

Abstract
Methylmercury, a potent neurotoxin, is able to pass through the placenta, but its effects on the placenta itself have not been elucidated. Using an immortalized human trophoblast cell line, HTR8/SV-neo, we assessed the in vitro toxicity of methylmercury. We found that 1 μg/mL methylmercury decreased viability, proliferation, and migration; and it had effects on antioxidant genes similar to those seen in neural cells. However, methylmercury led to decreased expression of superoxide dismutase 1 and increased expression of surfactant protein D. HTR cells treated 0.01 or 0.1 μg/mL methylmercury had increased migration rates along with decreased expression of an adhesion gene, cadherin 3, suggesting that low doses of methylmercury promote migration in HTR cells. Our results indicate that trophoblast cells react differently to methylmercury relative to neural cell lines, and thus investigation of methylmercury toxicity in placental cells is needed to understand the effects of this heavy metal on the placenta.
Keywords: Trophoblast, Placenta, Toxicology, Methylmercury
1. Introduction
Methylmercury (MeHg) is an organometallic compound that enters the environment through a number of sources, both natural (i.e. volcanic eruption) and anthropogenic (i.e. combustion of fossil fuels). Human exposure generally comes from consumption of high trophic level seafood that has accumulated high levels of MeHg [1]. In the gastrointestinal tract, 95% of MeHg is absorbed and distributed throughout the body by the blood [2]. The brain and the kidney are the primary target organs of MeHg [3], and much of the published literature is devoted to the effects of MeHg on the central nervous system (CNS). The ability of MeHg to form a complex with L-cysteine allows it to utilize neutral amino acid carriers to cross the blood-brain barrier as well as the placenta [4–8]. It has been well established that MeHg is especially toxic to the developing fetus, resulting in complications such as spontaneous abortions, stillbirths, physical malformations, motor impairment, cognitive delay, and behavioral abnormalities [9–14]. Thus, women that consume higher levels of MeHg may be at risk for pregnancy complications due to fetal toxicity. A study using data from the National Health and Nutritional Examination Survey (NHANES) from 2011–2012 showed that Asian women of reproductive age and women that consume 2 or more fish meals per week tend to have higher blood concentrations of MeHg than other subpopulations [15]. 3.4 μg/L is presumed to be the lower limit of maternal blood MeHg that is associated with accumulation in the fetus [16], and 23% of Asian-American women of reproductive age had MeHg levels higher than 3.5 μg/L according to the NHANES study [15]. Reference doses (RfD) using developmental neurotoxicity as an endpoint for MeHg exposure vary from 0.1 μg/kg/d to 0.2 μg/kg/d, while RfD based on adult exposures are around 0.5 μg/kg/d [17]. These RfD were determined using epidemiological studies from widespread poisoning events in Japan [18] and Iraq [19], as well as long-term studies in Pacific Island populations (Seychelles: [20], Faroe Islands: [21], New Zealand: [22,23]). While much work has been done to examine the effects of MeHg on the fetus and on the CNS, very little work has examined the direct action of MeHg on the placenta. MeHg has been directly measured in human placental tissue [24–26], and in vitro studies have shown that trophoblast cells take up MeHg through neutral amino acid carriers [5,7,8]. MeHg is a potent cytotoxin that could potentially interfere with placental integrity and function in addition to causing damage to the developing fetus. Therefore, understanding how MeHg affects the placenta itself is an important factor in predicting how MeHg exposure may affect pregnancy. Using an in vitro model, we assessed how MeHg affects an immortalized first trimester human trophoblast cell line, the HTR8/SV-neo cells. This cell line was chosen because it contains normal trophoblast cells rather than placental choriocarcinoma cells. We predicted this cell population would show significant sensitivity to MeHg, and we measured this sensitivity using assays for viability, proliferation, migration, and mRNA and protein expression.
2. Materials and Methods
2.1 Cell culture and MeHg treatment
HTR8/SV-neo cells were obtained from Dr. Richard Leach [27] and underwent short tandem repeat (STR) profiling at ATCC to authenticate their identity as trophoblast cells. While a recent publication [28] reported that HTR8/SV-neo cells are a mixed population of cells consisting of primarily stromal cells and some trophoblast cells, this group did not perform any type of DNA fingerprinting to verify the identity of their line. The HTR cells were cultured in RPMI 1640 (University of Illinois Cell Media Facility) containing 5% Fetal Bovine Serum (S11150, Atlanta Biologicals, Atlanta, GA), 1% Penicillin/Streptomycin (30-002-CL, Corning Inc., Corning, NY) at 37°C, 5% CO2. Cells were removed from culture dishes when necessary by incubation with 0.05% Trypsin (25-025-CL, Corning Inc., Corning, NY) at 37°C for 7 minutes, neutralization with RPMI 1640, and centrifugation at 63 x g for 5 minutes. Methylmercury chloride in water (33553, Alfa Aesar, Haverhill, MA) was diluted from a stock concentration of 1000 μg/mL to desired concentrations in RPMI 1640 using serial dilutions of 1:10. The final experimental concentrations were 0.01, 0.1, and 1 μg/mL MeHg. MeHg-treated medium was added directly to the HTR cells, and cells receiving 0 μg/mL MeHg (i.e., only culture medium) were used as the control treatment for all experiments. All assays were repeated in three biological replicates (i.e., independently thawed cell populations assayed on separate days, hereafter referred to as ‘replicates’) with two technical replicates (i.e., two wells per treatment, hereafter referred to as ‘duplicates’) each unless otherwise specified [29].
2.2 Median lethal dose (LC50)
HTR cells were treated in 12-well culture plates (TP92012, MidSci, St. Louis, MO) with serial dilutions of MeHg between 0 and 5 μg/mL for 24 hours. Cells were stained with Trypan blue (0.4%, T8154-100ML, Sigma Aldrich, St. Louis, MO), which preferentially stains dead cells [30]. A hemocytometer was used to count the total number of cells as well as the number of dead cells.
2.3 Proliferation assay
HTR cells in 6-well culture plates (TP92006, MidSci, St. Louis, MO) were treated with 0, 0.01, 0.1 and 1 μg/mL MeHg. The cells were harvested by trypsinization and centrifugation as described above prior to counting with a hemocytometer at 0, 24, 48, and 72 hours.
2.4 Migration assays
Short-term migratory ability was assessed using a gap closure migration assay kit (CBA-125, Cell Biolabs, Inc., San Diego, CA). HTR cells were grown to confluency in a 6-well culture plate that had been pretreated with a hydrogel circle in the middle of each well. This hydrogel prevented cells from growing in the center of the wells. The hydrogel was then removed, allowing cells to migrate into the remaining open area of the circle. Cells were then immediately treated with 0, 0.01, 0.1, and 1 μg/mL MeHg, and images of the circle were taken at 0, 1, 3, 6, 9, and 12 hours. The area of the circle was measured using ImageJ (Version 1.5, National Institutes of Health, Bethesda, MD) [31].
Long-term migratory ability was assessed using the scratch migration assay. HTR cells in 6-well culture plates were grown to confluency and a pipette tip was used to make a scratch through the middle of the culture well. The scratched area was gently washed with RPMI 1640 before the cells were treated with 0, 0.01, 0.1, and 1 μg/mL MeHg. This experiment was repeated with a total of 4 replicates with 2 duplicates each. Images of the scratched area were taken every 12 hours for 48 hours and the area of the scratch was measured using ImageJ.
