

AI-2–mediated quorum sensing of E. coli is directly connected to sugar metabolism via the phosphocarrier HPr protein of PTS.
Abstract
Quorum sensing (QS), a bacterial process that regulates population-scale behavior, is mediated by small signaling molecules, called autoinducers (AIs), that are secreted and perceived, modulating a “collective” phenotype. Because the autoinducer AI-2 is secreted by a wide variety of bacterial species, its “perception” cues bacterial behavior. This response is mediated by the lsr (LuxS-regulated) operon that includes the AI-2 transporter LsrACDB and the kinase LsrK. We report that HPr, a phosphocarrier protein central to the sugar phosphotransferase system of Escherichia coli, copurifies with LsrK. Cocrystal structures of an LsrK/HPr complex were determined, and the effects of HPr and phosphorylated HPr on LsrK activity were assessed. LsrK activity is inhibited when bound to HPr, revealing new linkages between QS activity and sugar metabolism. These findings help shed new light on the abilities of bacteria to rapidly respond to changing nutrient levels at the population scale. They also suggest new means of manipulating QS activity among bacteria and within various niches.
INTRODUCTION
Bacteria, despite being unicellular organisms, are capable of coordinating population-level behavior through a process termed quorum sensing (QS). In this process, bacteria secrete chemical signaling molecules called autoinducers (AIs), which accumulate as cell density increases. Once the AI level reaches a threshold, signaling a “quorum” of cells, the AI signals are transported intracellularly, where they activate gene expression and enable coordinated phenotypic responses in the population. The importance of QS in biofilm formation and maintenance (1), bacterial persistence (2), and pathogenicity (3) has appeared in many review articles. Further, the interplay between the signaling components that comprise QS systems has been the subject of many studies in metabolic engineering and synthetic biology, wherein genetic networks have been developed to enable “programmed” design and control of metabolic pathways and bacterial phenotype (4, 5). For example, researchers have exploited QS circuitry for the design and implementation of “smart” bacteria that target and destroy cancers and pathogens (6, 7). Some of these systems depend on the “orthogonality” of the signaling system with the metabolic activity of the host organism. For instance, Saeidi et al. (8) engineered bacteria that sense acyl-homoserine lactone QS signals produced by Pseudomonas aeruginosa and subsequently release toxins to eradicate the pathogen. Other systems rely on the interdependence of QS activity and host metabolism. For example, an autoinduction system was constructed by Tsao et al. (9), in which Escherichia coli secrete AI-2 [dihydroxy-pentane-dione (DPD)] and, at the appropriate time for gene expression, self-induce expression of a recombinant protein by amplifying expression from the native lsr (LuxS-regulated) promoter using T7 polymerase. Further mechanistic understanding of how the cell regulates QS processes, for example, based on the availability of substrates like glucose, will further enable researchers to exploit these QS systems for the design of synthetic biology systems with new capabilities.
There is evidence that the AI-2–mediated QS system is partially regulated by substrate availability and cell metabolism. LuxS synthesizes AI-2 as a by-product of the activated methyl cycle (3), after which AI-2 accumulates extracellularly. AI-2 is imported by LsrACDB (10) and phosphorylated by the kinase LsrK, sequestering it within the cell (11). Phosphorylated AI-2 relieves LsrR-mediated repression of the lsr operon (12), allowing transcription of the lsr genes and acceleration of AI-2 uptake. It has recently been reported that another key bacterial process, chemotaxis, is linked to AI-2 signaling via LsrB and enhances not only self-aggregation of E. coli but also its coaggregation with Enterococcus faecalis (13, 14). Several studies suggest that the bidirectional lsr operon, in addition to being regulated by LsrK and LsrR, is also subject to carbon catabolite repression (CCR). For instance, activation of the lsr promoter does not occur in the presence of glucose (10) or glycerol (15) and requires the global regulators cyclic adenosine monophosphate (cAMP) and cAMP receptor protein (CRP) (10). Binding sites for cAMP-CRP exist between the lsrR and lsrACDB promoters (12), and binding of cAMP-CRP likely modulates the promoter activity (16). Here, we propose a new mechanism linking cell metabolism to the AI-2 QS system. Specifically, we have discovered that the activity of LsrK is regulated by the phosphoenolpyruvate (PEP)–dependent sugar phosphotransferase system (PTS) protein HPr.
PTS is important for sugar uptake and regulation of carbohydrate metabolism. It comprises three units—EI, HPr, and the EII protein complex—that sequentially transfer a phosphoryl group from PEP to the transported carbohydrate. The active transport of PTS sugars affects the phosphorylation state of each of the PTS components. Although EI and HPr are general PTS proteins, EII is specific to the carbohydrate being transported, and one of the most commonly studied is a subunit involved in glucose transport, EIIAGlc. The phosphorylation state of EIIAGlc regulates the activity of adenylate cyclase, which synthesizes cAMP, a global regulator within the cell (17). As discussed above, the cAMP-CRP complex regulates transcription from the lsr promoter. There is evidence to suggest that the phosphorylation state of the other general PTS proteins also regulates AI-2 QS activity. Pereira et al. (18) demonstrated that phosphorylated EI is required for the initial uptake of AI-2, although the mechanism behind this has not been elucidated.
Our research demonstrates that LsrK is tightly bound to the PTS protein HPr, and that HPr directly influences LsrK activity. LsrK was previously demonstrated to have a range of substrate specificities (19), perhaps illustrating this enzyme’s involvement in additional functions besides QS. Moreover, we demonstrate that uptake of PTS carbohydrates has direct involvement in signaling and show for the first time that PTS regulates AI-2 QS not only through the global regulator cAMP but also directly through specific interactions with LsrK. This finding suggests that bacteria have evolved sophisticated mechanisms for incorporating information about substrate availability and cell metabolism into QS processes. This discovery is of fundamental importance, as phenomena such as pathogenicity, bacterial persistence, and biofilm formation have previously been shown to be influenced by nutrient availability and QS. Here, we suggest that these processes may be more closely linked than previously thought.
RESULTS
Characterization of a copurified E. coli protein, HPr, with LsrK
Our original objective in this work was to develop a structural basis for the role of LsrK in the processes of AI-2–mediated QS, having recently elucidated the crystal structures of the LsrR transcriptional regulator (20). This structural insight provides a basis for efforts targeting AI-2 QS activity in metabolic engineering, device creation, and biofilm modulation, among others. The LsrK protein was expressed mostly as an insoluble form [the glutathione S-transferase (GST)–LsrK fusion protein] or as a soluble aggregate (the N-terminal His-tagged LsrK) in E. coli; thus, the LsrK protein used here for crystallization was obtained by a specific expression protocol: (i) Luria-Bertani (LB) medium containing 0.5 M NaCl and 0.4% glucose and (ii) heat shock at 42°C before the isopropyl-β-D-thiogalactopyranoside (IPTG) induction at 20°C (21). The single crystal of the purified LsrK protein also contained an unknown E. coli protein. We identified this protein by N-terminal sequencing and mass analysis after trypsin in-gel digestion (fig. S1), revealing that it is the phosphocarrier protein HPr. We suspected that our method (0.5 M NaCl and 0.4% glucose; heat and osmotic shock) used for the expression of LsrK seemed to be related to the amount of the in vivo HPr protein; we then tried to coexpress an N-terminal His-tagged LsrK (HisLsrK) with HPr. The yield of the HisLsrK/HPr complex protein was greatly increased by coexpression. The HisLsrK and HPr protein yields increased from ~1.5 to ~7 mg per 1 liter of LB culture. We also tried to express the GST-LsrK protein in an E. coli (ΔptsH) strain following our previously reported protocol (21), but it was not obtained in soluble form. Because optimal expression of LsrK seemed to be strongly dependent on the presence of HPr, we coexpressed HisLsrK/HPr for further studies.
Crystal structure of the LsrK/HPr protein complex
The presence of an additional N-terminal His-tag in the purified HisLsrK/HPr protein did not appreciably change the crystallization conditions used for the previous LsrK/HPr construct (21). Statistics for our x-ray data collection and structure refinements are summarized in table S1 [selenomethionine-labeled HisLsrK/HPr, HisLsrK/HPr, HisLsrK/HPr/ADP (adenosine 5′-diphosphate), and HisLsrK/HPr/ATP (adenosine 5′-triphosphate)]. Structures of HisLsrK/HPr were determined by SAD (single-wavelength anomalous diffraction) phasing using the anomalous signal of selenomethionine and additionally by molecular replacement (MR) using HPr coordinates [Protein Data Bank (PDB) code, 1POH]. The crystal asymmetric unit contained two molecules of the HisLsrK/HPr complex (Fig. 1A), in which two monomer units had almost identical structures (not shown). We suggest that the functioning unit of the HisLsrK/HPr protein complex seemed to be a tightly bound monomeric form consisting of both the HisLsrK and HPr proteins because, in size exclusion chromatography with inline multi-angle light scattering (SEC-MALS) analyses, we found that the HisLsrK/HPr protein existed as one peak at concentrations of 1 to 2 mg/ml (fig. S2). Crystal structures of HisLsrK/HPr, HisLsrK/HPr/ADP, and HisLsrK/HPr/ATP were very similar (fig. S3A), revealing again that HPr formed a tight complex with the LsrK protein (Fig. 1). We do note, however, that the asymmetric unit of the crystals had very high water content (67%), and many parts of the LsrK protein were not identified in the electron density map owing to the highly increased flexibility (14 residues of the N-terminal His-tag region, M1-E9, L46-S54, D364-W371, and W505-L530).
