

Abstract
A general protocol is described for inducing enantioselective halolactonizations of unsaturated carboxylic acids using novel bifunctional organic catalysts derived from a chiral binaphthalene scaffold. Bromo- and iodolactonization reactions of diversely substituted, unsaturated carboxylic acids proceed with high degrees of enantioselectivity, regioselectivity, and diastereoselectivity. Notably, these BINOL-derived catalysts are the first to induce the bromo- and iodolactonizations of 5-alkyl-4(Z)-olefinic acids via 5-exo mode cyclizations to give lactones in which new carbon–halogen bonds are created at a stereogenic center with high diastereo- and enantioselectivities. Iodolactonizations of 6-substituted-5(Z)-olefinic acids also occur via 6-exo cyclizations to provide δ-lactones with excellent enantioselectivities. Several notable applications of this halolactonization methodology were developed for desymmetrization, kinetic resolution, and epoxidation of Z-alkenes. The utility of these reactions is demonstrated by their application to a synthesis of precursors of the F-ring subunit of kibdelone C and to the shortest catalytic, enantioselective synthesis of (+)-disparlure reported to date.
Graphical Abstract

INTRODUCTION
Cyclizations via halofunctionalization of olefins 1 represent an important class of reactions for the construction of heterocyclic compounds with carbon-halogen bonds (C–X) outside (exo) or inside (endo) the newly formed ring as in 2 and 3, respectively (Equation 1). Consequently, the development of catalytic, enantioselective halocyclization reactions is a burgeoning field, and, although there have been a plethora of advances over the past six years,1 significant gaps in the methodology remain.
 |
(1) |
Enantioselective halocyclizations of α,ω-hydroxy alkenes 1 (Y = H2; Z = O)2 and α,ω-amino alkenes 1 (Y = H2; Z = NR′)3 have been reported, although the majority of the work in the area of enantioselective halofunctionalizations of olefins has been focused on enantioselective halolactonization reactions (Y = O, Z = OH).4–6 Borhan described the first highly enantioselective (up to 95:5 er) chlorolactonizations of a series of 4-aryl-substituted-4-pentenoic acids to generate the corresponding chlorolactones using the commercially-available, cinchona alkaloid derivative (DHQD)2PHAL (4) as the catalyst (Figure 1).4a There have been only two other catalysts capable of effecting enantioselective chlorolactonizations. Tang reported catalyst 5, which promoted chlorolactonizations 4-aryl-substituted-4-pentenoic acids,4b and Zhou reported a cinchonine-squarimide catalyst that promotes chlorolactonizations of vinylbenzoic acids.4d
Figure 1.