2.5 RNA isolation, cDNA synthesis, and RT-PCR
To determine oxidative stress-related genes that were most likely to be affected by MeHg, we assessed gene expression using the TaqMan Human Antioxidant Mechanisms Array Plate (4414119, ThermoFisher Scientific, Waltham, MA). HTR cells in 75 cm2 vented flasks (TP90075, MidSci, St. Louis, MO) were treated with either 0 or 1 μg/mL MeHg for 24 hours and RNA was isolated using TRIzol (15596062, Invitrogen, Carlsbad, CA) [32]. The resulting RNA was reverse-transcribed into cDNA using the Maxima First-Strand cDNA Synthesis Kit (K1671, ThermoFisher Scientific, Waltham, MA). TaqMan Universal Master Mix (4304437, ThermoFisher Scientific, Waltham, MA) and cDNA were added to the array plate, which contained the necessary primers and probes at the bottom of the wells. Standard conditions [33] were used for RT-PCR (Quant Studio 7 Flex Real-Time PCR System, Applied Biosystems, Foster City, CA). 12 housekeeping genes were included on the plate, and hmbs, pgk1, and ppia were chosen to calculate relative gene expression as described in Gong et al. [34]. Three replicates were completed without duplicates for this assay because the array plate was designed by the manufacturer to contain only one well/gene.
Further RT-PCR analysis was carried out to assess changes in two genes associated with cell migration and adhesion: catenin beta 1 (ctnnb1, primer/probe: Hs00355045_m1, ThermoFisher Scientific, Waltham, MA) and cadherin 3 (cdh3, primer/probe: Hs00999915_m1, ThermoFisher Scientific, Waltham, MA). Expression of superoxide dismutase 1 (sod1, primer/probe: Hs00533490_m1, ThermoFisher Scientific) and surfactant protein D (sftpd, primer/probe: Hs01108490_m1, ThermoFisher Scientific, Waltham, MA) at all three experimental doses was also assessed. 18s ribosomal RNA (18s, 4333760T, ThermoFisher Scientific, Waltham, MA) and glyceraldehyde 3-phosphatase dehydrogenase (gapdh, 4333764T, ThermoFisher Scientific, Waltham, MA) were used as housekeeping genes. For these assays, HTR cells in 25 cm2 vented flasks (TP90025, MidSci, St. Louis, MO) were treated with 0, 0.01, 0.1, or 1 μg/mL MeHg for 24 hours. RNA isolation, cDNA synthesis, and RT-PCR was carried out as described above.
2.6 Protein isolation and western blotting
Levels of superoxide dismutase 1 (SOD1) protein and surfactant protein D (SFTPD) in MeHg-treated cells were quantified through Western blot. HTR cells that had been treated with 0, 0.01, 0.1, and 1 μg/mL MeHg were washed with cold 1X PBS (21-040-CV, Mediatech Inc., Manassas, VA) and lysed with ice-cold NP-40 lysis buffer containing a protease inhibitor cocktail (04693159001, Roche, Basel, Switzerland) through repeated pipetting. The cell lysate was agitated at 4°C for 30 minutes and then centrifuged at 12,000 x g for 20 minutes. The protein concentration of the resulting supernatant was determined using a commercially available BCA assay kit (23225, ThermoFisher Scientific, Waltham, MA).
Proteins were resolved on a 10% polyacrylamide gel (4561036, BioRad, Hercules, CA) and transferred to a polyvinylidene difluoride (PVDF) membrane. Transfer was confirmed with Ponceau S (P7170-1L, Sigma-Aldrich, St. Louis, MO). Membranes were blocked in 5% bovine serum albumin (BSA) in PBS overnight at 4°C prior to incubation with primary antibody at a concentration of 1:1000 (SOD1: PA5-27240, ThermoFisher Scientific, Waltham, MA; SFTPD: ab97849, Abcam, Cambridge, UK; Beta-Actin [loading control]: A5060, Sigma-Aldrich, St. Louis, MO) overnight at 4°C. The membranes were washed with PBS before incubation with secondary antibody (Anti-rabbit IgG, HRP-linked, #7074, Cell Signaling Technology, Danvers, MA) at a concentration of 1:5000 for one hour at room temperature. Detection of protein bands was accomplished using the SuperSignal West Pico PLUS Chemiluminescent Substrate (34580, ThermoFisher Scientific, Waltham, MA) and membranes were digitized on an ImageQuant 4000 (GE Healthcare Life Sciences, Marlborough, MA) using the automatic exposure setting. The background for each lane was corrected individually using the baseline subtraction method [35], and the relative densities of loading controls and protein of interest (POI) bands were calculated using ImageJ. The relative densities of POI bands were adjusted by the relative density of the loading control band from the same lane.
2.7 Statistical analyses
Nested ANOVA was used to verify that there were no significant differences between biological replicates for each assay. To determine the LC50, the number of live cells was divided by the number of total cells to yield a percent viability at each dose. The median lethal dose was interpolated using a linear regression model. For both the proliferation assay (total cell number) and the migration assay (percent circle or scratch coverage), values from the MeHg groups were compared directly to the control group using ANOVA and Tukey-Kramer’s post-hoc test. RT-PCR results for both the oxidative stress gene array and the additional RT-PCR assays were analyzed using the ΔΔCt method [33], followed by one-way ANOVA and Tukey’s post-hoc test. Western blot results were analyzed by one-way ANOVA and Tukey’s post-hoc test. All results are presented as the mean ± the standard error of the mean (S.E.M.) unless otherwise specified.
3. Results
The median lethal dose at 24 hours was calculated to be 1.69 ± 0.12 μg/mL (Figure 1). Survival was not affected by doses of MeHg lower than 0.43 μg/mL at 24 hours. Doses higher than 3 μg/mL resulted in complete mortality.
Figure 1.
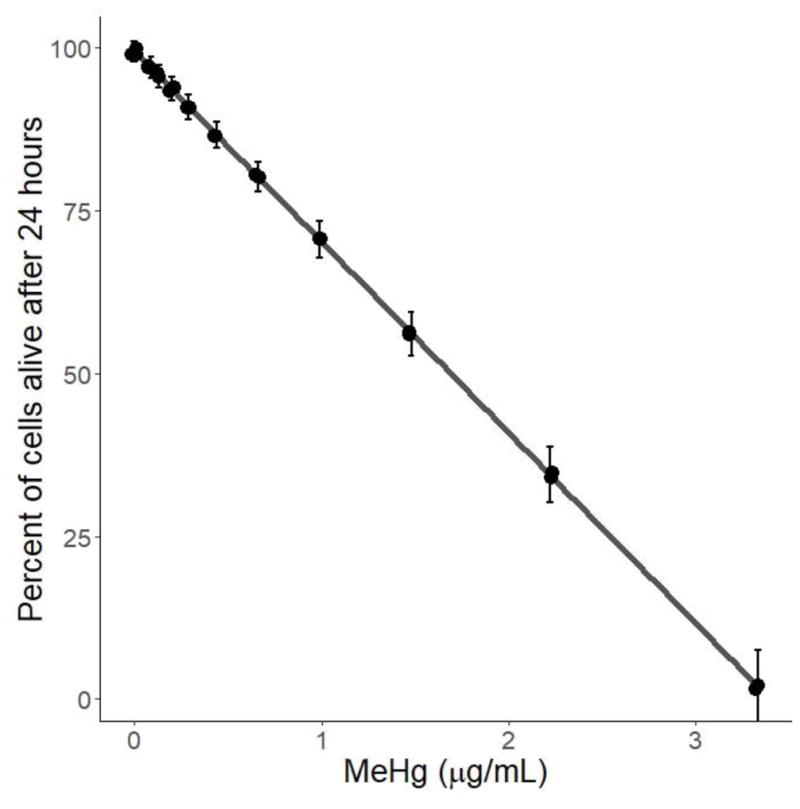
Results from LC50 assay. HTR8/SV-neo cells were treated with serial dilutions between 0 μg/mL and 5 μg/mL for 24 hours (N=3 replicates x 2 duplicates). The percent of live cells are displayed relative to the dose of MeHg. The median lethal dose (LC50) was calculated to be 1.65±0.12 μg/mL MeHg. Viability was not affected at doses below 0.43 μg/mL MeHg, and at concentrations above 3 μg/mL, viability was 0%.
