

Abstract
To gain insight into how mutant huntingtin (mHtt) CAG repeat length modifies Huntington’s disease (HD) pathogenesis, we profiled mRNA in over 600 brain and peripheral tissue samples from HD knock-in mice with increasing CAG repeat lengths. We find repeat length dependent transcriptional signatures are prominent in the striatum, less so in cortex, and minimal in the liver. Co-expression network analyses reveal 13 striatal and 5 cortical modules that are highly correlated with CAG length and age, and that are preserved in HD models and some in the patients. Top striatal modules implicate mHtt CAG length and age in graded impairment of striatal medium spiny neuron identity gene expression and in dysregulation of cAMP signaling, cell death, and protocadherin genes. Importantly, we used proteomics to confirm 790 genes and 5 striatal modules with CAG length-dependent dysregulation at both RNA and protein levels, and validated 22 striatal module genes as modifiers of mHtt toxicities in vivo.
Huntington’s disease (HD) is a dominantly inherited neurodegenerative disorder characterized by movement disorder, cognitive and psychiatric symptoms1. The hallmark of HD neuropathology is selective degeneration of striatal medium spiny neurons (MSNs) and, to a lesser extent, cortical pyramidal neurons (CPNs)2. Motor symptom onset for HD has a broad range but is usually in middle age. HD is relentlessly progressive, and patients die from complications of the disease about 10-20 years after onset. Currently there are no therapies to prevent the onset or slow the progression of HD1.
HD is caused by a CAG trinucleotide repeat expansion encoding an elongated polyglutamine (polyQ) stretch of the huntingtin (HTT) gene3. Unaffected individuals have fewer than 36 repeats, whereas HD individuals have 36 to 250 CAG repeats. HD belongs to a group of nine neurodegenerative disorders caused by CAG repeat expansion in distinct polyQ proteins4. Despite broad expression of these proteins, polyQ disorders present different clinical and pathological phenotypes that are attributed to distinct polyQ protein contexts.
A pivotal human genetic clue to pathogenesis of all polyQ disorders is that the length of the CAG repeat is inversely correlated with the age of disease onset4,5. Patients with HTT CAG lengths in the 40s often have motor onset in the fourth decade of life, while repeat lengths greater than 60 lead to juvenile onset1. In contrast to age of onset, the influence of CAG length on disease progression is much more modest1,6, suggesting an important effect of CAG length in early disease pathogenesis6. Recent imaging studies of HD patients suggest that CAG length correlates with caudate atrophy7, and that combined CAG length and age is a useful predictor of many clinical outcomes in HD1. Overall, HD patient studies underscore a critical role of CAG length in the early stages of pathogenesis.
The central role of CAG length on the age of HD motor symptom onset led to the “polyQ molecular trigger” hypothesis that suggests polyQ expansion in HTT leads to repeat-length-dependent, dominantly acting pathogenic changes in the vulnerable neurons to initiate the disease5. To date, the search for mutant HTT (mHTT) CAG length-sensitive molecular pathogenic changes has been limited to studies in cultured cells8,9, and very little is known about genes and pathways that are dysregulated by the expanded CAG mutant protein in the HD-vulnerable and pathogenically-relevant brain regions, namely the striatum and cortex10. Here we undertook a multi-step, integrative systems biology study of both the transcriptome and proteome of HD allelic series knock-in mice to identify murine mutant huntingtin (mHtt) CAG length- and age-dependent molecular networks, which is complemented by functional validation studies in a Drosophila model expressing a mHTT fragment. We also created a new interactive online resource (www.HDinHD.org) to disseminate our data.
RESULTS
Longitudinal RNA-sequencing analyses of mHtt allelic series knockin mice
We reasoned that transcriptomic study of HD-vulnerable brain regions (i.e. striatum and cortex) and a relatively disease-resistant peripheral tissue (i.e. liver) from an allelic series of HD knockin mice at distinct ages (Fig. 1a) could provide crucial systems-level insight into CAG length-dependent selective pathogenesis in HD. Using deep mRNA sequencing (RNA-seq), we profiled the striatum, cortex, and liver of 2-, 6-, and 10-month (denoted “m”) old mice that express one wildtype endogenous Htt allele and a second Htt allele with knock-in of human mHTT exon 1 carrying one of six different CAG lengths (denoted Q; i.e. Q20, Q80, Q92, Q111, Q140, and Q175)11. These Htt knockin mice were chosen to include those that exhibit progressive disease phenotypes (Q140 and Q175), as well as those with modest (Q111) and no overt phenotypes but with molecular signature changes (Q80, Q92, and Q111)12-14. We sequenced 8 animals per genotype and age for each tissue, with a total of 432 RNA-seq samples. Differential gene expression analyses showed robust age- and CAG length-dependent increases in aberrantly expressed genes in the striatum and to a lesser degree in the cortex, while such CAG length-dependent gene expression is not a feature of the liver transcriptome (Fig. 1b; Supplementary Table 1).
Figure 1. Workflow and differential expression analysis with respect to Htt CAG length.
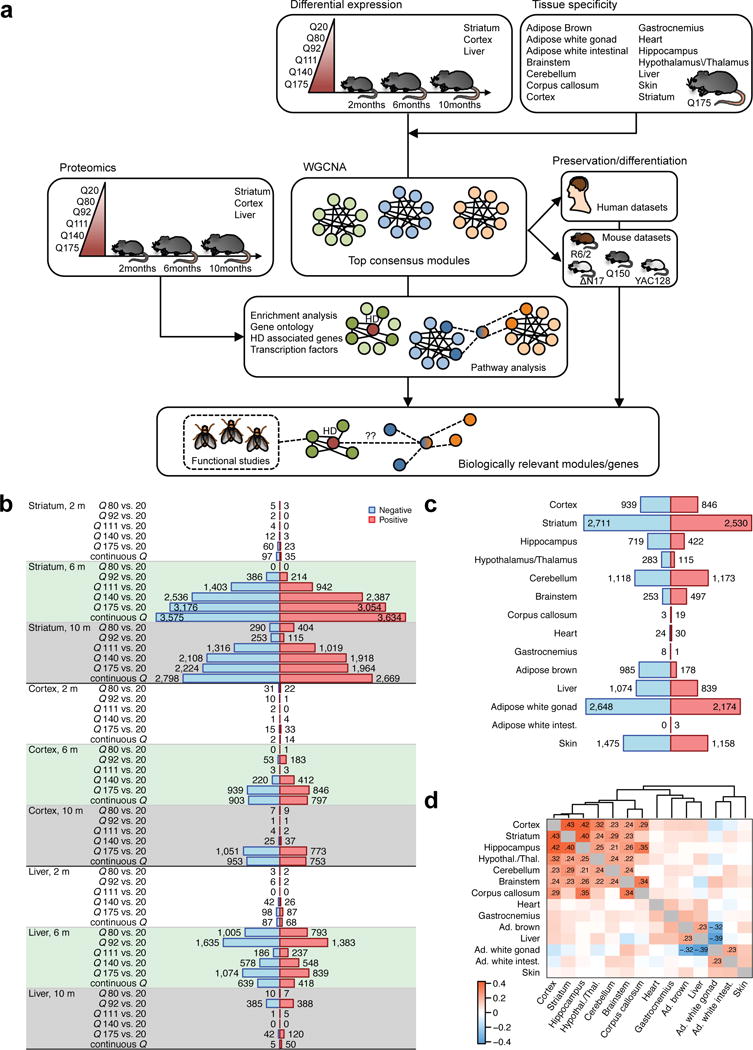
(a) Overview of experiment design and analysis strategy. (b) Numbers of significantly (FDR<0.05) differentially expressed (DE) genes in striatum, cortex and liver. Blue (red) bars represent genes significantly down- (up-) regulated with increasing CAG length (Q). (c) Numbers of DE genes in the 14 tissues for which we profiled Q175 samples and controls including striatum, cortex, liver, and 11 additional tissues. The screening in the cortex and liver corresponds exactly to that presented in panel (b); the numbers for striatum are slightly different since the analysis in panel (b) used Q20 controls, while the one in panel (c) used WT controls. (d) Heatmap shows correlation of differential expression Z statistics of individual genes across the 14 data sets. Correlations whose absolute value is at least 0.2 are shown explicitly. Ad., adipose; intest., intestinal; Hypothal./Thal., hypothalamus/thalamus.
Brain and peripheral tissue transcriptomes in Q175 mice
To evaluate whether CAG length-dependent genes are also dysregulated in other brain regions and peripheral tissues, we generated a second set of RNA-seq data (173 samples) to survey the transcriptome of five additional brain tissues and six peripheral tissues from Q175 and wild-type (WT) mice at 6m of age (Fig. 1c; Supplementary Table 2). Although this tissue survey used WT controls carrying murine Htt (Q7) and not the Q20 controls used in the full series, we found very few if any differentially expressed genes between Q20 and WT mice (Supplementary Fig. 1). For consistency with the rest of the tissue survey, we also carried out differential analysis between striatum Q175 and WT samples (WT samples for liver and cortex were not available). Our tissue survey confirmed that the striatum, the brain region most vulnerable in HD, has the largest number of differentially expressed (DE) genes (Fig. 1c). Intriguingly, white gonadal fat, a tissue not previously implicated in HD but that is adjacent to testes that do show degeneration in HD15, showed the second highest number of DE genes. The tissue survey also replicated previously reported results such as the lack of gene expression changes in the heart of HD mice16. To gain insight into global tissue similarities and differences of transcriptomic response to polyQ expansion within Htt, we evaluated the concordance of differential expression among the brain and peripheral tissues (Fig. 1d). We observe that the brain tissues cluster relatively tightly together, while peripheral tissues form two looser clusters. None of the peripheral tissues is correlated with brain tissue at or above the correlation coefficient threshold of r = 0.20, suggesting that Q175 mice exhibit brain-specific differential transcriptome changes at 6m of age.
Huntingtin CAG length- and age-dependent gene co-expression network
We reasoned that a consensus network analysis17 across the 3 time-points would allow us to define modules of genes that are co-expressed at all 3 time-points and study their CAG repeat-dependent variation across all three ages. The 3 consensus analyses (one per tissue) identified 37 striatal, 35 cortical, and 34 liver consensus modules (Supplementary Table 3) across a total of 140 striatum, 142 cortex and 141 liver samples. The modules range in size from 38 to nearly 1,900 genes and contain a total of 12,654 genes in the striatum, 11,325 genes in the cortex, and 11,590 genes in the liver (each gene can belong to only one module or can be unassigned). In this manner, network analysis reduced thousands of genes across 3 ages to a relatively small number of coherent gene modules that represent distinct transcriptional responses to the varying CAG length alleles in the murine Htt locus. To quantify the overall relationship between a module and CAG length across all 3 ages, we used a meta-analysis of correlations of module eigengenes (summary expression profiles) with CAG length. We identified 13 striatal and 5 cortical modules that pass a meta-analysis significance Z statistic threshold of 5 (corresponding to P ≈ 10−7, Fig. 2 and Supplementary Table 3). None of the liver modules satisfied this criterion.
Figure 2. Consensus coexpression network analysis of striatum and cortex identifies multiple CAG length-dependent modules.
