

Abstract
For decades oncogenic RAS proteins were considered undruggable due to a lack of accessible binding pockets on the protein surfaces. Seminal early research in RAS biology uncovered the basic paradigm of post-translational isoprenylation of RAS polypeptides, typically with covalent attachment of a farnesyl group, leading to isoprenyl-mediated RAS anchorage at the plasma membrane and signal initiation at those sites. However, the failure of farnesyltransferase inhibitors to translate to the clinic stymied anti-RAS therapy development. Over the past ten years, a more complete picture has emerged of RAS protein maturation, intracellular trafficking, and location, positioning and retention in subdomains at the plasma membrane, with a corresponding expansion in our understanding of how these properties of RAS contribute to signal outputs. Each of these aspects of RAS regulation presents a potential vulnerability in RAS function that may be exploited for therapeutic targeting, and inhibitors have been identified or developed that interfere with RAS for nearly all of them. This review will summarize current understanding of RAS membrane targeting with a focus on highlighting development and outcomes of inhibitors at each step.
Keywords: RAS, plasma membrane, lipid rafts, therapeutics, Galectin
1. Introduction
The RAS homologues most prominently associated with cancers, H-, N-, and KRAS, are ubiquitously expressed with overlapping yet non-redundant functions1–3. RAS propagates growth factor signaling, most prominently the MAPK mitogenic pathway (Raf/MEK/ERK) and PI3K/mTOR survival pathways 1, 4, 5. Constitutively active (CA) RAS mutations are highly transforming and tumorigenic 6. Combined, CA RAS mutations are associated with up to ~30% of all human malignancies 7. HRAS mutations occur most prominently in cervix (9%), salivary gland (15%), and urinary tract (11%) malignancies, while NRAS mutations are associated with skin (18%) and hematopoietic cancers (>10%), as well as many other cancer types. KRAS CA mutations are associated with many adenocarcinomas, including >50% of pancreatic cancers (isotype-specific cancer mutation rates reviewed in detail in 7). The molecular mechanisms of isotypic RAS oncogenesis are still not completely understood, and RAS inhibition remains an important anti-cancer strategy 1, 4, 5. However, RAS has been a stubbornly obstinate drug target in cancer therapeutics. This is primarily due to the globular nature of small (~21 kDa) RAS proteins. Despite constant efforts to target RAS directly, a lack of binding pockets on the protein surface has deemed the protein notoriously ‘undruggable’.
RAS proteins undergo a complex series of post-translational modifications and organelle shuttling in their pathways to becoming mature proteins, which promote mitogenic signaling via interactions at the plasma membrane (PM). Unsurprisingly, point mutations associated with increased (and unchecked) RAS signaling facilitate oncogenic signaling. The trafficking of RAS to the plasma membrane and RAS localization and interactions at the membrane have been explored for many years as vulnerabilities in RAS oncogenesis and oncogene addiction in cancer8. A majority of RAS-driven cancer cases in the United States (e.g., in lung, pancreas, and colorectal cancers) are driven by, or associated with, mutant KRAS 7. Direct targeting of KRAS has proven clinically intractable due to a lack of drug-binding pockets, and drugs against the membrane anchors have generally not been successful 9. However, alternative strategies to modulate KRAS anchorage to the plasma membrane may prove viable for combating KRAS-driven cancer progression. MEK inhibition alone seems to have variable outcomes in KRAS tumors 10. New advances in our understanding of RAS PM targeting and membrane anchorage, as well as development and analysis of novel drugs targeting each step, together set the stage for potentially viable clinical approaches to disrupt RAS-driven cancer progression with improved anti-cancer efficacies. This review will describe 1) the processes of RAS maturation; 2) trafficking to the plasma membrane; 3) localization and retention in membrane microdomains and associated effects on RAS signaling; and 4) current developments in therapeutic strategies designed to disrupt RAS at each stage.
2. RAS membrane targeting
RAS proteins are globular, cytoplasmic polypeptides, which undergo a series of irreversible lipid modifications within the C-terminal targeting domains (tDs) that increase the hydrophobicity of the C-termini and support anchorage of the RAS proteins to endoplasmic reticulum membranes and subsequent transport to the Golgi, prior to delivery to the plasma membrane 7. In addition to a covalently-linked prenylation group at the extreme C-terminus (after subsequent processing steps detailed below), RAS proteins require a secondary lipid binding motif. In the case of KRAS (KRAS4B splice variant, described henceforth as “KRAS” unless otherwise indicated), the tD contains Lys-rich sequences comprising a bipartite polybasic domain which enhances PM interaction based on the charged residues 8. In contrast, the tDs in H-, N- and RRAS also contain adjacent palmitoylation target sites via thioester linkage (C181/184 in HRAS, C181 in NRAS, C213 in RRAS) 9, 10. RAS isotypes have distinct distributions in PM microdomains, driven by protein conformation (either GDP- or GTP-bound) and post-translational modifications (farnesylated and/or palmitoylated) 11–14. H- and NRAS are anchored to the lipid ordered (i.e., lipid raft) membrane while GDP loaded, and shuttled to the lipid ordered/lipid disordered border upon GTP loading 15–17. RRAS, a conserved RAS paralogue with limited mitogenic signaling properties, preferentially anchors within the lipid ordered domain, regardless of activation state 16, 18. However, RRAS subfamily paralogue RRAS2 (TC21) is associated with multiple cancer types, and recently RRAS1 (hereafter referred to as RRAS) has been found to have more profound roles in cancer than previously understood 11–18. RAS proteins are postulated to exclusively signal from the plasma membrane, and must reach the PM from either the Golgi or other sites. Approximately 40% of RAS proteins assemble at the PM into small, transient nanoclusters consisting of 6 or 7 RAS proteins per cluster, and the size and duration of RAS nanoclusters also contributes to signal output19. Therefore, regulators of RAS transit to the PM and nanoclustering represent possible modes of therapeutic targeting in cancer. In addition, the lateral distribution of RAS proteins within the plane of the plasma membrane plays a critical role in RAS signaling. Recent advances in our understanding of how RAS PM microdomain targeting, shuttling and retention are controlled allow new considerations for drug targeting of RAS function in cancer treatment.
2.1. Post-translational Modifications and vulnerabilities in RAS protein processing
Each of the steps in RAS protein maturation and its intracellular trafficking to reach the plasma membrane, represent potential target spots for RAS interference. These are described below and summarized in Figure 1.
Figure 1. RAS protein maturation processes and inhibitors.

Posttranslational modifications of RAS polypeptides mediate RAS trafficking through the endoplasmic reticulum and Golgi, and subsequent trafficking to the plasma membrane as described in the text. In the specific case of KRAS4B, chaperone protein PDE6δ (PDE) is critical for cytosolic trafficking; NRAS appears to be chaperoned by VPS35 binding to its farnesyl group. Small molecule inhibitors discussed in the text are shown for each step in RAS membrane targeting.
2.1.1. Iso-prenylation & CaaX motif
The final 23/24 amino acids of RAS constitute the so-called hypervariable region (HVR), a poorly conserved domain which dictates the RAS isotype-specific post-translational modifications 20. Notably, RAS proteins end in a cysteine-aliphatic-aliphatic-X (CaaX) motif, in which the X is usually serine, methionine, or glutamine. After being translated via cytosolic ribosomes, the RAS isotype (H-, K-, NRAS and TC21) is enzymatically isoprenylated with a 15-carbon farnesyl group by farnesyl transferase (Table 1). The farnesyl transferase binds RAS within the cytosol and attaches the lipid moiety to the cysteine residue of the CaaX motif 21. The weakly mitogenic paralogue, RRAS contains a cysteine-aliphatic-aliphatic-Leucine (CaaL) motif (Table 1). With only this single amino acid difference, a CaaL motif preferentially drives covalent addition of a 20-carbon geranylgeranyl lipid group to the cysteine residue via geranylgeranyl transferase 22. TC21, distinguished in the CaaX motif from RRAS only by the two terminal residues, has been experimentally demonstrated to undergo either farnesylation or geranylgeranylation 23; the ability of TC21 to be farnesylated may be the root of TC21 as the more oncogenic isotype 24. Both farnesyl transferase and geranylgeranyl transferase attach the respective lipid group via an irreversible thioether bond. Covalently linked lipid moieties have been shown to account for specific RAS isotype activation by distinct guanine nucleotide exchange factors. For example, RAS-GRF1 and RAS-GRF2 can activate HRAS, while RAS-GRF2 is unable to activate RRAS specifically because of the geranylgeranyl attachment 25, 26.