For the proliferation assay, the number of cells was significantly decreased in the 1 μg/mL group when compared to the 0 μg/mL group at 24, 48, and 72 hours (p<0.001 at 24 and 48 hours, p<0.01 at 72 hours, Figure 2). Proliferation in the 0.01 and 0.1 μg/mL groups was not significantly affected.
Figure 2.

Effect of MeHg on proliferation. HTR8/SV-neo cells were treated with 0, 0.01, 0.1, and 1 μg/mL MeHg and the total number of cells was counted at 0, 24, 48, and 72 hours (N=3 replicates x 2 duplicates). At 24, 48, and 72 hours, the number of cells in the 1 μg/mL MeHg treatment group was significantly less than the number of cells in the 0 μg/mL group (*** denotes p<0.001). Cell number was not affected by 0.01 or 0.1 μg/mL MeHg at any time point.
For both the short-term and long-term migration assays, 0.01 and 0.1 μg/mL MeHg showed stimulatory effects on migration while the 1 μg/mL dose was inhibitory when compared to the control group (Figure 3). The 1 μg/mL dose had more prominent effects on migration over a longer period, as the scratch coverage at this dose differed significantly from all other doses (p<0.001, Figure 3B). On the shorter time scale, only 1 μg/mL and 0.01 μg/mL were significantly different from each other (Figure 3A). For both assays, the interaction effect between time and dose was not significant (p=0.36 and p=0.85 for the short- and long-term assays, respectively). Coverage of the circle or the scratch within a dose were significantly different between each time-point in the long-term assay (p<0.01 between 36 and 48 hours, p<0.0001 for all other time-points), and only between hours 1–3 and 6–9 for the short-term assay (p<0.05 for both time-points).
Figure 3.

Effect of MeHg on migration. For the short-term migration assay (A–B), the rate of circle closure after treatment with 0, 0.01, 0.1, or 1 μg/mL MeHg was measured at 0, 1, 3, 6, 9, and 12 hours (N=3 replicates x 2 duplicates). Data is presented as the percent of the circular area covered at each time point (A). Migration of cells treated with 0.01 μg/mL and 1 μg/mL were significantly different (** p<0.01). Representative images of each treatment group at 0 hours and 12 hours are provided in (B). For the long-term migration assay (C–D), the area of a scratch after treatment with 0, 0.01, 0.1, or 1 μg/mL MeHg was measured at 0, 12, 23, 36, and 48 hours (N=4 replicates x 2 duplicates). Scratch coverage over time (F) was significantly reduced for the 1 μg/mL MeHg group compared to all other treatments (*** denotes p<0.001), as shown in representative images of all treatment groups at 0 hours and 24 hours (D).
Out of the 85 genes tested in the antioxidant mechanisms array, 59 oxidative stress-related genes were consistently expressed in HTR cells (Table 1). 10 genes were significantly down-regulated (Superoxide dismutase 1 [sod1] and serine/threonine kinase 25 [stk25] p<0.0001; thioredoxin reductase 1 [txnrd1], superoxide dismutase 2 [sod2], thioredoxin reductase 2 [txnrd2], and sirtuin 2 [sirt2] p<0.001; selenoprotin P [sepp1], serine/threonine kinase 2 [sgk2], and sulfiredoxin 1 [srxn1] p<0.01; glutathione peroxidase 1 [gpx1] p<0.05, Figure 4). 9 genes were significantly up-regulated (peroxiredoxin 3 [prdx3], chemokine ligand 5 [ccl5], scavenger receptor class A 3 [scara3], alternative oxidase 1 [aox1], epoxide hydrolase 2 [ephx2], cytochrome b-245 alpha chain [cyba], selenoprotein S [sels], peroxiredoxin 4 [prdx4], and surfactant protein D [sftpd] p<0.05, Figure 4).
Table 1.
Oxidative stress gene array results. HTR8/SV-neo cells were treated with either 0 μg/mL or 1 μg/mL MeHg for 24 hours (N=3 replicates x 1 duplicate). Fold-change in expression for each gene was determined using the ΔΔCt method relative to the endogenous control genes (hmbs, pgk1, and ppia) and the control treatment of 0 μg/mL. The genes below are grouped by function, and significant values (p<0.05) are shown in bold.
| Gene | 2−ΔΔCt | SEM | t | p | Gene | 2−ΔΔCt | SEM | t | p | Gene | 2−ΔΔCt | SEM | t | p |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| REGULATORY | gsr | 1.116 | 0.134 | 0.864 | 0.479 | oxsr1 | 0.874 | 0.105 | −1.196 | 0.354 | ||||
| Protein Degradation | gpx7 | 0.810 | 0.097 | −1.957 | 0.190 | sgk2 | 0.335 | 0.040 | −16.522 | 0.004 | ||||
| sels | 2.518 | 0.302 | 5.023 | 0.037 | gpx1 | 0.463 | 0.056 | −9.679 | 0.011 | stk25 | 0.081 | 0.010 | −95.054 | 0.0001 |
| rnf7 | 0.989 | 0.119 | −0.094 | 0.934 | Peroxidases | Oxidases/Oxidase Activators | ||||||||
| Cell Cycle Regulation | prdx4 | 2.572 | 0.309 | 5.093 | 0.037 | cyba | 2.439 | 0.293 | 4.916 | 0.039 | ||||
| gft2i | 0.815 | 0.098 | −1.887 | 0.200 | prdx3 | 2.115 | 0.254 | 4.393 | 0.048 | aox1 | 2.230 | 0.268 | 4.595 | 0.044 |
| Transcription Regulation | prdx6 | 1.539 | 0.185 | 2.918 | 0.100 | ncf2 | 1.672 | 0.201 | 3.350 | 0.078 | ||||
| foxm1 | 1.220 | 0.146 | 1.500 | 0.2724 | prdx2 | 1.484 | 0.178 | 2.718 | 0.113 | nox5 | 1.320 | 0.158 | 2.018 | 0.181 |
| Translation Regulation | prdx1 | 1.432 | 0.172 | 2.515 | 0.128 | Hydrolases | ||||||||
| csde1 | 1.050 | 0.126 | 0.393 | 0.7323 | prdx5 | 1.134 | 0.136 | 0.982 | 0.430 | ephx2 | 2.297 | 0.276 | 4.704 | 0.042 |
| Pro-Apoptotic | pxdn | 0.940 | 0.113 | −0.534 | 0.688 | nudt1 | 1.386 | 0.166 | 2.320 | 0.146 | ||||
| bnip3 | 1.485 | 0.178 | 2.720 | 0.1128 | ptgs2 | 0.826 | 0.099 | −1.756 | 0.221 | Phosphatases | ||||
| CELL SIGNALING | ptgs1 | 0.684 | 0.082 | −3.849 | 0.061 | dusp1 | 1.124 | 0.135 | 0.918 | 0.456 | ||||
| Cell Adhesion | Reductases | Deacetylases | ||||||||||||
| pdlim2 | 1.434 | 0.172 | 2.522 | 0.1278 | msra | 1.211 | 0.145 | 1.453 | 0.283 | sirt2 | 0.211 | 0.025 | −31.074 | 0.001 |
| Cellular Signaling | oxr1 | 1.090 | 0.131 | 0.691 | 0.561 | OTHER | ||||||||
| ccl5 | 2.119 | 0.254 | 4.400 | 0.0480 | dhcr24 | 1.066 | 0.128 | 0.517 | 0.657 | Copper Binding | ||||
| nos2 | 1.670 | 0.200 | 3.342 | 0.0791 | srxn1 | 0.390 | 0.047 | −13.036 | 0.006 | prnp | 1.879 | 0.225 | 3.898 | 0.060 |
| gpr156 | 1.579 | 0.190 | 3.056 | 0.0925 | txnrd2 | 0.136 | 0.016 | −52.904 | 0.001 | atox1 | 1.057 | 0.127 | 0.447 | 0.699 |
| prex1 | 0.911 | 0.109 | −0.811 | 0.5025 | txrnd1 | 0.094 | 0.011 | −80.621 | 0.001 | ccs | 0.759 | 0.091 | −2.646 | 0.118 |
| ENZYMATIC | Free Radical Detoxifiers | Metallothionein | ||||||||||||