Fig. 1. Crystal structure of the HisLsrK/HPr/ATP complex.
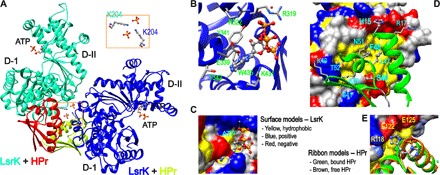
(A) The crystal asymmetric unit contained two molecules of the HisLsrK/HPr complex, in which the contact between two HisLsrK/HPr units is maintained by the ionic interaction mediated by two phosphate and LsrK-K204 residues (boxed region). (B) The ATP molecule seems to bind LsrK mainly via the hydrophobic interaction mediated by the adenine base, and the phosphate groups participate in additional hydrogen bonds and ionic interactions. The presence of hydrogen bonds is indicated with cyan lines, and the slightly longer distances than the limit of a hydrogen bond are indicated with orange lines. (C) The base of ATP fits into the hydrophobic pocket of LsrK. (D) The residues of HPr that participate in the interaction with LsrK are represented in the green ribbon model. F48 fits into the deep hydrophobic pocket of LsrK, and the neighboring L47 also participates in this hydrophobic interaction. HPr-H15 is located in the interface of the HisLsrK/HPr complex. (E) A structural overlay of free HPr and that in the HisLsrK/HPr complex suggests that H15 has structural flexibility, and the steric hindrance induced by the phosphorylated H15 may not be stringent. The phosphate group is simply attached to the H15-ND1 atom of the free HPr form for more clear comparison. The residues of LsrK that are located close to HPr-H15 are labeled yellow (R118, 4.15 Å; E122, 2.65 Å; E125, 4.50 Å).
We found that the structure of LsrK can be divided into two domains (I and II), in which HPr and ATP bind to domains I and II, respectively (Fig. 1A). The ADP molecule did not strongly bind to HisLsrK/HPr, as the electron density 2FoFc map of ADP in HisLsrK/HPr/ADP was less clear than that of ATP in HisLsrK/HPr/ATP (fig. S3, C and D). We tried to obtain HisLsrK/HPr crystals containing either DPD or an AI-2 antagonist, isopropyl-DPD (19), by cocrystallization or via the crystal soaking method, but we were unsuccessful. We further note that cocrystallization and soaking with both adenosine 5′-O-(3-thiotriphosphate) (ATPγS) and either DPD or isopropyl-DPD produced the HisLsrK/HPr crystals containing only ATPγS. We therefore suggest that the structure revealed for the HisLsrK/HPr complex is likely an inactive form in that it does not bind DPD, the QS substrate for LsrK. Because the structure of HisLsrK/HPr/ATPγS (2.9 Å) was identical to that of HisLsrK/HPr/ATP (2.7 Å), a low-resolution structure of HisLsrK/HPr/ATPγS was not reported here. In addition, structure-based sequence alignments showed that the residues of the ATP-binding site (Fig. 1B) are well conserved among six selected Gram-negative and Gram-positive bacteria (Fig. 2). Upon examination of the HisLsrK/HPr complex, we found that HPr-F48 fits within the deep hydrophobic pocket of LsrK consisting of L123, I148, L151, L152, A155, Y162, M210, and A211, and further that the neighboring HPr-L47 also participates in the hydrophobic interaction (Fig. 1D). Although hydrogen bonds and ionic interactions exist between the HisLsrK and HPr proteins, we suggest that the HisLsrK/HPr complex is maintained principally by strong hydrophobic interactions.
Fig. 2. Structure-based alignments of various LsrK and HPr sequences.

Various LsrK (A) and HPr (B) sequences of Gram-negative [Escherichia coli (E. coli), Salmonella typhimurium LT2 (S. typh), Yersinia pestis (Y. pest), and Klebsiella pneumoniae subsp. pneumoniae (K. pneu)] and Gram-positive [Bacillus subtilis (B. subt), Bacillus thuringiensis (B. thur), and Streptococcus sp. oral taxon 056 (Str. sp)] bacteria were aligned on the basis of the determined structure of the HisLsrK/HPr/ATP complex. The E. coli LsrK residues that contact with ATP (marked with A) and HPr (marked with numbers) are indicated in the bottom of the aligned sequences. The letters and numbers are colored following the characteristics of the interactions (hydrophobic, red; hydrogen bond, green; ionic interaction, blue), and the other neighboring residues remain as black.
Functionally, HPr transfers an activated phosphate group from EI to EIIA, where it is eventually attached to an imported sugar molecule via the EIIB/EIIC transporter. The phosphorylation state of HPr is therefore dependent on the rate of sugar import by PTS, and concomitantly, the population of native HPr increases during active glucose uptake. Our results indicate that HPr-H15 is a key residue mediating the phosphate transfer and that phosphorylated HPr (p-HPr) carries the phosphate group at the H15-ND1 atom. HPr-H15 is located at the interface of the HisLsrK/HPr complex, close to LsrK-E122 (Fig. 1D). A structural overlay of free HPr (PDB code, 1POH) to HPr bound in an HisLsrK/HPr complex reveals that the front parts of α helix 1 and α helix 2 display additional degrees of freedom, in which the positions of the imidazole ring of H15 are different from each other (Fig. 1E). That is, the HPr-H15 residue seems to have motional freedom in the HisLsrK/HPr complex; the average B factors of total HPr and HPr-H15 are 66 and 114, respectively, and the simulated annealing omit map of the region including HPr-H15 was also weak (fig. S3B, right). Therefore, a steric clash and unfavorable ionic interactions induced by the phosphate group of p-HPr could inhibit the formation of an LsrK/HPr complex. We suggest, however, that such inhibition would be less stringent than that imposed by a strict steric hindrance mechanism.
Finally, we note that the F48 and L47 residues of HPr are completely conserved in Gram-negative bacteria, and the corresponding residues (I47 and M48) are observed in three Gram-positive bacteria (Fig. 2). Further, because (i) M48 is classified as a hydrophobic residue and (ii) the hydrophobic residues of LsrK used for the interaction to HPr are relatively well conserved in both Gram-negative and Gram-positive bacteria (Fig. 2), we hypothesize that although lsrK is not present in all Gram-positive bacteria, the LsrK/HPr complex may be present in some.
The inhibitory effect of p-HPr on LsrK activity is less than that of native HPr
Because the HisLsrK/HPr crystals grown with both ATPγS and DPD did not include the DPD molecule, the activity of LsrK (phosphorylating imported DPD) may be inhibited by the bound HPr protein. Therefore, we studied the kinetics of LsrK via a coupled assay system using pyruvate kinase (PK) and lactate dehydrogenase (LDH) (22). That is, the enzyme activity of LsrK was determined as a function of HPr concentration in experiments using an increasing concentration of DPD in the presence of excess ATP (0.75 mM) (Fig. 3A). Because the purified HisLsrK contained HPr, the reference activity of LsrK was already affected by the equivalent amount of HPr. Lineweaver-Burk plots of LsrK activities with different concentrations of HPr showed that HPr inhibited LsrK activity in a mixed manner of competitive and uncompetitive inhibition (Km, 58 ± 11 μM; kcat, 17 ± 1 s−1; Ki1, 0.08 ± 0.01 μM; Ki2, 0.27 ± 0.03 μM), not via competitive or uncompetitive inhibition alone (Fig. 3, top). That is, mixed inhibition should also have the equilibrium process between ATP/LsrK/HPr and ATP/LsrK/DPD/HPr. The measured inhibition constants (Ki1 and Ki2) showed that the inhibition effect of HPr on LsrK activity was very strong, which is in strong agreement with the binding strengths inferred from the HisLsrK/HPr complex structure.
Fig. 3. Enzyme activity of LsrK is inhibited by HPr.
