

Abstract
Neuroprotective strategies using diazoxide (DZX) have been demonstrated to limit ischemia/reperfusion (I/R)-induced injury and cell apoptosis. In type 2 diabetes mellitus, DZX has been reported to improve β-cell function and reduce their apoptosis, through suppressing endoplasmic reticulum (ER) stress. However, the effects of DZX on ER stress during I/R-induced neuronal apoptosis in the hippocampus remains to be elucidated. In the present study, pretreatment of hippocampal neurons with DZX was revealed to inhibit oxygen-glucose deprivation (OGD)-stimulated apoptosis in vitro and to alleviate I/R-induced hippocampal injury and behavioral deficits in rats in vivo. Furthermore, OGD and I/R were demonstrated to induce ER stress via upregulating the expression of ER stress-associated proteins, including C/EBP homologous protein, 78 kDa glucose-regulated protein and caspase-12, whereas the exogenous administration of DZX effectively downregulated ER stress-associated protein expression following OGD and I/R. In addition, DZX was revealed to significantly increase the protein expression of B-cell lymphoma (Bcl)-2 and suppress the expression of caspase-3 and Bcl-2-associated X protein. These findings suggested that DZX may protect cells against apoptosis via regulating the expression of ER stress-associated proteins in vitro and in vivo, thus enhancing the survival of hippocampal cells. The present results suggested a novel mechanism that may underlie the protective effect of DZX administration in the central nervous system.
Keywords: diazoxide, endoplasmic reticulum stress, oxygen-glucose deprivation, cerebral ischemia-reperfusion, apoptosis
Introduction
Ischemia/reperfusion (I/R) injury is defined as the serious tissue damage caused by the restoration of blood circulation in ischemic tissues, and is a considerable cause of morbidity and mortality worldwide (1). Multiple subclinical I/R incidents may lead to cumulative tissue injury, which has been implicated in chronic degenerative disorders, including vascular dementia, which is a heterogeneous group of brain disorders characterized by cognitive impairment due to cerebrovascular pathologies (2). Ischemic tolerance may have potential as a neuroprotective strategy against cerebral I/R injury. Ischemic tolerance is defined as induced resistance to a severe ischemic episode through preconditioning with sub-lethal ischemic stimuli; pharmacological agents may also provide neuroprotection, a strategy defined as pharmacological preconditioning (3,4). However, the precise mechanisms underlying the development of ischemic tolerance in the human brain remain to be elucidated.
Neuroprotective strategies using diazoxide (DZX) have been demonstrated to limit cerebral I/R injury and apoptosis. DZX is a K+ channel agonist and the mitochondrial ATP-dependent K+ (mKATP) channels have been suggested to mediate its neuroprotective effects (5). In rat and mouse models of I/R injury, DZX preconditioning has been demonstrated to reduce the volume of brain infarcts induced by middle cerebral artery occlusion (5,6). On a subcellular level, previous studies have suggested that the stabilization of the mitochondrial membrane potential and the preservation of mitochondrial integrity may contribute to the anti-apoptotic actions of DZX in neurons (7–9). On a molecular level, the neuroprotective effects of DZX may be mediated by the inhibition of caspase-3 activation and mitochondrial cytochrome c release, and the upregulation of Kir6.1 expression, which is an mKATP channel subunit enriched in brain mitochondria (10,11). Although the neuroprotective effects of DZX have previously been demonstrated, the detailed cellular mechanisms underlying its actions against I/R-induced brain injury remain to be elucidated.
The molecular mechanisms linking endoplasmic reticulum (ER) stress to apoptosis in various brain regions have previously been investigated; failure of the physiological ER function may initiate apoptosis, which has been implicated in the pathophysiology of several neurodegenerative diseases (12–14). A previous study reported that DZX improved the function and reduced the apoptosis of type 2 diabetes pancreatic β-cells by reducing ER stress (15), thus suggesting that the modulation of ER stress may be implicated in the cytoprotective effects of DZX in β-cells. Therefore, it may be hypothesized that the reduction of ER stress is involved in the mechanisms underlying the protective effects of DZX against I/R-induced apoptosis in hippocampal neurons, which are particularly vulnerable to ischemic injury and die during the first days of reperfusion (16–19). The present study aimed to identify the anti-apoptotic effects of DZX in rat hippocampal neurons during the development of I/R injury, and investigate the involvement of ER stress-associated mechanisms using an oxygen-glucose deprivation (OGD) model in vitro and occlusion of the middle cerebral artery to confirm the findings in vivo.
Materials and methods
Animals
A total of 26 adult female Wistar rats (age, 8–12 weeks; weight, 200–220 g) were purchased and housed in an environmentally-controlled breeding room of Tianjin 4th Center Hospital (Tianjin, China), at a temperature of 20±2°C, a relative humidity of 60±5% and under a 12-h light/dark cycle. Rats were fed standard laboratory chow with water access ad libitum. They were maintained in accordance with internationally accepted principles for laboratory animal use. All work was conducted in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The experimental protocols were approved by the Institutional Animal Care of Tianjin 4th Center Hospital (approval no. 2012009).
