

Abstract
Background
Mutations in the promoter region of the TERT gene have been detected in a variety of cancers. These mutations can potentially lead to unlimited cell divisions and result in poor clinical prognosis.
Objective
To determine the role and relevance of TERT promoter region mutations in both clear cell (ccRCC) and non–clear cell (nccRCC) renal cell carcinoma using ultra-deep and whole-genome sequencing methods on primary tumor samples.
Design, setting, and participants
DNA from 281 kidney tumors (147 ccRCC and 134 nccRCC) was sequenced between 2013 and 2015, and clinical outcomes for these patients from a single institution were retrospectively analyzed.
Outcome measurements and statistical analysis
Differences in patient characteristics and mutational status were tested using Fisher’s exact test for categorical variables and the Wilcoxon rank sum test for continuous variables. Survival times were estimated using the Kaplan-Meier method and differences were tested using the log-rank test.
Results and limitations
TERT mutations occurred in 12.2% of ccRCC and 10.4% of nccRCC cases. In >80% of the cases, mutations were located at C228T and were found to co- occur only rarely with other relevant RCC driver genes. The median follow-up among survivors overall was 2.5 yr (range 0.1–18.3). TERT promoter mutations were significantly associated with cancer-specific survival in ccRCC (hazard ratio 2.68, 95% confidence interval 1.19–6.01; p = 0.013). In nccRCC, TERT mutations were significantly associated with larger tumors and metastatic development. Assessment of further relevant clinical associations was precluded in the nccRCC group by the heterogeneous and small sample size.
Conclusions
Our data suggests that TERT mutational status reflects a distinct pathogenesis with an aggressive disease course in RCC. Stratifying patients with this unique tumorigenesis that leads to poor clinical outcomes could be a putative target for novel therapeutics.
Patient summary
We show a previously unrecognized frequency of TERT promoter mutations in both clear cell and non–clear cell renal cell carcinoma. TERT promoter mutations were associated with some worse outcomes in patients with clear cell renal cell carcinoma.
Keywords: Renal cell carcinoma, Sequencing analysis, TERT promoter, Mortality, Prognosis
1. Introduction
Renal cell carcinoma (RCC) represents a heterogeneous group of malignancies comprising multiple histopathologic entities associated with a different landscape of molecular alterations in each subtype. Clear cell RCC (ccRCC) is the most common subtype, accounting for approximately 75% of all RCCs. The remaining subtypes are often grouped together as non–clear cell RCC (nccRCC), consisting of papillary RCC (15%), chromophobe RCC (5–10%), unclassified RCC (5%), and other rare entities [1].
Despite advances in our understanding of the genomic underpinnings of RCC, the full spectrum of cancer drivers in different histological subgroups is not yet completely understood. We sought to characterize the prevalence of further relevant somatic mutations in RCC, and focused specifically on the TERT gene, located on chromosome 5p. Up to 90% of human cancers have high levels of TERT RNA expression, and consequently high TERT enzyme activity. The TERT enzyme is responsible for addition of telomeric repeats to chromosomal ends [2]. Telomeres, located at the end of every chromosome, function to protect chromosomes from recombination and degradation. Telomeres are gradually shortened after every cell division, which leads to senescence and apoptosis of cells after a given number of divisions [3]. Telomerases, with their core unit TERT, are expressed and active in proliferating cells, and are downregulated in differentiated cells [4,5] because of transcriptional silencing [2].
The role of TERT in telomere and telomerase dynamics is the basis behind its oncogenic potential, whereny overactive TERT can maintain telomere length, enabling malignant cells to divide indefinitely and resulting in immortalization of cells [4,6]. Cancer-specific TERT transcription is putatively caused by mutations upstream of the translational start site of TERT, in the TERT promoter region [7]. Mutations in this crucial regulatory element allow TERT expression, leading to the creation of a novel binding site for the ETS family of transcription factors, which ultimately increases TERT transcriptional activity.
Overexpression of the TERT gene, and consequently high TERT activity, has been described in many cancers including melanoma, glioblastoma, and thyroid and urothelial tumors [8–12], and TERT promoter region mutations have been associated with advanced disease course and poor outcome [9,13–15].
It has previously been reported that the mutational status of the TERT promoter in RCC is only 6–10% [7,16–18]. However, most groups studied only ccRCC and the results were limited by the detection methods used, namely polymerase chain reaction and Sanger sequencing [17], which may underestimate the prevalence of TERT promoter alterations. Furthermore, the largest genomic characterization of ccRCC, performed by the Cancer Genome Atlas network [19], did not include the TERT gene in their analysis. The only comprehensive study that used next-generation sequencing to detect TERT promoter and gene aberrations investigated chromophobe RCC [18]. While TERT promoter mutations were detected in <5% of the cases analyzed, a wide range of breakpoints were observed within the TERT promoter region and were associated with TERT upregulation; however, the association with clinical outcomes was not established.