Halolactonization catalysts.
Enantioselective bromo- and iodolactonization reactions have attracted greater attention than any other cyclization involving halofunctionalization of olefins. For example, the C3-symmetric trisimidazoline catalyst 6 was first reported to induce 6-exo bromolactonizations of 5-aryl-substituted-5-hexenoic acids to deliver δ-lactones,5f and it has since been shown to work equally well with tri- and tetrasubstituted olefinic acids.5g In 2010 Yeung showed that the quinidine-derived thiocarbamate 7 catalyzed enantioselective bromolactonizations of 4-aryl-substituted-4-pentenoic acids,5b and by making slight changes in the quinidine catalyst, he was able to induce the enantioselective bromolactonizations of a variety of other substrates.5c–e Yeung later developed several proline derived catalysts 8 for the enantioselective bromolactonizations of 4-substituted-4-pentenoic acids and 5-substituted-5-hexenoic acids, but each substrate class required catalyst optimization.
Jacobsen’s tertiary aminourea catalyst 9 promoted enantioselective iodolactonizations of 5-substituted-5-hexenoic acids with moderate to excellent selectivities.6a Subsequently, Hansen disclosed that the squarimide variant 10 also induced iodolactonization reactions, although the selectivities were lower than with 9.6b The catalyst 11, which was developed by Johnston, effected the iodolactonizations of 5-substituted-5-hexenoic acids with generally high enantioselectivities.6c Ishihara reported that iodolactonizations of benzyl-substituted-4-pentenoic acids in the presence of the binaphthyl 12 formed γ-butyrolactones with good enantioselectivities.6h
The first example of a catalytic, enantioselective fluorolactonization was reported by Rueping, who utilized a combination of (DHQ)2PHAL (4) and Selectfluor® to generate a fluorinated isobenzofuran in modest yield (50%) and with low enantioselectivity (27% ee).7a The only catalytic, highly enantioselective fluorolactonization using an electrophilic fluorine source reported thus far was achieved using the binaphthyl derivative 13, although the substrate scope was somewhat limited.7b The methylene spacer between the naphthol and hydroxyl group was found to be critical because the naphthol based catalyst gave racemic product. Jacobsen recently reported a method for asymmetric fluorolactonization using a chiral aryl iodide catalyst and a nucleophilic fluoride source that delivered fluorinated isochromanones with generally high enantioselectivity.7c
Several chiral Lewis acid-derived catalysts have also been used to promote enantioselective halolactonizations,6e,f,g but these catalysts have some notable limitations. Specifically, halolactonizations of olefinic acids bearing an alkyl group on the double bond often led to lower enantioselectivities compared to aryl-substituted olefinic acids, and most catalytic systems appear to be limited to a specific halogen atom.
Despite the many advances in catalysts that have been developed to promote enantioselective halolactonizations, the substrate scope of each is typically limited, especially for chloro- and iodolactonizations. Although there were several gaps in the methodology when we initiated our work, a major deficiency in the contemporaneous art was the lack of any example of a halolactonization that proceeded via an exo mode of ring closure to give a lactone bearing a new C–X bond at a stereogenic secondary carbon atom (e.g., 2, R = alkyl, aryl; Z = Y = O). This was somewhat surprising because several examples of the corresponding exo halocyclizations of hydroxy alkenes 1 (R = alkyl or aryl; Y = H2, Z = O) to furnish the corresponding tetrahydrofurans were known.2a–d The sole example of a halolactonization that created a stereogenic carbon atom bearing a halogen atom involved an endo cyclization mode.5c It should be noted that during the course of our studies, Yeung reported the use of 4 to catalyze the bromolactonization of several 5-substituted-4(Z)-pentenoic acids in an exo fashion to give stereogenic C–X bonds at secondary carbons.5d Herein we report the details of our methodological studies directed toward the design and development of novel organic catalysts for enantioselective bromo- and iodolactonizations,8 as well as the applications of these enantioselective halolactonizations to solving several synthetic problems in this account.9
RESULTS AND DISCUSSION
Catalyst Design and Conditions: Bromolactonizations
The difficulty associated with developing an enantioselective bromocyclization reaction arises, in part, from the reversibility of bromonium ion formation and its propensity to transfer Br+ to another olefin prior to intramolecular capture by a pendant nucleophile.10 In order to address this problem, most successful known catalysts are bifunctional containing both Lewis/Brønsted acid and Lewis/Brønsted base functionalities (Figure 1). This critical attribute enables the catalyst to coordinate with the substrate as well as with the brominating reagent or bromonium ion. When we began designing a catalyst for bromolactonizations, we envisioned that established bifunctional motifs found in some of the known catalysts (e.g., 4–9, Figure 1) could be mounted on a chiral binaphthyl backbone, which despite its near ubiquity in the field of enantioselective reactions had not yet been applied to halocyclization reactions.
The initial goal was to develop analogs of the bifunctional catalyst 17, which bear amidine and thiocarbamate groups as the requisite Lewis base and acid motifs appended to a BINOL-derived scaffold. An amidine group was selected rather than a tertiary amine because bromine was well-known to rapidly oxidize benzylic tertiary amines.11 Having a phenyl group at the 3-position of 17 was designed to increase steric bulk around the proposed catalophore, a tactic known to enhance the enantioselectivity of reactions catalyzed by BINOL derivatives.12
Toward the synthesis of 17, the known (R)-BINOL 1413 was first converted into 15 by monotriflation followed by nickel-mediated cross-coupling with potassium cyanide (Scheme 1). Reduction of the nitrile and amidine formation provided 16. Unfortunately, all of our efforts to convert the phenol moiety into the thiocarbamate 17 were unsuccessful.
Scheme 1.