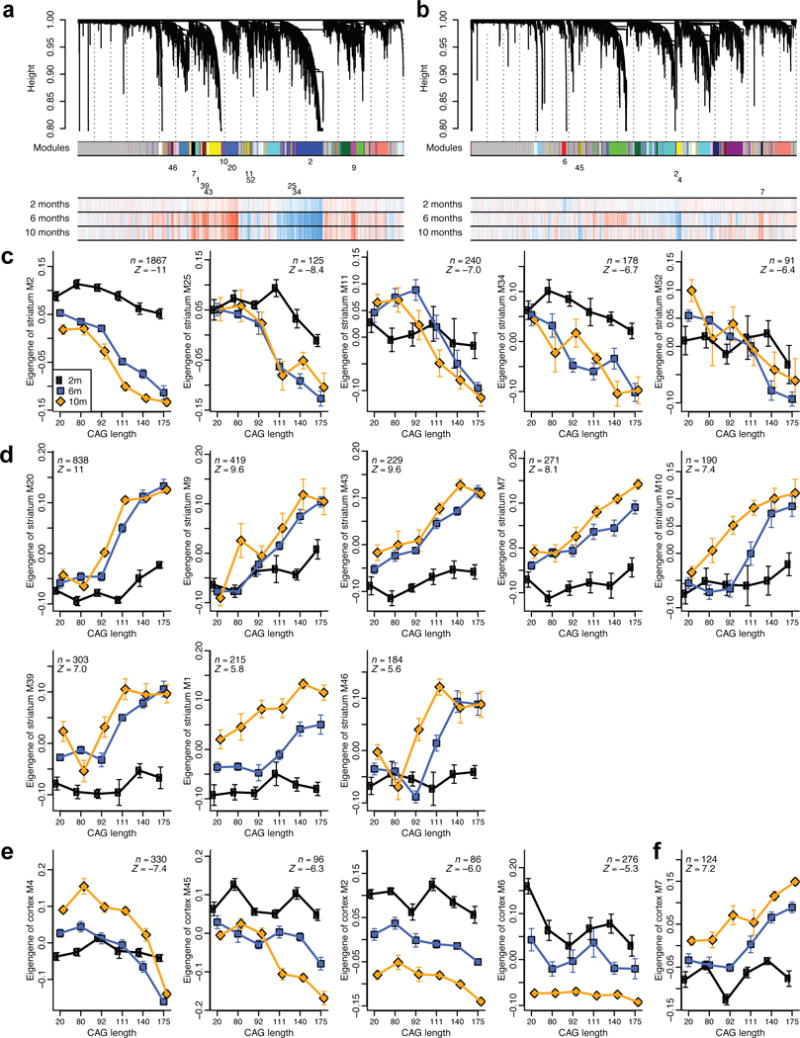
(a and b) Striatum (a) and cortex (b) gene clustering trees. Each module (cluster) is labeled by a unique non-grey color, shown below the tree in the first color band, and a numeric label. For clarity, the numeric labels are shown only for modules that are strongly associated with CAG length Q (meta-analysis Z statistic |Z| > 5). The three-row heatmap below the tree indicates associations of individual genes with Q at each timepoint (2, 6, 10 months); blue and red indicate genes down- and up-regulated, respectively, with increasing Q. (c-f) Variation of module eigengenes with Q at each of the 3 ages (black: 2 months, blue: 6 months, orange: 10 months). Points represent means of eigengene “expression” across samples with a single CAG length. Error bars give SEM. Inset gives the number of genes in the module (n) and the meta-analysis association Z statistics for Q. Only modules with |Z| > 5 are shown. Eigengene expression values for the 6 month samples were shifted so that their mean equals the mean of the Q20 samples across 2 and 10 months. Panels (c) and (d) show eigengenes of striatum modules and (e) and (f) of cortex modules negatively and positively associated with Q, respectively.
The network analyses provide several layers of information. First, the strength and significance of associations between modules and CAG length are strongest in the striatum, suggesting that CAG length affects entire co-expression modules more strongly in the striatum than in the cortex or liver. Second, the meta-analysis significance statistics allow us to rank modules by their overall association with CAG length (Fig. 2c-f). Third, a major output of network analysis is a continuous (“fuzzy”) measure of module membership (MM) for all genes in all modules (Supplementary Table 3). The module membership measures how similar the gene expression profile is to the eigengene (summary profile) of the module. Genes whose profiles are highly similar to the eigengene can be identified as intramodular hub genes18; such genes are often useful for implicating relevant biological pathways and prioritizing genes for functional studies19.
To explore the biological implications of the CAG length related modules, we carried out gene functional enrichment studies (Supplementary Tables 4-7). Striatal module M2 has the strongest negative association with CAG length (Fig. 2c) and is highly enriched with genes involved in cAMP signaling, postsynaptic density proteins, and caudate (i.e. striatum) marker genes (Supplementary Tables 4 and 7). M11 and M52 appear to be glia-related, and M25 is enriched for glutamate receptor signaling. Arguably, one of the most interesting modules is M34, which is down-regulated in a CAG length-dependent manner even at 2m, and is highly enriched with transcription and chromatin factors.
Among the 8 striatal modules that have positive correlation with CAG length, M20 has the strongest correlation and is enriched for P53 signaling, cell division, and protocadherin genes. M7, M39, and M46 are involved in stress responses, including cell death (M7), DNA damage repair (M39), and glucocorticoid signaling (M46). Intriguingly, DNA damage repair has been implicated in modifying pathogenesis in HD mice20 and age-at-onset in HD patients21. Other up-regulated striatal modules reveal the impact of mutant CAG repeats on mitochondria (M9, M43) and proteostasis (M1, M10, M39) genes, which have already been implicated in HD22,23.
Among the cortical modules correlated with CAG length (Fig. 2e, 2f), M4 has the most significant negative correlation and is enriched in genes related to calcium signaling, synapses, and glutamatergic neurons (Supplementary Tables 4-6). These pathways are consistent with a pathogenic role of cortical neurons in HD10. Cortical M45 is a glial module involved in FGF signaling, ensheathment of neurons, and fatty acid biosynthesis, and M6 and M7 are enriched with axon guidance genes. The latter suggests certain neurodevelopmental pathways could be dysregulated by mHtt.
Preservation of module-CAG length associations in independent HD mouse model and patient datasets
We next studied whether genes in the 18 CAG length-dependent modules are dysregulated in other brain regions or peripheral tissues of HD mice. We found that average module-CAG length correlations are more preserved among the brain tissues; none of the modules strongly change with genotype in the peripheral tissues in Q175 mice (Fig. 3a and Supplementary Table 8). Interestingly, striatal M34 and M43 modules appear striatum-specific, as they show few correlations with genotype in other brain regions. Three modules enriched with glial genes (M11, M20, and M45) exhibit high correlation across multiple brain regions, suggesting that glial gene changes may be more widespread in HD mouse brains.
Figure 3. Association of modules with genotype in mouse and disease status in human data and enrichment in human HD-dependent genes.
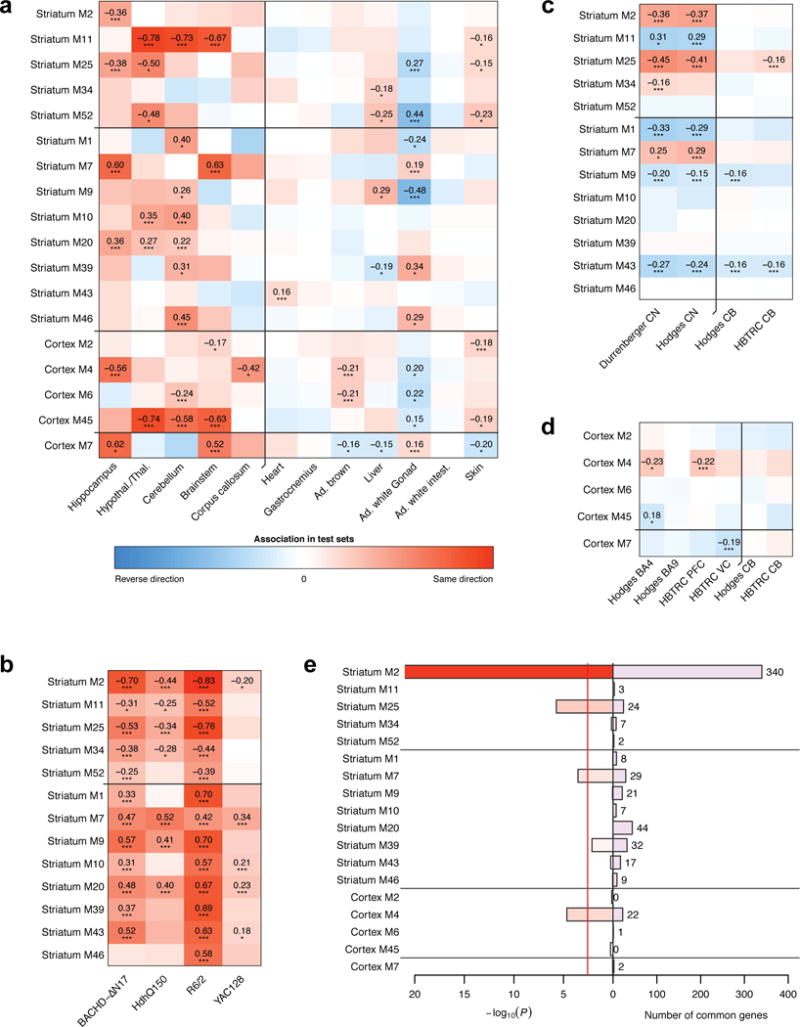
(a-d) Weighted mean correlations (Methods) of module genes with genotype (mouse data) or HD status (human data). Statistical significance was determined from a permutation test and is indicated by the stars below each mean (*, P<0.05; **, P<0.01; ***, P<0.001; the p-values are listed explicitly in Supplementary Table S8). Red and blue color indicates the same and opposite direction, respectively, of differential expression in the discovery (striatum or cortex) and test set. (e) Enrichment of the CAG length-dependent striatal and cortical modules in genes that change consistently and significantly across human CN and cortex data (Methods). Bars show hypergeometric test p-values and numbers of common genes. Red line indicates Bonferroni-corrected threshold of 0.05. The enrichment p-value axis is truncated to 10−20 for clarity.
We next asked whether the CAG length dependence of these modules derived from allelic series knockin mice are preserved in other HD mouse models (Fig. 3b; see Methods and Supplementary Table 9 for a description of the data). We find that the majority of the module-genotype associations are preserved in R6/2 (13 of 13) and BACHD-∆N17 mice (12 of 13, Fig. 3b), both of which express neuropathogenic fragments of mHTT, and in two other full-length mHtt mouse models (Q150 knockin and YAC128).
Finally, three striatal modules (M2, M25, and M7) are significantly preserved in two human post-mortem HD caudate datasets24,25, and one cortical module (M4) is preserved in two out of four HD cortical datasets24,26 (Fig. 3c and 3d). These modules are mostly not preserved in cerebellar datasets, consistent with selective vulnerability in HD. Furthermore, we found 543 of our striatal CAG length-dependent module genes are significantly altered (FDR < 0.05) in at least one of the publicly available HD caudate gene expression datasets24,25, and importantly, 431 of these genes are significantly altered in both patient datasets (Supplementary Table 10). Similarly, we found 25 CAG length-dependent cortical module genes that are significantly differentially expressed in at least one of the available HD patient cortical samples (Supplementary Table 10)24,26. As expected, these genes are significantly enriched in the three striatal modules (M2, M7 and M25) and one cortical module (M4), with M2 containing by far the largest number of such genes (Fig. 3e; Supplementary Table 10).
M2 module implicates mHtt CAG length in age-dependent impairment of striatal medium spiny neuron identity gene expression in HD mice
Our network analyses identified M2 as the module with the strongest association with CAG length and the largest number of dysregulated genes. Hub genes for module M2 (i.e. those with highest correlation with the M2 eigengenes; Fig. 4a) contain well-known striatal MSN marker genes (e.g. Ppp1r1b/Darpp-32, Drd1a, Drd2, Gpr6). Although striatal marker gene down-regulation has been shown before in HD mice and HD patients24,27, these studies could not rule out confounding effects such as neuronal loss (in postmortem brains), expression of only mHTT fragments, or transgene expression levels11.