| Glutathione Pathway | scara3 | 2.127 | 0.255 | 4.415 | 0.048 | mtl5 | 1.127 | 0.135 | 0.936 | 0.448 | ||||
| mgst3 | 2.040 | 0.245 | 4.248 | 0.051 | cat | 1.178 | 0.141 | 1.261 | 0.335 | Catabolism of Lipoproteins | ||||
| glrx2 | 1.994 | 0.239 | 4.153 | 0.053 | mpv17 | 0.821 | 0.098 | −1.822 | 0.210 | apoe | 1.257 | 0.151 | 1.702 | 0.231 |
| gstz1 | 1.889 | 0.227 | 3.921 | 0.059 | sod2 | 0.098 | 0.012 | −76.823 | 0.0002 | Extracellular Antioxidant | ||||
| gss | 1.522 | 0.183 | 2.859 | 0.104 | sod1 | 0.043 | 0.005 | −185.715 | 0.0001 | sepp1 | 0.252 | 0.030 | −24.791 | 0.002 |
| gpx4 | 1.303 | 0.156 | 1.938 | 0.192 | Kinases | Immune Function | ||||||||
| gpx3 | 1.229 | 0.147 | 1.553 | 0.261 | pnkp | 1.034 | 0.124 | 0.276 | 0.809 | sftpd | 7.052 | 0.846 | 7.151 | 0.019 |
Figure 4.

Effect of 1 μg/mL MeHg on selected oxidative stress genes. HTR8/SV-neo cells treated with 0 or 1 μg/mL MeHg for 24 hours (N=3 replicates) before RT-PCR. Only genes showing significant (p<0.05) or almost significant (0.05<p<0.07) fold-changes in expression are shown. The complete data are presented in Table 1. Expression levels are shown for each gene relative to control expression, which is shown as a horizontal line at 2−ΔΔCt=1. Expression levels below the horizontal line indicate decreased expression upon treatment with 1 μg/mL MeHg while levels above the horizontal line indicate increased expression. Asterisks denote significance: * p<0.05, **p<0.01, ***p<0.001, **** p<0.0001.
Superoxide dismutase 1 (SOD1) expression was also examined at lower doses of MeHg. mRNA expression of sod1 decreased upon treatment with all three doses of MeHg, but the decrease relative to the control group was not significant (Figure 5A). Protein levels of SOD1 also decreased in a dose-dependent manner with MeHg treatment (Figure 5B and 5C). The highest dose of 1 μg/mL MeHg caused a significant decrease in SOD1 protein levels (p<0.05).
Figure 5.

Effect of MeHg on SOD1 expression. HTR8/SV-neo cells were treated with 0, 0.01, 0.1, or 1 μg/mL MeHg for 24 hours (N=3 replicates x 2 duplicates). Expression of sod1 mRNA was quantified by RT-PCR (A). All three doses led to a decrease in sod1 expression relative to control expression (horizontal line), though none of the decreases were statistically significant. Western blot images for SOD1 and the loading control are shown in (B), and the associated quantification (C) revealed an overall dose-dependent decrease in SOD1 protein levels. 1 μg/mL MeHg treatment caused a significant decrease in SOD1 levels relative to 0 μg/mL MeHg (* denotes p<0.05).
Expression of surfactant protein D (sftpd) mRNA decreased slightly when cells were treated with 0.01 μg/mL MeHg (p<0.05, Figure 6). 1 μg/mL MeHg induced a dramatic increase in sftpd relative to the control group (p<0.0001). Intracellular protein levels of SFTPD were undetectable by western blot, most likely because it is a secreted protein.
Figure 6.

Effect of MeHg on sftpd expression. HTR8/SV-neo cells were treated with 0, 0.01, 0.1, or 1 μg/mL MeHg for 24 hours (N=3 replicates x 2 duplicates). Quantification of sftpd mRNA was carried out by RT-PCR. Relative to control expression (horizontal line), 0.01 μg/mL MeHg caused a slight decrease in sftpd expression (* denotes p<0.05) while 1 μg/mL MeHg caused a dramatic increase (**** denotes p<0.0001). Expression increased slightly with 0.1 μg/mL MeHg, but it was not statistically significant.
Cadherin 3 (cdh3) mRNA expression decreased when cells were exposed to 0.01 and 0.1 μg/mL MeHg, though only the cells treated with 0.01 μg/mL showed a statistically significant difference relative to the control (p<0.0001, Figure 7). Beta-catenin (ctnnb1) decreased in a dose-dependent manner, with 0.1 and 1 μg/mL MeHg causing significant decreases in expression (p<0.05, Figure 7).
Figure 7.

Effect of MeHg on migration-related genes. HTR8/SV-neo cells were treated with 0, 0.01, 0.1, or 1 μg/mL MeHg for 24 hours (N=3 replicates x 2 duplicates). Quantification of cdh3 and ctnnb1 by RT-PCR revealed decreased expression relative to control treatment (horizontal line). cdh3 was downregulated more by lower doses of MeHg, specifically 0.01 μg/mL (**** denotes p<0.0001). MeHg decreased expression of ctnnb1 in a dose-dependent manner, with the decreases caused by 0.1 and 1 μg/mL being significant (* denotes p<0.05).
4. Discussion
MeHg has variable effects on HTR8/SV-neo cells depending on the dose of MeHg used. We found that the LC50 of MeHg in trophoblast cells at 24 hours was 1.69 ± 0.12 μg/mL, and a dose of 1 μg/mL MeHg inhibits both proliferation and migration while also exhibiting a range of effects on oxidative stress genes, including significant decreases in expression of SOD1 at the mRNA and protein levels. High doses of MeHg also dramatically increased sftpd expression. We also found that lower doses of 0.01 and 0.1 μg/mL MeHg had stimulatory effects on HTR cell migration, evidenced by increased wound healing rates and altered expression of two migration related genes, cdh3 and ctnnb1.
Though we were not able to measure the concentrations of MeHg taken up by HTR cells from the cell culture medium, our experimental concentrations of 0, 0.01, and 1 μg/mL MeHg correspond to roughly 2.9, 29, and 290 times the lower limit of MeHg in maternal blood that is associated with accumulation in the fetus (3.5 μg/mL) according to the NHANES study [15]. A study by Straka et al. [7] determined the physiological amount of MeHg in human trophoblast cells isolated from placentae of healthy pregnancies to be between 3–4 ng Hg/μg of protein, and this amount also corresponded with in vitro exposure of about 6 μg/L MeHg in cell culture medium. Our experimental doses correspond to 1.7, 17, and 170 times this concentration, suggesting that our doses likely represent elevated MeHg levels for placental cells. In BeWo cells, MeHg uptake occurred in a dose-dependent manner via large neutral amino acid transporters (LAT1 and LAT2) when MeHg was bound with L-cysteine, and MeHg transport reached a steady state far later than endogenous amino acid transport [7,8]. Further study of the kinetics of MeHg into HTR cells could improve our understanding of how relevant MeHg concentrations affect this cell line in comparison to other cell lines, such as BeWo.