Organotypic hippocampal slice cultures and OGD
Organotypic hippocampal slice cultures were prepared based on the published protocol by Girard et al (20) with slight modifications. Briefly, rats were sacrificed by decollation, rat brains were removed using tweezers and embedded in 4% low-melting agarose (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 400-µm transverse sections were sliced using a vibrating microtome (Leica Microsystems, Ltd., Milton Keynes, UK). Hippocampi were dissected and transferred to 0.4 µm porous membrane inserts (EMD Millipore, Billerica, MA, USA). Four hippocampal sections were plated on 30 mm Transwell inserts (4 sections/insert) in a 6-well plate containing 1.2 ml artificial cerebrospinal fluid (Tocris Bioscience, Bristol, UK). Cultures were maintained in Dulbecco's modified Eagle medium (DMEM) (50%) with horse serum (25%) and Hanks' Balanced Salt solution (25%) (all Thermo Fisher Scientific, Inc.) at 37°C in a 5% CO2 atmosphere. On day 6 of culture, cultures were transferred to serum-free DMEM media with or without prior exposure to OGD. OGD induction was performed by changing medium to glucose free DMEM, which were then bubbled with N2 for 5 min prior to use. The plates were then maintained at a 5% CO2, 1% O2, 94% N2 atmosphere at 37°C in an OGD-chamber (Coy Laboratory Products, Inc., Grass Lake, MI, USA) for 2 h. Reperfusion was achieved by transferring the cultures to serum-free DMEM medium in a 5% CO2 atmosphere at 37°C for 12 h. Hippocampal sections without OGD treatment served as the control.
Electrophysiology
Neurons were visualized on an upright light microscope (BX50WI; Olympus Corporation, Tokyo, Japan) using infrared differential interference contrast optics. Whole cell voltage-clamp recordings were made using an EPC10 amplifier and PatchMaster 2.54 software (both HEKA Elektronik Dr. Schulze GmbH, Lambrecht, Germany). All experiments were performed at 23–26°C. Electrodes had a resistance of 3–4 MΩ when filled with the patch pipette solution. For patch clamp experiments, the internal solution that was used contained 140 mM KCl, 0.2 mM Na2ATP, 1 mM MgCl2, 10 mM EGTA and 5 mM HEPES (pH adjusted to 7.4 using KOH). The external recording solution contained 137 mM NaCl, 5 mM KCl, 1 mM CaCl2, 0.16 mM NaH2PO4, 3 mM NaHCO3, 10 mM glucose, 5 mM HEPES, 0.1 mM CdCl2 and 1 mM BaCl2 (pH adjusted to 7.4 using NaOH). The KATP current was recorded using ramp pulses of 10 sec duration from −120 to +80 mV from a holding potential of −40 mV, at 10-sec intervals as described previously (21). Following recording under normal external recording solution, the ATP-sensitive K+ (KATP) channel opener Pinacidil (100 µM; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) were applied in the solution through the continuously perfusion system. Amplitudes of KATP channel currents were measured at +80 mV using the software of PatchMaster 2.54 (HEKA Elektronik Dr Schulze GmbH) and Origin 7.0 (OriginLab, Northampton, MA, USA). The KATP channel current was obtained using the following equation: IKATP=Imax-Itest, where Imax is the current amplitude maximally activated by exposure to pinacidil, Itest is current amplitude during exposure to normal external recording solution.
In vivo cerebral I/R
Adult female rats were used for the induction of cerebral I/R through middle cerebral artery occlusion, using a modification of the intraluminal technique which has been described previously (18,22). Briefly, rats were exposed to ischemia by bilateral clamping of the middle cerebral artery. Following 2 h of ischemia, reperfusion was performed for 12 h by gently removing the arterial clamps. In sham-operated rats, similar procedures were followed, without middle cerebral artery occlusion. There were 3 rats in each group and the experiment was repeated 3 times.
DZX and 5-hydroxydecanoate (5-HD) pretreatment in vitro and in vivo
Prior to OGD, hippocampal slices were pretreated with 200 µM DZX for 1 h followed by treatment for 30 min with the blocker of pharmacological preconditioning 100 µM 5-HD, or with vehicle (PBS). Subsequently, hippocampal slices were cultured under OGD conditions, as aforementioned.
Prior to the induction of I/R, rats were intraperitoneally injected with 200 µl DZX (10 mg/kg) for 1 h, or DZX (10 mg/kg) plus 5-HD (5 mg/kg) for 1 h; rats in the control group were intraperitoneally injected with an equal volume of PBS 1 h prior to I/R induction.
Modified neurological scores
Modified neurological scores were used to grade the severity of neurological dysfunction and are presented in Table I (23). The neurological severity score is the result of the comprehensive evaluation of motor (muscle status, abnormal movement), sensory (visual, tactile and proprioceptive) and reflex tests. These tests aimed to model contralateral neglect in humans. During the calculation of the severity score of injury, 1 point is awarded for the inability to correctly perform the tasks that are tested, or for the failure of a tested reflex. There were 3 rats in each group and the experiment was repeated 3 times.
Table I.