The aim of this study was to use whole-genome and ultra-deep target sequencing methods to characterize the prevalence of TERT gene and promoter mutations in RCC from a large single-institutional cohort. In addition, we aimed to define the impact of TERT mutations on clinical outcomes, namely metastatic development, recurrence, and disease-specific survival, in affected patients.
2. Patients and methods
2.1. Patient selection
After approval by the institutional review board (IRB #12-245, 89-076, and 06-107), our institutional kidney cancer database was queried to identify patients with RCC who underwent genomic testing of their renal tumors between 2013 and 2015. Tumor samples were retrieved either by surgical resection or biopsy performed between 1999 and 2015. All tumor samples included were reviewed and classified by expert genitourinary pathologists to select areas of maximum tumor content for DNA extraction. Individuals with sequencing results from metastatic tissue were excluded from the study. Primary tumor samples from a total of 281 patients were included in the analysis (Supplementary Fig. 1).
2.2. Next-generation sequencing
All patients had previously provided consent for sequencing purposes. DNA from primary tumors and matched normal tissue was extracted according to a standard protocol and subjected to analysis on two next-generation sequencing platforms. Germline mutations were ruled out by analysis of normal blood or adjacent nontumoral tissue for every sample. In our cohort, samples from 270 (96%) patients were sequenced using the MSK-IMPACT assay [20], a hybridization capture-based next-generation sequencing assay for targeted deep sequencing (approx. 500×) of all exons and selected introns of 341 or 410 oncogenes, tumor suppressor genes, and members of pathways deemed actionable by targeted therapies. Details on the MSK-IMPACT panel of targeted genes are provided in Supplementary Table 5.
The remaining 11 (3.9%) samples were subjected to whole-genome sequencing in collaboration with the New York Genome Center on an Illumina HiSeq 2500 system to a mean haploid depth coverage of 87× and 91×.
2.3. Statistical analysis
A panel of nine genes (VHL, PBRM1, BAP1, SETD2, TERT, KDM5C, TP53, mTOR, PTEN) was selected a priori for analysis on the basis of previously published data for known driver mutations in RCC [18,21–23]. Patient and disease characteristics were summarized using the median for continuous and the number (percentage) for categorical variables. Differences in patient and disease characteristics by histology were assessed using Fisher’s exact test for categorical variables and the Wilcoxon rank sum test for continuous variables. Associations between gene mutations and TERT promoter mutations were assessed using Fisher’s exact test.
Cancer-specific survival (CSS) was evaluated for 279 patients with sufficient follow-up data. Recurrence-free survival (RFS) was evaluated for 185 patients who did not present with metastatic disease at the time of diagnosis. Follow-up time was calculated from the date of nephrectomy. Recurrence was defined as the date of pathologic confirmation of diagnosis. For patients with no pathological diagnosis, the date of the radiological examination was used instead. Patients with recurrence or stage IV disease and reported as deceased on last follow-up were considered to have died from disease. All analyses were stratified by histology. CSS and RFS were estimated using the Kaplan-Meier method. Between-group comparisons were carried out using the log-rank test. Univariable Cox regression was used to evaluate associations of patient and disease characteristics with CSS and RFS. Similarly, univariable Cox regression was used to evaluate associations between gene mutations and CSS and RFS. For the ccRCC cohort we investigated multivariable models adjusted for SSIGN score (stage, size, grade, and necrosis) [24] to assess the association between TERT promoter mutations and CSS or RFS.
Statistical significance was set at p < 0.05. All statistical analyses were conducted using R software version 3.2.5 (R Core Development Team, Vienna, Austria).
3. Results
3.1. Overview
A total of 281 patients were analyzed, 147 (52.3%) with ccRCC and 134 (47.7%) with nccRCC. Baseline characteristics are listed in Table 1. The median patient age was 56.6 yr and 65.8% were male. ccRCC patients were older (p = 0.011), had higher grade (p = 0.001) and American Joint Committee on Cancer stage (p = 0.004) disease, and had a higher number of tumors with sarcomatoid features (p = 0.001) when compared to nccRCC patients. The study populations differ significantly in terms of demographics, grade and stage, and sarcomatoid features, so we stratified our analyses by ccRCC and nccRCC. Gene mutation frequencies for the nine genes selected (VHL, PBRM1, BAP1, SETD2, TERT, KDM5C, TP53, mTOR, PTEN) and the presence or absence or metastases at last follow-up are described in Figure 1A and Figure 1A for the ccRCC and the nccRCC groups, respectively. In 81(29%) patients, no genomic alterations in the panel studied were detected. Further mutations assessed with target or whole-genome sequencing did not reach frequencies >2%.