Attempted synthesis of catalyst 17
The initial disappointment notwithstanding, we queried whether 16, which has an acidic phenolic hydroxyl group, might itself serve as a suitable catalyst for bromolactonizations.8a The first step toward testing this hypothesis involved identifying reaction conditions that gave minimal amounts of background reaction. In our initial screenings, we discovered that stirring solutions of 5-phenyl-4(E)-pentenoic acid (18) in PhMe/CH2Cl2 (1:1) containing 2,4,4,6-tetrabromobenzo-quinone (TBCO) at temperatures less than −40 °C for 14 h gave negligible amounts of racemic lactones 19a and 19b.
Having discovered conditions under which a background reaction was insignificant, a solvent screen was performed for the bromolactonization of the olefinic acid 18 promoted by TBCO in the presence of 16 (10 mol %) (Table 1) to identify conditions that provided 19a with high regioselectivity and enantiomeric ratio (er). Although reactions in pure toluene or THF proceeded with low conversion and regioselectivity (Table 1, entries 1 and 2), use of CH2Cl2 led to furnished 19a in good yield and with good enantioselectivity (Table 1, entry 3). We then examined mixtures of toluene and CH2Cl2 (Table 1, entries 4–6) and discovered that a mixture (2:1) of toluene and CH2Cl2 provided 19a with excellent regioselectivity (15:1) and a high enantioselectivity (97:3 er) (Table 1, entry 5). Notably, similar combinations of a polar solvent (CH2Cl2 or CHCl3) and a nonpolar solvent (toluene or hexane) have been used in a number of other enantioselective halolactonizations.5,6
Table 1.
Solvent Screening for Bromolactonizationa

| ||||
|---|---|---|---|---|
| Entry | Solvent | Yield (%)b | 19a:19b | erd |
| 1 | PhMe | trace | 4:1 | ND |
| 2 | THF | ca. 20 | 1:1 | ND |
| 3 | CH2Cl2 | 64 | 15:1 | 91:9 |
| 4 | PhMe/CH2Cl2 (1:1) | 75 | 15:1 | 96:4 |
| 5 | PhMe/CH2Cl2 (2:1) | 95 | 15:1 | 97:3 |
| 6 | PhMe/CH2Cl2 (4:1) | 50 | 15:1 | 97:3 |
Reactions run on 0.1 mmol scale.
Yield of mixture of 19a,b.
Regioselectivity determined by 1H NMR spectra of the crude reaction mixtures.
er determined by chiral HPLC; absolute stereochemistry of 19a was assigned based upon correlation of optical rotation with reported value;5c the absolute stereochemistry of 19b was assigned based upon working model (vide infra);
ND = not determined.
Subsequent to these exploratory studies, we found that N-bromosuccinimide (NBS) and 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) could also be employed as brominating agents to furnish 19a with similarly high enantioselectivities and yields. The fact that the enantioselectivity of bromolactonization with catalyst 16 is independent of the brominating reagent is unlike what is observed with other organic catalysts.5 Moreover, this observation has mechanistic implications and suggests that the intermediate bromonium ion might be formed by delivery of Br+ from an N-bromo amidine that is generated in situ rather than directly from the original brominating reagent (vide infra).14 When we sought to recover 16 for reuse, we were somewhat surprised to isolate the 6-bromo analog 20 rather than the original catalyst 16. Because 16 underwent facile bromination to give 20 under the conditions for bromolactonization, we believe that 20 is the dominant active catalyst in bromolactonization reactions. This fortuitous finding led to the identification of 20 as an effective organic catalyst to promote enantioselective brominations, although we rarely prepared and used it for this purpose, and iodolactonizations (vide infra).8b
 |
(2) |
Modification of Original Catalyst Design
Although we established that 16 catalyzed the bromolactonization of 18 to give 19a with high regio- and enantioselectivity, we found that it promoted the bromolactonization of 21, a common test substrate for bromolactonizations,5 to give 22 with reduced enantioselectivity (86:14 er) (Table 2, entry 1). Perhaps not surprisingly, we also showed that preformed 20 catalyzed the bromolactonization of 21 with the same enantioselectivity as 16 (Table 2, entry 2). In order to identify analogs of 16 and 20 that might be superior catalysts, we prepared the series of analogs 23a–i (Figure 2), and surveyed their suitability for promoting the enantioselective bromolactonization of 21.
Table 2.
Effects of Catalyst Modificationsa

| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | R1 | R2 | R3 | R4 | R5 | erb |
| 1 | 16 | Ph | H | Me | H | H | 86:14 |
| 2 | 20 | Ph | H | Me | H | Br | 86:14 |
| 3c | 23a | Ph | H | Me | H | NO2 | NR |
| 4 | 23b | H | H | Me | H | H | 84:16 |
| 5 | 23c | Ph3Si | H | Me | H | H | 74:26 |
| 6 | 23d | TRIPd | H | Me | H | H | 50:50 |
| 7 | 23e | Ph | H | Me | Ph | H | 85:15 |
| 8 | 23f | Ph | H | Ph | H | H | 82:18 |
| 9 | 23g | H | H | Ph | H | H | 87:13 |
| 10 | 23h | H | H | t-Bu | H | H | 62:38 |
| 11 | 23i | H | Me | Me | H | H | 50:50 |
Figure 2.

