serial dilutions of mouse serum with the Cloud-Clone ELISA kit for α-1 antitrypsin (SEB697Hu, 96 tests) according to the manufacturer’s instructions.
Animal Studies
C57BL/6 male mice (6–8 weeks old) were purchased from Jackson Laboratories. All animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Schepens Eye Research Institute.
Mice were anesthetized with ketamine/xylazine intraperitoneally. Each animal was injected retro-or-bitally (100 μL) with 1.00E+11 vector genomes (VG)/mouse of the following vectors: Anc82.CB7.CI. EGFP.FF2A.hA1AT.RBG and Anc82DI.CB7.CI. EGFP.FF2A.hA1AT.RBG. Blood was collected via submandibular bleeds using GoldenRod animal lancets (MEDIpoint) prior to injection and 3, 7, 15, and 28 days after injection. Samples were centrifuged at 8,000 rpm for 7.5 min, and the serum was collected.
Animals were euthanized, and livers were collected and submerged in 4% paraformaldehyde solution (Electron Microscopy Sciences) for 30 min and then placed in 30% sucrose overnight. The next day, the liver was mounted in Tissue-Tek O.C.T. compound (Sakura Finetek) and flash-frozen in cool isopentane.
Tissue Histology
To visualize EGFP expression in the liver, 15-μm sections were mounted with Vectashield Hard Set mounting medium with DAPI (H-1500) and imaged with a Zeiss Axio Imager M2 at the same gain and intensity across all sections.
Molecular Representations
All molecular representations in this study were generated using PyMOL and PDB: 2QA0 (AAV8) and PDB: 3UX1 (AAV9).
Production and Purification of AAV3, AAV3s, AAV9, and AAV9s
Vectors were purified by affinity chromatography using either AVB Sepharose HP (25-4112-11, GE Healthcare) (AAV3 and AAV3s) or POROS CaptureSelect AAV9 affinity resin (Thermo Fisher Scientific) according to the manufacturer’s instructions (AAV9 and AAV9s). For more details, please see the Supplemental Experimental Procedures.
Statistical Methods
All data were analyzed using R prior to normalization for reporting in the main figures. The p values are reported in Table S2. Viral titers were compared using a paired one-tailed Student’s t test, and RNA levels were compared using a paired two-tailed Student’s t test.
For expression constructs, protein lysate preparation, degradation studies, immunoprecipitations, crosslinking, RNA quantification, TEM, and western/dot blotting, see the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Dependence on AAP for capsid assembly varies widely across 21 AAV variants
AAP-dependent capsid proteins are subject to multiple rapid degradation pathways
AAP promotes interactions between capsid proteins
Specific capsid residues at trimer interface influence dependence on AAP
Acknowledgments
We thank Dirk Grimm, Itay Koren, and Steve Elledge for discussions and reagents; Ru Xiao and the Grousbeck Gene Therapy Center Gene Transfer Vector Core; Phil Seifert for technical expertise with TEM studies; Amanda Dudek for technical assistance; Eric Zinn for discussions and providing draft graphics and values for Figure 3E; and Mohammadsharif Tabebordbar and Connie Cepko for discussions and feedback. This work was supported by NIH National Eye InstituteCore Grant P30EY003790, the NIH Common Fund (5DP1EY023177-03), Giving/Grousbeck, and Lonza Houston (to L.H.V.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.04.026.
AUTHOR CONTRIBUTIONS
A.C.M. conceived, designed, and performed all experiments (except for animal work, AAV-ID, and TEM), sequence analyses, and molecular modeling; collected and analyzed all data; prepared all figures; and wrote the manuscript. S.P. purified vector preparations and performed AAV-ID and TEM. A.K.C.D. performed statistical analyses. J.B. injected mice, harvested serum, and wrote the Experimental Procedures for animal experiments. E.A.-M. designed animal experiments, injected mice, harvested livers, and gave feedback on manuscript drafts. L.H.V. secured funding, helped with drafting the manuscript, and supervised all experiments.
DECLARATION OF INTERESTS
L.H.V. is an inventor of AncAAV and other AAV technologies, which are licensed to various biotechnology and pharmaceutical entities. L.H.V. is a consultant to a number of companies with gene therapy interests, including Selecta Biosciences and Lonza Houston, licensees of Anc-AAV technology. This work was in part supported by Lonza Houston.