Table 1.
Demographic data and baseline clinical characteristics
| Overall | ccRCC (n = 147) | nccRCC (n = 134) | p value | |
|---|---|---|---|---|
| Age, yr (range) | 56.6 (19.1 – 86.3) | 58.0 (31.2 – 81) | 53.5 (19.1 – 86.3) | 0.011 |
| Tumor size, cm (range) | 8.0 (1.2 – 29.0) | 8.0 (2.0 – 22.2) | 8.4 (1.2 – 29) | 0.320 |
| Gender, n (%) | 0.315 | |||
| Female | 96 (34.2) | 46 (31.3) | 50 (37.3) | |
| Male | 185 (65.8) | 101 (68.7) | 84 (62.7) | |
| Grade, n (%) | 0.001 | |||
| G2 | 24 (8.5) | 22 (15.0) | 2 (1.5) | |
| G3 | 96 (34.2) | 61 (41.5) | 35 (26.1) | |
| G4 | 73 (26.0) | 62 (42.2) | 11 (8.2) | |
| NA | 88 (31.3) | 2 (1.4) | 86 (64.2) | |
| Sarcomatoid, n (%) | 0.001 | |||
| No | 205 (73.0) | 96 (65.3) | 109 (81.3) | |
| Yes | 70 (24.9) | 49 (33.3) | 21 (15.7) | |
| NA | 6 (2.1) | 2 (1.4) | 4 (3.0) | |
| T stage, n (%) | 0.007 | |||
| T1 | 64 (22.7) | 29 (19.8) | 35 (26.1) | |
| T2 | 41 (14.6) | 14 (9.6) | 27 (20.2) | |
| T3 | 157 (55.8) | 96 (65.3) | 61 (45.6) | |
| T4 | 11 (3.9) | 5 (3.4) | 6 (4.5) | |
| NA | 8 (2.8) | 3(2.0) | 5 (3.7) | |
| N stage, n (%) | 0.271 | |||
| N0 | 106 (37.7) | 61 (41.5) | 45 (33.6) | |
| N1 | 35 (12.5) | 14 (9.5) | 21 (15.7) | |
| N2 | 10 (3.6) | 4 (2.7) | 6 (4.5) | |
| Nx | 122 (43.4) | 65 (44.2) | 57 (42.5) | |
| NA | 8 (2.8) | 3 (2.0) | 5 (3.7) | |
| M stage, n (%) | <0.001 | |||
| M0 | 193 (68.7) | 93 (63.3) | 100 (74.6) | |
| M1 | 87 (31) | 54 (36.7) | 33 (24.6) | |
| NA | 1 (0.4) | 0 (0) | 1 (0.7) | |
| AJCC | 0.004 | |||
| I | 52 (18.5) | 22 (15.0) | 30 (22.4) | |
| II | 28 (10.0) | 8 (5.4) | 20 (14.9) | |
| III | 105 (37.4) | 56 (38.1) | 49 (36.6) | |
| IV | 96 (34.2) | 61 (41.5) | 35 (26.1) |
RCC = renal cell carcinoma; cc = clear cell; ncc = non–clear cell; NA = not available; AJCC = American Joint Committee on Cancer.
Fig. 1.
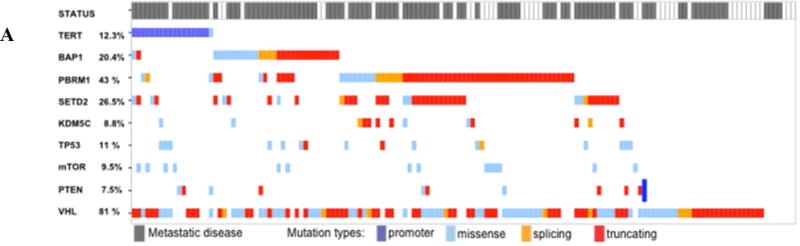
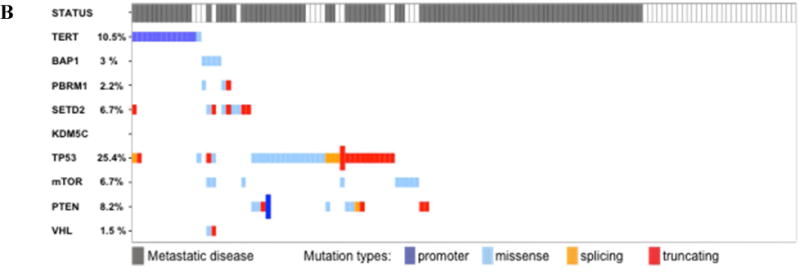
Overview of concordance and mutual exclusivity of TERT promoter mutations and nine selected known renal cell carcinoma (RCC) driver genes in (A) clear cell RCC and (B) non–clear cell RCC. The presence of metastatic disease at last follow-up is highlighted in grey in the top row.