Figure 4. Striatal markers in module M2 undergo early and progressive CAG length-dependent changes.
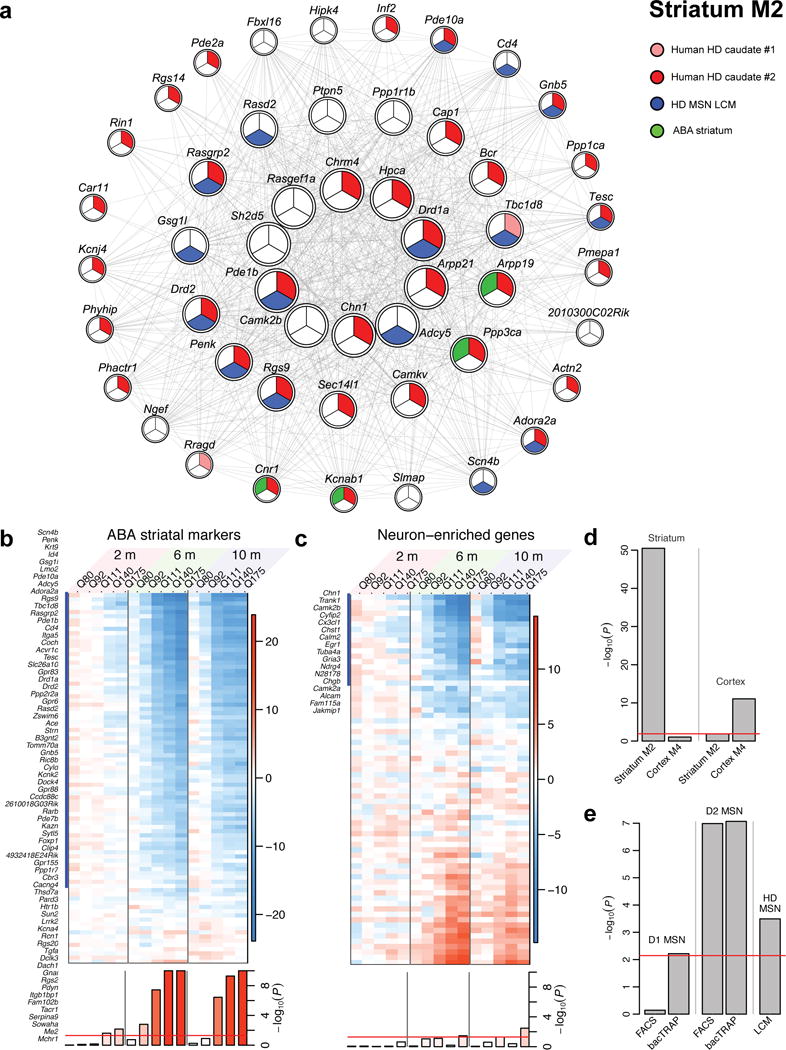
(a) Network of the top 50 hub genes in M2. Strong red, weak red or white color indicates significant change (FDR<0.1) in both, one of two or none of the human CN datasets; blue color indicates membership in top 100 ABA striatal markers; green indicates significant association with HD status in laser capture microdissection (LCM) data. (b, c) Heatmaps of striatum differential expression Z statistics of top ABA striatum neuronal marker genes (b) and ABA general neuronal marker genes (c). Genes that overlap with module M2 are marked with a blue bar and listed. Blue and red colors represent genes under- and over-expressed in higher CAG length compared to Q20, respectively. Bottom row shows the permutation test significance of the average of the Z statistics in each column. The y-axis range is restricted to 10 for clarity. Red line represents the significance threshold P=0.05. (d) Hypergeometric enrichment p-values of striatum module M2 and cortex module M4 in top ABA striatal and cortical markers. Red line indicates the Bonferroni-corrected threshold of 0.05. (e) Hypergeometric enrichment p-values of striatum module M2 in D1- and D2-MSN specific genes determined by FACS and bacTRAP, and in genes significantly associated with HD status in LCM data.
Our current study directly addresses whether striatal marker gene dysregulation is an early and CAG length-dependent pathogenic event in HD mice with endogenous levels of full-length mHtt expression. To assess striatal marker gene dysregulation, we used a collection of striatum-specific marker genes ranked by Allen Brain Atlas (ABA)28 that are also marker genes of the striatal MSNs29. Remarkably, M2 contains 70 of the 88 top striatum-specific ABA marker genes that are present in our striatal transcriptome datasets (hypergeometric p-value 3.3×10−51; Fig. 4b; Supplementary Table 11). These striatal marker genes exhibit striking patterns of down-regulation that are both CAG length- and age-dependent (Fig. 4b), which is not observed for ABA pan-neuronal marker genes in the striatum (Fig. 4c; Supplementary Table 11). This finding reveals surprisingly that Htt CAG length expansion selectively impairs age-dependent maintenance of striatal marker gene expression.
To further strengthen this observation, we asked specifically whether CAG expansion differentially impacts striatal direct-pathway MSNs (i.e. D2-MSNs) that are affected earlier in HD compared to indirect pathway MSNs (i.e. D1-MSNs) that are affected later in HD30. Indeed, the M2 module is significantly enriched with genes in two independently generated D2-MSN gene sets while such enrichment is absent for D1-MSN gene sets31,32 (Fig. 4e; Supplementary Table 11). This latter finding provides fresh evidence that mHtt CAG length selectively affects D2-MSN enriched genes. Finally, module M2 genes are also well preserved in laser-captured HD patient striatal MSNs24, suggesting MSN identity genes may also be affected in HD patients (Fig. 4e; Supplementary Table 11).
Emerging concepts suggest that operationally defined neuronal identity genes are the genes that are relatively unique to, and stably expressed in, a group of neurons throughout their lifetime and subserve critical functions33,34. Our finding that the M2 module contains the most known MSN marker genes (e.g. ABA) implies that mHtt CAG expansion may impair age-dependent maintenance of striatal MSN identity gene expression. Importantly, we found that mHtt CAG length may also modestly affect the ABA cortical marker genes (i.e. M4 module; Fig. 4d); Supplementary Table 11, suggesting that mHtt CAG expansion may impair age-dependent maintenance of striatal and likely cortical neuronal identity genes, but with the magnitude of dysregulation much more robust in the striatum.
M7 module reveals up-regulation of cell death signaling genes in HD mouse striatum
Annotation of the striatal M7 module surprisingly reveals the CAG length- and age-dependent up-regulation of 17 cell death related genes in HD mouse striata (Fig. 5a, 5b). The cell death genes in M7 are differentially expressed in brain but not peripheral tissues in Q175 mice, and their differential expression is also preserved in other HD mouse and patient striata (Fig. 5c-5e; Supplementary Table 12). This finding is surprising, since heterozygous HD knockin mice do not show striatal MSN cell loss up to 12m of age13,14. We used unbiased stereology to confirm that Q175 heterozygous mice do not show changes in the striatal neuron and GFAP+ astrocyte cell numbers up to 12m of age (Supplementary Fig. 2). Thus, we conclude that the up-regulation of cell death genes in M7 as well as down-regulation of MSN marker genes in M2 are unlikely due to cellular composition changes (e.g. MSN loss or proliferation of astrocytes); instead, they likely reflect CAG length-dependent increase in the vulnerability of MSNs to degeneration (akin to a prodegenerative state) in HD mice.
Figure 5. Cell death genes in striatum module M7.
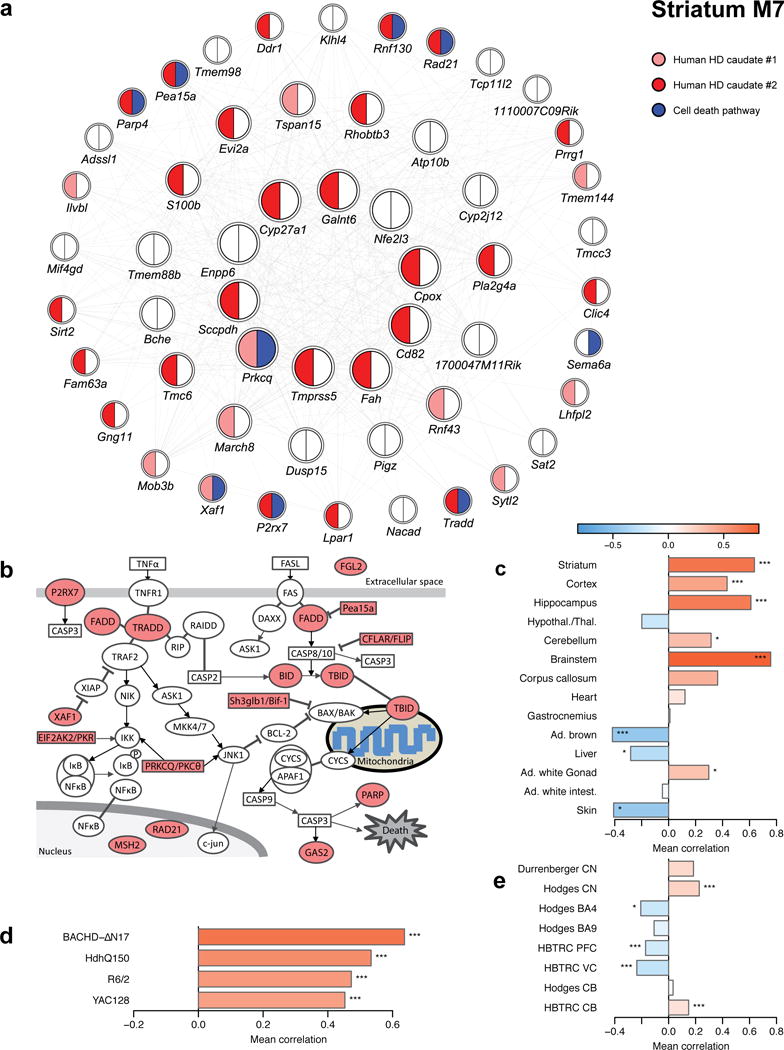
(a) Network plot of the top 50 hub genes in striatum module M7. Strong red, weak red and white color indicate significant (FDR<0.1) and consistent DE between HD samples and controls in both, one or none of the human CN datasets; blue color indicates genes implicated in cell death. (b) Graphical representation of the cell death pathway with dysregulated genes from striatum module M7 highlighted in red. (c-e) Association of cell death genes in striatum module M7 with genotype or HD status in other mouse striatum data (c), the tissue survey (d), and human post-mortem data (e). Bars show the weighted average correlation of the cell death M7 genes with the relevant genotype or HD status; stars show the corresponding permutation test significance (*, P<0.05; **, P<0.01; ***, P<0.001). The correlations and p-values are also listed in Supplementary Table S12.
Dysregulation of protocadherin clusters in striatal modules
Our results raise the question of how relatively subtle mutation changes (i.e. CAG repeat expansion) in Htt lead to graded transcriptional changes among the 18 modules. A potential clue to this question is that four of the 18 modules (M20, M34, M39, and M46) are highly enriched with cadherin and protocadherin genes (Pcdhs; Fig. 6). Cadherins and protocadherins function in neurodevelopment, intercellular signaling, adhesion, synaptic function, and neuronal survival35, with a combinatory code of protocadherins thought to underlie single neuron identity in vivo36. Prior studies have unraveled critical roles of transcriptional and chromatin factors, Ctcf, Rad21 and Rest, in regulating the expression of Pcdhs in neurons37,38. Our current study reveals striking dysregulation of 37 out of 58 clustered Pcdhs (Fig. 6). Intriguingly, among the known Pcdh transcriptional regulators, REST is already known to be dysregulated by mHTT in vitro and in vivo39, and both Ctcf and Rad21 are present in prominent CAG length-dependent modules (M34 and M7, respectively). Bioinformatic analyses reveal that the two highest Htt CAG length dependent modules (M2 and M20) are significantly enriched with known Ctcf binding sites in the brain (Supplementary Fig. 3). Together, our findings suggest the study of Pcdh gene clusters may be a tractable route to dissect the upstream regulatory mechanisms underlying mHtt CAG length-dependent molecular networks in HD mouse brains.