Modified neurological severity scores.
| Tests | Points |
|---|---|
| Raising the rat by the tail | |
| Flexion of forelimb | 1 |
| Flexion of hindlimb | 1 |
| Head moving more than 10° (vertical axis) | 1 |
| Placing the rat on the floor | |
| Inability to walk straight | 1 |
| Circling toward the paretic side | 1 |
| Falling down to the paretic side | 1 |
| Abnormal movements | |
| Immobility and staring | 1 |
| Tremor (wet-dog shakes) | 1 |
| Myodystonia, irritability, seizures | 1 |
| Sensory tests | |
| Visual and tactile placing | 1 |
| Proprioceptive test (deep sensory) | 1 |
| Reflexes (blunt or sharp stimulation), when absent | |
| Pinna reflex | 1 |
| Corneal reflex | 1 |
| Startle reflex | 1 |
| Maximum points | 14 |
Flow cytometric analysis of apoptosis
Following OGD, hippocampal neurons were harvested in buffer containing 0.05% trypsin, 0.02% EDTA and 0.05% glucose, to prepare single-cell suspensions. Subsequently, cells (1×106 cells/ml) were sequentially washed in PBS and resuspended in binding buffer containing 10 mM HEPES/NaOH (pH 7.4), 140 mM NaCl and 2.5 mM CaCl2.
Subsequently, FITC Annexin V Dead Cell Apoptosis kit (Invitrogen; Thermo Fisher Scientific, Inc.) was used to stain cells, 5 µl of FITC Annexin V (Component A) and 1 µl of the 100 µg/ml PI working solution were added to each 100 µl cell suspension and incubated at room temperature for 15 min. Stained cells were analyzed using a FACStar Plus flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). The ratio of fluorescence intensity, following excitation at 488 nm, was monitored at an emission wavelength of 515 nm for FITC and 560 nm for PI. Data analysis was performed using BD Cell Quest software (version 7.5.3; BD Biosciences).
Immunohistochemical staining of 78 kDa glucose-regulated protein (GRP78) and C/EBP homologous protein (CHOP)
Hippocampal slices were blocked in PBS containing 10% normal goat serum (Invitrogen; Thermo Fisher Scientific, Inc.) and 0.3% Triton X-100 for 1 h at room temperature, and then incubated for 12 h at 4°C with the following primary antibodies: GPR78 polyclonal antibody (1:200; cat. no. ab21685), CHOP monoclonal antibody (1:200; cat. no. ab11419; both Abcam, Cambridge, UK). Subsequently, they were incubated with goat-anti-mouse (1:500; cat. no. ab6788) and goat-anti-rabbit immunoglobulin G H&L secondary antibodies (1:400; cat. no. ab6720; both Abcam) for 1 h at room temperature, washed with PBS and incubated with 3,3′-diaminobenzidine (DAB) for 5 min at room temperature. Omission of the primary antibody was used as a negative control for non-specific staining.
Histological evaluation
Hippocampal slices were fixed in 4% paraformaldehyde overnight at room temperature and then transferred to 70% ethanol. Subsequently, they were embedded in paraffin, frozen in liquid nitrogen-cooled isopentane, and sectioned at 4-µm thickness. For histopathological analysis, paraffin-embedded sections were stained with hematoxylin for 15 min and eosin for 2 min at room temperature. Scoring was performed under magnification, ×40 (upright light microscope; Leica Microsystems GmbH, Wetzlar, Germany), following the examination of >40 consecutive fields of view per slide (24).
TTC staining
Following decapitation, brains were removed and sectioned into 2-mm thick slices. These slices were kept in the dark and stained with 2% TTC (Sigma-Aldrich; Merck KGaA) for 30 min at room temperature, and then fixed with 4% PFA for 20 min at room temperature (Thermo Fisher Scientific, Inc.). Subsequently images were captured under magnification, ×4 and ×40 (Leica Microsystems GmbH; Upright Microscope). The infarct volume was evaluated by Image Pro-Plus 5.1 analysis system (Media Cybernetics, Inc., Rockville, MD, USA).
Western blot analysis
Hippocampal neurons were lysed for 30 min using lysis buffer containing 0.5% SDS, 1% NP-40, 1% sodium deoxycholate, 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), and protease inhibitors. Lysates were centrifuged at 13,000 × g for 30 min at 4°C and supernatants were collected. Equal quantity of protein (50 µg per lane) was separated by 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes using transfer buffer containing 195 mM glycine, 25 mM Tris-HCl and 20% (v/v) methanol. Transfer efficiency was confirmed following staining with Ponceau S for 3 min at room temperature. Membranes were blocked for 1 h at room temperature with 3% (w/v) bovine serum albumin (Sigma-Aldrich; Merck KGaA) in TBS (50 mM Tris pH 8.0 and 150 mM NaCl) containing 0.05% Tween-20 (TBST). Membranes were then incubated with primary antibodies at 4°C overnight, including Anti-GRP78 BiP polyclonal antibody (1:200; cat. no. ab21685); CHOP monoclonal antibody (1:200; cat. no. ab11419; both Abcam); caspase-12 polyclonal antibody (1:300, cat. no. PA5-19963); caspase-3 monoclonal antibody (1:400; cat. no. 43-7800); Bcl-2 polyclonal antibody (1:200; cat. no. PA5-11379); Bax monoclonal antibody (1:200; cat. no. MA5-14003; all Invitrogen; Thermo Fisher Scientific, Inc.); GAPDH (1:800; cat. no. SAB2103104-50UG; Sigma-Aldrich; Merck KGaA). Then incubated with secondary antibodies at room temperature for 1 h, including goat anti-rat (1:500; cat. no. ab7010), Donkey anti-Rabbit (1:600, cat. no. ab98489), Goat Anti-Mouse (1:600; cat. no. ab7067; all Abcam). Protein bands were visualized using the Fujifilm LAS-3000 Imaging system (Fujifilm Corporation, Tokyo, Japan).