Table 1.
RAS Lipid Modifications
| RAS isotype | C-Terminal prenylation motif | Isoprenylation | Secondary Lipid Binding Motif | Palmitoylation | Oncogenic |
|---|---|---|---|---|---|
| HRAS | CVLS | Farnesyl | Palmitoylation | 2 sites | X |
| NRAS | CVVM | Farnesyl | Palmitoylation, hydrophobic residues | 1 site | X |
| KRAS4A | CVIM | Farnesyl | Palmitoylation, hydrophobic residues in exon 4A | 1 site | X |
| KRAS4B | CVIM | Farnesyl | Electrostatic interaction | none | X |
| RRAS | CVLL | Geranylgeranyl | Palmitoylation | 1 site | weak |
| TC21 | CVIF | Farnesyl or geranylgeranyl | Palmitoylation | 1 site | X |
Therapeutics designed to interfere with this first stage of RAS prenylation can be divided into at least two groups. One strategy is development of peptidomimetics that compete with unmodified Ras for farnesyltransferase. Another would be nonpeptidomimetics, such as farnesylpyrophosphate (FPP) analogs, which compete for binding to the farnesyltransferase protein 27–29. Collectively, small molecule inhibitors which inactivate the enzymatic function of farnesyltransferase are labeled FTIs (farnesyltransferase inhibitors). Treatment with FTIs (such as SCH 66336) inhibit cell growth in a variety of cancer cell lines when treated in vitro and in vivo tumor xenografts 30. Subsequently, many studies focused on the effect of FTIs on HRAS, showing great efficacy in disrupting membrane association, and blunting colony formation in soft agar (a classical measurement for cellular transformation) 31, 32. Despite a wealth of data showing blunted cancer growth using various in vitro systems, clinical trials using FTIs alone have had disappointingly poor outcomes 30, 33, 34. Further investigation revealed that cells treated with FTIs can yield alternatively prenylated mutant KRAS or NRAS, by attachment of a geranylgeranyl group 35, 36. This led to a new approach for developing a class of inhibitors for Geranylgeranyltransferase (GGTIs), though monotherapy or in conjunction with FTIs are not effective due to toxicity issues 37. A class of inhibitors targeting both farnesyltransferase and geranylgeranyltransferase (such as L-778, 123) were developed and failed to make it through phase I clinical trials. Despite dual inhibition, similar to treatment with FTIs, Ras activity was not inhibited 38. Although FTIs continue to be explored in anti-RAS therapies, combinatorial treatments with drugs targeting other aspects of RAS processing or signaling may be needed.
2.1.2 Proteolytic cleavage & Carboxymethylation
Once the RAS proteins are isoprenylated by either a farnesyl (H-,K-,N-, TC21) or geranylgeranyl (R-, M-) group the protein hydrophobicity is increased, thus causing higher affinity to the endoplasmic reticulum for subsequent modification. Thereafter, the CaaX or CaaL sequence targets RAS to the cytosolic surface of the endoplasmic reticulum where RAS and a-factor converting enzyme (RCE1), proteolytically removes the –aaX tripeptide 39. This proteolytic step is required for further steps in RAS processing. Multiple approaches have been taken to target this potential vulnerable spot in RAS protein maturation. A first-generation class of RCE1 inhibitors, including NCS1011, as well as peptide-based inhibitors showed Ras mislocalization in yeast 40–42. Natural RCE inhibitors have also been discovered but their efficacy in blocking RAS targeting and function are unknown 43. Newer libraries of NCS1011-based RCE1 inhibitors were recently developed, which disrupted plasma membrane targeting of RAS - particular KRAS although H- and NRAS were also mistargeted - and were more effective in this regard than FTIs 44. This processing step continues to represent a potential avenue for RAS functional blockade in cancer and merits further development.
Following –aaX proteolysis, the newly C-terminal prenylcysteine is targeted by isoprenylcysteine carboxyl methyltransferase (ICMT), which methyl-esterifies the α carboxyl group 45. ICMT inhibition represents another potential area for RAS blockade along its maturation route, and efforts to inhibit ICMT-mediated RAS processing have shown promising in vivo results in animal models. Several types of ICMT inhibitors have been developed, all of which interfere with RAS targeting, although not all have shown greater efficacies than FTIs 46. Cysmethynil is an indole-structured ICMT inhibitor that has demonstrated anti-tumor properties, including induction of cell death by autophagy, and reduction of xenograft tumor growth with prostate cancer cells 47. A recently devised amino derivative of cysmethynil, called compound 8.12, showed an improved IC50. Treatment with compound 8.12 facilitated cell cycle arrest, autophagy, apparent apoptosis in prostate and liver cancer cell lines, and importantly, had stronger anti-tumor effects than cysmethynil in xenograft studies, with minimal off-target toxicity. Synergistic effects were also observed in combination with inhibition of epidermal growth factor receptor (EGFR, upstream RAS activator) 48. These combined in vitro and in vivo results underscore ICMT inhibition as a promising strategy in anti-RAS therapeutics.
2.1.3. Secondary lipid membrane binding
The isoprenylated RAS proteins weakly bind endomembranes, yet a second motif within the HVR strengthens further membrane interaction, trafficking, and proper PM microdomain localization. A common secondary motif is the reversible addition of a 16 carbon palmitate group 49. Palmitoyl groups are added to H-,N-,R-, and splice variant K(A)-RAS. The more physiologically predominant splice variant K(B)-RAS contains a polybasic (mostly lysine) sequence which allows an electrostatic interaction with the acidic headgroups of lipid bilayers (Table 1). KRAS traffics through a Golgi-independent route, described in more detail below 50, 51. HRAS is palmitoylated on two Cysteine residues, while N- and K-(A)RAS are monopalmitoylated, and NRAS requires a third HVR motif for plasma membrane association, which consists of a stretch of hydrophobic residues 52 (Table 1). RAS palmitoylation (H-, N-, K(A)-) is performed via a heterodimeric complex consisting of Palmitoyltransferase ZDHHC9 (DHHC9) and Golgin subfamily A member 7 (GCP16) 53. DHHC9 is one member of a family of DHHC-motif containing protein S-acyltransferases (PATs) 53. Alternatively, the precise function of GCP16 is unclear. GCP16 is required for the heterodimeric complex localization, and plays a factor in DHHC9 protein stability 54. Another DHHC family member, DHHC19, is responsible for palmitate transfer to RRAS, but not H-, N-, or KRAS(4A) 55. Various broad spectrum PAT inhibitors have been identified or developed; one of these, 2-bromopalmitate, has been demonstrated experimentally to promote Golgi retention of CA HRAS, although potential effects on HRAS oncogenesis require further study56, 57. To date, selective DHHC9 or GCP16 inhibitors have not been explored for RAS inhibition.
Palmitoylated RAS proteins mainly reside in recycling endosomes, which function as a shuttle along the post-Golgi exocytic pathway to the plasma membrane. Palmitate groups are attached via a thioester bond. This reversible addition of a lipid moiety allows for spatio-temporal regulation of RAS proteins. Depalmitoylating enzymes, such as acyl protein thioesterase I (APT1) or FKBP12, cleave the thioester bond between the Cysteine residue and the palmitate. Palmitate removal reduces the protein’s affinity for the plasma membrane, which triggers the RAS to recycle back to the Golgi where it can be palmitoylated numerous times during the half-life of the protein 58–60. An APT1 inhibitor has been shown to reduce ERK phosphorylation (a key mitogenic event in RAS downstream signaling), but anti-RAS and anti-tumor effects have not been thoroughly explored 61. Moreover, APTs and FKBP12 have many protein substrates, making design of specific targeting approaches difficult.
Palmitoylation is required for recycling via endosomal targeting, and the lack of proper palmitoylation leads to improper PM localization 62. In addition to H-, N-, and KRAS4A, RRAS also requires palmitoylation to exit from the Golgi, and properly traffic anterograde via vesicles to the plasma membrane 63. NRAS retention at the PM is < 5 min, much shorter than that for HRAS (< 20 min), apparently as a result of one versus two palmitate anchors 64. However, the distinct palmitoylation profiles of H- and NRAS are also responsible for differential targeting within the Golgi: HRAS across the whole Golgi stack, whereas NRAS has only limited localization at the trans Golgi 65. Effects of this differential Golgi sub-compartment distribution, based on distinct palmitoylation, on RAS PM microdomain distribution, longevity and signaling remain unclear, but upon further elucidation these properties of H- and NRAS may represent new targeting vulnerabilities.