TERT mutations were present in 12.2% (18/147) of ccRCC and 10.4% (14/134) of nccRCC cases. Of these alterations, the promoter region was affected in 17/18 (94.4%) ccRCC and 13/14 (92.8%) nccRCC patients.
3.2. ccRCC cohort
The genes most commonly mutated in the 147 ccRCC patients were located on chromosome 3p: VHL (81.0%), PBRM1 (42.9%), SETD2 (27.9%), and BAP1 (20.4%). TERT alterations were the fifth most frequently assessed mutations, at 12.2% (n = 18; Fig. 1A). Among those, 17 patients (94.4%) had alterations detected in the promoter region, and one (5.6%) was diagnosed with a TERT gene mutation and was excluded from further association analyses. TERT promoter alterations were mutually exclusive in all cases: 13 (76.5%) were at site C228T, two (11.8%) at C250T, and two (11.8%) at C898T.
VHL mutations co-occurred in 70.6% of the cases with TERT promoter mutations. Among patients with TERT promoter mutations, 88.2% were wild-type for PBRM1 (p = 0.008). TERT promoter mutations were not significantly associated with any of the other mutations studied (Fig. 1A and Supplementary Table 1). Nevertheless, the low mutation frequencies might have low power for detecting associations.
A total of 54 patients with metastatic disease at baseline and six patients who did not have surgery were excluded from the RFS analysis. The median follow-up for ccRCC patients was 3.1 yr for those who did not die from cancer and 3.2 yr for patients without recurrence. During follow-up, 32 patients died from RCC and 42 patients experienced disease recurrence. Univariable Cox regression results for the association with CSS and RFS are presented in Table 2. Higher tumor size and grade, sarcomatoid status, and SSIGN score [24] were significantly associated with higher risks of disease recurrence and death from cancer (Table 2). The survival analysis showed that patients with TERT promoter mutations had significantly shorter CSS (hazard ratio [HR] 2.68, 95% confidence interval [CI] 1.19–6.01; p = 0.013; Fig. 2A). However, there was no significant association between TERT promoter mutations and RFS (HR 1.35, 95% CI 0.48–3.79; p = 0.567; Fig. 2B). Analysis of associations between other mutations and CSS and RFS revealed that BAP1 mutations were associated with a higher risk of cancer-specific death, whereas SETD2 mutations were associated with a risk of tumor recurrence (Table 3).
Table 2.
Univariable Cox regression results for the association of patient and disease characteristics with cancer-specific and recurrence-free survival among patients with clear cell renal cell carcinoma
| Cancer - specific survival | Recurrence - free survival | |||
|---|---|---|---|---|
|
| ||||
| HR (95% CI) | p value | HR (95% CI) | p value | |
| Age | 1.01 (0.97 – 1.04) | 0.644 | 0.99 (0.96 – 1.02) | 0.526 |
| Tumor size (cm) | 1.12 (1.05 – 1.20) | 0.001 | 1.24 (1.13 – 1.36) | <0.001 |
| Gender | 0.388 | 0.892 | ||
| Female | 1 | 1 | ||
| Male | 0.73 (0.36 – 1.49) | 0.96 (0.51 – 1.81) | ||
| Grade | <0.001 | <0.001 | ||
| G2 | 1 | 1 | ||
| G3 | 0.59 (0.10 – 3.54) | 4.35 (1.29 – 14.69) | ||
| G4 | 9.11 (2.14 – 38.9) | 11.74 (3.41 – 40.45) | ||
| Sarcomatoid | <0.001 | 0.011 | ||
| No | 1 | 1 | ||
| Yes | 11.45 (4.89 – 26.78) | 2.45 (1.23 – 4.9) | ||
| SSIGN score | 1.26 (1.14 – 1.40) | <0.001 | 1.16 (1.07 – 1.27) | 0.001 |
HR = hazard ratio; CI = confidence interval.
Fig. 2.