Our results indicate that HTR cells are able to tolerate MeHg exposure at very low concentrations. We observe a lowest observable mortality dose (LOMD) of 0.43 μg/mL at 24 hours, which corresponds to roughly 71 times the physiological level of MeHg in human trophoblast cells [7]. Using rat cerebellar granular cells, Castoldi et al. [36] found that the LOMD after 24 hours was about 0.22 μg/mL MeHg and significant cell death occurred at concentrations higher than 1.08 μg/mL. Microglia cells had a viability of 78% when treated with 1 μg/mL MeHg for 24 hours and survival dropped off sharply at concentrations above 3 μg/mL [37]. Based on our results, the viability of HTR cells exposed to MeHg appears to be intermediate between the two neural cell lines. BeWo cells showed a 30% reduction in cell number when treated with 0.65 μg/mL MeHg for 24 hours [7], but we did not see similar effects until concentrations reached 1 μg/mL MeHg. The HTR cell line could potentially be more robust to MeHg than the BeWo cell line, though further study would be needed to confirm this hypothesis. Since we wanted to observe the cytotoxic effects of MeHg on HTR cells at a subapoptotic dose, we chose our experimental doses of 0.01, 0.1, and 1 μg/mL based on this goal and the results of our LC50 assay. We expected that our highest dose of 1 μg/mL MeHg would cause about 30% cell death after 24 hours, leaving approximately 70% of the total cells for cytotoxicity analysis. The inhibition of proliferation and migration that we observed at 1 μg/mL MeHg was likely enhanced by increased cell death at this dose, and this is supported by upregulation of bnip3, a pro-apoptotic gene, in our oxidative stress gene array. However, continued proliferation and migration of HTR cells treated with 1 μg/mL beyond 24 hours confirms that the rate of cell death was not enough to completely halt these processes.
The inhibition of proliferation after treatment with 1 μg/mL MeHg is consistent with literature from in vitro neural assays. Work with neural cells has shown that MeHg disrupts microtubule formation, leading to cell cycle arrest and accumulation in the G2/M phase at 0.75 μg/mL MeHg [38]. Furthermore, doses of MeHg higher than 0.75 μg/mL directly suppress the activity of Cyclin E, an important regulator of cell division, in granule cells [39]. Results from our oxidative stress gene array also showed decreased expression of a cell cycle regulation gene, gft2i, after treatment with 1 μg/mL MeHg, supporting the notion that MeHg promotes cell cycle arrest.
MeHg had different effects on migration depending on the dose, though the dose-dependent effects differed from those of MeHg on neuronal migration. In cerebellar granule cells (CGCs), migration is inhibited by MeHg at concentrations higher than 0.2 μg/mL over the course of 3 days and concentrations higher than 0.1 μg/mL over the course of 7 days [40]. Neonatal cerebellar cells showed inhibition of migration over the course of 48–58 hours by a low concentration of 0.02 μg/mL MeHg [41]. Cytotoxicity of MeHg in neuronal cells is often attributed to calcium (Ca2+) dysregulation [42], and a decreased number of calcium spikes in CGCs has been associated with inhibited cell migration [43]. Furthermore, inhibition of migration by MeHg on CGCs can be reversed by compounds which increase internal Ca2+, such as caffeine, NMDA, Rp-CAMPS, 9CP-Ade, and IGF1 [43]. Interestingly, our lower doses of MeHg (0.01 and 0.1 μg/mL) increased migration in HTR cells, with more drastic increases seen at longer time points (>12 hours). The migration assay results were supported by RT-PCR results. Cadherin 3 (cdh3) is a cell-adhesion gene, and it is down-regulated during cell migration and invasion [44]. We saw significant decreases in cdh3 expression at our lowest dose of MeHg, which was also the dose that promoted the highest rate of migration. Beta-catenin (ctnnb1) is a critical component of the Wnt signaling pathway, and is known to be a potent instigator of migration, especially in cancerous cells [45–47]. Overall, we saw decreases in this gene relative to control expression, though we saw more significant decreases in ctnnb1 expression at our two highest doses of MeHg, which had lower rates of migration. It is likely that other migration-related genes are also important in allowing MeHg-treated HTR cells to migrate faster than control HTR cells. Placental cells must display a certain degree of plasticity to adapt to the maternal environment during pregnancy, especially in response to changes in pO2 which regulate trophoblast function and differentiation [48]. Early pregnancy presents a hypoxic environment, which often induces oxidative stress in the placenta [49]. It is possible that relative to neuronal cells, trophoblast cells are capable of a more robust response to intracellular ion changes as a result of oxidative stress due to the highly variable environmental conditions to which trophoblast cells must respond. Trophoblast cells could potentially respond to gradients in oxidative stress and intracellular ions to promote different phenotypes, such that a low level of oxidative stress might promote migration (i.e., in HTR cells treated with 0.01 and 0.1 μg/mL MeHg) while a higher level of oxidative stress induces cytotoxicity (i.e., HTR cells treated with 1 μg/mL MeHg). However, both increased and decreased migration of trophoblast cells could cause problems in pregnancy. Inhibited migration, as seen in HTR cells treated with 1 μg/mL MeHg, could interfere with trophoblast invasion or angiogenesis. Overexpression of a migratory phenotype could prevent trophoblast cells from fusing and differentiating.
It is unlikely that the differences in migration at our longer time points are due to differences in proliferation. A dose of 1 μg/mL MeHg clearly inhibits both proliferation and migration, and these effects occur quickly. While our lower doses of 0.01 and 0.1 μg/mL MeHg showed significant increases in migration after 12 hours, these doses did not promote significant increases in proliferation across the same amount of time. Thus, the effect of our low doses on migration is likely due primarily to actual effects on migration.
The oxidative stress gene array was conducted with our highest dose, 1 μg/mL MeHg, to detect patterns in oxidative stress-related gene expression as a result of MeHg treatment and to select those genes which were most affected by MeHg for further analysis. Oxidative stress is defined as an unbalance of reactive oxygen species (ROS) and antioxidant capacity, and it can arise from either an overabundance of ROS or an inhibition of antioxidant capacity. MeHg has the ability to both increase ROS and inhibit antioxidant mechanisms, making it a particularly potent toxin [50]. MeHg is a reactive electrophilic compound which often interacts with neutrophilic compounds, specifically those containing sulfhydryl groups and selenohydryl groups [50]. Cysteine and glutathione (GSH) are two important biological non-protein thiols that commonly form complexes with MeHg. This interaction is thought to be a primary driver of cellular damage, as both cysteine-MeHg complexes are able to mimic methionine for entry into the cell as well as into the mitochondria, and GSH bound to MeHg is no longer available for use by the glutathione pathway to detoxify ROS in the cell [50]. Many antioxidant enzymes also contain residues that bind with MeHg, preventing them from engaging in antioxidant activity. This includes selenoproteins such as glutathione peroxidases (GPX) and selenoprotein S (SELS), and enzymes with cysteine residues such as the peroxiredoxins (PRDX) [51]. These enzymes are often upregulated in response to ROS in the cell, and inhibition of these enzymes by MeHg concurrently inhibits the reduction of ROS, which promotes further mRNA expression. While inhibiting antioxidant enzymes, MeHg also increases the levels of ROS in the cell by causing an increase in cytosolic and mitochondrial Ca2+, which in turn disrupts the mitochondrial electron transport chain (METC) and promotes the production of ROS through upregulation of nitric oxide synthase (NOS) [48].