References
- Bell CL, Gurda BL, Van Vliet K, Agbandje-McKenna M, Wilson JM. Identification of the galactose binding domain of the adeno-associated virus serotype 9 capsid. J Virol. 2012;86:7326–7333. doi: 10.1128/JVI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleker S, Pawlita M, Kleinschmidt JA. Impact of capsid conformation and Rep-capsid interactions on adeno-associated virus type 2 genome packaging. J Virol. 2006;80:810–820. doi: 10.1128/JVI.80.2.810-820.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMattia MA, Nam HJ, Van Vliet K, Mitchell M, Bennett A, Gurda BL, McKenna R, Olson NH, Sinkovits RS, Potter M, et al. Structural insight into the unique properties of adeno-associated virus serotype 9. J Virol. 2012;86:6947–6958. doi: 10.1128/JVI.07232-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley LF, Kawano Y, Adachi K, Sun XX, Dai MS, Nakai H. Identification and characterization of nuclear and nucleolar localization signals in the adeno-associated virus serotype 2 assembly-activating protein. J Virol. 2015;89:3038–3048. doi: 10.1128/JVI.03125-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley LF, Powers JM, Adachi K, Baumgart JT, Meyer NL, Xie Q, Chapman MS, Nakai H. Adeno-associated Virus (AAV) Assembly-Activating Protein Is Not an Essential Requirement for Capsid Assembly of AAV Serotypes 4, 5, and 11. J Virol. 2017;91:e01980–16. doi: 10.1128/JVI.01980-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel N, Hareendran S, Sen D, Gadkari RA, Sudha G, Selot R, Hussain M, Dhaksnamoorthy R, Samuel R, Srinivasan N, et al. Bioengineering of AAV2 capsid at specific serine, threonine, or lysine residues improves its transduction efficiency in vitro and in vivo. Hum Gene Ther Methods. 2013;24:80–93. doi: 10.1089/hgtb.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Vandenberghe LH, Wilson JM. New recombinant se-rotypes of AAV vectors. Curr Gene Ther. 2005;5:285–297. doi: 10.2174/1566523054065057. [DOI] [PubMed] [Google Scholar]
- Girod A, Wobus CE, Zádori Z, Ried M, Leike K, Tijssen P, Kleinschmidt JA, Hallek M. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J Gen Virol. 2002;83:973–978. doi: 10.1099/0022-1317-83-5-973. [DOI] [PubMed] [Google Scholar]
- Grieger JC, Snowdy S, Samulski RJ. Separate basic region motifs within the adeno-associated virus capsid proteins are essential for infectivity and assembly. J Virol. 2006;80:5199–5210. doi: 10.1128/JVI.02723-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Zolotukhin S. E Pluribus Unum: 50 Years of Research, Millions of Viruses, and One Goal–Tailored Acceleration of AAV Evolution. Mol Ther. 2015;23:1819–1831. doi: 10.1038/mt.2015.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse S, Penaud-Budloo M, Herrmann AK, Börner K, Fakhiri J, Laketa V, Krämer C, Wiedtke E, Gunkel M, Ménard L, et al. Relevance of Assembly-Activating Protein for Adeno-associated Virus Vector Production and Capsid Protein Stability in Mammalian and Insect Cells. J Virol. 2017;91:e01198–17. doi: 10.1128/JVI.01198-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern A, Schmidt K, Leder C, Müller OJ, Wobus CE, Bettinger K, Von der Lieth CW, King JA, Kleinschmidt JA. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J Virol. 2003;77:11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet. 2014;15:445–451. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landegger LD, Pan B, Askew C, Wassmer SJ, Gluck SD, Galvin A, Taylor R, Forge A, Stankovic KM, Holt JR, Vandenberghe LH. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat Biotechnol. 2017;35:280–284. doi: 10.1038/nbt.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochrie MA, Tatsuno GP, Christie B, McDonnell JW, Zhou S, Surosky R, Pierce GF, Colosi P. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J Virol. 2006;80:821–834. doi: 10.1128/JVI.80.2.821-834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam HJ, Lane MD, Padron E, Gurda B, McKenna R, Kohlbrenner E, Aslanidi G, Byrne B, Muzyczka N, Zolotukhin S, Agbandje-McKenna M. Structure of adeno-associated virus serotype 8, a gene therapy vector. J Virol. 2007;81:12260–12271. doi: 10.1128/JVI.01304-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumer M, Sonntag F, Schmidt K, Nieto K, Panke C, Davey NE, Popa-Wagner R, Kleinschmidt JA. Properties of the adeno-associated virus assembly-activating protein. J Virol. 