Figure 6. Protocadherin dysregulation across multiple modules.
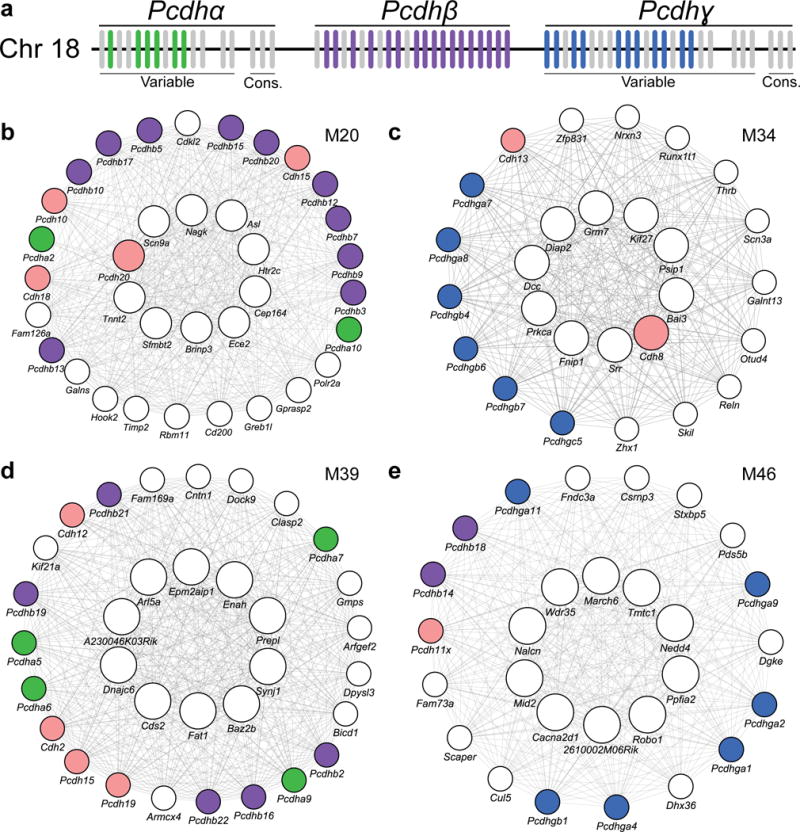
(a) Schematic of the clustering of mouse protocadherins. Each vertical bar represents one of the protocadherin genes. Colored bars show protocadherins that are members of CAG length-dependent allelic series consensus modules. (b-e) Network plots of top 10 hub genes (center ring), and other hub genes and protocadherins (outer ring) in striatal modules M20 (b), M34 (c), M39 (d), and M46 (e). Colored circles indicate clustered (colors corresponding to schematic above: green, Pcdhα; purple Pcdhβ; blue, Pcdhγ) and unclustered protocadherins (pink).
Proteomic validation of CAG length-dependent genes and modules
An important step in a transcriptome profiling study is to validate transcriptional changes by orthogonal methods, ideally at the protein level. Although we (Supplementary Fig. 4) and others12 have verified more than a dozen genes in the M2 module that are dysregulated in Q175 mice; we believe more relevant evidence for validation of CAG length-dependent transcriptome changes is unbiased validation at the proteome level. To this end, we utilized striatal tissues from the same 6-month mouse brains used for RNA-seq to perform quantitative proteomic analyses with mass spectrometry40 (MS, Fig. 7a). Prior studies have shown that such proteomic studies allow the identification and quantification of a substantial proportion of all proteins expressed in the investigated cells41. Furthermore such studies can be used as input for WGCNA to build unbiased protein networks42. In the current study, we quantified relative protein abundance in 45 of the 48 striatal samples using an accurate label-free quantitation method (MaxLFQ43; Supplementary Table 13) that permits application of standard statistical tests. In total, 254,543 unique peptides and 10,047 proteins were identified according to an accepted FDR of less than 1% on the protein and peptide level after removing contaminants (36) and reverse hits (250). Not all proteins could be quantified in every sample, but on average 7,774 proteins were identified and quantified per sample.
Figure 7. High-throughput proteomic analysis confirms CAG length-dependent changes in 6-month striatum of HD mice.
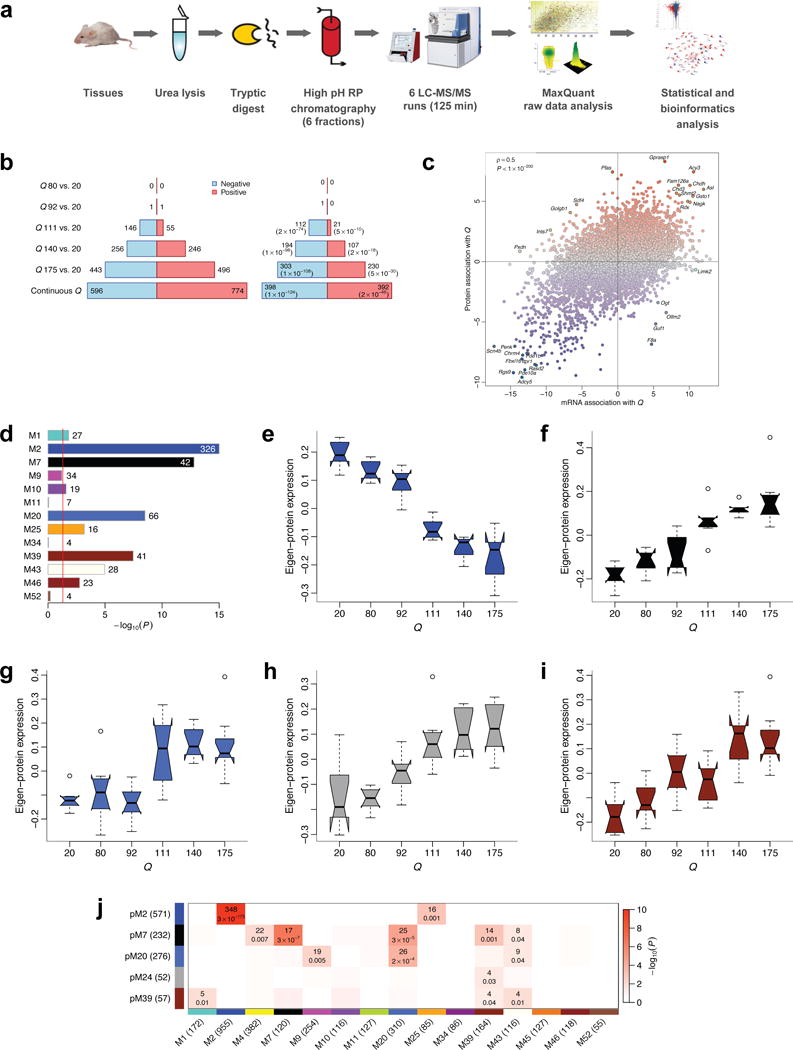
(a) Workflow of proteomic profiling. (b) Left barplot represents numbers of proteins significantly (FDR<0.1) associated with CAG length (Q). Blue (red) bars denote proteins down- (up-) regulated with increasing CAG length. Right barplot represents numbers of significant proteins whose mRNA also changes significantly (FDR<0.1) and in the same direction. Values in brackets are hypergeometric p-values of overlaps of significant protein and gene mRNA profiles. (c) Z statistics for protein association with Q vs. the corresponding mRNA Z statistics. Blue (red) dots represent proteins whose abundance decreases (increases) with increasing CAG length. Selected concordant and discordant genes are labeled. (d) Bars show hypergeometric enrichment p-values of mRNA module genes in proteins that are significantly differentially abundant in the same direction as the module. Numbers give the corresponding gene counts. For clarity, the p-value axis is truncated to 10−15. (e-i) Summary profiles of the 5 protein network modules with strongest association with Q, as a function of Q. Boxes indicate the median, interquartile range and confidence interval for the median. Whiskers indicate the range of data up to 1.5 of the inter-quartile range; points beyond the range of whiskers (if any) are shown individually. (j). Numbers of common genes and hypergeometric overlap p-values among selected protein (rows) and mRNA (columns) modules. Row and column labels indicate module sizes within the 7,039 genes common to both mRNA and protein network analyses.
Across the 45 common striatal samples and 7,039 protein-mRNA pairs present in both filtered protein data (Methods) and mRNA network analysis, the correlation of protein LFQ intensities and variance-stabilized mRNA data was 0.42. This agrees with other high-throughput proteomic studies44 which have found similar correlations.
Using the quantitative protein readouts from MaxLFQ, we found CAG length-dependent increases in dysregulated proteins in Q111, Q140, and Q175 mice, with a total of 1,370 proteins found to be CAG length correlated. Moreover, we confirmed 790 proteins with continuous CAG length-dependent changes in both RNA and protein levels in the striatum, with 133 proteins in Q111 mice, 301 proteins in Q140 mice, and 533 proteins in Q175 mice (Fig. 7b; Supplementary Tables 14 and 15). We have validated a substantial number of genes that are altered at both the RNA and protein level in a CAG length-dependent manner, hence presenting a valuable list of candidate genes for future functional studies in HD model organisms.
Intriguingly, although the majority of proteins that are significantly correlated with CAG length are positively correlated with their mRNA (i.e. of the 1,370 CAG length-correlated proteins identified at the FDR level of 0.1, 1,023 or 75% have positive correlation with their mRNA), a small number of proteins (29) with significant CAG length correlation have a negative and nominally significant (P < 0.05) correlation with their mRNA levels (Fig. 7c and Supplementary Table 15). This subset of genes may reflect the impact of mHtt CAG expansion on post-transcriptional regulation of a subset of proteins (e.g. translation, protein stability, or clearance). Among these proteins, F8a (or Hap40) is a known HTT interactor that mediates vesicular trafficking45. F8a is down-regulated at protein levels but up-regulated at RNA levels with increasing CAG length. Our data are consistent with a previously observed reduction of Hap40 in the synaptosome of Q175 mice46, suggesting the need to study how mHtt may regulate Hap40 as well as other proteins (e.g. Ogt, Golgb1; Fig. 7c) with opposing variation in RNA and protein levels and whether such regulation could be pathologically relevant.
The advantage of performing high-throughput proteomic validation, instead of validating individual proteins, is the ability to examine CAG length-dependent molecular networks at both RNA and protein levels. We first used enrichment analyses to show that 8 CAG length-dependent transcriptome modules are significantly enriched with differentially expressed proteins in HD mice, with the M2 and M7 modules having most of such proteins (Fig. 7d; Supplementary Table 14). Moreover, by performing WGCNA directly on the proteomic dataset using label-free quantitative protein inputs42,43, we defined protein network modules (pMs), with several modules showing association with CAG length (Supplementary Table 15). Summary profiles (eigen-proteins) of 5 protein modules that pass a stringent significance threshold |Z| > 4.5 (P ≈ 10−5) are shown in Figures 7e-7i. Impressively, these independently created CAG length-dependent protein modules show remarkable overlap with the transcriptome modules (i.e. pM2 with M2, pM7 with M7; Fig. 7j; Supplementary Table 16), demonstrating the robustness of these CAG length-dependent molecular (RNA and protein) networks and strengthening the confidence of selecting hub genes from such modules for further functional studies.