Probability of (A) cancer-specific survival (CSS) and (B) recurrence-free survival (RFS) by TERT status among patients with clear cell renal cell carcinoma. The median follow-up was 3.1 yr (range 0.1–11.2) for CSS and 3.2 yr (range 0.1–11.2) for RFS. wt = wild type; mut = mutant.
Table 3.
Univariable Cox regression results for the association of mutations with cancer-specific and recurrence-free survival among patients with clear cell renal cell carcinoma Wild-type (wt), Mutant (mut)
| Gene | Cancer - specific survival | Recurrence - free survival | ||
|---|---|---|---|---|
|
|
||||
| HR (95% CI) | p value | HR (95% CI) | p value | |
| VHL | 0.227 | 0.080 | ||
| Wild type | 1.00 | 1.00 | ||
| Mutant | 0.59 (0.25 – 1.38) | 0.53 (0.26 – 1.08) | ||
| PBRM1 | 0.475 | 0.407 | ||
| Wild type | 1.00 | 1.00 | ||
| Mutant | 0.77 (0.38 – 1.58) | 0.77 (0.42 – 1.42) | ||
| SETD2 | 0.453 | 0.001 | ||
| Wild type | 1.00 | 1.00 | ||
| Mutant | 1.32 (0.64 – 2.73) | 3.09 (1.63 – 5.86) | ||
| BAP1 | 0.002 | 0.141 | ||
| Wild type | 1.00 | 1.00 | ||
| Mutant | 3.10 (1.52 – 6.30) | 1.74 (0.83 – 3.65) | ||
| KDM5C | 0.255 | 0.682 | ||
| Wild type | 1.00 | 1.00 | ||
| Mutant | 0.31 (0.04 – 2.31) | 1.28 (0.39 – 4.15) | ||
| TP53 | 0.567 | 0.053 | ||
| Wild type | 1.00 | 1.00 | ||
| Mutant | 0.66 (0.15 – 2.79) | 2.37 (0.99 – 5.71) | ||
| mTOR | 0.701 | 0.34 | ||
| Wild type | 1.00 | 1.00 | ||
| Mutant | 1.23 (0.42 – 3.58) | 0.5 (0.12 – 2.07) | ||
| PTEN | 0.097 | 0.333 | ||
| Wild type | 1.00 | 1.00 | ||
| Mutant | 2.25 (0.86 – 5.88) | 1.66 (0.59 – 4.68) | ||
HR = hazard ratio; CI = confidence interval.
Multivariable Cox regression models were adjusted for SSIGN score to assess the association between mutations and CSS and RFS (Supplementary Table 2). TERT promoter (HR 3.55, 95% CI 1.37–9.20; p = 0.009) and BAP1 mutations (HR 2.93, 95% CI 1.28–6.71; p = 0.011) were significantly associated with higher risk of cancer-specific death. Only TP53 mutations were significantly associated with higher risk of recurrence (HR 2.17, 95% CI 1.07–4.41; p = 0.033).
3.3. nccRCC cohort
A total of 134 tumors, consisting of a variety of tumor histologies (Supplementary Table 3), were analyzed in the nccRCC cohort. The genes most frequently mutated among those evaluated for analysis were TP53 (25.4%), TERT (10.4%), PTEN (8.2%), and mTOR (6.7%; (Fig. 1B). A TERT gene mutation was observed for one patient (E441del). All 13 non-exonic alterations affected the promoter region, of which 11 alterations involved hotspot C228T (84.6%).
TERT promoter mutations showed no association with the incidence of mutations in the other genes evaluated. Moreover, they were mutually exclusive to mutations in all the other genes, except for TP53 in two cases and SETD2 in one case (Fig. 2B and Supplementary Table 4).
Univariable Cox regression analyses were performed to test the association of patient and disease characteristics with CSS and RFS (Table 4). Two patients were excluded from the CSS analysis because of unknown cause of death. A total of 33 patients with metastatic disease at presentation and three patients who did not undergo surgery were excluded from the RFS analysis. The median follow-up among nccRCC patients was 2.2 yr for those who did not die from cancer and 2.3 yr for patients who did not experience recurrence. During follow-up, 38 patients died from RCC and 51 patients had disease recurrence.
Table 4.