2.2. RAS protein trafficking and chaperones
2.2.1. KRAS, PDE6δ and GPR31
Whereas H- and NRAS typically traffic to the PM via Golgi-dependent vesicular transport, KRAS4B does not. The mechanism by which KRAS is able to traffic through the cytosol to reach the plasma membrane has largely remained a mystery until recently. Bastiaens and colleagues considered the model of RhoGDI, a cytosolic chaperone for prenylated RHO proteins (RAS subfamily proteins involved in cell motility), which protects the hydrophobic geranylgeranyl group on RHO, allowing the protein to be extracted from Golgi membranes. They recently established that cGMP phosphodiesterase type 6 δ (PDE6δ) subunit, with a similar structure to RhoGDI, plays a complementary role as a cytosolic chaperone for KRAS and is important for the ability of KRAS to transit between membranes and for its oncogenic signaling 66, 67. PDE6δ also binds HRAS and NRAS and may be a chaperone for a variety of prenylated proteins; however, in this case the degree of palmitoylation appears to counter PDE6δ binding, with NRAS binding better than HRAS 67. PDE6δ silencing blocks KRAS function and tumor cell growth, suggesting that this molecule may be a novel target in KRAS cancer therapeutic development. Moreover, the fact that PDE6δ-null mice are viable and generally healthy indicates that molecular targeting of this protein may have limited deleterious side effects 68. Indeed, small molecule inhibitors of PDE6δ (Deltarasin and Deltazinone 1) have shown promising anti-KRAS oncogenic results 69, 70. In addition, KRAS was recently shown to couple to a G protein-coupled receptor, GPR31, in a farnesyl-dependent fashion, and this interaction is important for KRAS trafficking from endoplasmic reticulum to PM, KRAS-mediated macropinocytosis, and tumor cell growth 71. However, it is not clear if this represents a ubiquitous mechanism of KRAS regulation across multiple cancer types. It will be interesting to observe pre-clinical developments based on these angles in KRAS inhibition.
2.2.2. NRAS and VPS35
A newly discovered mechanism of NRAS-specific trafficking may provide a partial explanation for differential signaling and targeting relative to HRAS, and may also indicate a novel therapeutic target in NRAS-driven cancers. Zhou and colleagues used affinity purification to identify a retromer vesicle coat protein, VPS35, as an NRAS-binding protein that requires the farnesyl group, but neither palmitoylation nor GTP coupling, for interaction. VPS35 knockdown inhibits NRAS PM targeting, mitogenic signaling, and results in limited growth in NRAS mutant melanoma cells. Moreover, VPS35 does not display any of these properties with respect to HRAS or KRAS4B 72. Thus, as an apparent NRAS-specific transport chaperone, a therapeutic targeting VPS35-NRAS coupling may be a novel target in disrupting NRAS.
3. Plasma membrane microdomain targeting of RAS and potential therapeutics
3.1. Plasma membrane microdomains
Lipid bilayers are comprised of subdomains that contain increased concentrations of cholesterol and glycosphingolipids, which are referred to as the lipid ordered domain, or lipid rafts. The fatty-acid side chains present in lipid ordered membranes tend to be more saturated than those in the surrounding membrane. Due to the presence of cholesterol and fatty acid saturation, a lipid ordered domain exhibits less fluidity than the surrounding plasma membrane. These microdomains are distinct regions of the membrane that are characterized by displaying a resistance to extraction with nonionic detergents. A multitude of proteins involved in cell signaling have been shown to reside within the lipid ordered microdomain, contributing to both a positive and negative role in cell signaling. Lipid rafts may positively regulate signal transduction by responding to agonist stimulation causing cluster formation, therefore leading to downstream signal activation. Alternatively, they may negatively regulate signal transduction by spatially segregating proteins, leading to reduced downstream activation 73. RAS proteins segregate in lipid ordered or disordered domains, or at the microdomain borders, by isotype, GTP loading, and palmitoylation state, and these states modulate specificity and duration of RAS signal outputs. The regulatory steps of RAS signaling and anchorage in microdomains and potential target sites are described below, and summarized in Figure 2.
Figure 2. RAS signaling and tumorigenesis in response to inhibition of Galectins and mTOR.

a) HRAS-GAL1 and KRAS-GAL3 coupling support downstream RAS oncogenic signaling. b) Schematic of predicted effects of anti-Galectin treatments in H- and KRAS cancers. Galectin-1 inhibition using OTX008 or other GAL1 inhibitors results in loss of HRAS at the lipid ordered/disordered domain borders, and disruption of MAPK mitogenic signaling. GAL3 inhibition with modified citrus pectin, GSC-100, or other inhibitors blocks MAPK signaling by destabilizing KRAS membrane retention. Some PI3K signal output is retained by these treatments. c) Predicted effects of dual inhibition of Galectins and mTOR. Rapamycin or rapalog treatment potently reduces mTOR survival signaling. Combinatorial pathway inhibition blocks MAPK and PI3K signaling by RAS, resulting in an additive anti-tumor effect over either monotherapy.
3.2. RAS microdomain targeting
The HVR of different RAS proteins dictate the trafficking to distinct sub-membranous domains. Data collected from a variety of experimental techniques including biophysical, biochemical, and electron microscopy studies show that interactions of RAS with the plasma membrane proteins are dynamic 74–76. Biochemical and electron microscopy studies suggest that approximately 50% of the inactive form of HRAS (GDP bound) is localized to lipid ordered domains, while activated HRAS (GTP bound) exits lipid ordered domains and preferentially resides in the lipid disordered membrane 77. Recently, semi-atomic in silico simulations provided evidence which indicates GTP bound HRAS resides within the border between the lipid ordered/lipid disordered domains 78. Similar to HRAS, inactive (GDP bound) NRAS is found in both lipid ordered and lipid disordered membranes. Atomic force microscopy has shown GTP bound activated NRAS is likely at the lipid ordered/lipid disordered boundary, where it may help reduce line tension at the phase boundary 79, 80. The independent studies publishing the trafficking of H-and NRAS show a similar trend. However NRAS PM localization is more controversial, as experiments looking at fluorescence recovery after photobleaching (FRAP) have reported GTP-bound NRAS to reside within lipid rafts 75, 81, which underscores the importance in further investigation of NRAS PM localization. KRAS traffics to the lipid disordered subdomain, regardless of activity state, trafficking directly from the endoplasmic reticulum without modification from the Golgi. Despite the electrostatic force generating the association with the plasma membrane, approximately 85% of KRAS is found to localize within the lipid disordered domain. Importantly, KRAS generates signals from the lipid disordered membrane that is spatially distinct from HRAS 76, 82. Collectively, the farnesyl lipid moiety appears to preferentially localize RAS proteins to the lipid disordered domain, while the palmitate group prefers the lipid ordered domain. This trend accounts for similar trafficking of H- and NRAS to the lipid raft border, and for the nonpalmitoylated KRAS to preferentially propagate signals from the lipid disordered domain. However, membrane lipid binding of KRAS and subsequent signaling is further dictated by distinct preferences of arginine and lysine residues in the polybasic domain, as well as the length of the attached prenyl group. Interestingly, KRAS nanocluster composition based on these distinctions was found to relate directly to signal output; for example, KRAS that selectively forms nanoclusters enriched for PIP2, the substrate for PI3K, showed elevated PI3K signaling (AKT phosphorylation) at the expense of reduced RAF signaling (MEK phosphorylation) 83. RRAS is geranylgeranylated, not farnesylated, and palmitoylated, and found to be localized to lipid ordered microdomains in both active and inactive forms (GTP or GDP bound) 78, 84. We recently found that the distinctions in RRAS and HRAS microdomain targeting account for their distinct vesicle trafficking from Golgi to the PM, and oncogenic and signaling effects, and that these properties could be exchanged by switching the targeting domains 63, 85, suggesting similar regulation of HRAS oncogenesis by subtle changes in RAS microdomain targeting, nanoclustering, and orientation towards the membrane.
3.3. KRAS4A
For decades KRAS4B was considered the major splice variant and this isoform occupied most of the attention in KRAS signaling studies. The Philips group recently used splice junction priming to screen cancer cell lines and human colorectal tumors, and found that KRAS4A was not only detected in multiple lines but was expressed at levels on par with the 4B variant 86. They found that KRAS4A, which has both a bipartite polybasic region (like –4B) as well as a palmitoylation site contained in the 4A exon which is not spliced out, can reach the PM using either of these targeting motifs. While KRAS4A does not bind PDE6δ, it is possible this variant traffics via Golgi targeting in similar fashion to the other palmitoylated RAS isotypes, and hence KRAS4A would not require the chaperone. Thus, this highly expressed KRAS variant may not only be subject to therapeutic intervention through these dual membrane targeting motifs, but KRAS4B blockade alone may not be sufficient to inhibit KRAS oncogenesis. Because of its dual targeting motifs, KRAS4A is likely to require combinatorial approaches to interfere with both motifs. Much more work needs to be done to determine whether this variant is expressed as widely in other cancers, as well as additional mechanistic details and effects of available drugs that inhibit the putative trafficking pathways.