Statistical analysis
Data are expressed as the mean ± standard deviation. The statistical significance of the differences between groups was assessed using Student's t-test for pair-wise comparisons or one-way analysis of variance followed by Fisher's least significance difference test. Statistical analysis was performed using SPSS version 13.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Role of mKATP channels in OGD-treated hippocampal cells
Following treatment with or without DZX and 5-HD, organotypic hippocampal cultures were subjected to OGD and then used for cell apoptosis analysis by flow cytometry. Using the increase in the intensity of Annexin V fluorescence as an index for enhanced apoptosis, hippocampal cells subjected to OGD exhibited a significant increase in apoptosis compared with the control group (P<0.01; Fig. 1A). Treatment with DZX, which is an agonist of mKATP channels, was revealed to suppress the OGD-stimulated apoptosis of hippocampal cells, whereas 5-HD, which is an mKATP direct antagonist, appeared to reverse the anti-apoptotic actions of DZX (P<0.01; Fig. 1A).
Figure 1.

Anti-apoptotic effects of DZX in OGD-treated hippocampal cells. (A) Pretreatment of hippocampal cells with DZX attenuates cell apoptosis following exposure to OGD, as indicated by flow cytometric analysis. (B) Pretreatment with DZX decreases the positive currents in OGD-treated hippocampal cells, as revealed by whole-cell patch clamp recordings. Data are expressed as the mean ± standard deviation. *P<0.05, **P<0.01, as indicated. DZX, diazoxide; OGD, oxygen-glucose deprivation; PI, propidium iodide; 5-HD, 5-hydroxydecanoate.
To further explore the role of the plasma membrane KATP channels in the protective effects of DZX in OGD-treated hippocampal cells, the K+ currents of the hippocampal cells were examined using electrophysiology. As presented in Fig. 1B, OGD-treated cells exhibited higher positive currents compared with control cells (P<0.01). Pretreatment with DZX appeared to suppress the currents of OGD-stimulated cells (P<0.01), whereas 5-HD appeared to enhance the positive currents in DZX-treated hippocampal cells following OGD (P<0.05; Fig. 1B). Collectively, the present findings suggested that pretreatment of hippocampal neurons with DZX may suppress OGD-induced cell apoptosis, through directly targeting the plasma membrane KATP channel.
DZX regulates the expression of ER stress-associated proteins in hippocampal cells following OGD
In order to investigate the role of ER stress in the anti-apoptotic effects of DZX, the protein expression of the ER stress-associated proteins CHOP and GRP78 (also termed binding immunoglobulin protein) was examined using immunohistochemistry and western blot analysis. The present results demonstrated that hippocampal cells in the control group exhibited limited GRP89 and moderate CHOP expression (Fig. 2A). Conversely, the expression of GRP78 and CHOP was significantly upregulated following OGD, thus suggesting that OGD may induce ER stress in hippocampal cells (P<0.05; Fig. 2A). Pretreatment with DZX resulted in a significant decrease in CHOP and GRP78 expression in OGD-exposed hippocampal cells, which was revealed to be abolished by the administration of 5-HD (Fig. 2A).
Figure 2.

DZX regulates the expression of CHOP, GRP78, caspase-12 and caspase-3 in OGD-treated hippocampal cells. (A) Immunohistochemical staining demonstrated the distribution of GRP78 and CHOP in hippocampal cells exposed to OGD. (B) Western blot analysis was used to detect the expression of GRP78 and CHOP in hippocampal cells exposed to OGD. (C) Protein expression levels of caspase-12 and caspase-3 in hippocampal cells exposed to OGD were detected using western blotting. Data are expressed as the mean ± standard deviation. *P<0.05, as indicated. DZX, diazoxide; CHOP, C/EBP homologous protein; GRP78, 78 kDa glucose-regulated protein; OGD, oxygen-glucose deprivation; 5-HD, 5-hydroxydecanoate.
Western blot analysis also demonstrated a similar expression pattern for CHOP and GRP78 (Fig. 2B) and suggested the implication of ER stress in the mechanism of action of DZX in hippocampal cells. Caspase-12 and caspase-3 activation is involved in ER stress-mediated apoptosis in various cell types (25,26); therefore, their expression was investigated in hippocampal cells following DZX pretreatment. OGD was revealed to markedly upregulate the protein expression of caspase-12 and caspase-3, whereas DZX suppressed the OGD-induced caspase-12 and caspase-3 upregulation; 5-HD counteracted the effects of DZX and appeared to increase the expression of caspase-12 and caspase-3 in OGD-stimulated hippocampal cells (Fig. 2C). These findings suggested that DZX may inhibit hippocampal cell apoptosis by suppressing ER stress and subsequently the activation of proapoptotic caspases.