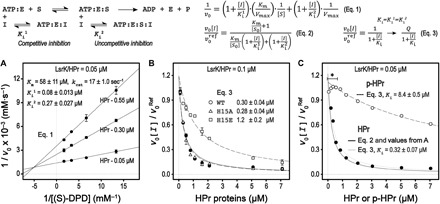
An enzymatic model of the LsrK and HPr interactions is summarized at the top. (A) Enzyme kinetic studies of LsrK were performed with a fixed ATP concentration (0.75 mM). The Lineweaver-Burk plots of LsrK activities in the presence of the HPr protein show that HPr inhibited LsrK activity via a combined manner of competitive and uncompetitive inhibition, as noted. Competitive inhibition (Ki1) was more substantive than uncompetitive inhibition (Ki2). (B) The relative activity of LsrK was measured as increasing the concentration of the wild-type (WT) and mutant HPr proteins. The concentrations of (S)-DPD and ATP were fixed at 3 and 1 mM, respectively. The reaction reference already contained the copurified HPr at a molar equivalent of LsrK (0.1 μM). Although the inhibition curves were not completely fitted by a simple inhibition equation (Eq. 3), inhibition activities of the HPrH15E and HPrH15A mutants were about five times less than and similar to that of the WT HPr, respectively. (C) The relative inhibition of LsrK activity by the native HPr and p-HPr proteins was assessed with 0.75 mM (S)-DPD and ATP. The relative inhibition of LsrK activity by the native HPr protein was described using the parameters determined in (A) (Eq. 2). The comparison of the relative inhibition activities between HPr and p-HPr was analyzed by using a simple inhibition equation (Eq. 3). The inhibition activity of p-HPr was much lower (more than 25 times) than that of the native HPr. The higher LsrK activity in the case of the p-HPr inhibition (marked with an asterisk) likely resulted from the phosphorylation of the copurified HPr with the LsrK protein by the EI enzyme that was retained in the p-HPr solution.
The ratio of HPr and p-HPr in E. coli depends on the rate of the sugar import by PTS, and p-HPr is predominant in glucose-depleted conditions (for example, stationary growth phase). Accordingly, we assessed the effects of H15 mutations (H15A and H15E) on LsrK activity. Although inhibition of LsrK activity by HPr did not follow simple competitive inhibition (Fig. 3A), we found that the relative inhibition of the mutant HPr proteins could be compared to the wild type. That is, we found that HPr-mediated inhibition of H15A mutants was nearly identical to that of the wild type (Fig. 3B), suggesting that the presence of the H15 residue was not critical for the interaction between LsrK and HPr. However, the H15E mutant displayed several times lower inhibition compared to the wild type, but it still had a low Ki value (1.2 ± 0.2 μM). We further note that this may be explained by noting that p-HPr carries a large and negative phosphate group at the H15-ND1 atom and, hence, should have less binding affinity for LsrK than the HPrH15E mutant. Eventually, we determined the effect of p-HPr on LsrK activity through the same coupled assay system as above, because (i) the EI uses PEP for the phosphorylation of HPr, (ii) the rate of the PEP consumption by EI is directly coupled to HPr phosphorylation (23), and (iii) additional PEP (less than 10%) from the reaction mixture of the EI and HPr to the coupled assay mixture did not perturb the reaction rate of PK. In addition, the complete phosphorylation of 15N-labeled HPr (15NHPr) by EI was confirmed by the 1H/15N heteronuclear single-quantum coherence (HSQC) nuclear magnetic resonance (NMR) experiments (fig. S4); the phosphorylated 15NHPr (p-15NHPr) (0.15 mM) was maintained for at least 5 hours in the presence of 3.0 mM PEP, regardless of the different ratios of HPr to EI (100 and 50). We note that the HSQC spectrum of p-15NHPr was similar to a previously reported HSQC spectrum (24). p-HPr displayed a much reduced ability to inhibit LsrK (Ki, 8.4 ± 0.5 μM) than did the native HPr (Ki, 0.32 ± 0.07 μM) (Fig. 3C). The Ki value of p-HPr, while high, was not so high as to preclude its hypothetical role in inhibiting LsrK. This is in agreement with our conclusion from the structural analysis of the HisLsrK/HPr complex that showed the presence of additional degrees of freedom in the HPr-H15 residues (Fig. 1 and fig. S3B). The presence of a strict steric clash during the protein-protein interaction normally causes complete loss of binding. In sum, these results project a hypothesis that the phosphorylation state of HPr affects the in vivo activity of LsrK during AI-2–mediated QS signaling.
LsrK activity is increased in the stationary phase
To investigate the in vivo activity of LsrK in the presence of the wild-type HPr or HPrH15A, Miller assay experiments were performed. LsrK activity was accessed indirectly via LacZ expression levels in the presence or absence of 40 μM synthetic AI-2. For this, we transformed the mid-copy pSkunk plasmid harboring the wild-type HPr, its mutant HPrH15A, or the empty plasmid into PH02, which is a ptsH and luxS knockout strain (table S2). In addition, a reporter pLW11 plasmid was cotransformed into PH02, because this plasmid carries the lacZ reporter gene under the lsr operon promoter (10). In the absence of the wild-type HPr or HPrH15A, we observed an increase in LacZ expression in the presence of 40 μM AI-2 when compared to the same condition but in the absence of AI-2 (Fig. 4A). Next, the effects of the wild-type HPr and HPrH15A on the in vivo activity of LsrK were accessed according to different E. coli growth stages. To evaluate whether the wild-type HPr and its mutant HPrH15A affected cell growth, we measured the PH02 growth rate under different conditions. PH02 transformed with pSkunk-HPr grew faster than the same strain carrying pSkunk-HPrH15A or pSkunk-empty (no HPr). That is, the growth rate observed in the absence of HPr (pSkunk-empty) was similar to that of the HPr mutant (pSkunk-HPrH15A) (Fig. 4B). Because HPr is involved in sugar transport, it is probable that the H15 mutation abolishes HPr activity as a phosphocarrier in PTS, affecting the use of sugars and other carbon sources that affect the phosphorylation state of PTS. The presence of HPrH15A decreased the lacZ activity irrespective of the different growth phases. On the other hand, the lacZ activity of the E. coli strain carrying pSkunk-HPr was suppressed during exponential growth but was increased at the late stationary growth phase likely due to the increased population of p-HPr (Fig. 4A). Although the strains carrying pSkunk-empty and pSkunk-HPrH15A took longer to reach an OD (optical density) of 4.0, the increased lacZ activity of E. coli carrying pSkunk-HPr became more apparent as the culture OD was increased.
Fig. 4. E. coli lsr promoter activity during cell growth (indirect measurement of LsrK activity).
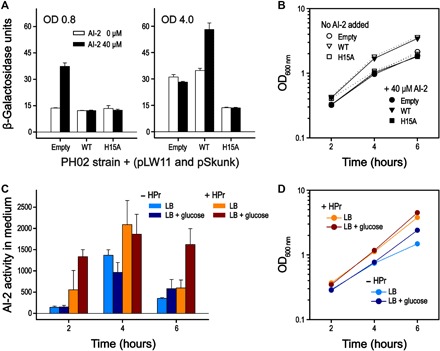
(A) E. coli PH02 strain (ΔptsH and ΔluxS) carrying the reporter plasmid pLW11 (lacZ gene under lsr operon) and the pSkunk-empty, pSkunk-HPr, or pSkunk-HPrH15A plasmid. Cells were cultivated in LB medium in the presence or absence of 40 μM AI-2. The culture aliquots were collected for the measurement of β-galactosidase activity at 0.8 and 4.0 OD600 nm. The data showed representative experiments performed independently. Data are means ± SDs of technical triplicates. (B) Growth curves of the E. coli PH02 strains cotransformed with pLW11 and pSkunk-empty (circle), pSkunk-HPr (triangle), or pSkunk-HPrH15A (square) were measured in the absence or presence of 40 μM AI-2, as indicated. Aliquots were collected for the measurement of OD at 600 nm at different time points during cell growth. The growth rates of the E. coli strains in the absence of AI-2 were only slightly higher than those in the presence of 40 μM AI-2. (C) E. coli PH01 (ΔptsH) strains cotransformed with pLW11 and either pSkunk-empty or pSkunk-HPr were inoculated into LB medium with or without 0.8% glucose at t = 0. Samples were taken every 2 hours for the measurement of the extracellular AI-2 activity. Cultures with pSkunk-empty or pSkunk-HPr are indicated as “− HPr” or “+ HPr,” respectively. Data are the average of technical duplicates. (D) Growth curves of the E. coli PH01 strains cotransformed with pLW11 and either pSkunk-empty or pSkunk-HPr in the presence or absence of 0.8% glucose.