2012;86:13038–13048. doi: 10.1128/JVI.01675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolson SC, Samulski RJ. Recombinant adeno-associated virus utilizes host cell nuclear import machinery to enter the nucleus. J Virol. 2014;88:4132–4144. doi: 10.1128/JVI.02660-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacouret S, Bouzelha M, Shelke R, Andres-Mateos E, Xiao R, Maurer A, Mevel M, Turunen H, Barungi T, Penaud-Budloo M, et al. AAV-ID: A Rapid and Robust Assay for Batch-to-Batch Consistency Evaluation of AAV Preparations. Mol Ther. 2017;25:1375–1386. doi: 10.1016/j.ymthe.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillay S, Meyer NL, Puschnik AS, Davulcu O, Diep J, Ishikawa Y, Jae LT, Wosen JE, Nagamine CM, Chapman MS, Carette JE. An essential receptor for adeno-associated virus infection. Nature. 2016;530:108–112. doi: 10.1038/nature16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa-Wagner R, Porwal M, Kann M, Reuss M, Weimer M, Florin L, Kleinschmidt JA. Impact of VP1-specific protein sequence motifs on adeno-associated virus type 2 intracellular trafficking and nuclear entry. J Virol. 2012;86:9163–9174. doi: 10.1128/JVI.00282-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D, Gadkari RA, Sudha G, Gabriel N, Kumar YS, Selot R, Samuel R, Rajalingam S, Ramya V, Nair SC, et al. Targeted modifications in adeno-associated virus serotype 8 capsid improves its hepatic gene transfer efficiency in vivo. Hum Gene Ther Methods. 2013;24:104–116. doi: 10.1089/hgtb.2012.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag F, Schmidt K, Kleinschmidt JA. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc Natl Acad Sci USA. 2010;107:10220–10225. doi: 10.1073/pnas.1001673107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag F, Köther K, Schmidt K, Weghofer M, Raupp C, Nieto K, Kuck A, Gerlach B, Böttcher B, Müller OJ, et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J Virol. 2011;85:12686–12697. doi: 10.1128/JVI.05359-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach S, Wistuba A, Bock T, Kleinschmidt JA. Assembly of adeno-associated virus type 2 capsids in vitro. J Gen Virol. 1997;78:1453–1462. doi: 10.1099/0022-1317-78-6-1453. [DOI] [PubMed] [Google Scholar]
- Thomas CE, Storm TA, Huang Z, Kay MA. Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J Virol. 2004;78:3110–3122. doi: 10.1128/JVI.78.6.3110-3122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe LH, Wilson JM, Gao G. Tailoring the AAV vector capsid for gene therapy. Gene Ther. 2009;16:311–319. doi: 10.1038/gt.2008.170. [DOI] [PubMed] [Google Scholar]
- Wistuba A, Weger S, Kern A, Kleinschmidt JA. Intermediates of adeno-associated virus type 2 assembly: identification of soluble complexes containing Rep and Cap proteins. J Virol. 1995;69:5311–5319. doi: 10.1128/jvi.69.9.5311-5319.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wistuba A, Kern A, Weger S, Grimm D, Kleinschmidt JA. Subcellular compartmentalization of adeno-associated virus type 2 assembly. J Virol. 1997;71:1341–1352. doi: 10.1128/jvi.71.2.1341-1352.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Xiao W, Conlon T, Hughes J, Agbandje-McKenna M, Ferkol T, Flotte T, Muzyczka N. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J Virol. 2000;74:8635–8647. doi: 10.1128/jvi.74.18.8635-8647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Zak R, Luxton GW, Ritchie TC, Bantel-Schaal U, Engel-hardt JF. Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. J Virol. 2002;76:2043–2053. doi: 10.1128/jvi.76.5.2043-2053.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Li B, Jayandharan G, Mah CS, Govindasamy L, Agbandje-McKenna M, Herzog RW, Weigel-Van Aken KA, Hobbs JA, Zolotukhin S, et al. Tyrosine-phosphorylation of AAV2 vectors and its consequences on viral intracellular trafficking and transgene expression. Virology. 2008;381:194–202. doi: 10.1016/j.virol.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinn E, Pacouret S, Khaychuk V, Turunen HT, Carvalho LS, Andres-Mateos E, Shah S, Shelke R, Maurer AC, Plovie E, et al. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 2015;12:1056–1068. doi: 10.1016/j.celrep.2015.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.