Modifiers of mHTT toxicity in Drosophila
An important step to explore the functional significance of the CAG length-dependent modules is to genetically perturb top hub genes in an established HD animal model system. To this end, we tested 49 striatal module hub genes (Supplementary Table 17) as genetic modifiers in a Drosophila model expressing a mHTT fragment, using age-dependent climbing deficits as a sensitive readout of mHTT-induced motor dysfunction42,47. Impressively, we found 22 modifier genes using such an in vivo assay, 11 of which are in the M2 module, including the top hub genes Arpp21, Camk2b, and Chn1 (Fig. 8a, 8b; Supplementary Figs. 5 and Supplementary Tables 18 and 19). Four M7 hub genes and hub genes from M20, M34, M43 and M9 are also modifiers in this assay. In support of our hypothesis that the hub genes of M34, such as Ctcf, may be critical regulators of mHtt CAG-dependent transcriptional deficits and neuronal toxicities, we have found that two independent heterozygous loss-of-function alleles of the Drosophila Ctcf gene can significantly, albeit partially, ameliorate climbing deficits in this Drosophila model (Fig. 8a, 8b; Supplementary Tables 18 and 19). Moreover, 11 of our validated modifier genes are those shown to be CAG length dependent at both RNA and protein levels (Supplementary Table 18), providing particularly strong evidence that these genes may be relevant for future study in mammalian HD models. Although one may not simply translate fly modifier results to those in mammalian models, we reason that loss-of-function mutants may mimic genes in modules with negative CAG length correlation (e.g. M2) and overexpression mutants may mimic the positively correlated modules (e.g. M7 and M20). Based on such estimation, it is intriguing that 9 out of 11 modifiers in M2 module are LOF suppressors, suggesting their down-regulation in HD mice may represent compensatory changes to mitigate mHtt toxicities. On the contrary, two genes (Camkv and Nagk) showed CAG-dependent changes that may result in enhanced toxicities. Future genetic analyses of CAG length-dependent module genes in both fly and mouse models of HD will help to systematically validate genes that are modifiers of HD pathogenesis in vivo.
Figure 8. Genetic perturbation studies in a fly model expressing mHTT fragment.
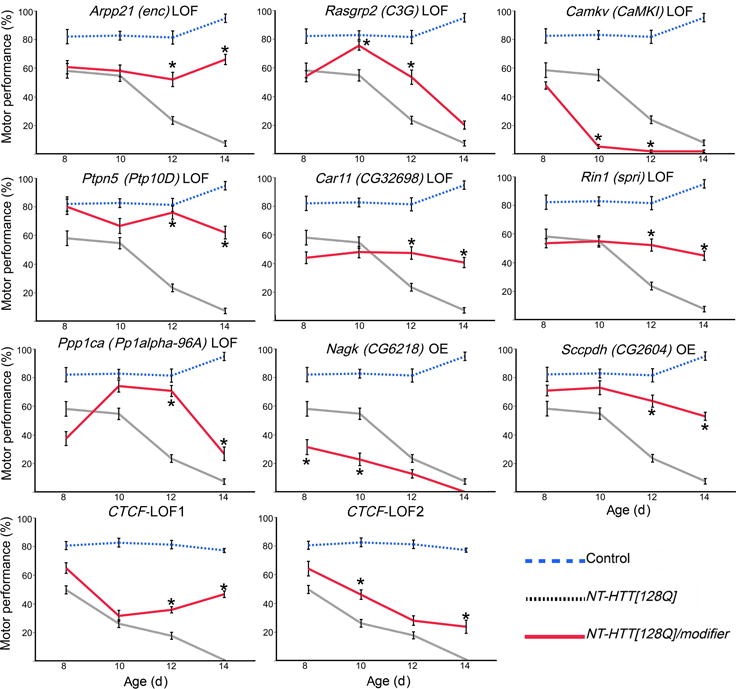
Summary of motor performance tests for selected modifiers. Blue line represents controls, black line represents transgenic (NT-HTT[128Q]) flies, and red line represents transgenic flies with an active modifier. Each data point represents two replicates of 15 age-matched females per genotype and 10 trials (n=20). Error bars indicate SEM. Dunnett’s test following ANOVA was used to quantify statistical significance. Supplementary Figure 6 lists the relevant test statistics (ANOVA F values and Dunnett’s test p-values). Stars indicate pairs of means that are significantly different between NT-HTT[128Q] and NT-HTT[128Q]/modifier (p<0.05).
HDinHD: an online resource for HD research
Raw and processed data, as well as network analysis plots and phenotypic data related to the allelic series HD mice, are hosted on a dedicated website and server at www.HDinHD.org (see Methods). Currently, HDinHD comprises three sections: 1) a data repository and gene expression browser, allowing access to genomic and proteomic data from the allelic series, including raw and normalized RNA-seq counts and relative protein expression values (MaxLFQ) from MS; 2) a network browser for online mining of the WGCNA analysis reported here; 3) a suite of analysis tools that allow the user to save, share and annotate gene lists, which can be compared with existing gene sets, either published or generated by other HDinHD users. A long-term goal of HDinHD is to compile a structured catalog of the HD-related genomic proteomic, and phenotypic datasets. We envision that HDinHD will help access the large-scale HD allelic series RNA-seq and proteomic data and consensus network analysis results, and will provide a collaborative environment for HD researchers to access and share both published and unpublished datasets. Online analysis tools will allow the user to annotate gene lists and to test hypotheses in silico in order to facilitate mechanistic and therapeutic research for HD.
DISCUSSION
A key advance of our study is the application of extensive genome-wide transcriptomics with deep RNA-seq using mice with CAG repeat expansion in the endogenous gene, followed by strategic proteomics-based validation studies, to elucidate for the first time the CAG length-dependent molecular networks in disease-relevant brain regions. One novel finding in our study is the number of CAG length-dependent genes and modules appearing to correlate with known vulnerabilities in HD (i.e. striatum more than cortex and none in liver), hence supporting the hypothesis that CAG length-dependent molecular changes are likely relevant to the underlying vulnerability in the disease.
Our study identified 13 striatal and 5 cortical gene co-expression modules that are strongly associated with Htt CAG length. These modules highlight several biological pathways impacted by Htt CAG repeat expansion in the striatum and cortex. A continuous measure of module membership identifies module hub genes that, at least from a statistical point of view, deserve prioritization for genetic perturbation studies. The HD relevance of these CAG length-dependent modules is strengthened by the preservation of their association with mHtt expression in multiple HD mouse and HD patient datasets. Moreover, our proteomics data provide high confidence validation of about 790 proteins as being Htt CAG length dependent at both RNA and protein levels in the striatum of HD mice. We show that five independently generated, CAG length-dependent protein modules (e.g. M2 and M7) are highly preserved with the transcription modules, and are also preserved in other HD models and patients. Hence, the hub genes in such modules should be prioritized for further target validation studies in HD mice.
A major insight from this study is that CAG repeat expansion in endogenous Htt impairs the age-dependent maintenance of striatal MSN identity gene expression. While previous studies have shown that a subset of MSN marker genes are dysregulated in HD mice and patients24,27, these studies were not designed to assess the impact of endogenous mHtt CAG length and evaluated only a subset of MSN identity genes. Our comprehensive transcriptomic analyses of Htt knockin allelic series mice allow us to uncover, for the first time, the graded effect of mHtt CAG length and age in selective down-regulation of the vast majority of MSN identity genes, but not general neuronal marker genes, in the striatum. Remarkably, module M2 also is enriched with genes from D2-MSNs, the most vulnerable neuronal cell type in HD. Our results are consistent with the emerging concept that neuronal cell type identity genes need to be actively maintained in postnatal brains by a specific set of transcription and chromatin factors33,34. Disruption of such a program likely leads to progressive and selective loss of neuronal identity followed by neurodegeneration, which has been found in animal models34. Our study highlights the importance of future studies to unravel the molecular program involved in maintaining striatal MSN identity gene expression, and to elucidate how mHtt CAG expansion may cause age-dependent interference of such a program. Ultimately, we need to test whether restoring such a program could prevent disease onset or progression in HD.
An important long-term goal of our systems level study of HD pathogenesis is to identify the precise molecular mechanisms linking mHTT polyQ expansion to transcription and chromatin dysregulation. To this end, several clues have already emerged from our analyses. First, the dysregulation of a large number of clustered Pcdh genes in four of our modules (Fig. 6) suggests that known regulatory factors for Pcdh expression (i.e. Ctcf, Rad21 and Rest) may also be involved in mHtt-induced transcriptional dysregulation. Indeed, Rest is a known polyQ length dependent interactor of mHttthat is involved in age-dependent neuronal transcription48. Hence it would be interesting to study the role of Rest in Htt CAG length dependent gene expression in vivo. Similarly, Ctcf and Rad21 are critical chromatin loop regulators49, with Ctcf known to be involved in cell identity gene expression50. Since Ctcf binding sites are enriched in the promoter region of M2 and M20 genes (Supplementary Fig. 3), and two independent heterozygous null alleles of Ctcf can ameliorate behavioral deficits in a Drosophila model of mHTT fragment toxicities (Fig. 8), it is now important to investigate whether Ctcf is dysregulated in HD striatum and contributes to mHtt CAG length-dependent transcriptionopathy, including MSN identity gene down-regulation. Finally, M34 is not only enriched with transcription and chromatin factors (Supplementary Tables 3 and 4) but is also altered in a CAG length-dependent manner at an early age (i.e. 2m). Hence, M34 module genes (e.g. Ctcf and Ezh2) should be prime candidates to study early and causal transcriptional regulators that underlie CAG length- and age-dependent modules in HD mouse striatum.
In summary, our study provides a large-scale, comprehensive transcriptomic and proteomic characterization of the effects of CAG repeat expansion in endogenous murine Htt in vivo, identifying a consistent set of genes (both RNA and proteins) and networks that are dysregulated in a CAG length- and age-dependent manner in HD mouse brains. We provide integrative genomic evidence to show converging molecular networks that are perturbed in both HD mice and patients, and provide proof-of-concept that 22 genes from such networks modify mHTT fragment toxicities in Drosophila. Together, our integrative systems findings and online database (HDinHD) constitute a rich and novel resource to facilitate the discovery of mHtt CAG length-dependent pathogenic mechanisms and novel therapeutic targets for HD.
METHODS
Animal breeding and husbandry
We analyzed 6 heterozygous Htt KI lines expressing CAG repeats of 20, 80, 92, 11151, 14052, and 17512. For each one of the 6 lines, male heterozygous (HET) mice were crossed with C57BL/6J female mice at Jackson Laboratory (Bar Harbor, ME). For each line, animals born within 3-4 days from litters having 4 to 8 pups were identified by ear tags, tail sampled for genotyping and weaned at around 3 weeks of age. HET mice were selected based on the CAG repeat to allow a Gaussian distribution of CAG repeats in the experimental cohort to avoid skewed distributions. Best Gaussian fit was judged by eye. Body weight cut off: experimental animals had to weigh more than 11 g (females) and more than 13 g (males) by 5 weeks of age. Animals presenting any anomaly were excluded. Unacceptable anomalies were cataracts, malocclusion, missing/small eye, ear infection, unreadable or missing tag. Mice were housed in cages enriched with two play tunnels, a plastic bone and enviro-dri® (Shepherd Specialty Papers). Animal cage changes occurred weekly. The cages were maintained on a 12:12 light/dark cycle. Water and food were freely available at all times.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals, NRC (2010). The protocols were approved by the Institutional Animal Care and Use Committee of PsychoGenics, Inc., an AAALAC International accredited institution (Unit #001213).