The gene from the oxidative stress gene array that was most downregulated by 1 μg/mL MeHg were superoxide dismutase 1 (sod1). To assess the effect of lower doses of MeHg on expression of sod1, we conducted further RT-PCR assays and Western blots. The results from these assays supported the data from the oxidative stress gene array, as expression of sod1 mRNA as well as SOD1 protein decreased with MeHg treatment. SOD1 is a very important antioxidant enzyme, but its specific interactions with MeHg are not well understood. Some studies have shown that SOD proteins can be inhibited by hydrogen peroxide [52], so it is possible that an increase in intracellular hydrogen peroxide caused indirectly by MeHg may inhibit generation of SOD. It should be noted that mRNA levels of sod2 also decreased, though we did not conduct further analysis on this gene. Further investigation of the interactions between MeHg and SOD proteins is needed to elucidate the inhibition of this protein by MeHg. It is also possible that MeHg directly interacts with the active site of SOD1, but with this type of interaction it would also be expected that mRNA expression would increase to counteract disruption of SOD1 proteins [53].
The dramatic upregulation of sftpd in our oxidative stress array was a surprising result, and we carried out further RT-PCR and Western blot experiments to assess the effect of lower doses of MeHg on this protein. SFTPD is a C-type lectin that is highly expressed in the lung as well as in other epithelial tissues [54]. The placenta was identified as a site of minor SFTPD synthesis in humans [55], and Leth-Larson et al. [56] showed that SFTPD is expressed throughout the female reproductive tract as well as within the villous and extravillous trophoblast cells of the placenta. SFTPD is considered to be an important antimicrobial protein as it is able to recognize pathogens and promote an immune response [57]. It functions as an immunomodulator during pregnancy [58]. Most studies investigating SFTPD have focused on its role as an antimicrobial peptide, but our results suggest that it also responds to MeHg-induced injury. Treatment with 1 μg/mL MeHg caused significant increases in sftpd in both our oxidative stress gene array as well as our additional RT-PCR experiments. We were unable to detect SFTPD by Western blots of cell lysate, and this is potentially due to rapid secretion of the protein after synthesis in the cell. In the lung, SFTPD is secreted into the pulmonary airspace after synthesis in alveolar cells [59], so localization of SFTPD is also potentially extracellular in the placenta. To our knowledge, we provide the first evidence of SFTPD as a potential actor in responding to MeHg toxicity in the placenta. The role of this protein in relation to heavy metal toxicity in the placenta, as well as in other tissues, necessitates further investigation.
The health of the placenta is critical to the health of the developing fetus. Much study of MeHg in the placenta has been related to trans-placental transport between the mother and the fetus [17]. Our results indicate that MeHg also has detrimental effects on the placenta itself. Oxidative stress and disruption of the cell cycle can interfere with normal function of the placenta, especially during the first trimester when it is growing and differentiating [60]. Further studies should continue to investigate how MeHg at a variety of doses interacts with the placenta at the tissue level, as well as how maternal characteristics affect placental toxicity.
5. Conclusions
The roles of MeHg as a neurotoxin and a developmental toxin have been well-characterized in the literature. It is known that MeHg is able to cross the placenta, but very little work has been done to examine how MeHg affects the placenta itself. Through our in vitro model, we demonstrate that first trimester human trophoblast cells exposed to 1 μg/mL MeHg are vulnerable to cell cycle disruption and oxidative stress similar to that seen in neural cell lines, and lower doses were able to increase migration. We also provide evidence that MeHg inhibits SOD1 in trophoblast cells, and that SFTPD is synthesized in these cells in response to MeHg. Based on our results, it is evident that the placenta is at risk to MeHg-induced injury.
HIGHLIGHTS.
The effects of methylmercury on first trimester trophoblast cells are examined
1 μg/mL methylmercury inhibits proliferation and migration, and has many effects on antioxidant genes
0.01 μg/mL and 0.1 μg/mL promote proliferation and decrease expression of a cell adhesion gene
Trophoblast cells display unique responses to methylmercury in relation to neural cell lines
Acknowledgments
We would like to acknowledge Dr. Richard Leach from Michigan State University for providing the HTR8/SV-neo cells, and Dr. Bob Hudson from the University of Illinois at Urbana-Champaign for providing our methylmercury stock solution. Funding: This work was funded by the National Institutes of Health (R21 ES026388) and the Jonathan Baldwin Turner Graduate Fellowship from the University of Illinois College of Agricultural and Environmental Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gribble MO, Karimi R, Feingold BJ, Nyland JF, O’Hara TM, Gladyshev MI, Chen CY. Mercury, selenium and fish oils in marine food webs and implications for human health. J Mar Biol Assoc United Kingdom. 2015;96 doi: 10.1017/S0025315415001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agency for Toxic Substances and Disease Registry. Toxicological profile for mercury. Atlanta, GA: 1999. https://permanent.access.gpo.gov/gpo31588/tp46.pdf. [Google Scholar]
- 3.Gupta RC. CHAPTER 32 - Mercury BT - Veterinary Toxicology. Academic Press; Oxford: 2007. pp. 442–448. https://doi.org/10.1016/B978-012370467-2/50129-2. [Google Scholar]
- 4.Kerper LE, Ballatori N, Clarkson TW. Methylmercury transport across the blood-brain barrier by an amino acid carrier. Am J Physiol. 1992;262:R761–5. doi: 10.1152/ajpregu.1992.262.5.R761. [DOI] [PubMed] [Google Scholar]
- 5.Kajiwara Y, Yasutake A, Adachi T, Hirayama K. Methylmercury transport across the placenta via neutral amino acid carrier. Arch Toxicol. 1996;70 doi: 10.1007/s002040050279. [DOI] [PubMed] [Google Scholar]
- 6.Yin Z, Jiang H, Syversen T, Rocha JBT, Farina M, Aschner M. The methylmercury-L-cysteine conjugate is a substrate for the L-type large neutral amino acid transporter. J Neurochem. 2008;107:1083–1090. doi: 10.1111/j.1471-4159.2008.05683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Straka E, Ellinger I, Balthasar C, Scheinast M, Schatz J, Szattler T, Bleichert S, Saleh L, Knöfler M, Zeisler H, Hengstschläger M, Rosner M, Salzer H, Gundacker C. Mercury toxicokinetics of the healthy human term placenta involve amino acid transporters and ABC transporters. Toxicology. 2016;340:34–42. doi: 10.1016/j.tox.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Balthasar C, Stangl H, Widhalm R, Granitzer S, Hengstschläger M, Gundacker C. Methylmercury Uptake into BeWo Cells Depends on LAT2-4F2hc, a System L Amino Acid Transporter. Int J Mol Sci. 2017;18:1730. doi: 10.3390/ijms18081730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsumoto H, Koya G, Takeuchi T. Fetal minamata disease: A neuropathological study of two cases of intrauterine intoxication by a methyl mercury compound. J Neuropathol Exp Neurol. 1965;24:563–574. https://www.scopus.com/inward/record.uri?eid=2-s2.0-0000185397&partnerID=40&md5=2aa4580e892954d98968e0e0eca7c8f0. [PubMed] [Google Scholar]