The apparent HPr-mediated inhibition of LsrK was also studied by measuring extracellular AI-2 levels of the PH01 strain, which is a ptsH knockout strain that still carries luxS. Figure 4C shows the extracellular AI-2 activity of PH01 pSkunk pLW11, where pSkunk either is the empty plasmid or encodes ptsH. The cells were grown in LB medium with or without 0.8% added glucose. Uptake of glucose normally decreases lsr-mediated expression, significantly reducing AI-2 uptake and resulting in prolonged accumulation of extracellular AI-2. This was seen in the strain with ptsH. This is likely partially due to down-regulation of cAMP in the presence of glucose, which is required for activation of the lsr promoter (10). However, in the cell line with the empty plasmid, AI-2 uptake is not inhibited even in the presence of glucose, indicating that activation of the lsr operon was occurring. Given the dependence of lsr on cAMP-CRP, one could infer that deleting HPr results in up-regulation of cAMP, but a previous study indicates that ptsH knockout strains actually have lower levels of cAMP than wild-type strains (25). Thus, we suggest that these findings further support the hypothesis that HPr inhibits LsrK activity and that LsrK activity is higher in the absence of HPr.
The HisLsrK/HPr complex binds less DPD than ATPγS
One-dimensional (1D) NMR saturation transfer difference (STD) experiments were used to roughly estimate the binding affinities of small molecules, ATP and DPD, to the target proteins (with large molecular weight), in which the 1H saturation pulses were only applied to the methyl proton region of the protein [usually −1 to 1 part per million (ppm)]. The binding exchange process of a small ligand to the protein amplifies the transferred saturation from the protein to the ligand, and the resulting 1D difference spectrum between off-saturation (30 ppm) and on-saturation (0.5 ppm) only produces the 1D peaks of the ligand exhibiting the binding exchange process (26). If both ATPγS (1 mM) and DPD (10 mM racemic mixture) molecules have similar binding exchange processes to the HisLsrK/HPr protein, then the 1D peaks of DPD in the STD spectrum should be much higher than those of ATPγS. However, the STD spectra in the presence or absence of the HisLsrK/HPr protein show that the HisLsrK/HPr protein binds to ATPγS, but much less to DPD (Fig. 5). The peak intensities of the ATPγS molecule in the STD spectrum are greatly amplified compared to those of the excess DPD. The addition of more HPr protein specifically reduced the peak intensities of DPD but not ATPγS, which shows that HPr binding decreases the binding exchange process of DPD with the HisLsrK/HPr complex (Fig. 5B, middle). We suggest that the DPD peak identified in the STD experiments might be attributed to trace amounts of the free LsrK protein or to the trace binding ability of the HisLsrK/HPr complex to DPD.
Fig. 5. 1D STD NMR experiments of ATPγS and DPD in the presence of LsrK and HPr proteins.
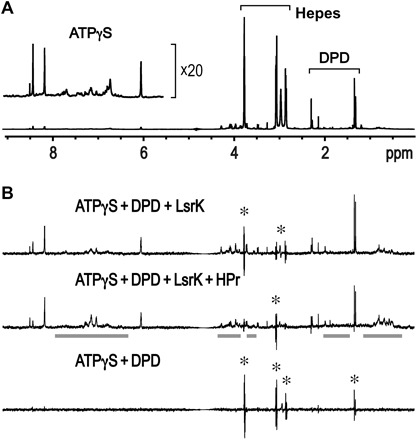
(A) The peak intensities of the 1D NMR spectrum reflect the relative concentrations of ATPγS (1 mM) and DPD (10 mM racemic mixture). (B) A small molecule displaying a binding exchange process with target proteins produces positive peaks in the 1D STD spectra. An incomplete cancellation due to slightly different peak shapes between two 1D spectra obtained with on- and off-saturation at 0.5 and 30 ppm, respectively, resulted in the peak spikes with both positive and negative signs (marked with an asterisk). Overall integration of the peak spike is zero, and thus, the molecule corresponding to the peak spikes is shown not to bind to LsrK. The residual protein peaks in the 1D STD spectrum are indicated with bold gray lines. The saturation transfer efficiency from the LsrK/HPr protein to DPD was much lower than that of ATPγS (top), which means that the LsrK/HPr protein exhibited much less binding to DPD than to ATPγS. The presence of an additional HPr further decreased the peak intensities of DPD (middle). The 1D STD spectrum in the absence of the proteins did not produce any positive peaks (bottom).
Inferred kinetic mechanism of LsrK and HPr from the structurally homologous sugar kinase
Structural comparison of the LsrK protein by the Dali server (27) revealed several homologous structures [xylulose kinase (XK), glycerol kinase (GK), and rhamnulose kinase], with high Z scores ranging from 35 to 40. We note that sequence identities between LsrK and these proteins are not high, 19.0% and 20.8% for E. coli XK (ecXK) (28) and E. coli GK (ecGK) (29), respectively, and thus, it was very difficult to find the presence of an LsrK homolog. Nonetheless, structural similarities among these sugar kinases verified that LsrK belongs to the sugar kinases/heat shock protein 70 (Hsp70)/actin superfamily consisting of two domain folds (30). Enzymes of this superfamily are known to have large interdomain motions for catalysis, and the domain motion was well characterized for ecGK (29) and human XK (hXK) (31). The structural overlay of the HisLsrK/HPr protein to each of the ecGK/EIIAGlc (32) and ecXK (28) complexes shows that the structure of the HisLsrK/HPr protein corresponds to an open and inactive form (Fig. 6). The xylulose and ADP are located in domains I and II, respectively, in the open forms of hXK and ecXK, and a large interdomain movement is required for the enzyme activity (28, 31). It has been suggested that the EIIAGlc binding to domain II of ecGK inhibits the glycerol phosphorylation activity via long-range conformational changes and that the phosphorylation of EIIAGlc can disrupt the interaction with ecGK (32). EIIAGlc is mostly dephosphorylated at high glucose concentrations, which allows EIIAGlc to inhibit various metabolic enzymes including adenylate cyclase and GK and various sugar permease via protein-protein interaction. This links the relevant biological activities to PTS (17). On the other hand, HPr forms a tight complex with domain I of LsrK, instead of domain II. We suggest that HPr plays a similar role to EIIAGlc for ecGK, in which p-HPr has a lower affinity to LsrK and inhibits LsrK activity much less than the unphosphorylated HPr.
Fig. 6. Structural comparison of HisLsrK with ecGK and ecXK.
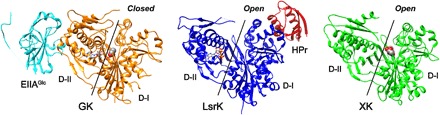
GK and XK are the members of the sugar kinases/Hsp70/actin superfamily and consist of two domains, in which the substrate sugar and ATP molecules bind to domain I (D-I) and domain II (D-II), respectively. ADP and ATP molecules in GK and HisLsrK are shown in the ball-and-stick model, and the glycerol and xylulose in GK and XK are represented as spheres. The structure of ecGK (PDB code, 1GLB) corresponds to the closed form, which allows the reaction of the phosphate transfer from the ATP to the glycerol. The native EIIAGlc binds to domain I and has been proposed to inhibit the enzyme activity through long-range conformational changes. On the other hand, the structures of HisLsrK and ecXK (PDB code, 2ITM) are open form, in which the distance between the ATP and the substrate molecules is too far for the enzyme reaction. HPr binds domain I of HisLsrK, instead of domain II.
DISCUSSION
The presence of de novo regulation of LsrK mediated by HPr was inductively revealed from structural studies of the LsrK/HPr complex. That is, we discovered that the yield of purified LsrK using the previous complicated expression methodology (21) seemed to be critically dependent on the in vivo quantity of the HPr protein. We showed that the structure of the HisLsrK/HPr complex is likely to be an inactive form, as the absence of DPD in the HisLsrK/HPr crystals grown with both ATPγS and DPD is, in part, explained by the following: (i) Crystallization of the HisLsrK protein already contained the HPr protein, and (ii) HPr is more tightly bound to ATP:LsrK (Ki1, 0.08 ± 0.01 μM) than to ATP:LsrK:DPD (Ki2, 0.27 ± 0.03 μM). Although the exact mechanism by which HPr inhibits LsrK activity is still unknown, the absence of DPD in the HisLsrK/HPr complex structure and the 1D STD NMR data of DPD in the presence of the HisLsrK/HPr complex also suggest that a structural change of domain I induced by HPr binding is not adequate for recruiting the DPD molecule. The in vivo ratio of HPr and p-HPr is closely related to the rate of sugar uptake, and we note that about 25-fold lower inhibition of LsrK activity by p-HPr, compared to unphosphorylated HPr, likely enables the QS process to be dependent on the E. coli sugar metabolism. Although a strict steric hindrance during protein-protein interactions generally results in an all-or-none response, the less stringent steric hindrance imported by the phosphorylated H15 during the interaction between LsrK and HPr likely enables fine-tuning of the AI-2–mediated QS according to both in vivo concentration and the ratio of HPr and p-HPr.