3.4. Galectins regulate RAS membrane localization
The process which regulates membrane microdomain shuttling is largely unknown, though at least one type of scaffold protein, Galectins - a family of carbohydrate-binding proteins with high affinity for β-galactosides - have been identified as a critical part of this process. Galectin overexpression – notably Galectin-1 (GAL1) and Galectin-3 (GAL3) - have been observed in several tumor types, and have been associated with tumor progression 87, 88. GTP-bound HRAS has been demonstrated to selectively bind GAL1, and GTP-bound KRAS selectively binds GAL3 89, 90. While GAL3 does not directly bind to NRAS, increased GAL3 expression simultaneously increases KRAS signaling while decreasing NRAS activation 91. This is due to an interaction of the N-terminus of GAL3 with RAS exchange factor, RASGRP4, which diminishes NRAS GTP loading 92. Coincidently, no Galectin proteins have currently been shown to interact with NRAS specifically.
3.4.1. GAL1 and HRAS
In the case of HRAS, GAL1 contains a prenyl-binding pocket, which interacts with the farnesyl group in GTP-HRAS, independent of lectin function. This interaction is thought to alter the orientation of the HRAS globular domain with respect to the plasma membrane, and thereby regulate lateral segregation of HRAS and promote MAPK signaling90, 93. Indeed, ectopic GAL1 overexpression or suppression increases or abrogates GTP-bound HRAS nanoclustering, respectively 94. Thus, upon GTP-loading, the conformational shift of HRAS promotes affinity for Galectin binding, which subsequently allows lateral movement at the plasma membrane, allowing for distinct signaling platforms93. Moreover, ectopic expression of GAL1 has been previously demonstrated to divert HRAS signaling to the Raf/MEK/ERK effector pathway, at the expense of PI3K 95. NRAS also appears to be able to be oriented at the membrane in a manner supporting GAL1 interaction, suggesting NRAS signaling may also be regulated by a similar mechanism 96. Thus, GAL1 plays a key role in maintaining H- and NRAS in the active state by preventing shuttling away from the lipid disordered domain border. We found that GAL1 inhibition with small molecule inhibitor OTX008 sensitized HRAS-driven tumorigenesis to blockade with mTOR inhibition by rapamycin 97, suggesting one mechanism of potential dual pathway targeting based on HRAS microdomain localization. Interestingly, inhibitors of mTOR signaling (rapalogs, next generation analogs of rapamycin) were recently found to specifically promote HRAS nanoclustering through a GAL1-dependent mechanism, modulating HRAS tumorigenic function 98. Thus, GAL1 inhibition may have potent effects on HRAS oncogenesis through interfering with HRAS at multiple levels.
3.4.2. GAL3 and KRAS tumor inhibition
Whereas HRAS localization is mediated by GAL1, PM anchorage of activated KRAS is supported by the GAL3 scaffold, which binds the farnesyl group on KRAS 99–104. GAL3 over-expression leads to chronic KRAS activation, potentiation of RAS signaling, tumor cell activation including increased proliferation and migration, and tumor progression 100, 105, 106. KRAS nanoclustering and other biophysical aspects may be in play in these aspects of GAL3 function 101, 104, 107, 108. GAL3-mediated KRAS activation is associated with ERK but not PI3K signaling. There is substantial evidence for GAL3 inhibition as a potential tumor blocker, via GSC-100, a small molecule inhibitor of GAL3 109–116. A soluble vitamin, modified citrus pectin (MCP), also acts as a selective and potent GAL3 inhibitor 117, can be provided in drinking water, and has shown efficacy in anti-tumor treatments 110, 112, 114–116, 118–122. Small molecule inhibitors of GAL3 have shown efficacy against acute myeloid leukemia (AML) and are in clinical trials for other diseases 109, 117, 123. Dual MAPK/PI3K inhibition has shown promise in blocking KRAS mutant tumor progression 124. We have found that Lewis lung carcinoma cells (LLC) in our lab harbor one mutant Kras allele encoding CA KrasG12C (Fig. 3a). A single CA KRAS allelic mutation is common in KRAS-driven cancers, and tumor progression appears to result from cooperative signaling between the mutant and WT proteins 125. This led us to test whether: 1) Gal3 inhibition with MCP would inhibit tumor growth in these cells, and; 2) mTOR inhibition, coupled with Gal3 inhibition with MCP, would yield additive anti-tumor effects on Kras mutant LLC tumors in mice. Growth of allograft ectopic tumors made by subcutaneous implantation of LLCs was partially inhibited by either MCP or rapamycin treatment alone. Dual inhibition with MCP and rapamycin together yielded stronger tumor inhibitory effects (Fig. 3b). These effects correlated with reduced Erk phosphorylation (Thr202/Tyr204), assessed by blotting lysates of resected tumors, in MCP-treated tumors, and reduced S6 (Thr389) phosphorylation in rapamycin-treated tumors. In addition, both Erk and S6 activation (mTOR downstream target) were blocked with dual inhibition, reflecting the stronger effects on tumor growth (Fig. 3c). These results indicate that Gal3 inhibition selectively blocks the Erk pathway in these CA Kras tumor cells, consistent with a role for GAL3 in RAS/RAF/MEK/ERK signaling in human cancers99. Further experimentation is needed to explore this mechanism and putative translational applications in KRAS cancer therapies.
Figure 3. Dual inhibition of mTOR and Gal3 in Kras mutant LLC tumor growth and Ras signaling.
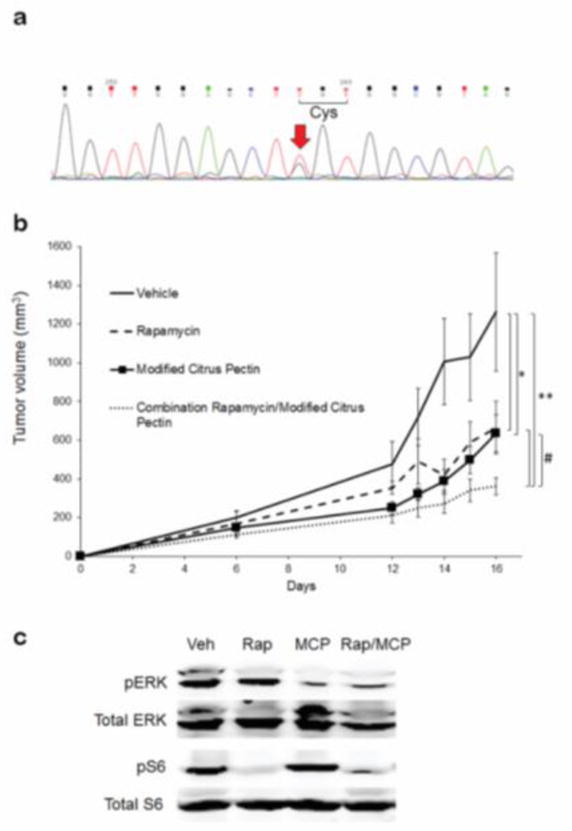
a) Genomic sequencing of exon1 of Kras in LLC cells using flanking primers revealed a G->T SNP resulting in G12C codon substitution. n = 3, sequenced both forward and reverse. b) LLC allograft tumor growth in C57Bl/6 mice. Mice were shaved and injected i.p. with 5 mg/kg rapamycin, and/or 100 mg/kg MCP, or vehicle as indicated, every 48 hours beginning at day 0 of tumor implantation in the flanks. Tumor volumes were measured with calipers. *, p < 0.03. **, p < 0.001. #, p < 0.05. n = 10. c) Ras pathway signaling in LLC tumor lysates after mono or dual drug treatment. Resected tumors from (b) were lysed and proteins blotted for total Erk, total S6, phospho-Erk (pERK), and phospho-S6 (pS6) as indicated. Veh, vehicle; Rap, rapamycin; MCP, modified citrus pectin. Blots representative of 4 independent experiments.