- 10.Choi BH, Lapham LW, Amin-Zaki L, Saleem T. Abnormal neuronal migration, deranged cerebral cortical organization, and diffuse white matter astrocytosis of human fetal brain: A major effect of methylmercury poisoning in utero. J Neuropathol Exp Neurol. 1978;37:719–733. doi: 10.1097/00005072-197811000-00001. https://www.scopus.com/inward/record.uri?eid=2-s2.0-0018256847&partnerID=40&md5=cd4ce316bdd99656a1c9f515f410063c. [DOI] [PubMed] [Google Scholar]
- 11.Willes RF, Truelove JF, Nera EA. Neurotoxic response of infant monkeys to methylmercury. Toxicology. 1978;9:125–135. doi: 10.1016/0300-483X(78)90037-9. [DOI] [PubMed] [Google Scholar]
- 12.Fuyuta M, Fujimoto T, Kiyofuji E. Teratogenic effects of a single oral administration of methylmercuric chloride in mice. Acta Anat (Basel) 1979;104:356–362. doi: 10.1159/000145084. https://www.scopus.com/inward/record.uri?eid=2-s2.0-0018749237&partnerID=40&md5=95bb897a4afd47f6a29d8cc9965e218e. [DOI] [PubMed] [Google Scholar]
- 13.Fuyuta M, Fujimoto T, Hirata S. Embryotoxic effects of methylmercuric chloride administered to mice and rats during organogenesis. Teratology. 1978;18:353–365. doi: 10.1002/tera.1420180310. [DOI] [PubMed] [Google Scholar]
- 14.Domingo JL. Metal-induced developmental toxicity in mammals: a review. J Toxicol Environ Heal. 1994;42:123. doi: 10.1080/15287399409531868. http://search.ebscohost.com/login.aspx?direct=true&db=asn&AN=8428831&site=ehost-live. [DOI] [PubMed] [Google Scholar]
- 15.Buchanan S, Anglen J, Turyk M. Methyl mercury exposure in populations at risk: Analysis of NHANES 2011–2012. Environ Res. 2015;140:56–64. doi: 10.1016/j.envres.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Mahaffey KR, Clickner RP, Bodurow CC. Blood organic mercury and dietary mercury intake: National Health and Nutrition Examination Survey, 1999 and 2000. Environ Health Perspect. 2004;112:562–570. doi: 10.1289/ehp.6587. https://www.scopus.com/inward/record.uri?eid=2-s2.0-7944226409&partnerID=40&md5=60821e2aa1448c9965556b3f57d27217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.National Research Council. Toxicological Effects of Methylmercury. The National Academies Press; Washington, DC: 2000. [DOI] [PubMed] [Google Scholar]
- 18.Methyl mercury in fish. A toxicologic-epidemiologic evaluation of risks. Report from an expert group. Nord Hyg Tidskr Suppl. 1971;4(Suppl 4):1–364. https://www.scopus.com/inward/record.uri?eid=2-s2.0-0014993033&partnerID=40&md5=0a8212ab12d478431be4ec3b3bddf467. [PubMed] [Google Scholar]
- 19.Marsh DO, Clarkson TW, Cox C, Myers GJ, Amin-Zaki L, Al-Tikriti S. Fetal methylmercury poisoning. Relationship between concentration in single strands of maternal hair and child effects. Arch Neurol. 1987;44:1017–1022. doi: 10.1001/archneur.1987.00520220023010. [DOI] [PubMed] [Google Scholar]
- 20.Davidson P, Myers G, Cox C, Axtell C, Shamlaye C, Sloane-Reeves J, Cernichiari E, Needham L, Choi A, Wang Y, Berlin C, Clarkson TW. Effects of prenatal and postnatal methylmercury exposure from fish consumption on neurodevelopment: outcomes at 66 months of age in the Seychelles Child Development Study. 1998 doi: 10.1001/jama.280.8.701. [DOI] [PubMed] [Google Scholar]
- 21.Grandjean P, Weihe P, White RF, Debes F, Araki S, Yokoyama K, Murata K, SØRENSEN N, Dahl R, JØRGENSEN PJ. Cognitive Deficit in 7-Year-Old Children with Prenatal Exposure to Methylmercury. Neurotoxicol Teratol. 1997;19:417–428. doi: 10.1016/s0892-0362(97)00097-4. http://dx.doi.org/10.1016/S0892-0362(97)00097-4. [DOI] [PubMed] [Google Scholar]
- 22.Kjellstroem T, Kennedy P, Wallis S, Mantell C. Physical and mental development of children with prenatal exposure to mercury from fish. Stage 1: Preliminary tests at age 4, Rapp. - Naturvaardsverket. 1986 [Google Scholar]
- 23.Kjellstroem T, Kennedy P, Wallis S, Stewart A, Friberg L, Lind B, Wutherspoon T, Mantell C. Physical and mental development of children with prenatal exposure to mercury from fish. Stage 2. Interviews and psychological tests at age 6. Rapp - Naturvaardsverket (Sweden) No 3642. 1989 [Google Scholar]
- 24.Ask K, Akesson A, Berglund M, Vahter M. Inorganic mercury and methylmercury in placentas of Swedish women. [accessed February 27, 2018];Environ Health Perspect. 2002 110:523–6. doi: 10.1289/ehp.02110523. http://www.ncbi.nlm.nih.gov/pubmed/12003757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capelli R, Minganti V, Semino G, Bertarini W. The presence of mercury (total and organic) and selenium in human placentae. [accessed February 27, 2018];Sci Total Environ. 1986 48:69–79. doi: 10.1016/0048-9697(86)90154-3. http://www.ncbi.nlm.nih.gov/pubmed/3945797. [DOI] [PubMed] [Google Scholar]
- 26.Soria ML, Sanz P, Martínez D, López-Artíguez M, Garrido R, Grilo A, Repetto M. Total mercury and methylmercury in hair, maternal and umbilical blood, and placenta from women in the Seville area. [accessed February 27, 2018];Bull Environ Contam Toxicol. 1992 48:494–501. doi: 10.1007/BF00199063. http://www.ncbi.nlm.nih.gov/pubmed/1504492. [DOI] [PubMed] [Google Scholar]
- 27.Graham CH, Hawley TS, Hawley RC, MacDougall JR, Kerbel RS, Khoo N, Lala PK. Establishment and Characterization of First Trimester Human Trophoblast Cells with Extended Lifespan. Exp Cell Res. 1993;206:204–211. doi: 10.1006/excr.1993.1139. http://dx.doi.org/10.1006/excr.1993.1139. [DOI] [PubMed] [Google Scholar]
- 28.Abou-Kheir W, Barrak J, Hadadeh O, Daoud G. HTR-8/SVneo cell line contains a mixed population of cells. Placenta. 2017;50:1–7. doi: 10.1016/j.placenta.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 29.Hurlbert SH. Pseudoreplication and the Design of Ecological Field Experiments. Ecol Monogr. 1984;54:187–211. doi: 10.2307/1942661. [DOI] [Google Scholar]
- 30.Strober W. Trypan Blue Exclusion Test of Cell Viability. Curr Protoc Immunol. 2015;111 doi: 10.1002/0471142735.ima03bs111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Justus CR, Leffler N, Ruiz-Echevarria M, Yang LV. In vitro Cell Migration and Invasion Assays. J Vis Exp. 2014:51046. doi: 10.3791/51046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rio DC, Ares M, Jr, Hannon GJ, Nilsen TW. Purification of RNA using TRIzol (TRI Reagent) Cold Spring Harb Protoc. 2010;5 doi: 10.1101/pdb.prot5439. [DOI] [PubMed] [Google Scholar]