Different Km values of LsrK proteins from E. coli and Salmonella typhimurium LT2
The previously reported Km values of hXK (31) for d-xylulose and of ecGK (29) for glycerol are 24 ± 3 and 16 ± 5 μM, respectively, and the kcat of hXK is 35 ± 5 s−1. The kinetic parameters of the Salmonella typhimurium (S. typhimurium) LT2 LsrK protein for DPD (Km, 1.0 ± 0.2 mM; kcat, 7.6 ± 0.6 s−1 at 0.5 mM ATP) (22) are different from our kinetic values of the E. coli LsrK (Km, 58 ± 11 μM; kcat, 17 ± 1 s−1 at 0.75 mM ATP). Perhaps, the higher Km values noted for the S. typhimurium LT2 LsrK might be a result of the assembly of an LsrK/HPr complex, because (i) the Km values of GK and XK for their substrate sugars are similar to those of the E. coli LsrK for DPD, (ii) the apparent effect of HPr binding mimics the increased Km value of LsrK activity (−1/Km is the x intercept in the Lineweaver-Burk plot) (Fig. 3A), and (iii) the HPr-binding sequence of S. typhimurium LT2 is almost identical to that of E. coli (Fig. 2). On the other hand, the different Km values of LsrK between E. coli and S. typhimurium likely suggest that a critical concentration of AI-2 for the initiation of the AI-2–mediated QS signaling may vary with different bacterial species. Resolution to these two possibilities requires further studies.
QS is integrated to sugar metabolism via HPr of PTS
PTS is a key bacterial system that monitors the available sugars in the environment, regulating inherent metabolic processes. The phosphorylation status of the PTS components plays an important role via protein-protein interactions. The phosphorylation state of EIIAGlc is a key determinant for the CCR, also known as glucose effect in E. coli (17). In the CCR of Gram-positive bacteria, phosphorylation at S46 of HPr plays a central role (33). As HPr-S46 is located in the binding surface for LsrK, we suggest that inhibition of LsrK activity by HPr carrying the phosphorylated S46 could be abolished (Fig. 1D), perhaps indicating differences in QS regulation among Gram negatives and Gram positives. Although the dephosphorylated EIIAGlc directly inhibits many metabolic enzymes (adenylate cyclase, GK, lactose permease, and maltose permease) through the direct protein-protein interaction (17, 34), there have been several reports for the interaction between HPr and non-PTS proteins in E. coli: (i) Both HPr and p-HPr bind glycogen phosphorylase (GP), but only HPr binding appreciably increases GP activity (35); (ii) HPr, but not p-HPr, directly antagonizes the activity of Rsd, a negative regulator of the primary sigma factor (σ70), which enables the σ70-RNA polymerase holoenzyme to occupy the promoter regions of various housekeeping genes (34). The involvement of HPr, not EIIAGlc, in the regulation of GP and Rsd (anti-sigma factor) seems to be more reasonable, because these two functions are not sugar-specific but, instead, are global regulatory functions of E. coli. The in vivo concentration of HPr is also higher than that of EIIAGlc in E. coli (36). Because the AI-2–mediated QS signaling accompanies the changes of bacterial global regulation, it seems reasonable that the upstream PTS component, HPr, would be a better regulator than EIIAGlc.
A model for LsrK/HPr interplay between QS and PTS
At low cell density, secreted AIs diffuse away and otherwise do not accumulate. Rather, as cells grow and continuously secrete AIs, the accumulative production of AIs at high density initiates their detection and consecutive responses. Individual cell behaviors, in turn, become properties of the collective. This transition, called QS, enables persistence in biofilms and many other phenotypes. Abstractions of the signal transduction mechanisms have led to the design and implementation of many application-specific functions. The synchronous responses of bacteria at high density therefore provide great benefits for concerted QS signaling. QS is generally thought to be under control of transcriptional regulation, exemplified by bacteria-specific QS phenomena such as bioluminescence, biofilm formation, and virulence factor secretion. The inhibition of LsrK by HPr discovered here provides another regulatory mechanism of QS. It is particularly noteworthy that regulation of enzyme activities typically occurs at much faster characteristic times than regulation of activities involving gene expression. This will have ramifications on the design of new synthetic biology constructs. We depict a simplified view of the regulatory structure of LsrK and HPr and their involvement in PTS and QS (Fig. 7). That is, the phosphorylation status of HPr enables the QS of bacteria to be directly linked to their metabolic state; the previous report showing the involvement of ptsI in the import of AI-2 and the transcription of lsr operon (18) can now be explained by the direct interaction of HPr and p-HPr with the LsrK protein. The de novo regulation of LsrK activity by HPr and p-HPr that uses a protein-protein interaction and, at the same time, enables self-identification of a bacterium’s metabolic state could provide additional ways to synchronize the responses of bacteria to AI-2 and, more importantly, to differentiate the QS of certain bacteria in mixed microbial flora according to their metabolic states.
Fig. 7. Summarized model for the role of HPr between two distinct bacterial processes: QS and PTS.
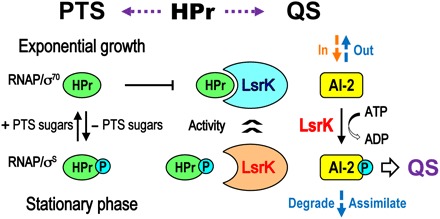
QS signaling should be strongly related to a bacterial metabolic status, because QS accompanies a transition from planktonic to sessile growth. General transcriptional regulation is under control of σ70 and σS during the exponential growth and stationary phase, respectively. In the PTS sugar transport system, HPr delivers the activated phosphate group from EI to EIIA and then the phosphate is eventually transferred to imported glucose (PTS sugars) via the transporter (EIIB and EIIC). The native HPr is dominant during the exponential growth, but p-HPr is accumulated in the stationary phase largely due to the absence of sugar import. The Ki1 (0.08 μM) and Ki2 (0.27 μM) values of the HPr protein were very low, and thus, LsrK inhibition by HPr is likely to be released during the late stationary phase when the accessible HPr is extremely limited. In addition, at these later times, AI-2 is taken up by the Lsr transporter and more LsrK is expressed. In the current scenario, LsrK activity is modulated directly via glucose metabolism and by transcriptional regulation via AI-2 QS. The former is presumably far quicker than the latter, indicating that QS activity and sugar metabolism are tightly linked, and hence, population-scale behavior can be influenced rapidly by rapid changes in nutrient availability within a particular niche.
MATERIALS AND METHODS
Cloning for protein expression
The E. coli lsrK gene contained the sequence of the Bam HI restriction enzyme site, and thus, the lsrK gene was amplified from the genomic DNA of the E. coli K strain using the 5′- and 3′-primers with the Bgl II and Xho I restriction enzyme sites, respectively. The polymerase chain reaction (PCR) product was cloned into the pGEX-4T-1 expression vector (GE Healthcare) using the Bam HI and Xho I sites (21). For the coexpression of the N-terminal His-tagged LsrK and the native HPr proteins simultaneously, the lsrK and ptsH genes were cloned into the pACYCDuet-1 expression vector (Novagen). The lsrK gene was inserted into the first multiple cloning site (MCS) of the vector by using the Bam HI and Not I sites, and the ptsH gene was inserted into the second MCS by using the Nde I and Xho I sites, respectively.
To express the E. coli EI and HPr proteins as an intact form, the ptsI and ptsH genes were cloned into the pET-11a vector (37). The ptsH gene was additionally cloned into the pET-24a vector using the Nde I and Xho I sites to express the C-terminal six-His–tagged (C-His) HPr protein. The mutations resulting in the mutants H15E and H15A were introduced using the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
Coexpression and purification of the HisLsrK/HPr protein complex
It was difficult to achieve the coexpression of GST-LsrK and HPr by double transformation of the pGEX-4T-1 and pET-24a plasmids that included the lsrK and ptsH genes, respectively. Therefore, we constructed the pACYCDuet-1 plasmid and transformed it into E. coli BL21 (DE3). The transformed cells were grown in LB medium supplemented with chloramphenicol (34 μg/ml), 0.4% glucose, and an additional 250 mM NaCl at 37°C. When the culture OD at 600 nm (OD600) reached ~0.5, the culture flasks were immediately moved to a different shaking incubator (42°C) for an hour. The culture flasks were cooled on ice for 30 min, and then IPTG induction (1.0 mM) was carried out for an additional 20 hours in a shaking incubator (20°C). To obtain the l-selenomethionine (Se-Met)–labeled proteins, the cells were grown in M9 minimal medium containing Se-Met (50 μg/ml, TCI), 19 other amino acids (each 100 μg/ml), and 250 mM NaCl. The powders of Se-Met and amino acids were added into the cultures 30 min before the IPTG induction at 0.7 to 0.8 OD. The yield of the HisLsrK/HPr protein was not sufficient when the selected amino acids (Thr, Phe, Leu, Ile, Lys, and Val) that suppress the methionine biosynthesis were only supplied, as in the previous case of the LsrK expression (20).