General design for analogs of catalyst 16.
We first evaluated the effect of placing an electron withdrawing group on the binaphthyl scaffold, and we found that 23a, the 6-nitro analog of 16, did not catalyze the bromolactonization of 21 (Table 2, entry 3). We then probed the consequence of varying the steric requirements of substituents at the 3- and 3′-positions of the binaphthyl moiety as well as on the amidine group (Figure 2). In our initial design of catalyst 16, the phenyl ring was incorporated at C3 to help create a sterically biased catalytic pocket, which we believed would be important for obtaining high enantioselectivities. However, we discovered that bromolactonization of 21 with 23b (R1 = H, Table 2, entry 4), which lacks the phenyl group at the 3-position, gave 22 with nearly identical selectivity as 16 (84:16 er vs. 86:14 er) (Table 2, entry 3). On the other hand, increasing the size of the R1 substituent to triphenylsilyl (23c, Table 2, entry 5) or 2,4,6-triisopropylphenyl (23d, Table 2, entry 6) led to a significant erosion of enantioselectivity. Introducing a phenyl group at the 3-position adjacent to the amidine group led to 23e, which catalyzed the bromolactonization of 21 with selectivities comparable to that of the parent catalyst 16 (Table 2, entry 7)
We also briefly examined the effects of altering the substituent R3 on the amidine group upon the bromolactonization of 21. Although replacing the methyl group in 16 (Table 2, entry 8) and 23b (Table 2, entry 9) with a phenyl group did not significantly affect the enantioselectivities, the presence of a tert-butyl group 23h (R3 = t-Bu) led to significant drop in selectivity (62:38 er, Table 1, entry 10). In order to assess whether the bifunctional nature of the catalyst was critical for inducing enantioselectivity, the acidic phenolic group in 23b was methylated to give 23i. This modification led to complete loss of enantioselectivity (Table 2, entry 11), an observation that suggests that the acidic proton in the catalyst is imperative for enantioselectivity. Although screening a number of analogs of 16 led to several catalysts that promote the bromolactonization of 21 with comparable enantioselectivities, there were instances where 16 was superior, so it was used uniformly in subsequent experiments.
Application to Other Halolactonizations
At the time of our investigations, no catalyst was known to promote enantioselective halolactonizations with more than one halogen. We were thus inspired to ascertain whether 16 might catalyze other enantioselective halolactonizations. Chlorolactonization of 21 was first examined using 16 and 20 with N-chlorosuccinimide (NCS) and 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) in a variety of solvents. However, consistent with other literature reports, the reactions proceeded with low conversions and poor enantioselectivities.5b,d Owing to the poor enantioselectivity in these preliminary experiments, we did not further pursue chlorolactonizations.
We then turned our attention to iodolactonizations, and gratifyingly we discovered that the iodolactonization of 21 with N-iodosuccinimide (NIS) in the presence of 16 in a mixture (2:1) of toluene and CH2Cl2 at −40 °C delivered the iodolactone 24 (93:7 er). Although this reaction was sluggish, we found that the corresponding reaction with the brominated catalyst 20 was somewhat faster (Table 3, entry 1). Because of the operational advantage associated with using 20 as the catalyst, we screened several temperatures and conditions to identify a standard set of reaction parameters to establish the scope of this process. We discovered that iodolactonizations of 21 with NIS in the presence of 20 proceeded with comparable yields and enantioselectivities at temperatures between −10 and −40 °C, but there was a slight erosion in enantioselectivity when the reaction was performed at 0 °C (Table 3, entries 1–4). Not surprisingly, reaction times decreased significantly with increasing temperature. Because Jacobsen had reported that use of iodine as an additive had a beneficial effect on iodolactonizations using 9 as the catalyst,6a we tested the effects of adding catalytic amounts of iodine. However, the presence of iodine (1 or 10 mol %) had little effect on the enantioselectivity of the iodolactonization of 21 (Table 3, entries 6 and 7). It is notable, however, that the effect of iodine as an additive was not consistent, and some reactions benefited from the addition of iodine.
Table 3.
Optimization of Iodolactonizationa