Tissue selection and mRNA sequencing
Striatum, cortex, and liver were selected for full profiling. Specifically, at each of 3 time points (2, 6, 10 months), four female and four male heterozygous KI mice from each of the 6 Htt CAG repeat lengths were profiled, resulting in 48 samples from each tissue and each time point. Additional samples from wild type littermates from the Q20 line were profiled as well (striatum at all 3 ages, cortex and liver at 2 and 10m only). In addition to the fully profiled tissues, 11 other tissues were selected for a smaller study (referred to as the Tissue Survey) involving 8 (4 male and 4 female) wild type mice from the 6-month Q=20 line, and 8 (4 male and 4 female) heterozygous mice from the 6-month Q175 line, for a total of 16 samples per tissue. The 11 tissues include 5 brain regions (brainstem, cerebellum, hippocampus, hypothalamus/thalamus, and corpus callosum) and 6 peripheral tissues (white gonadal adipose, white intestinal adipose, brown adipose, skin, heart, and gastrocnemius muscle). Messenger RNA was extracted and prepared using the Illumina TruSeq RNA sample prep kit and sequenced on an Illumina HiSeq2000 sequencer using strand-specific, paired end, 50-mer sequencing protocols to a minimum read depth of 40 million reads per sample. The sequencing was performed in 2 separate batches (6-month samples in batch 1, 2- and 10-month samples in batch 2). Clipped reads were aligned to mouse genome mm9 using the STAR aligner53 using default settings. Read counts for individual genes were obtained using HTSeq54.
Data preprocessing
While differential expression and association testing in DESeq2 package uses raw counts, properly normalized and pre-processed data are necessary for downstream analyses such as WGCNA. For each tissue, we retained those mRNA profiles whose observed counts are 5 or more in at least 8 samples. This resulted in 17,197, 17,308 and 15,406 gene mRNA profiles retained for further analysis in the striatum, cortex and liver, respectively. We then applied the variance stabilizing transformation implemented in DESeq2 version 1.2.1055. Next, we removed variation due to gender using ComBat56 as well as possible outlier samples, identified as samples whose standardized sample connectivity is below −5 (that is, 5 standard deviations below the mean)57. Outlier identification was performed in an unsupervised manner, without regard to genotype or any other sample characteristics. All sample numbers after outlier removal are presented in Supplementary Table 20. At each step, we checked sample clustering trees for presence of strong clusters; if these were present, we reduced the inter-cluster variation using Empirical Bayes-moderated linear regression.
Weighted Gene Co-expression Network Analysis
Weighted Gene Co-expression Network Analysis (WGCNA)58,59 starts by constructing a matrix of pairwise correlations between all pairs of genes across the measured samples in a data set. To minimize effects of possible outliers, biweight midcorrelation60 was used with argument maxPOutliers=0.05. A “signed hybrid” pairwise gene co-expression similarity was then formed; it equals the gene-gene correlation if the correlation is positive, and equals zero otherwise. Next the co-expression similarity was raised to the power β=6 (WGCNA default) to arrive at the network adjacency. This procedure has the effect of suppressing low correlations that may be due to noise. The result is a network adjacency that is zero for negatively correlated genes and is positive for positively correlated genes. Adjacency of weakly correlated genes is nearly zero due to the power transformation.
Consensus Module Analysis
Consensus modules are defined as sets of nodes that are highly connected in multiple networks; loosely speaking, one could identify the consensus module in individual network analyses across multiple sets, so the module can be said to arise from a consensus of multiple data sets17. Within WGCNA, consensus modules are identified using a consensus dissimilarity that is used as input to a clustering procedure. To describe our definition of the consensus dissimilarity, we introduce the following component-wise quantile function for a set of k matrices A(1), A(2), …, A(k):
Thus, each component of the quantile matrix is the given quantile (0≤q≤1) of the corresponding components in the individual input matrices. Using this notation, we define the consensus network corresponding to input networks A(1), A(2), …, A(k) and quantile q as
where cTOM stands for calibrated Topological Overlap Measure (TOM). The calculation of cTOM starts with calculating the standard TOM58 in each input data set (network). The calibration aims to make TOM values comparable between different networks. In this work we use as calibration the quantile normalization implemented in the R package preprocessCore61. For purposes of quantile normalization we treat the independent components (say the lower triangle) of TOM for each input network as a vector of measurements corresponding to one “sample;” thus, quantiles of the calibrated TOM matrices in each network equal each other and equal the average of the corresponding quantiles in the original, uncalibrated TOM matrices.
Given the consensus network defined above, one defines the consensus dissimilarity ConsDissij as
The consensus dissimilarity is used as input to average-linkage hierarchical clustering. Modules are defined as branches of the resulting dendrogram and are identified using the Dynamic Tree Cut algorithm62. Modules are labeled by (in principle arbitrary) numeric labels and, for easier visualization, also by colors. Not all genes will be assigned to modules; the label 0 and color grey are reserved for genes not assigned to any module. This procedure results in exclusive module assignments: each gene can be assigned to at most one module (below we define a continuous measure of module membership that is non-exclusive). To make it easier to compare different network analyses (e.g., consensus analyses of different tissues), we have chosen the labels such that, whenever possible, modules from different analyses that overlap significantly in their gene content carry the same label. For example, striatum module 2 and cortex module 2 overlap significantly and hence carry the same label 2 and color blue.
Consensus module eigengenes
The module identification procedure results in modules containing genes with highly correlated expression profiles. It is useful to summarize such modules using a single expression profile per input data set. We use the module eigengene E, defined as the left-singular vector of the standardized expression matrix with the largest singular value58. Since consensus modules are defined across k independent data sets, one can form their summary profiles in each of the data sets. Thus, a consensus module gives rise to k eigengenes, one in each input data set, that provide a summary “expression value” for each sample in the data set. This allows one to relate consensus module eigengenes to other sample information, for example to disease status or other traits, in each data set, and study similarities and differences between the input data sets in terms of the module-trait associations.
Continuous measure of module membership
Module eigengenes lead to a natural measure of similarity (membership) of all individual genes to all modules. We define a continuous (“fuzzy”) measure of module membership of gene i in module I as
where xi is the expression profile of gene i and EI is the eigengene of module I. This definition is applicable to every individual network (data set). The value of module membership lies between −1 and 1. Higher MMiI indicate that the expression profile of gene i is similar to the summary profile of module I. Since we use signed networks here, we consider module membership near −1 low. The advantage of using correlation to quantify module membership is that the corresponding statistical significance (p-values) can be easily computed. Genes with highest module membership are called hub genes. Hub genes are centrally located inside the module and represent the expression profiles of the entire module. Some genes may have high continuous module membership in two or more modules and may, in this sense, be considered members of (or intermediate between) several modules.
Module membership in consensus modules
In a consensus module analysis, we calculate the fuzzy module membership MM for each gene in each data set. Thus, for each consensus analysis of 3 data sets there are 3 values for the module membership of each gene in each module. We then use meta-analysis to summarize the 3 module memberships into a single meta-analysis Z statistic19. Genes with the highest module membership meta-analysis Z statistics are called consensus hub genes. It has been shown that consensus hub genes can be useful in studying functional categories associated with clinical traits19.
Meta-analysis
Our analysis methods make extensive use of meta-analysis since we often pool association and module membership statistics across the 3 time points. A simple, yet powerful meta-analysis method relies on combining the Z statistics from individual data sets63,64. Specifically, for each gene i and data set a, one obtains a Z statistic Zia, for example, by the inverse normal transformation of the p-value. Next, a meta-analysis Zi statistic for each gene is calculated as
The meta-analysis statistic Zi is approximately normally distributed with mean 0 and variance 1; the corresponding p-value is then calculated using the normal distribution.
Matching of genes across data sets and organisms
To compare gene expression between different sets, we used the following gene matching procedure. We first transform all expression data to gene-level measurements. For microarray data where several probes or probe sets may represent a single gene, the probe-level data were turned into gene-level data using the function collapseRows65. For Affymetrix and Illumina microarrays we use the default settings of collapseRows (this implies selecting the probe with the highest mean expression as the representative for each gene); for two-color Agilent microarray data we select the most variant probe. For comparisons between different species, we map genes using the gene homology mappings provided by the Mouse Genome Informatics website, The Jackson Laboratory, Bar Harbor, Maine. World Wide Web (URL: http://www.informatics.jax.org/homology.shtml), retrieved April 2014.
Conversion between gene symbols and Entrez identifiers
We consistently used Entrez identifiers to unambiguously identify genes. Since many external resources identify genes by symbols, we used the Bioconductor package org.Mm.eg.db to convert gene symbols to Entrez identifiers. Typically not all gene symbols can be unambiguously mapped to an Entrez ID; genes with such ambiguous mappings were discarded. For example, from the top 100 ABA striatum markers28, only 88 could be mapped to an unambiguous Entrez ID.
CTCF enrichment analysis
A list of Ctcf target genes was created by scanning the canonical promoter region (1,000bp upstream of transcription start site) of Mus musculus RefSeq genes (assembly mm9) for the presence of Ctcf peaks in the “Cortex Adult 8 weeks CTCF TFBS Chip-Seq Peaks from ENCODE/LICR” UCSC track66. For each of the 18 consensus top modules, overlap with Ctcf target genes was determined, as well as the overlap of randomly generated RefSeq gene lists (with the same number of genes in the module) with the Ctcf Target Genes (10,000 permutations). Z-score was calculated as the number of standard deviations our observed value was above the mean of the observed values within the empirical null distribution.
Preservation studies in independent human and mouse data
To quantify module association with genotype or disease status in independent data, we downloaded the following 8 human and 4 mouse data sets (sample numbers below reflect our outlier removal):
Durrenberger (2011) CN (GSE26927)
19 human post-mortem caudate nucleus samples25, assayed on the Illumina HumanRef-8 v2.0 expression beadchip.
Hodges (2006) CN, BA4, BA9, CB (GSE3790)
Human post-mortem data from HD patients and controls from 4 different brain regions24, assayed on the Affymetrix U133A and B microarrays.
Harvard Brain Tissue Resource Center PFC, VC, CB
The roughly 800 individuals in these datasets are composed of approximately 400 Alzheimer’s disease (AD) cases, 230 Huntington’s Disease and 170 controls matched for age, gender, and post mortem interval (PMI). The tissue specimens for this study were provided by Harvard Brain Tissue Resource Center (HBTRC). Three brain regions (cerebellum (CB), visual cortex (VC), and dorsolateral prefrontal cortex (PFC) were profiled on a custom-made Agilent 44K microarray of 39,280 DNA probes uniquely targeting 37,585 known and predicted genes, including splice variants, miRNAs and high-confidence non-coding RNA sequences. Clinical outcomes available include age at onset, age at death, Braak scores (AD), Vonsattel scores (HD), regional brain enlargement/atrophy. The data can be accessed at http://www.synapse.org under accession ID syn4505. An analysis of AD samples and controls has been reported previously26; here we only used the HD and control (non-AD) samples.
BACHD-∆N17 (GSE64386)
Striatum samples from 4 wildtype and 4 BACHD-∆N17 mice at each of 2, 7, and 11 months of age67. For our analysis we discarded the 2-month data and treated the BACHD-∆N17 7- and 11-month mice as the transgenic group, with the 7- and 11-month wild type samples as controls.
R6/2 (GSE9857)
Striatum samples from 9 mice expressing a short N-terminal fragment of mutant huntingtin (R6/2) and 9 wild type controls27 assayed on the Affymetrix Mouse Genome 430 2.0 Array.
Q150 (GSE32417)
Striatal samples from 4 Hdh Q150 mice and 4 wild-type littermates each at 6, 12 and 18 months68, assayed on the Affymetrix Mouse Gene 1.0 ST Array. In our analysis of preservation of association with genotype we only used the 12- and 18-month samples.
YAC128 (GSE18551)
Striatal samples from 9 transgenic mice (4 at 12 months and 5 at 24 months) expressing human huntingtin with 120 CAG repeats (YAC128) and 9 wildtype littermates69, assayed on the Affymetrix Mouse Genome 430 2.0 Array.