The cultured cells were harvested by centrifugation at 4°C and then resuspended in buffer [50 mM tris-HCl (pH 7.5), 500 mM NaCl, 10 mM 2-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine, and 0.1% Tween 20]. The LsrK/HPr protein complex was purified by Ni+2 affinity column chromatography using the HisTrap FF column (GE Healthcare). The protein fractions were dialyzed against buffer [25 mM tris-HCl (pH 7.5), 50 mM NaCl, and 2 mM dithiothreitol (DTT)] and then applied to a Mono Q 5/50 GL column (GE Healthcare). The protein complex was eluted using a 200-ml NaCl gradient (0 to 1.0 M). The protein fractions were concentrated by ultrafiltration and then further purified by SEC using a HiLoad 16/26 Superdex 200 column (GE Healthcare) with buffer [50 mM Hepes (pH 7.5), 150 mM NaCl, and 2 mM DTT]. The purified LsrK/HPr complex protein was concentrated and then frozen using liquid nitrogen for storage in a −80°C refrigerator.
Expression and purification of the EI and HPr proteins
The native EI protein was expressed using BL21 (DE3) (Novagen) in LB medium. The cultured cells were resuspended in buffer [25 mM tris-HCl (pH 7.0), 1 mM DTT, 0.1% Tween 20, 1 mM benzamidine, and 1 mM PMSF]. After cell disruption by sonication, the supernatant was applied to a HiTrap Q FF column (GE Healthcare). Protein elution was performed using a 150-ml NaCl gradient with buffer [25 mM tris-HCl (pH 8.0), 1 M NaCl, and 1 mM DTT]. The fractions containing the EI protein were dialyzed into buffer [20 mM MES (pH 6.0) and 1 mM DTT] and then applied to a HiTrap-SP HP column. Elution was performed using a 150-ml NaCl gradient with buffer [25 mM MES (pH 6.0), 1 M NaCl, and 1 mM DTT]. The protein was further purified by SEC using a Superdex 200 column with buffer [5 mM Hepes (pH 7.5), 100 mM NaCl, and 1 mM DTT]. The purified EI protein was concentrated and then stored in a −80°C refrigerator.
The native HPr protein was also expressed using the BL21 (DE3) strain in LB medium. The 15N-labeled HPr (15NHPr) protein was expressed in M9 minimal medium supplemented with 15N-labeled ammonium chloride (1 mg/ml). The expressed HPr protein was purified by anion-exchange column chromatography using the HiTrap Q FF, and protein elution was carried out using a 150-ml NaCl gradient with buffer [50 mM tris-HCl (pH 8.0) and 1 M NaCl]. The fractions containing the HPr protein were concentrated and then further purified by SEC using Superdex 75 with buffer [5 mM Hepes (pH 7.5) and 100 mM NaCl].
The C-terminal His-tagged HPr proteins (wild type, H15A, and H15E) were expressed with the same method used for the native HPr proteins. The cell lysate was applied to a HisTrap FF column with buffer [25 mM tris-HCl (pH 8.0), 500 mM NaCl, and 5 mM imidazole]. The protein elution was performed using a 150-ml imidazole gradient with buffer [25 mM tris-HCl (pH 8.0), 500 mM NaCl, and 500 mM imidazole], and then the fractions containing the C-His HPr proteins were further purified by SEC using Superdex 75 with the same buffer used for the SEC of the native HPr protein. All the HPr proteins were concentrated and then also stored in a −80°C refrigerator.
Identification of an unknown protein that copurified with LsrK
Expression and purification of LsrK via the GST affinity column chromatography and thrombin digestion followed our previously published methods (21). To identify the copurified protein [that migrated between 10- and 15-kDa size markers during SDS–polyacrylamide gel electrophoresis (SDS-PAGE) (16%) analysis], we performed (i) mass spectrometric analysis and (ii) N-terminal sequencing. The band within SDS-PAGE containing the unknown copurified protein was excised and then in-gel–digested using N-tosyl-l-phenylalanine chloromethyl ketone (TPCK)–treated trypsin. The resulting peptides were analyzed by using an ultrafleXtreme MALDI-TOF mass spectrometer (Bruker Daltonics). Two peptide fragments (M1 to K24 and L50 to K79) were matched to the sequence of the HPr protein. For N-terminal sequencing, the same protein band in SDS-PAGE was transferred into a polyvinylidene difluoride membrane (Sigma-Aldrich), and then the first seven N-terminal amino acids from F2 to I8 of the HPr protein were confirmed using Edman degradation.
Crystallization of the HisLsrK/HPr protein
The presence of an additional N-terminal His-tag in the HisLsrK/HPr protein complex did not change the crystallization conditions of the previous LsrK protein expressed via GST fusion (20). Briefly, the HisLsrK/HPr complex crystals were grown by the hanging-drop vapor diffusion method, where the reservoir buffer contained 15 to 20% (v/v) PEG-400 (polyethylene glycol, molecular weight 400), 0.1 M sodium phosphate–citrate (pH 4.2), and 0.2 M lithium sulfate. To obtain protein crystals containing ATP, ADP, and ATPγS, each 5 mM nucleotide and MgCl2 were added to the HisLsrK/HPr protein (15 mg/ml) solution before the crystallization. The cryoprotectant solution included 30 to 35% 1,6-hexanediol in addition to the crystallization buffer, and each 5 mM nucleotide and MgCl2 were also included in the crystals of the HisLsrK/HPr/nucleotide complexes for flash freezing. The presence of 1,6-hexanediol in the cryoprotectant solution improved the diffraction resolution of the crystals by about 0.4 Å. The Se-Met–labeled HisLsrK/HPr complex crystals were dehydrated to improve the resolution of the x-ray diffraction. The crystals were grown in the presence of 5 mM ATPγS and MgCl2 using the same reservoir buffer as the native protein crystals. The crystals were moved to reservoir buffers including 10% higher concentrations of PEG-400 and then held at 4°C overnight. Finally, 30% (v/v) glycerol was added as a cryoprotectant.
X-ray diffraction data collection and model building
The x-ray diffraction data were collected under cold helium stream (100 K) and then processed using HKL-2000 software (HKL Research Inc.). The SAD data of the Se-Met–labeled HisLsrK/HPr/ATPγS complex crystal were collected at the BL-7A beamline of Pohang Accelerator Laboratory (PAL) at 3.2 Å resolution. The diffraction data of the HisLsrK/HPr, HisLsrK/HPr/ATP, HisLsrK/HPr/ADP, and HisLsrK/HPr/ATPγS crystals were collected at the BL-17A beamline of the Photon Factory and at the BL-5C beamline of PAL at 3.0, 2.7, 2.7, and 2.9 Å resolutions, respectively.
The SAD data were processed by using AutoSol, and automatic model building and refinement were carried out with AutoBuild in the PHENIX software package (38) to calculate the initial model structure of the HisLsrK/HPr complex. The initial model was improved by carrying out iterative cycles of model building and refinement using the COOT program (39) and the Phenix refinement program (40). The completed models based on the initial structure were accomplished with the best x-ray diffraction data of the HisLsrK/HPr/ATP complex by MR using the Phaser program in the CCP4 software package (41), and structure refinement was performed using the CNSsolve 1.3 program (42). The crystal structure of the E. coli HPr (PDB code, 1POH) was also used as a model for the MR process. Final refinements of all complex structures were finished using the Phenix program package. All structural visualizations were done by using Chimera (43). The structure-based sequence alignments of the LsrK and HPr homologs were performed with the ENDscript server (44) and our structure of the HisLsrK/HPr/ATP complex.
Enzyme activity of LsrK in the presence of HPr
The racemic mixture form of DPD and isopropyl-DPD was synthesized following the previous reported method (19). The concentration of the DPD racemic mixture was determined from the 1D NMR spectrum, in which the peak of 0.005% 3-(trimethylsilyl)propionic-2,2,3,3-d4 (TSP) acid was used as a concentration reference (1H, 2.892 mM). The concentration of the real substrate, (4S)-DPD, for LsrK activity was corrected to half the concentration of the DPD racemic mixture. The AI-2 kinase activity of the LsrK protein was performed at 25°C based on the previously reported method (22). The LsrK reaction buffer contained 50 mM Hepes (pH 7.5), 25 mM KCl, 75 mM NaCl, 3 mM MgCl2, 0.75 mM NADH (reduced form of nicotinamide adenine dinucleotide), 1.5 mM PEP, 0.75 mM ATP, LDH (10 U/ml), PK (7.5 U/ml), and bovine serum albumin (BSA) (1 mg/ml). The concentration of the HisLsrK protein was either 100 or 50 nM, and the concentrations of the DPD and HPr proteins were varied for different experiments. To measure the initial velocity of the HisLsrK enzyme reaction, the ultraviolet absorption at 340 nm was monitored using a 0.2-cm path length, and then the reaction rate was calculated using the extinction coefficient of NADH (6.22 mM−1 cm−1).