Substrate Scope of Halolactonizations
Having established that 16 and/or 20 catalyze highly regio- and enantioselective bromo- and iodolactonizations, we elucidated the scope of these reactions. 4-Substituted-4-pentenoic acids are commonly employed as test substrates to demonstrate the efficacy of enantioselective halolactonizations,5,6 so we examined the bromo- and iodolactonizations, respectively, of 21 and 25a–e (Table 4). Notably, the enantioselectivities obtained in these reactions with our new BINOL-derived catalysts are comparable to those obtained with other catalysts.5b,6a Although we did not fully evaluate electronic effects, the presence of an electron-withdrawing group on the aromatic ring appears to lead to a slight increase in enantioselectivity in bromolactonizations (Table 4, entries 1, 3, and 5); however, this effect was not observed for iodolactonizations (Table 4, entries 2 and 4). On the other hand, an electron-donating group (R = p-MeO-Ph, 25c) led to a significant reduction in enantioselectivity of the iodolactonization (Table 4, entry 6). This finding is consistent with literature reports that chlorolactonization4a and bromolactonization5b of 25c are even less enantioselective. In contrast to what we observed in the iodolactonization of 21 (vide supra) but consistent with the observations of Jacobsen,6a cyclization of 25c proceeded with somewhat higher enantioselectivity (er = 82:18) in the presence of iodine (Table 4, entry 6). This 4-Alkyl-4-pentenoic acids also underwent facile iodolactonizations, but steric effects are important, and the enantioselectivity is better for larger alkyl groups (Table 4, entries 7 and 8).
Table 4.
Halolactonization of 4-Substituted-4-Pentenoic Acidsa