Univariable Cox regression results for the association of patient and disease characteristics with cancer-specific and recurrence-free survival among patients with non–clear cell renal cell carcinoma patients
| Cancer - specific survival | Recurrence - free survival | |||
|---|---|---|---|---|
|
|
||||
| HR (95% CI) | p value | HR (95% CI) | p value | |
| Age | 1 (0.98 – 1.03) | 0.869 | 0.99 (0.97 – 1.01) | 0.534 |
| Tumor size (cm) | 1.02 (0.96 – 1.09) | 0.541 | 1.03 (0.99 – 1.08) | 0.168 |
| Gender | 0.932 | 0.454 | ||
| Female | 1.00 | 1.00 | ||
| Male | 1.03 (0.54 – 1.98) | 0.82 (0.48 – 1.39) | ||
| Sarcomatoid | 0.004 | 0.035 | ||
| No | 1.00 | 1.00 | ||
| Yes | 2.77 (1.38 – 5.55) | 2.11 (1.05 – 4.23) | ||
| TERT promoter | 0.191 | 0.261 | ||
| Wild type | 1.00 | 1.00 | ||
| Mutant | 1.8 (0.75 – 4.36) | 1.58 (0.71 – 3.51) | ||
HR = hazard ratio; CI = confidence interval.
Sarcomatoid differentiation was significantly association with higher risk of death from cancer (p = 0.004); no factors were significantly associated with RFS (Table 4).
Survival analysis for patients with and without TERT promoter mutations revealed a wild-type TERT advantage for CSS, but statistical significance was not reached (Fig. 3).
Fig. 3.
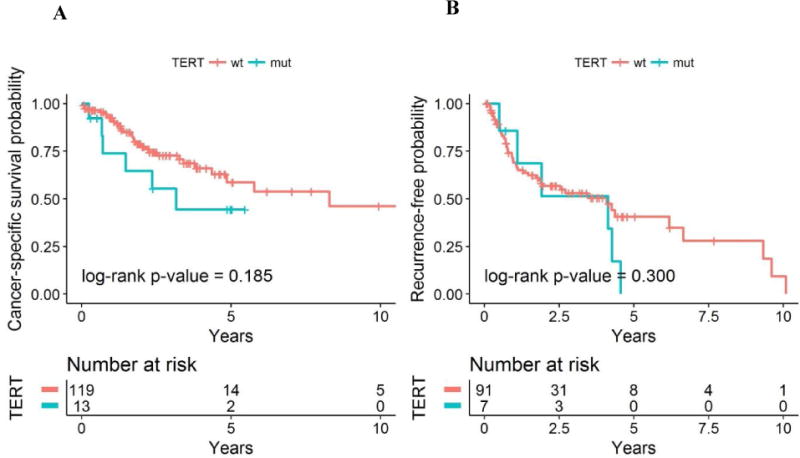
Probability of (A) cancer-specific survival (CSS) and (B) recurrence-free survival (RFS) by TERT status among patients with non–clear cell renal cell carcinoma. The median follow-up was 2.2 yr (range 0.1–18.3) for CSS and 2.3 yr (range 0.1–7.7) for RFS. wt = wild type; mut = mutant.
4. Discussion
The role of TERT in human mutagenesis was initially described in melanoma [10]. Subsequent studies showed that TERT promoter mutations are associated with worse survival outcomes in a variety of cancer types [25]. Our results show that TERT promoter mutations are common in ccRCC and are associated with poor clinical outcomes. As previously described, TERT promoter mutations were commonly detected in chromophobe RCC, but they also occurred in other nccRCC subtypes (papillary, unclassified and TFE3 translocation RCC).
Univariable analyses confirmed that TERT variations were associated with shorter CSS in only ccRCC. The lack of association in the nccRCC group may be due to the heterogeneous nature of the cohort.
The known hotspot mutation C228T was the most commonly mutated site in ~80% of our cases, followed by C250T. Previous studies have shown that these hotspot mutations lead to higher TERT expression and subsequent telomerase activity, which is ultimately associated with cancer progression and poor prognosis [26].
TERT promoter mutations allow the creation of ETS1 binding sites, leading to TERT transcription [27]. Recent data suggest that ETS transcription factors participate in the upregulation of hypoxia-inducible genes that are a hallmark of cells with defective VHL complex, as in ccRCC [28]. Furthermore, it has been shown that hypoxia increases ETS1 transcription and transactivation [29], which points to a cooperative effect with HIF. This supports the observation that TERT expression is enhanced in cases of concomitant VHL gene inactivation and TERT promoter mutation [16]. In our ccRCC cohort, we found that VHL mutations occurred in approximately 70% of patients carrying TERT promoter mutations. However, further co-occurrences could not be detected in ccRCC, possibly indicating the involvement of specific oncogenic pathways in tumors with TERT promoter mutations.
TERT promoter mutations were mutually exclusive to most genes tested in nccRCC. This suggests that TERT mutations are responsible for a unique pathway that leads to aggressive tumor behavior; however, the heterogeneous and incompletely unraveled genomic landscape of these entities poses difficulties in the identification of clear genomic drivers for cancer progression.