To compare the inhibition effects of the native and p-HPr proteins on HisLsrK, HPr was phosphorylated using the EI protein. The 15NHPr protein, not the nonlabeled HPr protein, was used for monitoring the phosphorylation status of the HPr protein by using the NMR HSQC spectrum. 15NHPr protein phosphorylation was performed at 25°C for 30 min in an HPr reaction buffer containing 50 mM Hepes (pH 7.5), 25 mM KCl, 75 mM NaCl, 3 mM MgCl2, 3 mM PEP, 0.15 mM 15NHPr, and 1.5 μM EI enzyme. HSQC spectra of 15NHPr confirmed that p-15NHPr could be maintained at least for an additional 5 hours at 25°C and that one-fourth of the amount of the EI protein was able to completely phosphorylate the same amount of 15NHPr. The concentration of the 15NHPr protein was adjusted using the same HPr reaction buffer without the EI protein, and then the same volume of the p-HPr reaction mixture was added to the LsrK reaction buffer without DPD. LsrK activity was monitored after the addition of 0.75 mM (S)-DPD. The same protocol was used for the native 15NHPr protein, but recall that the EI protein was absent.
Oligomeric state of LsrK
The oligomeric state of the LsrK protein was determined by using the MALS instrument (DAWN HELEOS II, Wyatt Technology) in the presence of buffer [10 mM Hepes (pH 7.5), 200 mM NaCl, and 10 mM β-mercaptoethanol]. The SEC-MALS analysis was performed using a TSKgel G3000SW column (300 mm × 7.5 mm, Tosoh) kept in a column oven (10°C). LsrK proteins (20 μl) with different concentrations were injected, and the absolute molecular weights were estimated using the values of BSA’s refractive index (dn/dc, 0.185 ml g−1) or the LsrK extinction coefficient (1.634 ml mg−1 cm−1) as reference.
STD NMR experiments of the HisLsrK/HPr complex
The concentration of the DPD stock solution was determined from the peak integral of 1D NMR spectrum by comparing the peak integral of the internal standard chemical TSP. All NMR measurements were carried out using a Bruker 800-MHz cryoprobed spectrometer. The STD experiments were performed with a Bruker pulse sequence (stddiffesgp.3) (26). The purified 50 μM LsrK/HPr proteins were prepared in buffer [50 mM Hepes (pH 7.5), 100 mM NaCl, 10 mM racemic mixture of DPD, 1 mM MgCl2, 1 mM ATPγS, and 5% D2O]. To identify the effect of HPr on the DPD binding of LsrK, an additional 75 μM HPr protein was added to the NMR sample and the same STD spectrum was recorded.
Chromosomal deletion of ptsH, cloning, and in vivo measurement of LsrK activity in the presence or absence of HPr and HprH15A
E. coli strains DH5 (New England Biolabs), LW7 (10), PH01, and PH02 (table S2) were grown in LB medium at 37°C for DNA manipulation and expression experiments. Miller assay and AI-2 uptake experiments were performed using ampicillin (50 μg/ml) and spectinomycin (25 μg/ml) to maintain the PH01 or PH02 strain transformed with pLW11 (10) and empty pSkunk, pSkunk-HPr, or pSkunk-HPrH15A (table S2).
To create the E. coli PH01 and PH02 strains, ptsH was knocked out from the ZK126 (45) and LW7 strain genomes (10), respectively, using the one-step replacement method described by Datsenko and Wanner (46). PCR was performed with the ptsHdel-F and ptsHdel-R primers using pKD3 as template (table S3). Deletion of the ptsH gene was confirmed by PCR with the ptsHout-F and ptsHout-R primers (table S3) using genomic DNA from recombinant colonies as template.
To perform in vivo assays, the wild-type HPr and HPrH15A were cloned into the pSkunk plasmid (47) using the ptsH–Bam HI and ptsH–Spe I primers (table S3). For the Miller assay experiments, E. coli PH02 (table S2) harboring the plasmid pLW11 (10) and empty pSkunk, pSkunk-HPr, or pSkunk-HPrH15A (table S2) were grown overnight in LB medium and then diluted 100-fold (OD600 = 0.05) into fresh LB medium supplemented with ampicillin (50 μg/ml) and spectinomycin (25 μg/ml). Cultures were incubated at 37°C with shaking at 250 rpm in flasks. When the OD600 reached approximately 0.2, the cultures were split into multiple 2-ml culture tubes and 40 μM synthetic AI-2 (DPD) was added. Cultures grew in the absence or presence of AI-2 and were sampled at OD600 = 0.8 and 4 for the measurement of β-galactosidase activity. Specific activity of β-galactosidase is expressed in Miller units (48).
To perform the AI-2 uptake experiments, PH01 harboring plasmids pLW11 and either empty pSkunk or pSkunk-HPr were grown overnight in LB medium and then diluted 100-fold into fresh LB medium or LB medium with 0.8% glucose. Cultures were supplemented with ampicillin (50 μg/ml) and spectinomycin (25 μg/ml). Every 2 hours, samples were taken and filtered through a 0.2-μm filter. A bioluminescent reporter strain, Vibrio harveyi BB170 (49), was used to measure the AI-2 activity of the conditioned medium samples. BB170 was grown at 30°C for 16 hours in AB medium and then diluted 5000-fold into fresh AB medium supplemented with kanamycin (10 μg/ml). Twenty microliters of the experimental samples was added to 180 μl of the diluted BB170 cells. Luminescence of cultures was recorded and presented as a fold change relative to a negative control, where fresh medium was added in place of an experimental sample.
Accession numbers
Three coordinates of the complexes have been deposited in the Research Collaboratory for Structural Bioinformatics PDB with the accession codes 5YA0 (HisLsrK/HPr), 5YA1 (HisLsrK/HPr/ATP), and 5YA2 (HisLsrK/HPr/ADP).
Supplementary Material
Acknowledgments
Funding: This research was supported by a National Research Foundation of Korea grant funded by the Korean government (no. 2016R1A2B4015436 to K.-S.R.) and by a Korea Basic Science Institute grant (T37412 to K.-S.R.). X.M. thanks Purdue University and China Scholarship Council (no. 201406140119) for financial support. Additionally, this work was supported, in part, by the U.S. Defense Threat Reduction Agency (HDTRA1-13-0037 to W.E.B.). Author contributions: J.-H.H. performed most experiments including the protein preparations and crystallizations. Y.E. and S.C. prepared additional protein samples. J.-H.H. and K.-S.R. determined the crystal structures. K.C. performed the mass identification. K.-S.R., M.J., and J.-Y.S. performed NMR and enzyme kinetic experiments. X.M. and H.O.S. synthesized AI-2 and its analogs. P.H., K.S., and W.E.B. performed all the cell-based experiments. J.-H.H., P.H., K.-S.R., W.E.B., and H.O.S. wrote the manuscript with help from all authors. K.-S.R., W.E.B., and H.O.S. supervised all of the research. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/6/eaar7063/DC1
fig. S1. Identification of copurified protein with LsrK.
fig. S2. SEC-MALS analysis of the purified LsrK/HPr protein complex.
fig. S3. Electron density maps in the region of the bound HPr, ATP, and ADP.
fig. S4. Monitoring the stability of the synthesized p-15NHPr using HSQC experiment.
table S1. X-ray data collection and refinement statistics.
table S2. Strains and plasmids used for Miller assay.
table S3. Primer sequences for the in vivo studies of the wild-type and mutant HPr proteins.