| |||||
|---|---|---|---|---|---|
| Entry | Acids | R | Product | Yield (%) | erb |
| 1 | 21 | Ph | 22 | 99 | 86:14 |
| 2 | 21 | Ph | 24 | 89 | 93:7 |
| 3 | 25a | p-NC-Ph | 26a | 92 | 94:6 |
| 4 | 25a | p-NC-Ph | 27a | 73 | 90:10 |
| 5 | 25b | m-NC-Ph | 26b | 89 | 91:9 |
| 6c | 25c | p-MeO-Ph | 27c | 90 | 74:26 |
| 7 | 25d | Me | 27d | 96 | 65:35 |
| 8 | 25e | t-Bu | 27e | 91 | 83:17 |
Reactions run on 0.10 mmol scale. Conditions for bromolactonization: 10 mol % 16, 1.2 equiv TBCO in PhMe/CH2Cl2 (2:1) at −50 °C. Conditions for iodolactonization: 10 mol % 20, 1.2 equiv NIS in PhMe/CH2Cl2 (2:1) at −20 °C.
er determined by chiral HPLC; absolute stereochemistry of 22 and 24 were assigned by correlation of optical rotations with previously reported values,5b,6a and other assignments are based upon analogy.
Results obtained with 10 mol % I2, 93% yield, 82:18 er.
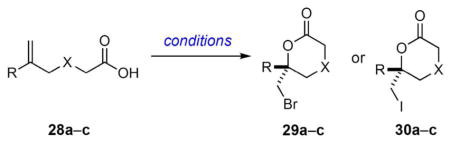
The next set of substrates examined was a small collection of 5-substituted-5-hexenoic acid derivatives that cyclize via 6-exo cyclizations to give δ-lactones (Table 5). Under the standard conditions, bromolactonization of 5-phenyl-5-hexenoic acid (28a) proceeded with 75% yield and 62:38 er (Table 5, entry 1), whereas the iodolactone 30a was obtained from 28a in slightly better enantioselectivity (76:24 er, Table 5, entry 2). Consistent with findings of Jacobsen,6a addition of iodine (10 mol %) led to a modest increase in enantioselectivity (85:15 er) to provide 30a. It should be noted at this juncture, however, that the presence of iodine did not always increase the selectivity of iodolactonizations catalyzed by 20 (vide infra). Although Jacobsen observed that the 6-exo cyclization of 28a and the 5-exo cyclization of 21 with 9 proceeded with opposite facial selectivities,6a use of 20 as a catalyst for these cyclizations gave products 24 and 30a that have the same absolute configuration. The 6-exo bromo- and iodolactonization of 28b delivered the bromolactone 29b and iodolactone 30b in 82:18 and 79:21 er, respectively (Table 5, entries 3 and 4); addition of iodine had little effect on the enantioselectivity of the iodolactonization. Halolactonization of 28c occurred to give bromo- and iododioxanones in 85:15 and 84:16 er, respectively, with a slight increase in selectivity being observed when iodine was present (Table 5, entries 5 and 6).
Table 5.
Halolactonization of 5-Substituted-5-Hexenoic Acidsa
| ||||||
|---|---|---|---|---|---|---|
| Entry | Acids | R | X | Product | Yield (%) | erb |
| 1 | 28a | Ph | CH2 | 29a | 78 | 62:38 |
| 2c | 28a | Ph | CH2 | 30a | 98 | 76:24 |
| 3 | 28b | Me | CH2 | 29b | 90 | 82:18 |
| 4d | 28b | Me | CH2 | 30b | 89 | 79:21 |
| 5 | 28c | Ph | O | 29c | 94 | 85:15 |
| 6e | 28c | Ph | O | 30c | 91 | 84:16 |
Reactions run on 0.10 mmol scale. Conditions for bromolactonization: 10 mol % 16, 1.2 equiv TBCO in PhMe/CH2Cl2 (2:1) at −50 °C. Conditions for iodolactonization: 10 mol % 20, 1.2 equiv NIS in PhMe/CH2Cl2 (2:1) at −20 °C.
er determined by chiral HPLC; absolute stereochemistry of 30a was assigned by correlation of optical rotation with previously reported value,6a and the other assignments are based upon analogy.
Results obtained with 10 mol % I2, 95% yield, 85:15 er.
Results obtained with 10 mol % I2, 90% yield, 80:20 er.
Results obtained with 10 mol % I2, 89% yield, 90:10 er.
In our initial exploratory studies, we discovered that 16 promoted the highly enantioselective cyclization of 18 to give 19a (Table 1). Although 19a contains a new stereocenter bearing a bromo group, the formation of similar chiral alkyl bromides via a 6-endo cyclization mode was known.5c However, enantioselective halolactonizations in which a new C–X bond is produced at a secondary carbon atom via a 5-exo cyclization was unprecedented. It is thus notable that the bromo- and iodolactonizations of a representative selection of 5-alkyl-4(Z)-pentenoic acids 31a–e catalyzed by 16 and 20 proceeded via a 5-exo cyclization to furnish the corresponding γ-lactones 32a–e and 33b–e in generally high yields and enantioselectivities (≥95:5 er) (Table 6, entries 1–9). The unsaturated acid 31a (R = Et, Table 6, entry 1) was the only substrate tested that cyclized with a modest er (85:15), suggesting that branching on the carbon atom attached directly to the double bond might play a role in determining selectivity. The γ-lactones 32a–e and 33b–e contain two contiguous stereogenic centers, one of which bears a carbon-halogen bond. The Z-olefin geometry appears to be an important prerequisite for high enantioselectivity because the corresponding 5-alkyl substituted-4(E)-pentenoic acids cyclized with only 50:50 to 78:22 er. The set of 5-aryl-4(Z)-pentenoic acids 31f–i also underwent facile iodolactonization via a 5-exo cyclization pathway to form γ-iodolactones 33f–i with high enantioselectivity (er ≥98:2) (Table 6, entries 10–13). Collectively, the cyclizations of 31a–i represent the first examples halolactonizations that occur via a 5-exo mode of ring closure to give products in which new C–O and C–X bonds are formed at contiguous stereogenic centers.
Table 6.
Halolactonization of 5-Substituted-4(Z)-Pentenoic Acidsa