Preprocessing of independent data
For Illumina and Agilent data we have started our pre-processing from the author-normalized data available online; for Affymetrix data sets we started from the raw data contained in CEL files and applied the Robust Multi-chip Average (RMA) normalization method61. We then applied the following steps: (1) removal of low-expressed probes whose expression indices are likely mostly noise; (2) log-transformation of the expression data (only for data which had not been log-transformed yet) which makes the data more amenable to standard statistical analysis; (3) removal of potential outliers identified using Sample Network methodology57; (4) adjustment for technical covariates, including batch effects, and for gender and age where necessary; (5) quantile normalization61 where there is evidence of significant correlation of quantiles of individual samples. Finally, to facilitate cross-platform and cross-organism comparisons, the probe-level data were turned into gene-level data using the function collapseRows. Although our aim was to pre-process all data sets in a uniform manner, the pre-processing differs by necessity somewhat from data set to data set depending on platform, availability of technical and biological sample information, sample number, and other factors.
Module-genotype association in independent data
We calculated weighted average correlations of module genes with mutant genotype in mouse data and HD patient status in human data. The weight of each gene equals its module membership Z score raised to power 4, thus emphasizing module hub genes. We further identified human genes that change in the same direction in allelic series striatum and in the 2 human CN datasets and pass the FDR threshold of 0.1 (suggestive significance) in each human CN dataset. For the cortex, we analogously identified genes that change in the same direction in allelic series cortex and human cortex data and pass the FDR threshold of 0.1 in 3 out the 4 human cortex datasets. Genes that pass the above criteria are summarized in Supplementary Table 9.
Proteomic sample preparation and profiling
Striata were lysed in 100μl lysis buffer (8M urea, 50mM Tris-HCl pH 8.2, 75mM NaCl, 5mM EDTA, 5mM EGTA, 10mM sodium pyrophosphate, protease inhibitor cocktail Complete Mini (Roche, Mannheim, Germany). Tissues were crushed using forceps and extracts were sonicated using the beaker resonator BR30 (Bandelin Electronic, Germany). Samples were centrifuged at >14,000g, 4°C for 20 min to remove tissue/cell debris and the protein concentration of the supernatant was determined by Bradford Assay.
For each sample, 200μg of protein was reduced with 10mM dithiothreitol for 30 min and alkylated with 55mM iodoacetamide for 30 min in the dark. Subsequently, endoproteinase Lys-C (Wako) was added at an enzyme-to-substrate ratio of 1:200 and incubated for 4 h at room temperature. Samples were thereafter diluted 1:4 with 20mM Tris-HCl pH 8.2 before adding trypsin (Promega) at an enzyme-to-substrate ratio of 1:100 followed by overnight incubation. The resulting peptide mixtures were acidified by addition of TFA to a final concentration of 0.5% and subsequently desalted using C18 Sep-Pak columns (100mg sorbent weight, Waters). Peptides were eluted with 50% ACN, 0.5% acetic acid, samples split into two aliquots (100μg each), snap-frozen in liquid nitrogen, and lyophilized.
Of each sample 100μg of peptides were subsequently fractionated by high pH reversed phase chromatography70. Briefly, lyophilisates were reconstituted in 20mM ammonium formate, pH 10 (buffer “A”), and loaded onto an XBridge C18 200 × 4.6 mm analytical column (Waters) operated with the Äkta Explorer system (GE Healthcare). Peptides were eluted and separated at a flow rate of 1mL per minute applying a linear gradient with increasing acetonitrile concentration from 7% to 30% buffer “B” (buffer “A” supplemented with 80% acetonitrile) over 15 min followed by an increase to 55% over 5 min, a washing phase for 5 min at 100% and a final equilibration phase at 0% B for 10 min. The collected 18 fractions of eluting peptides were combined in a concatenated way to generate 6 fractions for each individual sample70. Samples were frozen in liquid nitrogen and lyophilized. After desalting via C18 Sep-Pak columns (100mg sorbent weight, Waters), sample fractions were snap-frozen, lyophilized, and reconstituted in 0.1% FA for MS analysis.
Samples were loaded onto a reverse phase analytical column packed in-house with 1.9μM C18 beads (Dr. Maisch) by an EASY nLC1000 UPLC system (Thermo Fisher Scientific) at flow of 400nL/min at 95% buffer A (5% DMSO, 0.1% FA). Peptides were resolved by a linear gradient over 96 min from 10% to 30 % buffer (5% DMSO, 80% acetonitrile) followed by an increase over 50% B in 13 min to 60% B in 6 min. Eluting peptides were electrosprayed via a nanoelectrospray ion source into a Q Exactive mass spectrometer (Thermo Fisher Scientific). The Q Exactive mass spectrometer was operated in the data-dependent acquisition mode acquiring full scans at a resolution of 70,000 and fragmentation spectra (MS/MS mode) of the ten most abundant peptide ions at a resolution of 17,500 in the Orbitrap mass analyzer.
Processing of MS Data
All raw files acquired were processed with the MaxQuant software suite (version 1.5.2.10) using the Andromeda search engine for peptide and protein identification and quantification71. The experiments were collectively searched against a Uniprot mouse database (version 11/2014). Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionine and N-terminal acetylation were set as variable modifications. The minimum required peptide length was seven amino acids and up to two missed cleavages were allowed. A false discovery rate (FDR) of 1% was selected for both protein and peptide identifications and a posterior error probability (PEP) less or equal to 1% for each peptide-to-spectral match was required. The match between runs option was enabled for a time window of 0.5 min. For protein quantification, the MaxLFQ feature of MaxQuant was enabled and used with default parameters. In particular, the minimum LFQ ratio count was set to 2 as suggested in Cox et al.43.
The proteinGroups-output table of MaxQuant was used for further analysis. In particular this table contains the UniProt identifiers and the label-free quantification (LFQ) values for each sample analyzed. A protein group can potentially refer to several UniProt identifiers, if the detected peptides do not allow distinguishing between these identifiers. In these cases, the identifier of the protein that was identified with the most peptides (the “leading” protein) was used as main identifier. LFQ values equal to zero represent missing data and were replaced by NaN (not-a-number) flags. All protein groups containing proteins from the contaminant sequence database or the decoy sequence database were removed.
For differential and network analyses, we excluded one apparent outlier (normalized sample connectivity Zk<−4) and retained those of the 10,747 quantified proteins that had no more than 50% missing values across the 46 samples (i.e., at most 23 missing values). This resulted in 7,711 proteins for differential abundance and network analyses. For analysis of concordance between protein and mRNA data, we retained 7,039 protein-mRNA pairs that were retained in both protein and mRNA network analysis.
Statistical testing
All mRNA differential expression analyses were performed in R using the Bioconductor72 package DESeq2 version 1.4.5. We have removed samples that were identified as potential outliers in the pre-processing described above. We have used the default settings (Wald test with a beta prior), with gender as a covariate. We have disabled the independent filtering and Cooks distance cutoff options in DESeq2. We tested differential expression in two-sample tests of higher Q (80, 92, 111, 140, 175) vs. Q20 samples (this results in 5 comparisons), and we also tested association of the expression profiles with Q viewed as a continuous numeric variable. Modeling of RNA-seq data within the DESeq2 package explicitly models the dependence of variance on genotype and mean expression.
Differential expression and association testing for non-count data (module eigengenes, proteomic data and others) was carried out using a robust correlation test (Student t-based p-value based on the value of biweight mid-correlation, a robust estimator of correlation). For purposes of association testing, missing values, if any, were treated as missing at random, avoiding problems with imputation and/or estimation of censored correlation. We expect that our treatment makes the significance statistics conservative in the case of proteomic data, where missing values indicate no detected peaks. All non-count data were transformed, if necessary, so that the expected variance is approximately independent of value (e.g. variance stabilization within DESeq2 and log-transformation for other data).
Two-sided tests were used for all association and differential expression tests. For enrichment testing, one-sided tests (alternative of over-enrichment) were used.
The Benjamini-Hochberg False Dicovery Rate estimates73 was used to adjust significance p-values for multiple comparisons. For simplicity, the FDR estimates were computed separately for each association test.
Enrichment was tested using the Fisher exact (equivalently, hyper-geometric) test.
Code availability
R scripts used in the present work are available from the authors upon request.
Quantitative PCR Validation
An independent cohort of 6m Q175 and WT mice (n = 4 per genotype) was used to validate selected RNA-seq expression data from our network analyses as performed previously77. Statistical testing was performed using one-sided Student’s T tests. Primers used were: ActB For 5′-ATGCTCCCCGGGCTGTAT-3′ Rev 5′-CATAGGAGTCCTTCTGACCCATTC-3′, Pde10a For 5′-CTATCGGCGGGTTCCTTACC-3′ Rev 5′-TGGGAGAAGTGGTGTTGCTC-3′, Ddit4l For 5′-GCTTACAGATGCCCAGGTTCT-3′ Rev 5′-AGCCTCCTTCAGCCATTACT-3′, Penk For 5′-TTGCAGGTCTCCCAGATTTT-3′ Rev 5′-AGCCAGGACTGCGCTAAAT-3′, Cnr1 For 5′-CAGGCTCAACGTGACTGAGA-3′ Rev 5′-CCTTGTAGCAGAGAGCCAGC-3′, Gpx6 For 5′-CCTAAAGAACTCCTGCCCTCC-3′ Rev 5′-TGACCGAGTGGAACAAAGACA-3′, Gpr6 For 5′-ACACGCAGCCATGTGGCGTT-3′ Rev 5′-GGGCGGTGCTAGGCAATGCT-3′, Npas4 For 5′-AGCATTCCAGGCTCATCTGAA-3′ Rev 5′-GGCGAAGTAAGTCTTGGTAGGATT-3′, Darpp32 For 5′-TCCTCCTTCCCCTACCAGTG-3′ Rev 5′-AGCACAAACAAAACGCAGCA -3′, Drd1a For 5′-GGCCTCTTCCTGGTCAATC-3′ Rev 5′-GAGCGTAGTCTCCCAGATCG-3′, Drd2 For 5′-TGAACAGGCGGAGAATGG-3′ Rev 5′-CTGGTGCTTGACAGCATCTC-3′, Igfbp4 For 5′-GCAGGTGGGGGACTCAGTGC-3′ Rev 5′-GGCCAGAGGCAGACAGCCAG-3′, Actn2 For 5′-GGTGAAACAGCTGGTGCCGGT-3′ Rev 5′-GGCGCCGCAGACGCTCATTA-3′, Nfe2l3 For 5′-GGAAAACGAGGAAGGGGTGT-3′ Rev 5′-TGCTCAGAAAAGGTGGAATGT-3′, Gsto1 For 5′-ACCTGAAGAATAAGCCCGAGTG-3′ Rev 5′-GAGAATCCCCACCAAGGAAGC-3′, Il33 For 5′-TCCTTGCTTGGCAGTATCCA-3′ Rev 5′-ACCGTCGCCTGATTGACTTG-3′, Mobp For 5′-GTGGACGCCTGCTGATGTAA-3′ Rev 5′-CCAAAAGACCCGTTCCCTGA-3′, Mal For 5′-GTCAGCCCATCTTCCCCATT-3′ Rev 5′-TCAAGGTTCCCAGTTCCCAC-3′, Hdac1 For 5′-GTGCCTGCTTAGGAGCTCTG-3′ Rev 5′-CCTCCACCCTACAGAATTGG-3′, Onecut1 For 5′-CAGCCGATGTGAAGACTGGA-3′ Rev 5′-CGGTGGTGGTGGTGGTAATC-3′, and Wt1 For 5′ AAGGACACGACTGTGGATCTACATC-3′ Rev 5′-TTCCGGCAAACCTGATAGGA-3′.