Recently, Seguin and colleagues demonstrated that GAL3 supports KRAS-mediated macropinosome formation, which drives oncogene addiction by enhancing uptake of nutrients and reducing reactive oxygen species. This function of GAL3 is derived from direct interaction of GAL3 with αvβ3 integrin, which induced KRAS clustering pursuant to macropinosome formation126. Moreover, GAL3 inhibition with GCS-100 blocked the αvβ3 integrin-mediated clustering and downstream KRAS activation, and inhibited growth of KRAS-mutant lung and pancreatic tumors in mice 127. Thus, GAL3 association may represent a potent weak spot in KRAS-driven cancer progression, as GAL3 potentiates both tumor cell proliferation due to KRAS mitogenic signaling, and KRAS addiction and neutralization of reactive oxygen species. NRAS may also be sensitive to GAL3 inhibition128, 129, and there is some evidence for GAL3 modulation of N- and HRAS activation; however, these activation effects would be irrelevant in mutant H-/NRAS-driven cancers129. Interest in GAL3/RAS biology is an emerging topic, and further studies to explore therapeutic interventions are clearly warranted.
4. Conclusions
After many years of unsuccessful attempts to modulate oncogenic RAS in cancer, we now face the possibility of attacking RAS based on a wealth of new information on the maturation mechanisms and spatial regulation of RAS in cells. Therapeutics designed to target enzymes involved in RAS isoprenylation, palmitoylation, intracellular membrane anchorage, and trafficking, have shown some promise in blocking RAS oncogenesis. A challenge in this arena will be to avoid toxicities due to general inhibition of protein processing; however, some targets such as ICMT have been able to be inhibited with minimal toxic effects. Novel chaperone proteins may also be effective targets, and recent discoveries suggest that other RAS chaperones or associated molecules may yet be found. Recent work examining microdomain targeting and retention, as well as the structure of RAS tethered at the plasma membrane and their combined contributions to signal output, offer still more opportunities for interference. Finally, membrane proteins which support RAS microdomain association and/or nanoclustering have not only been identified as putative oncogenic drivers, but have been shown to be viable targets for anti-cancer therapies. Together, it is now firmly established that RAS maturation and subcellular targeting provide multiple vulnerabilities to exploit for precision medicine-based anti-RAS targeting in cancer treatment.
Acknowledgments
We thank Jeremy G.T. Wurtzel for technical assistance. This work was supported by NIH grant R01HL137207 to L.E.G.
Abbreviations used
- APT1
Acyl Protein Thioesterase 1
- CaaL motif
Cysteine-Aliphatic-Aliphatic-Leucine
- CaaX motif
Cysteine-Aliphatic-Aliphatic-X: X is any amino acid
- DHHC9
Palmitoyltransferase ZDHHC9
- EGFR
Epidermal Growth Factor Receptor
- ERK
Extracellular Signal Regulated Kinase
- GAL1
Galectin-1
- GAL3
Galectin-3
- GTP
Guanosine triphosphate
- HVR
Hypervariable Region
- MAPK
Mitogen Activated Protein Kinase
- mTOR
Mammalian Target of Rapamycin
- PIP2
Phosphatidylinositol 4,5-Bisphosphate
- PI3K
Phosphoinositide 3-Kinase
Footnotes
6. Conflict of Interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 2.Johnson L, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandez-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011;2:344–358. doi: 10.1177/1947601911411084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adjei A. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–1074. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- 5.Fernández-Medarde A, Santos E. Ras in Cancer and Developmental Diseases. Genes and Cancer. 2011;2:344–359. doi: 10.1177/1947601911411084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng CM, et al. Compartmentalized Ras proteins transform NIH 3T3 cells with different efficiencies. Mol Cell Biol. 2010;31:983–997. doi: 10.1128/MCB.00137-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox AD, Der CJ, Philips MR. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin Cancer Res. 2015;21:1819–1827. doi: 10.1158/1078-0432.CCR-14-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan J, Yeung SC. Recent advances in understanding the antineoplastic mechanisms of farnesyltransferase inhibitors. Cancer Res. 2005;65:9109–9112. doi: 10.1158/0008-5472.CAN-05-2635. [DOI] [PubMed] [Google Scholar]
- 10.Solit DB, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham SM, et al. Aberrant function of the Ras-related protein TC21/R-Ras2 triggers malignant transformation. Mol Cell Biol. 1994;14:4108–4115. doi: 10.1128/mcb.14.6.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graham SM, et al. TC21 causes transformation by Raf-independent signaling pathways. Mol Cell Biol. 1996;16:6132–6140. doi: 10.1128/mcb.16.11.6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graham SM, et al. TC21 and Ras share indistinguishable transforming and differentiating activities. Oncogene. 1999;18:2107–2116. doi: 10.1038/sj.onc.1202517. [DOI] [PubMed] [Google Scholar]
- 14.Sharma R, Sud N, Chattopadhyay TK, Ralhan R. TC21/R-Ras2 upregulation in esophageal tumorigenesis: potential diagnostic implications. Oncology. 2005;69:10–18. doi: 10.1159/000087283. [DOI] [PubMed] [Google Scholar]
- 15.Erdogan M, Pozzi A, Bhowmick N, Moses HL, Zent R. Signaling pathways regulating TC21-induced tumorigenesis. J Biol Chem. 2007;282:27713–27720. doi: 10.1074/jbc.M703037200. [DOI] [PubMed] [Google Scholar]
- 16.Macha MA, et al. Clinical significance of TC21 overexpression in oral cancer. J Oral Pathol Med. 2010;39:477–485. doi: 10.1111/j.1600-0714.2009.00854.x. [DOI] [PubMed] [Google Scholar]
- 17.Lee JH, et al. Greater expression of TC21/R-ras2 in highly aggressive malignant skin cancer. Int J Dermatol. 2011;50:956–960. doi: 10.1111/j.1365-4632.2010.04846.x. [DOI] [PubMed] [Google Scholar]
- 18.Liu WN, Yan M, Chan AM. A thirty-year quest for a role of R-Ras in cancer: from an oncogene to a multitasking GTPase. Cancer Lett. 2017;403:59–65. doi: 10.1016/j.canlet.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Zhou Y, Hancock JF. Ras nanoclusters: Versatile lipid-based signaling platforms. Biochim Biophys Acta. 2015;1853:841–849. doi: 10.1016/j.bbamcr.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 20.Henis YI, Hancock JF, Prior IA. Ras acylation, compartmentalization and signaling nanoclusters (Review) Mol Membr Biol. 2009;26:80–92. doi: 10.1080/09687680802649582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reiss Y, Goldstein Joseph L, Seabra’ Miguel C, Casey Patrick J, Michael S. Brown Inhibition of Purified p2lras Farnesyl: Protein Transferase by Cys-AAX Tetrapeptides. Cell. 1990;62:81–88. doi: 10.1016/0092-8674(90)90242-7. [DOI] [PubMed] [Google Scholar]
- 22.Wright LPaMRP. CAAX modification and membrane targeting of Ras. Journal of Lipid Research. 2006;47:883–891. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Hartman HL, Hicks KA, Fierke CA. Peptide specificity of protein prenyltransferases is determined mainly by reactivity rather than binding affinity. Biochemistry. 2005;44:15314–15324. doi: 10.1021/bi0509503. [DOI] [PubMed] [Google Scholar]
- 24.Graham SM, Rogers-Graham K, Figueroa C, Der CJ, Vojtek AB. Analyses of TC21/R-Ras2 signaling and biological activity. Methods Enzymol. 2001;333:203–216. doi: 10.1016/s0076-6879(01)33057-4. [DOI] [PubMed] [Google Scholar]
- 25.Oertli B, et al. The effector loop and prenylation site of R-Ras are involved in the regulation of integrin function [In Process Citation] Oncogene. 2000;19:4961–4969. doi: 10.1038/sj.onc.1203876. [DOI] [PubMed] [Google Scholar]
- 26.Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167–1177. doi: 10.1016/0092-8674(89)90054-8. [DOI] [PubMed] [Google Scholar]