DZX attenuates I/R-induced behavioral deficits in rats
Cerebral I/R induced severe behavioral deficits in rats, as demonstrated by the significant increase in neurological deficit scores in rats in the I/R group compared with control rats (P<0.01; Fig. 3). Following pretreatment with DZX, I/R rats exhibited a significant decrease in behavioral deficit scores (P<0.01; Fig. 3). Conversely, the administration of 5-HD prior to I/R abolished the beneficial effects of DZX and rats presented behavioral deficits similar to the I/R group. These findings suggested that DZX may protect rats against I/R-induced neurological damage.
Figure 3.
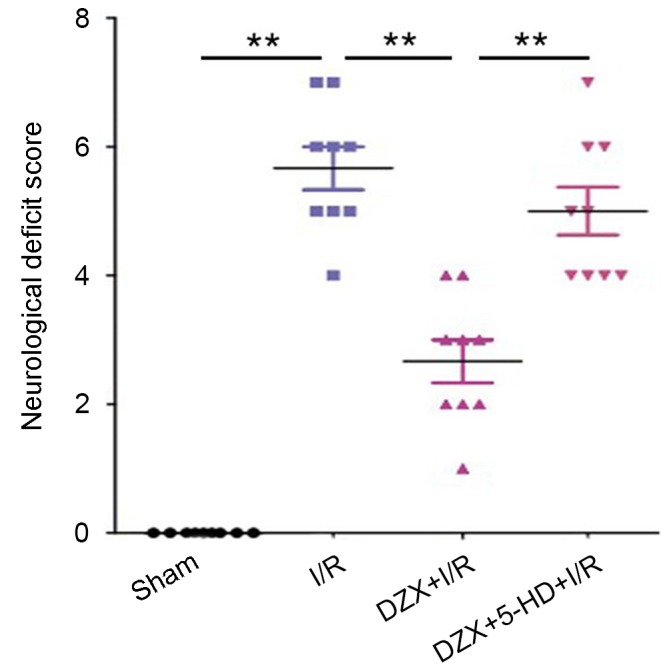
Pretreatment with DZX alleviates the I/R-induced behavioral deficits in rats in vivo. Rats were treated with DZX with or without 5-HD and subjected to cerebral ischemia for 2 h followed by 12 h of reperfusion. Sham rats were not subjected to cerebral ischemia. Data are expressed as the mean ± standard deviation. **P<0.01, as indicated. DZX, diazoxide; I/R, ischemia/reperfusion; 5-HD, 5-hydroxydecanoate.
DZX attenuates I/R-induced injury in rat hippocampus
Histological analysis was performed to investigate the effects of DZX pretreatment on infarct volumes and hippocampal morphology in rat hippocampal sections. TTC staining revealed that DZX markedly reduced infarct volumes following I/R (Fig. 4A). Conversely, 5-HD administration in DZX-treated rats resulted in an obvious increase in infarct volumes following I/R (Fig. 4A).
Figure 4.
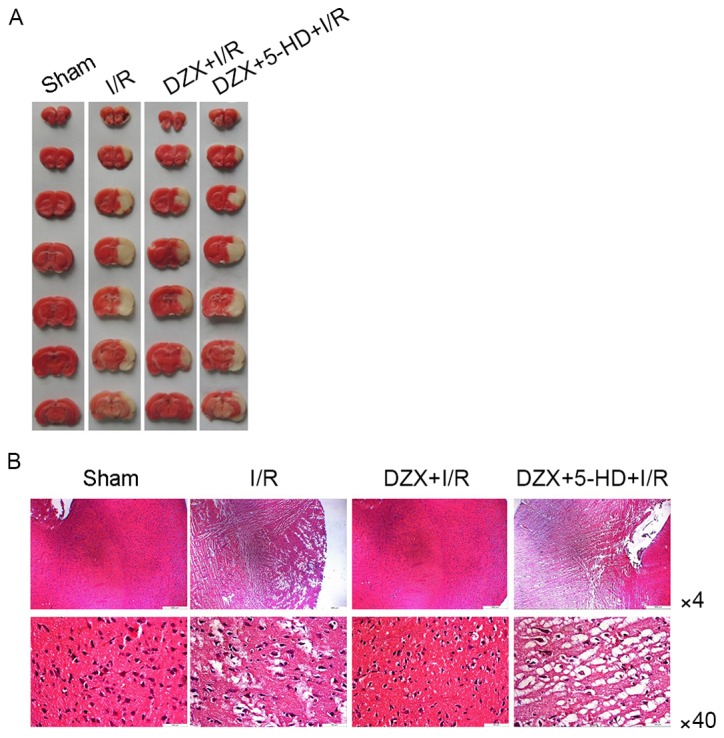
Pretreatment with DZX attenuates cerebral I/R injury in rats in vivo. (A) Effects of DZX pretreatment on cerebral infarct volumes in rats subjected to I/R. (B) Representative photomicrographs of rat hippocampal sections following histological evaluation demonstrate that pretreatment with DZX attenuated I/R-induced histopathological alterations. Rats were treated with DZX with or without 5-HD and subjected to cerebral ischemia for 2 h followed by 12 h of reperfusion. Sham rats were not subjected to cerebral ischemia. DZX, diazoxide; I/R, ischemia/reperfusion; 5-HD, 5-hydroxydecanoate.