Studies in bladder cancer with suppression of high TERT expression and telomerase activity showed promising results that may lead to treatments targeting tumors bearing TERT alterations [30]. We therefore believe that identifying TERT promoter alterations, as one of the most commonly mutated sites in kidney cancer, may reveal a valuable actionable target, in particular for patients lacking other alterations.
Our study has multiple limitations. Our cohort consisted of a high-risk population of patients treated at a tertiary referral center, which does not reflect the spectrum of RCC and potentially over-represents aggressive tumors bearing TERT promoter mutations. Furthermore, our cohort does not reflect the common histological landscape of nccRCC given the over-representation of chromophobe RCC, and may mask a higher incidence of TERT mutations in papillary RCC. In addition, systemic therapy data were not available. Larger validation studies with longer follow-up are needed to evaluate the impact of TERT promoter mutations in kidney cancer. Finally, tumor heterogeneity is a crucial factor that needs to be considered when assessing mutational status. Therefore, future analyses of multiple regions from primary tumors and matched metastatic sites are necessary to determine the relevance of TERT promoter mutations in RCC. Nevertheless, our findings are significant and help to expand knowledge of the genomic drivers of RCC.
5. Conclusions
Our data suggest that TERT promoter mutations are associated with worse CSS in ccRCC. Our work has highlighted a previously unrecognized frequency of TERT promoter mutations in both clear cell and non–clear cell RCC. The patterns of mutual exclusivity with other known cancer drivers in nccRCC suggest a unique tumorigenesis pathway in patients bearing TERT promoter mutations. Further studies are needed to validate our findings.
Supplementary Material
Take Home Message.
TERT mutational status reflects a distinct pathogenesis with an aggressive disease course in clear cell renal cell carcinoma. Stratifying patients with this unique tumorigenesis that leads to poor clinical outcomes could be a putative target for novel therapeutics.
Acknowledgments
Jozefina Casuscelli was sponsored by German Research Foundation grant CA1403/1-1. Brandon J. Manley and Mazyar Ghanaat were supported by Ruth L. Kirschstein National Research Service Award T3 to 2CA082088. Brandon J. Manley, Jonathan A. Coleman, Mazyar Ghanaat, and A. Ari Hakimi were supported by the Sidney Kimmel Center for Prostate and Urologic Cancers and NIH/NCI Cancer Center Support Grant P30 CA008748. James J. Hsieh was supported by the Jill and Jeffrey Weiss Fund for the Cure of Kidney Cancer and the J. Randall & Kathleen L. MacDonald Kidney Cancer Research Fund.
Funding/Support and role of the sponsor: None.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions: A. Ari Hakimi had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Hakimi, Casuscelli.
Acquisition of data: Casuscelli, Becerra, Manley, Tennenbaum, Russo, Coleman, Hakimi.
Analysis and interpretation of data: Casuscelli, Becerra, Manley, Zabor.
Drafting of the manuscript: Casuscelli, Becerra, Reznik.
Critical revision of the manuscript for important intellectual content: Hakimi, Hsieh, Russo, Coleman, Motzer, Voss, Feldman, Carlo, Stief.
Statistical analysis: Zabor.
Obtaining funding: None.
Administrative, technical, or material support: Redzematovic, Arcila, Reuter, Chen, Ghanaat, Kashan.
Supervision: Hakimi, Hsieh.
Other: None.
Financial disclosures: A. Ari Hakimi certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: None.