| |||||
|---|---|---|---|---|---|
| Entry | Acids | R | Product | Yield (%) | erb |
| 1 | 31a | Et | 32a | 90 | 85:15 |
| 2 | 31b | i-Pr | 32b | 94 | 97:3 |
| 3 | 31b | i-Pr | 33b | 93 | 97:3 |
| 4 | 31c | i-Bu | 32c | 87 | 95:5 |
| 5 | 31c | i-Bu | 33c | 94 | 98:2 |
| 6 | 31d | t-Bu | 32d | 97 | 97:3 |
| 7 | 31d | t-Bu | 33d | 99 | 97:3 |
| 8 | 31e | Cy | 32e | 94 | 98.5:1.5 |
| 9 | 31e | Cy | 33e | 97 | 98:2 |
| 10 | 31f | Ph | 33f | 93 | 98.5:1.5 |
| 11 | 31g | p-NC-Ph | 33g | 95 | 99:1 |
| 12 | 31h | p-Cl-Ph | 33h | 89 | 98:2 |
| 13 | 31i | 2-Np | 33i | 94 | 98:2 |
Reactions run on 0.10 mmol scale. Conditions for bromolactonization: 10 mol % 16, 1.2 equiv TBCO in PhMe/CH2Cl2 (2:1) at −50 °C. Conditions for iodolactonization: 10 mol % 20, 1.2 equiv NIS in PhMe/CH2Cl2 (2:1) at −20 °C.
er determined by chiral HPLC; absolute stereochemistry of 32d, 33d, and 33f were determined by X-ray crystallography, and other assignments are based upon analogy.
One might anticipate that the Z-alkenes 31f–i, like the E-alkene 18, would be electronically biased to cyclize via a 6-endo mode. However, the observation that cis-aryl olefinic acids 31f–i underwent selective exo cyclizations is consistent with the transition state 35 that is favored relative to 34, because the unfavorable steric interaction depicted in 34 prevents benzylic stabilization of the putative iodonium ion, or the nascent carbocation, by the π electrons of the aryl group (Figure 3).15 Cyclization of conformer 35 through the 5-exo mode is thus favored for steric and entropic reasons. The regiochemical outcome in these cyclizations are consistent with those observed by Denmark in related bromoetherifications.2d,f
Figure 3.

The 6-endo product 36 is disfavored because the phenyl ring in 34 cannot adopt a conformation that stabilizes the transition state leading to 36, thereby leading preferentially to the formation of the 5-exo product 37.
In a closely related set of experiments, we examined the iodolactonizations of 6-substituted-5(Z)-hexenoic acids 38a–d and found that the corresponding iodo δ-lactones 39a–d were produced in high yields and enantioselectivities of ≥98:2 (Table 7). These cyclizations are also notable in that two adjacent stereogenic centers, one of which is a C–I bond, are generated.
Table 7.
Iodolactonization of 6-Substituted-5(Z)-Hexenoic Acidsa

| |||||
|---|---|---|---|---|---|
| Entry | Acids | R | Product | Yield (%) | erb |
| 1 | 38a | Ph | 39a | 89 | 99:1 |
| 2 | 38b | p-NC-Ph | 39b | 88 | 99:1 |
| 3c | 38c | 2-Np | 39c | 93 | 98.5:1.5 |
| 4c | 38d | t-Bu | 39d | 98 | 98:2 |
Reactions run on 0.10 mmol scale.
er determined by chiral HPLC; absolute stereochemistry assigned based on analogy with 33d and 33f.
Results obtained after 38 h at −20 °C.
We then investigated halolactonizations of a set of 5-aryl-4(E)-pentenoic acids comprising 18 and 40a– d. Unsaturated acids 18, 40a, and 40b underwent bromolactonizations in the presence of 16 and TBCO by 6-endo cyclizations to form the δ-lactones 19a, 41a, and 41b with high yields and enantioselectivities (Table 8, entries 1–3). On the other hand, iodolactonization of 18 afforded an inseparable mixture (1:1.3) of 6-endo and 5-exo products 42a and 43a, respectively (Table 8, entry 4). Iodolactonization of 40c (Ar = p-MeO-Ph) selectively gave the 6-endo product 42c (>20:1) with moderate enantioselectivity (86.5:13.5 er, Table 8, entry 5), whereas iodolactonization of 40d (Ar = p-NC-Ph) afforded the 5-exo product 43d with poor enantioselectivity (Table 8, entry 6). With the exception of the iodolactonization of 18, the preferred regioselectivity in these cyclizations is consistent with electronic effects.
Table 8.
Halolactonization of 5-Aryl-4(E)-Pentenoic Acidsa