Representative H&E staining results of hippocampal sections isolated from rats of all experimental groups are presented in Fig. 4B. Hippocampal sections from control rats exhibited standard histology; however, hippocampal vacuolation, swelling and necrosis were observed in I/R rats. Pathological alterations appeared to be attenuated in rats treated with DZX prior to the induction of I/R injury. Conversely, co-administration of DZX and 5-HD prior to I/R resulted in the reappearance of pathological hippocampal morphology in I/R rats (Fig. 4B).
DZX regulates the expression of ER stress-associated proteins in rat hippocampus following I/R
The effects of DZX on the expression of ER stress-associated proteins in vivo were further investigated. As presented in Fig. 5, cerebral I/R appeared to markedly upregulate the protein expression levels of caspase-12, caspase-3, CHOP, GRP78 and the proapoptotic factor B-cell lymphoma (Bcl)-2-associated X protein (Bax) in the hippocampus. It is of note that pretreatment with DZX was revealed to suppress the expression of these proteins, whereas 5-HD abolished the effects of DZX in the rat hippocampus following I/R (Fig. 5). The expression of the anti-apoptotic factor Bcl-2 did not appear to differ between rats in the control and I/R groups. However, pretreatment with DZX appeared to increase Bcl-2 protein expression, whereas this effect was abolished following 5-HD administration (Fig. 5).
Figure 5.
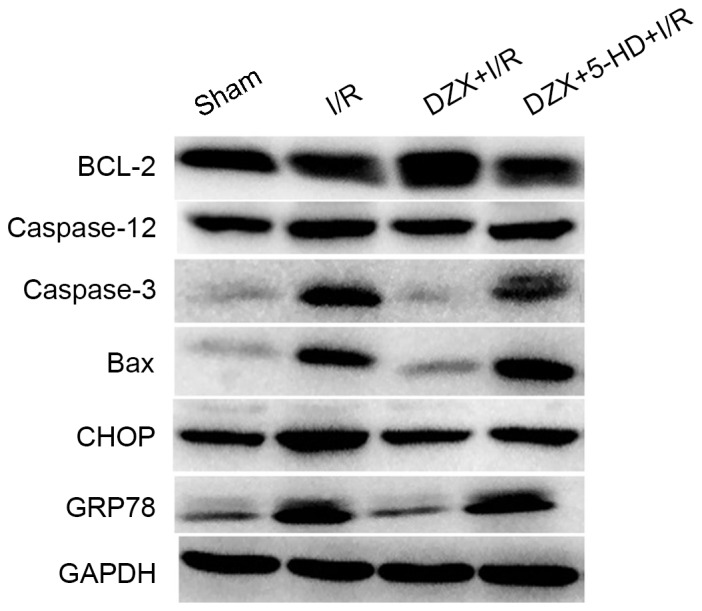
Pretreatment with DZX modulates the protein expression of Bcl-2, caspase-12, caspase-3, Bax, CHOP and GRP78 in rats subjected to I/R. Western blot analysis was used to detect the protein expression levels of Bcl-2, caspase-12, caspase-3, Bax, CHOP and GRP78 in hippocampal cells isolated from I/R-treated rats. Rats were treated with DZX with or without 5-HD and subjected to cerebral ischemia for 2 h followed by 12 h of reperfusion. Sham rats were not subjected to cerebral ischemia. DZX, diazoxide; Bcl-2, B-cell lymphoma-2; Bax, Bcl-2-associated X protein; CHOP, C/EBP homologous protein; GRP78, 78 kDa glucose-regulated protein; I/R, ischemia/reperfusion; 5-HD, 5-hydroxydecanoate.
Discussion
Reperfusion following transient cerebral ischemia leads to neuronal death, which aggravates ischemic brain injury (16,17,27). DZX is an agonist of mKATP channels which has been used to prevent cerebral I/R injury; however, the molecular mechanisms underlying DZX-induced neuroprotection and the effects of DZX on the hippocampus remain to be elucidated. In the present study, DZX was revealed to exert protective effects against OGD- and I/R-induced neuronal injury in the hippocampus. In addition, the present study investigated the molecular mechanisms underlying the anti-apoptotic effects of DZX.
Previous studies have suggested several mechanisms to explain the neuroprotective effects of DZX, including the stabilization of the mitochondrial membrane potential, the preservation of mitochondrial integrity (7–9), the inhibition of caspase-3 activation and mitochondrial cytochrome c release, the upregulation of Kir6.1 expression (10,11) and the activation of Bcl-2 and inactivation of Bax (28). The results of the present study demonstrated that DZX significantly suppressed hippocampal damage induced by I/R in vivo and by OGD in vitro. I/R- and OGD-induced apoptosis of hippocampal cells was revealed to be associated with excessive ER stress, as demonstrated by the upregulation in CHOP, GRP78, caspase-12, caspase-3, Bax expression. Pretreatment with DZX was revealed to attenuate hippocampal cell death in vitro and in vivo, via counteracting the OGD and I/R-induced mechanisms of cell injury.