- 27.End D, Smets G, Todd AV, Applegate TL, Fuery CJ, Angibaud P, Venet M, Sanz G, Poignet H, Skrzat S, Devine A, Wouters W, Bowden C. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res. 2001 Jan 1;61(1):131–137. [PubMed] [Google Scholar]
- 28.Yonemoto M, Satoh T, Arakawa H, Suzuki-Takahashi I, Monden Y, Kodera T, Tanaka K, Aoyama T, Iwasawa Y, Kamei T, Nishimura S, Tomimoto K. J-104,871, a novel farnesyltransferase inhibitor, blocks Ras farnesylation in vivo in a farnesyl pyrophosphate-competitive manner. Mol Pharmacol. 1998;54:1–7. doi: 10.1124/mol.54.1.1. [DOI] [PubMed] [Google Scholar]
- 29.Basso A, Kirschmeier P, Bishop WR. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res. 2006;47:15–31. doi: 10.1194/jlr.R500012-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Liu M, Bryant MS, Chen J, Lee S, Yaremko B, Lipari P, Malkowski M, Ferrari E, Nielsen L, Prioli N, Dell J, Sinha D, Syed J, Korfmacher WA, Nomeir AA, Lin CC, Wang L, Taveras AG, Doll RJ, Njoroge FG, Mallams AK, Remiszewski S, Catino JJ, Girijavallabhan VM, Bishop WR, et al. Antitumor activity of SCH 66336, an orally bioavailable tricyclic inhibitor of farnesyl protein transferase, in human tumor xenograft models and wap-ras transgenic mice. Cancer Res. 1998;58:4947–4956. [PubMed] [Google Scholar]
- 31.Ashar H, James L, Gray K, Carr D, McGuirk M, Maxwell E, Black S, Armstrong L, Doll RJ, Taveras AG, Bishop WR, Kirschmeier P. The farnesyl transferase inhibitor SCH 66336 induces a G(2) --> M or G(1) pause in sensitive human tumor cell lines. Exp Cell Res. 2001;262:17–27. doi: 10.1006/excr.2000.5076. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Makarewicz JM1, Knauf JA2, Johnson LK3, Fagin JA4. Transformation by Hras(G12V) is consistently associated with mutant allele copy gains and is reversed by farnesyl transferase inhibition. Oncogene. 2014;33:5442–5449. doi: 10.1038/onc.2013.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winquist E, Moore MJ, Chi KN, Ernst DS, Hirte H, North S, Powers J, Walsh W, Boucher T, Patton R, Seymour L. A multinomial Phase II study of lonafarnib (SCH 66336) in patients with refractory urothelial cancer. Urol Oncol. 2005;23:143–149. doi: 10.1016/j.urolonc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 34.Nakajima A, Tauchi T, Sumi M, Bishop WR, Ohyashiki K. Efficacy of SCH66336, a farnesyl transferase inhibitor, in conjunction with imatinib against BCR-ABL-positive cells. Mol Cancer Ther. 2003;2:219–224. doi: 10.4161/cbt.2.3.390. [DOI] [PubMed] [Google Scholar]
- 35.Whyte D, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, Bishop WR, Pai JK. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 36.Fiordalisi J, Johnson RL, 2nd, Weinbaum CA, Sakabe K, Chen Z, Casey PJ, Cox AD. High affinity for farnesyltransferase and alternative prenylation contribute individually to K-Ras4B resistance to farnesyltransferase inhibitors. J Biol Chem. 2003;278:41718–41727. doi: 10.1074/jbc.M305733200. [DOI] [PubMed] [Google Scholar]
- 37.Lobell R, Omer CA, Abrams MT, Bhimnathwala HG, Brucker MJ, Buser CA, Davide JP, deSolms SJ, Dinsmore CJ, Ellis-Hutchings MS, Kral AM, Liu D, Lumma WC, Machotka SV, Rands E, Williams TM, Graham SL, Hartman GD, Oliff AI, Heimbrook DC, Kohl NE. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 2001;61:8758–8768. [PubMed] [Google Scholar]
- 38.Lobell R, Liu D, Buser CA, Davide JP, DePuy E, Hamilton K, Koblan KS, Lee Y, Mosser S, Motzel SL, Abbruzzese JL, Fuchs CS, Rowinsky EK, Rubin EH, Sharma S, Deutsch PJ, Mazina KE, Morrison BW, Wildonger L, Yao SL, Kohl NE. Preclinical and clinical pharmacodynamic assessment of L-778,123, a dual inhibitor of farnesyl:protein transferase and geranylgeranyl:protein transferase type-I. Mol Cancer Ther. 2002;1:747–758. [PubMed] [Google Scholar]
- 39.Boyartchuk VL, Matthew N. Ashby, Jasper Rine Modulation of Ras and a-Factor Function by Carboxyl-Terminal Proteolysis. Science. 1997;275:1796–1800. doi: 10.1126/science.275.5307.1796. [DOI] [PubMed] [Google Scholar]
- 40.Manandhar SP, Hildebrandt ER, Jacobsen WH, Santangelo GM, Schmidt WK. Chemical inhibition of CaaX protease activity disrupts yeast Ras localization. Yeast. 2010;27:327–343. doi: 10.1002/yea.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manandhar SP, Hildebrandt ER, Schmidt WK. Small-molecule inhibitors of the Rce1p CaaX protease. J Biomol Screen. 2007;12:983–993. doi: 10.1177/1087057107307226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porter SB, et al. Inhibition of the CaaX proteases Rce1p and Ste24p by peptidyl (acyloxy)methyl ketones. Biochim Biophys Acta. 2007;1773:853–862. doi: 10.1016/j.bbamcr.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams DE, et al. Scalarane-based sesterterpenoid RCE-protease inhibitors isolated from the Indonesian marine sponge Carteriospongia foliascens. J Nat Prod. 2009;72:1106–1109. doi: 10.1021/np900042r. [DOI] [PubMed] [Google Scholar]
- 44.Mohammed I, et al. 8-Hydroxyquinoline-based inhibitors of the Rce1 protease disrupt Ras membrane localization in human cells. Bioorg Med Chem. 2016;24:160–178. doi: 10.1016/j.bmc.2015.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hrycyna CA, Sapperstein SK, Clarke S, Michaelis S. The Saccharomyces cerevisiae STE14 gene encodes a methyltransferase that mediates C-terminal methylation of a-factor and RAS proteins. EMBO J. 1991;10:1699–1709. doi: 10.1002/j.1460-2075.1991.tb07694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Judd WR, et al. Discovery and SAR of methylated tetrahydropyranyl derivatives as inhibitors of isoprenylcysteine carboxyl methyltransferase (ICMT) J Med Chem. 2011;54:5031–5047. doi: 10.1021/jm200249a. [DOI] [PubMed] [Google Scholar]
- 47.Wang M, et al. A small molecule inhibitor of isoprenylcysteine carboxymethyltransferase induces autophagic cell death in PC3 prostate cancer cells. J Biol Chem. 2008;283:18678–18684. doi: 10.1074/jbc.M801855200. [DOI] [PubMed] [Google Scholar]
- 48.Lau HY, et al. An improved isoprenylcysteine carboxylmethyltransferase inhibitor induces cancer cell death and attenuates tumor growth in vivo. Cancer Biol Ther. 2014;15:1280–1291. doi: 10.4161/cbt.29692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buss J, Sefton BM. Direct identification of palmitic acid as the lipid attached to p21ras. Mol Cell Biol. 1986;6:116–122. doi: 10.1128/mcb.6.1.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol. 2000;20:2475–2487. doi: 10.1128/mcb.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choy E, et al. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 1999;98:69–80. doi: 10.1016/S0092-8674(00)80607-8. [DOI] [PubMed] [Google Scholar]
- 52.Laude A, Prior IA. Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J Cell Sci. 2008;121:421–427. doi: 10.1242/jcs.020107. [DOI] [PubMed] [Google Scholar]
- 53.Swarthout JT, et al. DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J Biol Chem. 2005;280:31141–31148. doi: 10.1074/jbc.M504113200. [DOI] [PubMed] [Google Scholar]
- 54.Ohta E, Misumi Y, Sohda M, Fujiwara T, Yano A, Ikehara Y. Identification and characterization of GCP16, a novel acylated Golgi protein that interacts with GCP170. J Biol Chem. 2003;278:51957–51967. doi: 10.1074/jbc.M310014200. [DOI] [PubMed] [Google Scholar]
- 55.Baumgart F, Corral-Escariz M, Perez-Gil J, Rodriguez-Crespo I. Palmitoylation of R-Ras by human DHHC19, a palmitoyl transferase with a CaaX box. Biochim Biophys Acta. 2010;1798:592–604. doi: 10.1016/j.bbamem.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 56.Draper JM, Smith CD. Palmitoyl acyltransferase assays and inhibitors (Review) Mol Membr Biol. 2009;26:5–13. doi: 10.1080/09687680802683839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garant KA, et al. Oncolytic reovirus induces intracellular redistribution of Ras to promote apoptosis and progeny virus release. Oncogene. 2016;35:771–782. doi: 10.1038/onc.2015.136. [DOI] [PubMed] [Google Scholar]
- 58.Dekker FJ, et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat Chem Biol. 2010;6:449–456. doi: 10.1038/nchembio.362. [DOI] [PubMed] [Google Scholar]