REFERENCES AND NOTES
- 1.Teschler J. K., Zamorano-Sánchez D., Utada A. S., Warner C. J. A., Wong G. C. L., Linington R. G., Yildiz F. H., Living in the matrix: Assembly and control of Vibrio cholerae biofilms. Nat. Rev. Microbiol. 13, 255–268 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harms A., Maisonneuve E., Gerdes K., Mechanisms of bacterial persistence during stress and antibiotic exposure. Science 354, aaf4268 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Vendeville A., Winzer K., Heurlier K., Tang C. M., Hardie K. R., Making ’sense’ of metabolism: Autoinducer-2, LUXS and pathogenic bacteria. Nat. Rev. Microbiol. 3, 383–396 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Khalil A. S., Collins J. J., Synthetic biology: Applications come of age. Nat. Rev. Genet. 11, 367–379 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ford T. J., Silver P. A., Synthetic biology expands chemical control of microorganisms. Curr. Opin. Chem. Biol. 28, 20–28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hwang I. Y., Koh E., Wong A., March J. C., Bentley W. E., Lee Y. S., Chang M. W., Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nat. Commun. 8, 15028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Din M. O., Danino T., Prindle A., Skalak M., Selimkhanov J., Allen K., Julio E., Atolia E., Tsimring L. S., Bhatia S. N., Hasty J., Synchronized cycles of bacterial lysis for in vivo delivery. Nature 536, 81–85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saeidi N., Wong C. K., Lo T.-M., Nguyen H. X., Ling H., Leong S. S. J., Poh C. L., Chang M. W., Engineering microbes to sense and eradicate Pseudomonas aeruginosa, a human pathogen. Mol. Syst. Biol. 7, 521 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsao C.-Y., Hooshangi S., Wu H.-C., Valdes J. J., Bentley W. E., Autonomous induction of recombinant proteins by minimally rewiring native quorum sensing regulon of E. coli. Metab. Eng. 12, 291–297 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Wang L., Hashimoto Y., Tsao C.-Y., Valdes J. J., Bentley W. E., Cyclic AMP (cAMP) and cAMP receptor protein influence both synthesis and uptake of extracellular autoinducer 2 in Escherichia coli. J. Bacteriol. 187, 2066–2076 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xavier K. B., Miller S. T., Lu W., Kim J. H., Rabinowitz J., Pelczer I., Semmelhack M. F., Bassler B. L., Phosphorylation and processing of the quorum-sensing molecule autoinducer-2 in enteric bacteria. ACS Chem. Biol. 2, 128–136 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Xue T., Zhao L., Sun H., Zhou X., Sun B., LsrR-binding site recognition and regulatory characteristics in Escherichia coli AI-2 quorum sensing. Cell Res. 19, 1258–1268 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Laganenka L., Colin R., Sourjik V., Chemotaxis towards autoinducer 2 mediates autoaggregation in Escherichia coli. Nat. Commun. 7, 12984 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laganenka L., Sourjik V., Autoinducer 2-dependent Escherichia coli biofilm formation is enhanced in a dual-species coculture. Appl. Environ. Microbiol. 84, e02638–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xavier K. B., Bassler B. L., Regulation of uptake and processing of the quorum-sensing autoinducer AI-2 in Escherichia coli. J. Bacteriol. 187, 238–248 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graff S. M., Bentley W. E., Mathematical model of LsrR-binding and derepression in Escherichia coli K12. J. Bioinform. Comput. Biol. 15, 1650039 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Deutscher J., Aké F. M. D., Derkaoui M., Zébré A. C., Cao T. N., Bouraoui H., Kentache T., Mokhtari A., Milohanic E., Joyet P., The bacterial phosphoenolpyruvate: Carbohydrate phosphotransferase system: Regulation by protein phosphorylation and phosphorylation-dependent protein-protein interactions. Microbiol. Mol. Biol. Rev. 78, 231–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pereira C. S., Santos A. J. M., Bejerano-Sagie M., Correia P. B., Marques J. C., Xavier K. B., Phosphoenolpyruvate phosphotransferase system regulates detection and processing of the quorum sensing signal autoinducer-2. Mol. Microbiol. 84, 93–104 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Roy V., Smith J. A. I., Wang J., Stewart J. E., Bentley W. E., Sintim H. O., Synthetic analogs tailor native AI-2 signaling across bacterial species. J. Am. Chem. Soc. 132, 11141–11150 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Ha J.-H., Eo Y., Grishaev A., Guo M., Smith J. A. I, Sintim H. O., Kim E.-H., Cheong H.-K., Bentley W. E., Ryu K.-S., Crystal structures of the LsrR proteins complexed with phospho-AI-2 and two signal-interrupting analogues reveal distinct mechanisms for ligand recognition. J. Am. Chem. Soc. 135, 15526–15535 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ha J.-H., Eo Y., Ahn H.-C., Ryu K.-S., Increasing the soluble expression and crystallization of the Escherichia coli quorum-sensing protein LsrK. Acta Crystallogr. F Struct. Biol. Commun. 73, 253–258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu J., Hixon M. S., Globisch D., Kaufmann G. F., Janda K. D., Mechanistic insights into the LsrK kinase required for autoinducer-2 quorum sensing activation. J. Am. Chem. Soc. 135, 7827–7830 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waygood E. B., Steeves T., Enzyme I of the phosphoenolpyruvate: Sugar phosphotransferase system of Escherichia coli. Purification to homogeneity and some properties. Can. J. Biochem. 58, 40–48 (1980). [DOI] [PubMed] [Google Scholar]
- 24.van Nuland N. A. J., Boelens R., Scheek R. M., Robillard G. T., High-resolution structure of the phosphorylated form of the histidine-containing phosphocarrier protein HPr from Escherichia coli determined by restrained molecular dynamics from NMR-NOE data. J. Mol. Biol. 246, 180–193 (1995). [DOI] [PubMed] [Google Scholar]
- 25.Isaza M. P., Duncan M. S., Kaplan J. B., Kachlany S. C., Screen for leukotoxin mutants in Aggregatibacter actinomycetemcomitans: Genes of the phosphotransferase system are required for leukotoxin biosynthesis. Infect. Immun. 76, 3561–3568 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayer M., Meyer B., Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 123, 6108–6117 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Holm L., Laakso L. M., Dali server update. Nucleic Acids Res. 44, W351–W355 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Luccio E., Petschacher B., Voegtli J., Chou H.-T., Stahlberg H., Nidetzky B., Wilson D. K., Structural and kinetic studies of induced fit in xylulose kinase from Escherichia coli. J. Mol. Biol. 365, 783–798 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feese M. D., Faber H. R., Bystrom C. E., Pettigrew D. W., Remington S. J., Glycerol kinase from Escherichia coli and an Ala65→Thr mutant: The crystal structures reveal conformational changes with implications for allosteric regulation. Structure 6, 1407–1418 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Hurley J. H., The sugar kinase/heat shock protein 70/actin superfamily: Implications of conserved structure for mechanism. Annu. Rev. Biophys. Biomol. Struct. 25, 137–162 (1996). [DOI] [PubMed] [Google Scholar]
- 31.Bunker R. D., Bulloch E. M. M., Dickson J. M. J., Loomes K. M., Baker E. N., Structure and function of human xylulokinase, an enzyme with important roles in carbohydrate metabolism. J. Biol. Chem. 288, 1643–1652 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurley J. H., Faber H. R., Worthylake D., Meadow N. D., Roseman S., Pettigrew D. W., Remington S. J., Structure of the regulatory complex of Escherichia coli IIIGlc with glycerol kinase. Science 259, 673–677 (1993). [PubMed] [Google Scholar]
- 33.Saier M. H., Jr, Cyclic AMP-independent catabolite repression in bacteria. FEMS Microbiol. Lett. 138, 97–103 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Park Y.-H., Lee C.-R., Choe M., Seok Y.-J., HPr antagonizes the anti-σ70 activity of Rsd in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 110, 21142–21147 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seok Y.-J., Sondej M., Badawi P., Lewis M. S., Briggs M. C., Jaffe H., Peterkofsky A., High affinity binding and allosteric regulation of Escherichia coli glycogen phosphorylase by the histidine phosphocarrier protein, HPr. J. Biol. Chem. 272, 26511–26521 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Rohwer J. M., Meadow N. D., Roseman S., Westerhoff H. V., Postma P. W., Understanding glucose transport by the bacterial phosphoenolpyruvate: Glycose phosphotransferase system on the basis of kinetic measurements in vitro. J. Biol. Chem. 275, 34909–34921 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Schwieters C. D., Suh J.-Y., Grishaev A., Ghirlando R., Takayama Y., Clore G. M., Solution structure of the 128 kDa enzyme I dimer from Escherichia coli and its 146 kDa complex with HPr using residual dipolar couplings and small- and wide-angle X-ray scattering. J. Am. Chem. Soc. 132, 13026–13045 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D., Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J.-S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L., Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Robert X., Gouet P., Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Connell N., Han Z., Moreno F., Kolter R., An E. coli promoter induced by the cessation of growth. Mol. Microbiol. 1, 195–201 (1987). [DOI] [PubMed] [Google Scholar]
- 46.Datsenko K. A., Wanner B. L., One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wright R. C., Khakhar A., Eshleman J. R., Ostermeier M., Advancements in the development of HIF-1α-activated protein switches for use in enzyme prodrug therapy. PLOS ONE 9, e114032 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.J. H. Miller, Experiments in Molecular Genetics (Cold Spring Harbor Laboratory Press, 1972). [Google Scholar]
- 49.Bassler B. L., Greenberg E. P., Stevens A. M., Cross-species induction of luminescence in the quorum-sensing bacterium Vibrio harveyi. J. Bacteriol. 179, 4043–4045 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/6/eaar7063/DC1
fig. S1. Identification of copurified protein with LsrK.
fig. S2. SEC-MALS analysis of the purified LsrK/HPr protein complex.
fig. S3. Electron density maps in the region of the bound HPr, ATP, and ADP.
fig. S4. Monitoring the stability of the synthesized p-15NHPr using HSQC experiment.
table S1. X-ray data collection and refinement statistics.
table S2. Strains and plasmids used for Miller assay.
table S3. Primer sequences for the in vivo studies of the wild-type and mutant HPr proteins.