Mammalian cells have developed endogenous protective strategies to combat the toxic effects of ER stress, which are collectively termed the unfolded protein response (UPR). UPR is mediated by transmembrane ER receptors, including protein kinase RNA-like ER kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring enzyme 1 (IRE1); the UPR is a pro-survival response, but may switch to a pro-apoptotic mechanism if ER stress is not resolved (14). GRP78 maintains ER stress receptors in an inactive state, through protein-protein interactions (29). During ER stress receptor-mediated responses, CHOP is an important downstream target of PERK and ATF6 (29), whereas caspase-12 is a key downstream target of CHOP, which induces apoptosis (30). In the present study, treatment with DZX appeared to inhibit the expression of CHOP, GRP78 and caspase-12 in OGD and I/R-treated hippocampal cells; however, further studies are required to elucidate the exact mechanisms involved in the modulatory effects of DZX on CHOP, GRP78 and caspase-12 expression. Previous in vitro and in vivo studies have reported that CHOP-mediated apoptosis may be involved the activation of proapoptotic cytoplasmic Ca2+ signaling pathways (31–33). Excessive intracellular Ca2+ is transferred from the ER to the mitochondria, resulting in the activation of cell death cascades, involving nicotinamide adenine dinucleotide phosphate-oxidase-metabolized reactive oxygen species (ROS) (34); ROS are also part of a positive feedback cycle that enhances CHOP expression (31). Notably, DZX has been reported to attenuate mitochondrial Ca2+ overload during I/R (35). Therefore, it may be hypothesized that DZX may modulate the expression of ER stress-associated proteins, including CHOP, GRP78 and caspase-12, through mitochondria-mediated Ca2+ signaling pathways. Further studies are required to test and validate this hypothesis and elucidate the molecular mechanisms that may be involved.
In conclusion, the present study demonstrated that OGD and I/R directly induced an increase in ER stress in hippocampal neurons, via upregulating the expression of CHOP, GRP78 and caspase-12. DZX was revealed to protect against I/R-induced brain injury and hippocampal damage, possibly via modulating the expression of CHOP, GRP78 and caspase-12. Further studies are required to determine the effects of DZX on ER stress-associated proteins, and to investigate the implication of Ca2+ signaling pathways in the molecular mechanisms of DZX-induced neuroprotection.
Acknowledgements
Not applicable.
Funding
The present study was supported by grant from the National Natural Science Foundation of China (grant no. 81273040).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
YC designed this study and approved this submission. XL and LL performed all experiments. ZZ collected data and organized figures.
Ethics approval and consent to participate
All work was conducted in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The experimental protocols were approved by the Institutional Animal Care of Tianjin 4th Center Hospital (approval no. 2012009).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Cohn JN, Bristow MR, Chien KR, Colucci WS, Frazier OH, Leinwand LA, Lorell BH, Moss AJ, Sonnenblick EH, Walsh RA, et al. Report of the national heart, lung, and blood institute special emphasis panel on heart failure research. Circulation. 1997;95:766–770. doi: 10.1161/01.CIR.95.4.766. [DOI] [PubMed] [Google Scholar]
- 2.Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-I. [DOI] [PubMed] [Google Scholar]
- 4.Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- 5.Liu D, Lu C, Wan R, Auyeung WW, Mattson MP. Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of Bax translocation and cytochrome c release. J Cereb Blood Flow Metab. 2002;22:431–443. doi: 10.1097/00004647-200204000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Shimizu K, Lacza Z, Rajapakse N, Horiguchi T, Snipes J, Busija DW. MitoK(ATP) opener, diazoxide, reduces neuronal damage after middle cerebral artery occlusion in the rat. Am J Physiol Heart Circ Physiol. 2002;283:H1005–H1011. doi: 10.1152/ajpheart.00054.2002. [DOI] [PubMed] [Google Scholar]
- 7.Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2004;287:H841–H849. doi: 10.1152/ajpheart.00678.2003. [DOI] [PubMed] [Google Scholar]
- 8.Wu L, Shen F, Lin L, Zhang X, Bruce IC, Xia Q. The neuroprotection conferred by activating the mitochondrial ATP-sensitive K+ channel is mediated by inhibiting the mitochondrial permeability transition pore. Neurosci Lett. 2006;402:184–189. doi: 10.1016/j.neulet.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Busija DW, Katakam P, Rajapakse NC, Kis B, Grover G, Domoki F, Bari F. Effects of ATP-sensitive potassium channel activators diazoxide and BMS-191095 on membrane potential and reactive oxygen species production in isolated piglet mitochondria. Brain Res Bull. 2005;66:85–90. doi: 10.1016/j.brainresbull.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 10.He X, Mo X, Gu H, Chen F, Gu Q, Peng W, Qi J, Shen L, Sun J, Zhang R, Kj Y. Neuroprotective effect of diazoxide on brain injury induced by cerebral ischemia/reperfusion during deep hypothermia. J Neurol Sci. 2008;268:18–27. doi: 10.1016/j.jns.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Zhu QL, Wang GZ, Deng TZ, Chen R, Liu MH, Wang SW. The protective roles of mitochondrial ATP-sensitive potassium channels during hypoxia-ischemia-reperfusion in brain. Neurosci Lett. 2011;491:63–67. doi: 10.1016/j.neulet.2010.12.065. [DOI] [PubMed] [Google Scholar]