- 59.James G, Olson EN. Identification of a novel fatty acylated protein that partitions between the plasma membrane and cytosol and is deacylated in response to serum and growth factor stimulation. J Biol Chem. 1989;264:20998–21006. [PubMed] [Google Scholar]
- 60.Ahearn IM, et al. FKBP12 binds to acylated H-ras and promotes depalmitoylation. Mol Cell. 2011;41:173–185. doi: 10.1016/j.molcel.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zimmermann TJ, et al. Boron-based inhibitors of acyl protein thioesterases 1 and 2. Chembiochem. 2013;14:115–122. doi: 10.1002/cbic.201200571. [DOI] [PubMed] [Google Scholar]
- 62.Misaki R, et al. Palmitoylated Ras proteins traffic through recycling endosomes to the plasma membrane during exocytosis. J Cell Biol. 2010;191:23–29. doi: 10.1083/jcb.200911143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wurtzel JG, Kumar P, Goldfinger LE. Palmitoylation regulates vesicular trafficking of R-Ras to membrane ruffles and effects on ruffling and cell spreading. Small GTPases. 2012;3:139–153. doi: 10.4161/sgtp.21084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rocks O, et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- 65.Lynch SJ, et al. The differential palmitoylation states of N-Ras and H-Ras determine their distinct Golgi subcompartment localizations. J Cell Physiol. 2015;230:610–619. doi: 10.1002/jcp.24779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chandra A, et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol. 2011;14:148–158. doi: 10.1038/ncb2394. [DOI] [PubMed] [Google Scholar]
- 67.Philips MR. Ras hitchhikes on PDE6delta. Nat Cell Biol. 2012;14:128–129. doi: 10.1038/ncb2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang HT, et al. Anxiogenic-like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B) Neuropsychopharmacology. 2008;33:1611–1623. doi: 10.1038/sj.npp.1301537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zimmermann G, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 70.Papke B, et al. Identification of pyrazolopyridazinones as PDEdelta inhibitors. Nature communications. 2016;7:11360. doi: 10.1038/ncomms11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fehrenbacher N, et al. The G protein-coupled receptor GPR31 promotes membrane association of KRAS. J Cell Biol. 2017;216:2329–2338. doi: 10.1083/jcb.201609096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou M, et al. VPS35 binds farnesylated N-Ras in the cytosol to regulate N-Ras trafficking. J Cell Biol. 2016;214:445–458. doi: 10.1083/jcb.201604061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pike L. Lipid rafts: bringing order to chaos. Lipid Res. 2003;44:655–657. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 74.Niv H, Gutman O, Kloog Y, Henis YI. Activated K-Ras and H-Ras display different interactions with saturable nonraft sites at the surface of live cells. J Cell Biol. 2002;157:865–872. doi: 10.1083/jcb.200202009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roy S, Plowman S, Rotblat B, Prior IA, Muncke C, Grainger S, Parton RG, Henis YI, Kloog Y, Hancock JF. Individual palmitoyl residues serve distinct roles in H-ras trafficking, microlocalization, and signaling. Mol Cell Biol. 2005;25:6722–6733. doi: 10.1128/MCB.25.15.6722-6733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hancock J, Parton RG. Ras plasma membrane signalling platforms. Biochem J. 2005;389:1–11. doi: 10.1042/BJ20050231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Prior IA, et al. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat Cell Biol. 2001;3:368–375. doi: 10.1038/35070050. [DOI] [PubMed] [Google Scholar]
- 78.Janosi L, Li Z, Hancock JF, Gorfe AA. Organization, dynamics, and segregation of Ras nanoclusters in membrane domains. Proc Natl Acad Sci U S A. 2012;109:8097–8102. doi: 10.1073/pnas.1200773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nicolini C, Baranski J, Schlummer S, Palomo J, Lumbierres-Burgues M, Kahms M, Kuhlmann J, Sanchez S, Gratton E, Waldmann H, Winter R. Visualizing association of N-ras in lipid microdomains: influence of domain structure and interfacial adsorption. J Am Chem Soc. 2006;128:192–201. doi: 10.1021/ja055779x. [DOI] [PubMed] [Google Scholar]
- 80.Matallanas D, Arozarena Imanol, Berciano María T, Aaronson David S, Pellicer Angel, Lafarga Miguel, Crespo Piero. Differences on the Inhibitory Specificities of H-Ras, K-Ras, and N-Ras (N17) Dominant Negative Mutants Are Related to Their Membrane Microlocalization. The Journal of Biological Chemistry. 2002;278:4572–4581. doi: 10.1074/jbc.M209807200. [DOI] [PubMed] [Google Scholar]
- 81.Eisenberg S, Beckett AJ, Prior IA, Dekker FJ, Hedberg C, Waldmann H, Ehrlich M, Henis YI. Raft protein clustering alters N-Ras membrane interactions and activation pattern. Mol Cell Biol. 2011;31:3938–3952. doi: 10.1128/MCB.05570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4:373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 83.Zhou Y, et al. Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell. 2017;168:239–251 e216. doi: 10.1016/j.cell.2016.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hansen M, et al. C-terminal sequences in R-Ras are involved in integrin regulation and in plasma membrane microdomain distribution. Biochem Biophys Res Commun. 2003;311:829–838. doi: 10.1016/j.bbrc.2003.10.074. [DOI] [PubMed] [Google Scholar]
- 85.Michael JV, Wurtzel JGT, Goldfinger LE. Regulation of H-Ras-driven MAPK signaling, transformation and tumorigenesis, but not PI3K signaling and tumor progression, by plasma membrane microdomains. Oncogenesis. 2016;5:e228. doi: 10.1038/oncsis.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsai FD, et al. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A. 2015;112:779–784. doi: 10.1073/pnas.1412811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Astorgues-Xerri L, Riveiro ME, Tijeras-Raballand A, Serova M, Neuzillet C, Albert S, Raymond E, Faivre S. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat Rev. 2014;40:307–319. doi: 10.1016/j.ctrv.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 88.Henderson N, Sethi T. The regulation of inflammation by galectin-3. Immunol Rev. 2009;203:160–171. doi: 10.1111/j.1600-065X.2009.00794.x. [DOI] [PubMed] [Google Scholar]
- 89.Elad-Sfadia G, Haklai R, Balan E, Kloog Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J Biol Chem. 2004;279:34922–34930. doi: 10.1074/jbc.M312697200. [DOI] [PubMed] [Google Scholar]
- 90.Paz A, Haklai R, Elad-Sfadia G, Ballan E, Kloog Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene. 2001;20:7486–7493. doi: 10.1038/sj.onc.1204950. [DOI] [PubMed] [Google Scholar]
- 91.Shalom-Feuerstein R, Cooks T, Raz A, Kloog Y. Galectin-3 regulates a molecular switch from N-Ras to K-Ras usage in human breast carcinoma cells. Cancer Res. 2005;65:7292–7300. doi: 10.1158/0008-5472.CAN-05-0775. [DOI] [PubMed] [Google Scholar]
- 92.Shalom-Feuerstein R, Levy R, Makovski V, Raz A, Kloog Y. Galectin-3 regulates RasGRP4-mediated activation of N-Ras and H-Ras. Biochim Biophys Acta. 2008;1783:985–993. doi: 10.1016/j.bbamcr.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Belanis L, Plowman SJ, Rotblat B, Hancock JF, Kloog Y. Galectin-1 is a novel structural component and a major regulator of h-ras nanoclusters. Mol Biol Cell. 2008;19:1404–1414. doi: 10.1091/mbc.E07-10-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Prior I, Muncke C, Parton RG, Hancock JF. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol. 2003;160:165–170. doi: 10.1083/jcb.200209091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Elad-Sfadia G, Haklai R, Ballan E, Gabius HJ, Kloog Y. Galectin-1 augments Ras activation and diverts Ras signals to Raf-1 at the expense of phosphoinositide 3-kinase. J Biol Chem. 2002;277:37169–37175. doi: 10.1074/jbc.M205698200. [DOI] [PubMed] [Google Scholar]