References
- 1.Srigley JR, Delahunt B, Eble JN, et al. The International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia. Am J Surg Pathol. 2013;37:1469–89. doi: 10.1097/PAS.0b013e318299f2d1. [DOI] [PubMed] [Google Scholar]
- 2.Aubert G. Telomere dynamics and aging. Nat Genet. 2014;125:89–111. doi: 10.1016/B978-0-12-397898-1.00004-9. [DOI] [PubMed] [Google Scholar]
- 3.Counter CM, Avilion AA, LeFeuvre CE, et al. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–9. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM. Cancer-associated TERT promoter mutations abrogate telomerase silencing. eLife. 2015;4:e07198. doi: 10.7554/eLife.07918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell RJ, Rube HT, Xavier-Magalhaes A, et al. Understanding TERT promoter mutations: a common path to immortality. Mol Cancer Res. 2016;14:315–23. doi: 10.1158/1541-7786.MCR-16-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greider CW, Blackburn EH. Tracking telomerase. Cell. 2004;116(2 Suppl):S83–86. doi: 10.1016/s0092-8674(04)00053-4. [DOI] [PubMed] [Google Scholar]
- 7.Vinagre J, Almeida A, Populo H, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185. doi: 10.1038/ncomms3185. [DOI] [PubMed] [Google Scholar]
- 8.Jin L, Chen E, Dong S, et al. BRAF and TERT promoter mutations in the aggressiveness of papillary thyroid carcinoma: a study of 653 patients. Oncotarget. 2016;7:18346–55. doi: 10.18632/oncotarget.7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simon M, Hosen I, Gousias K, et al. TERT promoter mutations: a novel independent prognostic factor in primary glioblastomas. Neuro-oncology. 2015;17:45–52. doi: 10.1093/neuonc/nou158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–9. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowan M, Springer S, Nguyen D, et al. High prevalence of TERT promoter mutations in primary squamous cell carcinoma of the urinary bladder. Mod Pathol. 2016;29:511–5. doi: 10.1038/modpathol.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borah S, Xi L, Zaug AJ, et al. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science. 2015;347:1006–10. doi: 10.1126/science.1260200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heidenreich B, Rachakonda PS, Hosen I, et al. TERT promoter mutations and telomere length in adult malignant gliomas and recurrences. Oncotarget. 2015;6:10617–33. doi: 10.18632/oncotarget.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rachakonda PS, Hosen I, de Verdier PJ, et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci U S A. 2013;110:17426–31. doi: 10.1073/pnas.1310522110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang DS, Wang Z, He XJ, et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur J Cancer. 2015;51:969–76. doi: 10.1016/j.ejca.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Liu T, Liu L, et al. TERT promoter mutations in renal cell carcinomas and upper tract urothelial carcinomas. Oncotarget. 2014;5:1829–36. doi: 10.18632/oncotarget.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hosen I, Rachakonda PS, Heidenreich B, et al. TERT promoter mutations in clear cell renal cell carcinoma. Int J Cancer. 2015;136:2448–52. doi: 10.1002/ijc.29279. [DOI] [PubMed] [Google Scholar]
- 18.Davis CF, Ricketts CJ, Wang M, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–30. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–64. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Velasco G, Miao DD, Voss MH, et al. Tumor mutational load and immune parameters across metastatic renal cell carcinoma (mRCC) risk groups. Cancer Immunol Res. 2016;4:820–2. doi: 10.1158/2326-6066.CIR-16-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hakimi AA, Mano R, Ciriello G, et al. Impact of recurrent copy number alterations and cancer gene mutations on the predictive accuracy of prognostic models in clear cell renal cell carcinoma. J Urol. 2014;192:24–9. doi: 10.1016/j.juro.2014.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hakimi AA, Ostrovnaya I, Reva B, et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res. 2013;19:3259–67. doi: 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frank I, Blute ML, Cheville JC, Lohse CM, Weaver AL, Zincke H. An outcome prediction model for patients with clear cell renal cell carcinoma treated with radical nephrectomy based on tumor stage, size, grade and necrosis: the SSIGN score. J Urol. 2002;168:2395–400. doi: 10.1016/S0022-5347(05)64153-5. [DOI] [PubMed] [Google Scholar]
- 25.Yuan P, Cao JL, Abuduwufuer A, et al. Clinical characteristics and prognostic significance of TERT promoter mutations in cancer: a cohort study and a meta-analysis. PLoS One. 2016;11:e0146803. doi: 10.1371/journal.pone.0146803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang FW, Bielski CM, Rinne ML, et al. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis. 2015;4:e176. doi: 10.1038/oncsis.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vallarelli AF, Rachakonda PS, Andre J, et al. TERT promoter mutations in melanoma render TERT expression dependent on MAPK pathway activation. Oncotarget. 2016;7:53127–36. doi: 10.18632/oncotarget.10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salnikow K, Aprelikova O, Ivanov S, et al. Regulation of hypoxia-inducible genes by ETS1 transcription factor. Carcinogenesis. 2008;29:1493–9. doi: 10.1093/carcin/bgn088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiao N, Xu C, Zhu YX, Cao Y, Liu DC, Han X. Ets-1 as an early response gene against hypoxia-induced apoptosis in pancreatic beta-cells. Cell Death Dis. 2015;6:e1650. doi: 10.1038/cddis.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li C, Wu S, Wang H, et al. The C228T mutation of TERT promoter frequently occurs in bladder cancer stem cells and contributes to tumorigenesis of bladder cancer. Oncotarget. 2015;6:19542–51. doi: 10.18632/oncotarget.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.