- 12.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI0216886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 14.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sargsyan E, Ortsäter H, Thorn K, Bergsten P. Diazoxide-induced beta-cell rest reduces endoplasmic reticulum stress in lipotoxic beta-cells. J Endocrinol. 2008;199:41–50. doi: 10.1677/JOE-08-0251. [DOI] [PubMed] [Google Scholar]
- 16.Kirino T, Sano K. Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta Neuropathol. 1984;62:201–208. doi: 10.1007/BF00691853. [DOI] [PubMed] [Google Scholar]
- 17.Horn M, Schlote W. Delayed neuronal death and delayed neuronal recovery in the human brain following global ischemia. Acta Neuropathol. 1992;85:79–87. doi: 10.1007/BF00304636. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh A, Sarkar S, Mandal AK, Das N. Neuroprotective role of nanoencapsulated quercetin in combating ischemia-reperfusion induced neuronal damage in young and aged rats. PLoS One. 2013;8:e57735. doi: 10.1371/journal.pone.0057735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han L, Xu C, Jiang C, Li H, Zhang W, Zhao Y, Zhang L, Zhang Y, Zhao W, Yang B. Effects of polyamines on apoptosis induced by simulated ischemia/reperfusion injury in cultured neonatal rat cardiomyocytes. Cell Biol Int. 2007;31:1345–1352. doi: 10.1016/j.cellbi.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 20.Girard S, Brough D, Lopez-Castejon G, Giles J, Rothwell NJ, Allan SM. Microglia and macrophages differentially modulate cell death after brain injury caused by oxygen-glucose deprivation in organotypic brain slices. Glia. 2013;61:813–824. doi: 10.1002/glia.22478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oketani N, Kakei M, Ichinari K, Okamura M, Miyamura A, Nakazaki M, Ito S, Tei C. Regulation of K(ATP) channels by P(2Y) purinoceptors coupled to PIP(2) metabolism in guinea pig ventricular cells. Am J Physiol Heart Circ Physiol. 2002;282:H757–H765. doi: 10.1152/ajpheart.00246.2001. [DOI] [PubMed] [Google Scholar]
- 22.Nakka VP, Gusain A, Raghubir R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox Res. 2010;17:189–202. doi: 10.1007/s12640-009-9110-5. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Chen J, Wang L, Lu M, Chopp M. Treatment of stroke in rat with intracarotid administration of marrow stromal cells. Neurology. 2001;56:1666–1672. doi: 10.1212/WNL.56.12.1666. [DOI] [PubMed] [Google Scholar]
- 24.Verma R, Harris NM, Friedler BD, Crapser J, Patel AR, Venna V, McCullough LD. Reversal of the detrimental effects of post-stroke social isolation by pair-housing is mediated by activation of BDNF-MAPK/ERK in aged mice. Sci Rep. 2016;6:25176. doi: 10.1038/srep25176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 26.Hitomi J, Katayama T, Taniguchi M, Honda A, Imaizumi K, Tohyama M. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci Lett. 2004;357:127–130. doi: 10.1016/j.neulet.2003.12.080. [DOI] [PubMed] [Google Scholar]
- 27.Kondo F, Kondo Y, Makino H, Ogawa N. Delayed neuronal death in hippocampal CA1 pyramidal neurons after forebrain ischemia in hyperglycemic gerbils: Amelioration by indomethacin. Brain Res. 2000;853:93–98. doi: 10.1016/S0006-8993(99)02256-8. [DOI] [PubMed] [Google Scholar]
- 28.Zarch AV, Toroudi HP, Soleimani M, Bakhtiarian A, Katebi M, Djahanguiri B. Neuroprotective effects of diazoxide and its antagonism by glibenclamide in pyramidal neurons of rat hippocampus subjected to ischemia-reperfusion-induced injury. Int J Neurosci. 2009;119:1346–1361. doi: 10.1080/00207450802338721. [DOI] [PubMed] [Google Scholar]
- 29.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lakshmanan AP, Thandavarayan RA, Palaniyandi SS, Sari FR, Meilei H, Giridharan VV, Soetikno V, Suzuki K, Kodama M, Watanabe K. Modulation of AT-1R/CHOP-JNK-Caspase12 pathway by olmesartan treatment attenuates ER stress-induced renal apoptosis in streptozotocin-induced diabetic mice. Eur J Pharm Sci. 2011;44:627–634. doi: 10.1016/j.ejps.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Seimon TA, Obstfeld A, Moore KJ, Golenbock DT, Tabas I. Combinatorial pattern recognition receptor signaling alters the balance of life and death in macrophages. Proc Natl Acad Sci USA. 2006;103:19794–19799. doi: 10.1073/pnas.0609671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. 2009;119:2925–2941. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. 2010;191:1113–1125. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palomeque J, Rueda OV, Sapia L, Valverde CA, Salas M, Petroff MV, Mattiazzi A. Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ Res. 2009;105:1204–1212. doi: 10.1161/CIRCRESAHA.109.204172. [DOI] [PubMed] [Google Scholar]
- 35.Murata M, Akao M, O'Rourke B, Marbán E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: Possible mechanism of cardioprotection. Circ Res. 2001;89:891–898. doi: 10.1161/hh2201.100205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.