- 96.Abankwa D, Gorfe AA, Inder K, Hancock JF. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc Natl Acad Sci U S A. 2010;107:1130–1135. doi: 10.1073/pnas.0903907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Michael JV, Wurtzel JG, Goldfinger LE. Inhibition of Galectin-1 Sensitizes HRAS-driven Tumor Growth to Rapamycin Treatment. Anticancer Res. 2016;36:5053–5061. doi: 10.21873/anticanres.11074. [DOI] [PubMed] [Google Scholar]
- 98.Posada IMD, et al. Rapalogs can promote cancer cell stemness in vitro in a Galectin-1 and H-ras-dependent manner. Oncotarget. 2017;8:44550–44566. doi: 10.18632/oncotarget.17819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Elad-Sfadia G, Haklai R, Balan E, Kloog Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J Biol Chem. 2004;279:34922–34930. doi: 10.1074/jbc.M312697200. [DOI] [PubMed] [Google Scholar]
- 100.Levy R, Grafi-Cohen M, Kraiem Z, Kloog Y. Galectin-3 promotes chronic activation of K-Ras and differentiation block in malignant thyroid carcinomas. Mol Cancer Ther. 2010;9:2208–2219. doi: 10.1158/1535-7163.MCT-10-0262. [DOI] [PubMed] [Google Scholar]
- 101.Plowman SJ, Ariotti N, Goodall A, Parton RG, Hancock JF. Electrostatic interactions positively regulate K-Ras nanocluster formation and function. Mol Cell Biol. 2008;28:4377–4385. doi: 10.1128/MCB.00050-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shalom-Feuerstein R, et al. K-ras nanoclustering is subverted by overexpression of the scaffold protein galectin-3. Cancer Res. 2008;68:6608–6616. doi: 10.1158/0008-5472.CAN-08-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bhagatji P, Leventis R, Rich R, Lin CJ, Silvius JR. Multiple cellular proteins modulate the dynamics of K-ras association with the plasma membrane. Biophysical journal. 2010;99:3327–3335. doi: 10.1016/j.bpj.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tian T, Plowman SJ, Parton RG, Kloog Y, Hancock JF. Mathematical modeling of K-Ras nanocluster formation on the plasma membrane. Biophysical journal. 2010;99:534–543. doi: 10.1016/j.bpj.2010.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu KL, et al. Overexpression of galectin-3 enhances migration of colon cancer cells related to activation of the K-Ras-Raf-Erk1/2 pathway. J Gastroenterol. 2013;48:350–359. doi: 10.1007/s00535-012-0663-3. [DOI] [PubMed] [Google Scholar]
- 106.Song S, et al. Overexpressed galectin-3 in pancreatic cancer induces cell proliferation and invasion by binding Ras and activating Ras signaling. PLoS One. 2012;7:e42699. doi: 10.1371/journal.pone.0042699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cho KJ, et al. Raf inhibitors target ras spatiotemporal dynamics. Curr Biol. 2012;22:945–955. doi: 10.1016/j.cub.2012.03.067. [DOI] [PubMed] [Google Scholar]
- 108.Ashery U, et al. Spatiotemporal organization of Ras signaling: rasosomes and the galectin switch. Cellular and molecular neurobiology. 2006;26:471–495. doi: 10.1007/s10571-006-9059-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ruvolo PP, et al. Combination of galectin inhibitor GCS-100 and BH3 mimetics eliminates both p53 wild type and p53 null AML cells. Biochim Biophys Acta. 2016;1863:562–571. doi: 10.1016/j.bbamcr.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hossein G, Keshavarz M, Ahmadi S, Naderi N. Synergistic effects of PectaSol-C modified citrus pectin an inhibitor of Galectin-3 and paclitaxel on apoptosis of human SKOV-3 ovarian cancer cells. Asian Pac J Cancer Prev. 2013;14:7561–7568. doi: 10.7314/apjcp.2013.14.12.7561. [DOI] [PubMed] [Google Scholar]
- 111.Inohara H, Raz A. Effects of natural complex carbohydrate (citrus pectin) on murine melanoma cell properties related to galectin-3 functions. Glycoconj J. 1994;11:527–532. doi: 10.1007/BF00731303. [DOI] [PubMed] [Google Scholar]
- 112.Jiang J, Eliaz I, Sliva D. Synergistic and additive effects of modified citrus pectin with two polybotanical compounds, in the suppression of invasive behavior of human breast and prostate cancer cells. Integr Cancer Ther. 2013;12:145–152. doi: 10.1177/1534735412442369. [DOI] [PubMed] [Google Scholar]
- 113.Liu FT, et al. Modulation of functional properties of galectin-3 by monoclonal antibodies binding to the non-lectin domains. Biochemistry. 1996;35:6073–6079. doi: 10.1021/bi952716q. [DOI] [PubMed] [Google Scholar]
- 114.Liu HY, Huang ZL, Yang GH, Lu WQ, Yu NR. Inhibitory effect of modified citrus pectin on liver metastases in a mouse colon cancer model. World J Gastroenterol. 2008;14:7386–7391. doi: 10.3748/wjg.14.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nangia-Makker P, et al. Inhibition of human cancer cell growth and metastasis in nude mice by oral intake of modified citrus pectin. J Natl Cancer Inst. 2002;94:1854–1862. doi: 10.1093/jnci/94.24.1854. [DOI] [PubMed] [Google Scholar]
- 116.Pienta KJ, et al. Inhibition of spontaneous metastasis in a rat prostate cancer model by oral administration of modified citrus pectin. J Natl Cancer Inst. 1995;87:348–353. doi: 10.1093/jnci/87.5.348. [DOI] [PubMed] [Google Scholar]
- 117.Kolatsi-Joannou M, Price KL, Winyard PJ, Long DA. Modified citrus pectin reduces galectin-3 expression and disease severity in experimental acute kidney injury. PLoS One. 2011;6:e18683. doi: 10.1371/journal.pone.0018683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Blanchard H, Yu X, Collins PM, Bum-Erdene K. Galectin-3 inhibitors: a patent review (2008-present) Expert Opin Ther Pat. 2014;24:1053–1065. doi: 10.1517/13543776.2014.947961. [DOI] [PubMed] [Google Scholar]
- 119.Glinsky VV, Raz A. Modified citrus pectin anti-metastatic properties: one bullet, multiple targets. Carbohydr Res. 2009;344:1788–1791. doi: 10.1016/j.carres.2008.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hayashi A, Gillen AC, Lott JR. Effects of daily oral administration of quercetin chalcone and modified citrus pectin on implanted colon-25 tumor growth in Balb-c mice. Altern Med Rev. 2000;5:546–552. [PubMed] [Google Scholar]
- 121.Leclere L, et al. Heat-modified citrus pectin induces apoptosis-like cell death and autophagy in HepG2 and A549 cancer cells. PLoS One. 2015;10:e0115831. doi: 10.1371/journal.pone.0115831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yan J, Katz A. PectaSol-C modified citrus pectin induces apoptosis and inhibition of proliferation in human and mouse androgen-dependent and- independent prostate cancer cells. Integr Cancer Ther. 2010;9:197–203. doi: 10.1177/1534735410369672. [DOI] [PubMed] [Google Scholar]
- 123.Abu-Elsaad NM, Elkashef WF. Modified citrus pectin stops progression of liver fibrosis by inhibiting galectin-3 and inducing apoptosis of stellate cells. Can J Physiol Pharmacol. 2016;94:554–562. doi: 10.1139/cjpp-2015-0284. [DOI] [PubMed] [Google Scholar]
- 124.Yoon YK, et al. Combination of EGFR and MEK1/2 inhibitor shows synergistic effects by suppressing EGFR/HER3-dependent AKT activation in human gastric cancer cells. Mol Cancer Ther. 2009;8:2526–2536. doi: 10.1158/1535-7163.MCT-09-0300. [DOI] [PubMed] [Google Scholar]
- 125.Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013;3:112–123. doi: 10.1158/2159-8290.CD-12-0231. [DOI] [PubMed] [Google Scholar]
- 126.Seguin L, et al. An integrin beta(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat Cell Biol. 2014;16:457–468. doi: 10.1038/ncb2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Seguin L, et al. Galectin-3, a druggable vulnerability for KRAS-addicted cancers. Cancer Discov. 2017 doi: 10.1158/2159-8290.CD-17-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shalom-Feuerstein R, Cooks T, Raz A, Kloog Y. Galectin-3 regulates a molecular switch from N-Ras to K-Ras usage in human breast carcinoma cells. Cancer Res. 2005;65:7292–7300. doi: 10.1158/0008-5472.CAN-05-0775. [DOI] [PubMed] [Google Scholar]
- 129.Shalom-Feuerstein R, Levy R, Makovski V, Raz A, Kloog Y. Galectin-3 regulates RasGRP4-mediated activation of N-Ras and H-Ras. Biochim Biophys Acta. 2008;1783:985–993. doi: 10.1016/j.bbamcr.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]