- 33.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol. 2006;Chapter 15 doi: 10.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]
- 34.Gong H, Sun L, Chen B, Han Y, Pang J, Wu W, Qi R, Zhang TM. Evaluation of candidate reference genes for RT-qPCR studies in three metabolism related tissues of mice after caloric restriction. Sci Rep. 2016;6:38513. doi: 10.1038/srep38513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gassmann M, Grenacher B, Rohde B, Vogel J. Quantifying Western blots: Pitfalls of densitometry. Electrophoresis. 2009;30:1845–1855. doi: 10.1002/elps.200800720. [DOI] [PubMed] [Google Scholar]
- 36.Castoldi AF, Barni S, Turin I, Gandini C, Manzo L. Early acute necrosis, delayed apoptosis and cytoskeletal breakdown in cultured cerebellar granule neurons exposed to methylmercury. J Neurosci Res. 2000;59:775–787. doi: 10.1002/(SICI)1097-4547(20000315)59:6<775::AID-JNR10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 37.Garg TK, Chang JY. Methylmercury causes oxidative stress and cytotoxicity in microglia: Attenuation by 15-deoxy-delta 12, 14-prostaglandin J2. J Neuroimmunol. 2006;171:17–28. doi: 10.1016/j.jneuroim.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 38.Miura K, Koide N, Himeno S, Nakagawa I, Imura N. The involvement of microtubular disruption in methylmercury-induced apoptosis in neuronal and nonneuronal cell lines. Toxicol Appl Pharmacol. 1999;160:279–288. doi: 10.1006/taap.1999.8781. [DOI] [PubMed] [Google Scholar]
- 39.Burke K, Cheng Y, Li B, Petrov A, Joshi P, Berman RF, Reuhl KR, DiCicco-Bloom E. Methylmercury elicits rapid inhibition of cell proliferation in the developing brain and decreases cell cycle regulator, cyclin E. Neurotoxicology. 2006;27:970–981. doi: 10.1016/j.neuro.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mancini JD, Autio DM, Atchison WD. Continuous exposure to low concentrations of methylmercury impairs cerebellar granule cell migration in organotypic slice culture. Neurotoxicology. 2009;30:203–208. doi: 10.1016/j.neuro.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 41.Sass JB, Haselow DT, Silbergeld EK. Methylmercury-Induced Decrement in Neuronal Migration May Involve Cytokine-Dependent Mechanisms: A Novel Method to Assess Neuronal Movement in Vitro. Toxicol Sci. 2001;63:74–81. doi: 10.1093/toxsci/63.1.74. [DOI] [PubMed] [Google Scholar]
- 42.Roos D, Seeger R, Puntel R, Vargas Barbosa N. Role of calcium and mitochondria in MeHg-mediated cytotoxicity. J Biomed Biotechnol. 2012;2012:248764. doi: 10.1155/2012/248764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fahrion JK, Komuro Y, Li Y, Ohno N, Littner Y, Raoult E, Galas L, Vaudry D, Komuro H. Rescue of neuronal migration deficits in a mouse model of fetal Minamata disease by increasing neuronal Ca2+ spike frequency. Proc Natl Acad Sci U S A. 2012;109:5057–62. doi: 10.1073/pnas.1120747109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simpson KJ, Selfors LM, Bui J, Reynolds A, Leake D, Khvorova A, Brugge JS. Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat Cell Biol. 2008;10:1027–1038. doi: 10.1038/ncb1762. [DOI] [PubMed] [Google Scholar]
- 45.Müller T, Bain G, Wang X, Papkoff J. Regulation of epithelial cell migration and tumor formation by beta-catenin signaling. [accessed February 28, 2018];Exp Cell Res. 2002 280:119–33. doi: 10.1006/excr.2002.5630. http://www.ncbi.nlm.nih.gov/pubmed/12372345. [DOI] [PubMed] [Google Scholar]
- 46.Iwai S, Yonekawa A, Harada C, Hamada M, Katagiri W, Nakazawa M, Yura Y. Involvement of the Wnt-β-catenin pathway in invasion and migration of oral squamous carcinoma cells. [accessed February 28, 2018];Int J Oncol. 2010 37:1095–103. doi: 10.3892/ijo_00000761. http://www.ncbi.nlm.nih.gov/pubmed/20878057. [DOI] [PubMed] [Google Scholar]
- 47.Yang CM, Ji S, Li Y, Fu LY, Jiang T, Meng FD. β-Catenin promotes cell proliferation, migration, and invasion but induces apoptosis in renal cell carcinoma. Onco Targets Ther. 2017;10:711–724. doi: 10.2147/OTT.S117933. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Myatt L, Cui X. Oxidative stress in the placenta. Histochem Cell Biol. 2004;122 doi: 10.1007/s00418-004-0677-x. [DOI] [PubMed] [Google Scholar]
- 49.Burton GJ, Jauniaux E. Placental oxidative stress: From miscarriage to preeclampsia. J Soc Gynecol Investig. 2004;11 doi: 10.1016/j.jsgi.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 50.Farina M, Rocha JBT, Aschner M. Dev Neurotoxicology Res. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2010. Oxidative Stress and Methylmercury-Induced Neurotoxicity; pp. 357–385. [DOI] [Google Scholar]
- 51.Rhee SG. Overview on Peroxiredoxin. Mol Cells. 2016;39:1–5. doi: 10.14348/molcells.2016.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gottfredsen RH, Larsen UG, Enghild JJ, Petersen SV. Hydrogen peroxide induce modifications of human extracellular superoxide dismutase that results in enzyme inhibition. Redox Biol. 2013;1:24–31. doi: 10.1016/j.redox.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perry JJP, Shin DS, Getzoff ED, Tainer JA. The structural biochemistry of the superoxide dismutases. Biochim Biophys Acta. 2010;1804:245–62. doi: 10.1016/j.bbapap.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stahlman MT, Gray ME, Hull WM, Whitsett JA. Immunolocalization of surfactant protein-D (SP-D) in human fetal, newborn, and adult tissues. J Histochem Cytochem. 2002;50:651–660. doi: 10.1177/002215540205000506. [DOI] [PubMed] [Google Scholar]
- 55.Madsen J, Kliem A, Tornoe I, Skjodt K, Koch C, Holmskov U. Localization of lung surfactant protein D on mucosal surfaces in human tissues. [accessed February 27, 2018];J Immunol. 2000 164:5866–70. doi: 10.4049/jimmunol.164.11.5866. http://www.ncbi.nlm.nih.gov/pubmed/10820266. [DOI] [PubMed] [Google Scholar]
- 56.Leth-Larsen R, Floridon C, Nielsen O, Holmskov U. Surfactant protein D in the female genital tract. Mol Hum Reprod. 2004;10 doi: 10.1093/molehr/gah022. [DOI] [PubMed] [Google Scholar]
- 57.Yarbrough VL, Winkle S, Herbst-Kralovetz MM. Antimicrobial peptides in the female reproductive tract: A critical component of the mucosal immune barrier with physiological and clinical implications. Hum Reprod Update. 2015;21:353–377. doi: 10.1093/humupd/dmu065. [DOI] [PubMed] [Google Scholar]
- 58.Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, Bernal AL, Reid KBM, Madan T, Chakraborty T. Surfactant proteins SP-A and SP-D: Structure, function and receptors. Mol Immunol. 2006;43 doi: 10.1016/j.molimm.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 59.Crouch EC. Surfactant protein-D and pulmonary host defense. Respir Res. 2000;1:93–108. doi: 10.1186/rr19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu F, Tian FJ, Lin Y, Xu WM. Oxidative Stress: Placenta Function and Dysfunction. Am J Reprod Immunol. 2016;76:258–271. doi: 10.1111/aji.12454. http://10.0.4.87/aji.12454. [DOI] [PubMed] [Google Scholar]