Unbiased stereology
Unbiased stereological counting of the total numbers of NeuN+ neurons and GFAP+ astrocytes in striatum at 4.5 and 10 month WT and Q175 het mice (N=6 per age per genotype) were performed by MBF Labs (Williston, VT) using the optical fractionator method. Serial 60-μm thick coronal sections of the striatum were prepared and every 6th section was stained free-floating with anti-NeuN (1:100,000, Millipore MAB377) followed by horse anti-mouse (1:250, Vector BA-2001) or anti-GFAP (1:20,000, DAKO Z033401-2) followed by goat anti-rabbit (1:250, Vector BA-1000) for cell counting.
Screening in Drosophila
Mutant and overexpression alleles were obtained from the Bloomington Drosophila Stock Center at Indiana University (http://flystocks.bio.indiana.edu/). The inducible shRNAs were obtained from the Vienna Drosophila Resource Center (http://stockcenter.vdrc.at/control/main). Nervous system expression was achieved using elav-Gal4 (C155). The NT-HTT128Q (F33A) line used for this study has been previously reported74 and expresses human HTTN231Q128. Motor performance tests were carried out using two replicates of 15 age-matched females per genotype and 10 trials per time point as previously described42. All genes except Ctcf were tested in one batch (41 genotypes) against common controls; Ctcf was tested in a second batch of 30 genotypes. Dunnett’s test following ANOVA was used to quantify statistical significance of differences in motor performance. Supplementary Figure 6 lists the relevant test statistics (ANOVA F values and Dunnett’s test p-values).
HDinHD Database Access
Server, data sets, and network browser for the allelic series transcriptome study and consensus network analyses will be made available to the public at https://www.hdinhd.org.
Accession Codes
All of our transcription data are available at Gene Expression Omnibus (GSE65776) and our online tool (HDinHD). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD003442 (http://www.ebi.ac.uk/pride/archive/projects/PXD003442).
Supplementary Material
Acknowledgments
The research was supported by CHDI Foundation, Inc. HD research in the Yang lab is also supported by NINDS/NIH grants (R01NS074312, R01NS049501 and R01NS084298). X.W.Y is also supported by the David Weill fund from Semel Institute and Carol Moss Spivak Scholar in Neuroscience from Brain Research Institute at UCLA. We acknowledge the support of the NINDS Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691). We also thank PsychoGenics for help in breeding the knockin allelic series and dissecting the tissues as part of a contract research agreement with CHDI.
Footnotes
AUTHOR CONTRIBUTIONS
X.W.Y., P.L., S.H., G.C., J.R., and J.A. designed and supervised the study. D.H. and S.K. supervised allelic series HD KI mouse tissue collection, RNA-seq and stereological counting of MSNs and astrocytes in Q175 mice. F.G. and G.C. performed RNA-seq data processing. P.L. and S.H. performed WGCNA consensus module analyses, preservation studies, WGCNA analyses of proteomic datasets. J.C., N.W., X.L., and X.W.Y. contributed to analyses and generation of data and graphs used in Figs. 3-6, and Supplementary Table S4. J.C. performed studies for data shown in Supplementary Fig. S4. I.A.-R., K.E.-Z., and J.B. performed the mutant huntingtin Drosophila modifier study. D.C., Y.Z., and G. C. created the HDinHD database. E.M.R. and G.C. performed Ctcf enrichment analyses. A.T., C.S., and D.L. performed striatal tissue proteomic studies for the Htt KI mice. X.W.Y., P.L., S.H., and G.C. wrote the manuscript.
COMPETING FINANCIAL INTEREST
The authors declare that they have no competing interests that might be perceived to influence the results and/or discussion reported in this article.
References
- 1.Ross CA, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nature reviews. Neurology. 2014;10:204–216. doi: 10.1038/nrneurol.2014.24. [DOI] [PubMed] [Google Scholar]
- 2.Vonsattel JP, DiFiglia M. Huntington disease. Journal of neuropathology and experimental neurology. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 3.A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 4.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annual review of neuroscience. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 5.Gusella JF, MacDonald ME. Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nature reviews. Neuroscience. 2000;1:109–115. doi: 10.1038/35039051. [DOI] [PubMed] [Google Scholar]
- 6.Gusella JF, MacDonald ME. Huntington’s disease: seeing the pathogenic process through a genetic lens. Trends in biochemical sciences. 2006;31:533–540. doi: 10.1016/j.tibs.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 7.Aylward EH, et al. Regional atrophy associated with cognitive and motor function in prodromal Huntington disease. Journal of Huntington’s disease. 2013;2:477–489. doi: 10.3233/JHD-130076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biagioli M, et al. Htt CAG repeat expansion confers pleiotropic gains of mutant huntingtin function in chromatin regulation. Human molecular genetics. 2015;24:2442–2457. doi: 10.1093/hmg/ddv006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seong IS, et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Human molecular genetics. 2005;14:2871–2880. doi: 10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- 10.Wang N, et al. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nature medicine. 2014;20:536–541. doi: 10.1038/nm.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington’s disease. Nature reviews. Neuroscience. 2013;14:708–721. doi: 10.1038/nrn3570. [DOI] [PubMed] [Google Scholar]
- 12.Menalled LB, et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PloS one. 2012;7:e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menalled LB, et al. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington’s disease knock-in mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:8266–8276. doi: 10.1523/JNEUROSCI.22-18-08266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith GA, et al. Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington’s disease. Human molecular genetics. 2014;23:4510–4527. doi: 10.1093/hmg/ddu166. [DOI] [PubMed] [Google Scholar]
- 15.Van Raamsdonk JM, et al. Testicular degeneration in Huntington disease. Neurobiology of disease. 2007;26:512–520. doi: 10.1016/j.nbd.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Mielcarek M, et al. Dysfunction of the CNS-heart axis in mouse models of Huntington’s disease. PLoS genetics. 2014;10:e1004550. doi: 10.1371/journal.pgen.1004550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC systems biology. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horvath S, Dong J. Geometric interpretation of gene coexpression network analysis. PLoS computational biology. 2008;4:e1000117. doi: 10.1371/journal.pcbi.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langfelder P, Mischel PS, Horvath S. When is hub gene selection better than standard meta-analysis? PloS one. 2013;8:e61505. doi: 10.1371/journal.pone.0061505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu XH, et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Sci Transl Med. 2014;6:268ra178. doi: 10.1126/scitranslmed.3010523. [DOI] [PubMed] [Google Scholar]
- 21.Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell. 2015;162:516–526. doi: 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labbadia J, Morimoto RI. Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends in biochemical sciences. 2013;38:378–385. doi: 10.1016/j.tibs.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 24.Hodges A, et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Human molecular genetics. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 25.Durrenberger PF, et al. Common mechanisms in neurodegeneration and neuroinflammation: a BrainNet Europe gene expression microarray study. Journal of neural transmission. 2014 doi: 10.1007/s00702-014-1293-0. [DOI] [PubMed] [Google Scholar]
- 26.Zhang B, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuhn A, et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Human molecular genetics. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- 28.Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 29.Grange P, et al. Cell-type-based model explaining coexpression patterns of genes in the brain. Proc Natl Acad Sci U S A. 2014;111:5397–5402. doi: 10.1073/pnas.1312098111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reiner A, et al. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heiman M, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nature neuroscience. 2006;9:443–452. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]
- 33.Fishell G, Heintz N. The neuron identity problem: form meets function. Neuron. 2013;80:602–612. doi: 10.1016/j.neuron.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 34.Deneris ES, Hobert O. Maintenance of postmitotic neuronal cell identity. Nature neuroscience. 2014;17:899–907. doi: 10.1038/nn.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen WV, Maniatis T. Clustered protocadherins. Development. 2013;140:3297–3302. doi: 10.1242/dev.090621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toyoda S, et al. Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity. Neuron. 2014;82:94–108. doi: 10.1016/j.neuron.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 37.Guo Y, et al. CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc Natl Acad Sci U S A. 2012;109:21081–21086. doi: 10.1073/pnas.1219280110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monahan K, et al. Role of CCCTC binding factor (CTCF) and cohesin in the generation of single-cell diversity of protocadherin-alpha gene expression. Proc Natl Acad Sci U S A. 2012;109:9125–9130. doi: 10.1073/pnas.1205074109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zuccato C, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 40.Mann M, Kulak NA, Nagaraj N, Cox J. The coming age of complete, accurate, and ubiquitous proteomes. Molecular cell. 2013;49:583–590. doi: 10.1016/j.molcel.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 41.Geiger T, Wehner A, Schaab C, Cox J, Mann M. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Molecular & cellular proteomics: MCP. 2012;11 doi: 10.1074/mcp.M111.014050. M111 014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shirasaki DI, et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 2012;75:41–57. doi: 10.1016/j.neuron.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cox J, et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma K, et al. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015;18:1819–1831. doi: 10.1038/nn.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pal A, Severin F, Lommer B, Shevchenko A, Zerial M. Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. The Journal of cell biology. 2006;172:605–618. doi: 10.1083/jcb.200509091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valencia A, et al. Striatal synaptosomes from Hdh140Q/140Q knock-in mice have altered protein levels, novel sites of methionine oxidation, and excess glutamate release after stimulation. Journal of Huntington’s disease. 2013;2:459–475. doi: 10.3233/JHD-130080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaltenbach LS, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS genetics. 2007;3:e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu T, et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature. 2014;507:448–454. doi: 10.1038/nature13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dowen JM, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374–387. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jacobsen JC, et al. HD CAG-correlated gene expression changes support a simple dominant gain of function. Human molecular genetics. 2011;20:2846–2860. doi: 10.1093/hmg/ddr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. The Journal of comparative neurology. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- 53.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson WE, Rabinovic A, Li C. Adjusting batch effects in microarray expression data using Empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 57.Oldham MC, Langfelder P, Horvath S. Network methods for describing sample relationships in genomic datasets: application to Huntington’s disease. BMC systems biology. 2012;6:63. doi: 10.1186/1752-0509-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Statistical Applications in Genetics and Molecular Biology. 2005;4 doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 59.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langfelder P, Horvath S. Fast R Functions for Robust Correlations and Hierarchical Clustering. Journal of statistical software. 2012;46 [PMC free article] [PubMed] [Google Scholar]
- 61.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 62.Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut library for R. Bioinformatics. 2007 Nov;:btm563. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 63.Stouffer SA. The American soldier. Princeton University Press; Princeton: 1949. [Google Scholar]
- 64.Zaykin DV. Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. Journal of evolutionary biology. 2011;24:1836–1841. doi: 10.1111/j.1420-9101.2011.02297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller JA, et al. Strategies for aggregating gene expression data: the collapseRows R function. BMC bioinformatics. 2011;12:322. doi: 10.1186/1471-2105-12-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen Y, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gu X, et al. N17 Modifies Mutant Huntingtin Nuclear Pathogenesis and Severity of Disease in HD BAC Transgenic Mice. Neuron. 2015 doi: 10.1016/j.neuron.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giles P, et al. Longitudinal analysis of gene expression and behaviour in the HdhQ150 mouse model of Huntington’s disease. Brain research bulletin. 2012;88:199–209. doi: 10.1016/j.brainresbull.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 69.Becanovic K, et al. Transcriptional changes in Huntington disease identified using genome-wide expression profiling and cross-platform analysis. Human molecular genetics. 2010;19:1438–1452. doi: 10.1093/hmg/ddq018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Y, et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 2011;11:2019–2026. doi: 10.1002/pmic.201000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cox J, et al. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nature protocols. 2009;4:698–705. doi: 10.1038/nprot.2009.36. [DOI] [PubMed] [Google Scholar]
- 72.Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome biology. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met. 1995;57:289–300. [Google Scholar]
- 74.Al-Ramahi I, et al. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. The Journal of biological chemistry. 2006;281:26714–26724. doi: 10.1074/jbc.M601603200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.