| |||||
|---|---|---|---|---|---|
| Entry | Acids | Ar | Product | Yield (%) | erb |
| 1 | 18 | Ph | 19a | 94d | 98:2 |
| 2 | 40a | 2-Np | 41a | 97 | 96:4 |
| 3 | 40b | 2-thienyl | 41b | 92 | 94:6 |
| 4 | 18 | Ph | 42a+43a | 89e | 95:5e |
| 5 | 40c | p-MeO-Ph | 42c | 89 | 87:13 |
| 6f | 40d | p-NC-Ph | 43d | 94 | 58:42 |
Reactions run on 0.10 mmol scale. Conditions for bromolactonization: 10 mol % 16, 1.2 equiv TBCO in PhMe/CH2Cl2 (2:1) at −50 °C. Conditions for iodolactonization: 10 mol % 20, 1.2 equiv NIS in PhMe/CH2Cl2 (2:1) at −20 °C.
er determined by chiral HPLC; absolute stereochemistry of 19a, 41a, and 41b were assigned by correlation of optical rotations with previously reported values,5c whereas absolute stereochemistry of iodolactones assigned based on analogy.
The absolute stereochemistry of 43a and 43d (major enantiomer) were assigned based upon working model (vide infra).
Reaction run at −60 °C.
Combined yield; ratio of 1.0:1.3 for 42a/43a based upon 1H NMR analysis; er shown in parentheses is for 42a, and er for 43a is 52:48.
Reaction performed for 14 h at −20 °C and 48 h at −10 °C in CH2Cl2/PhMe (1:1).
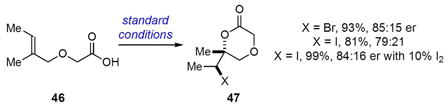
Inducing enantioselective halolactonizations of tri-substituted olefinic acids presents some significant challenges relative to 1,1- and 1,2-disubstituted substrates with only one report of a highly selective bromolactonization to date.5g For example, subjection of 44 and 46 to standard bromo- and iodolactonization conditions furnished the corresponding bromo- and iodolactones 45 (X = Br and I) and 47 (X = Br and I) in very good yields but with only moderate enantioselectivities (Equations 3 and 4). The enantioselectivity of the 6-exo cyclizations of 46 are slightly better than the 5-exo cyclizations of 44. The addition of 10 mol % iodine also led to modest increases in the enantioselectivities of the iodolactonizations of 44 and 46. The absolute stereochemistry of 45 and 47 is tentatively assigned based upon analogy with the cyclizations of other 4,4- and 5,5-disubsitituted olefinic acids (see Tables 4,5).
 |
(3) |
 |
(4) |
Working Model
It is evident from the foregoing discussions that we have established 16 and 20 as novel bifunctional catalysts to promote bromo- and iodolactonizations of a rather diverse set of substituted olefinic acids 48 to give the respective lactones 49–51, typically with excellent regio- and stereoselectivity (Scheme 2). Based on these results coupled with previous mechanistic proposals,1h,2f we have developed a tentative working model to rationalize the stereochemical outcomes for these processes.
Scheme 2.

Summary of enantioselective halolactonizations catalyzed by 16 and 20
As a starting point, we recognize that the bridged halonium ion intermediate could be stabilized by a Lewis acid/base interaction with either the basic amidine moiety or the naphthalenoxide ion of the catalyst 16 or 20. However, we are aware of no models for catalytic halocyclizations in which the halonium ion is stabilized by a phenoxide ion, whereas there are a number of mechanistic models wherein this ion is stabilized by a basic nitrogen atom. Indeed, amidines are known to transfer bromine from NBS to olefins.14 Accordingly, we presently prefer a model for halolactonizations catalyzed by 16 or 20 in which the halonium ion is stabilized by the amidine moiety, and there is a Bronsted acid/base interaction between the carboxylate group of the olefinic acid and the naphthol moiety. Assuming that these are the key ionic interactions in the transition states for the halolactonization, one may envision the competing transition states for the enantioselective cyclizations that deliver 49–51 as those shown in Figure 4A–C, respectively.
Figure 4.

Tentative working stereochemical model for halolactonization reactions of 48 (see Scheme 2) catalyzed by 16 and 20 showing adverse steric interactions (red double headed arrow) in disfavored transition states. (A) Competing transition states for halolactonizations of acids 48 (n = 1,2; R1 = R2 = H). (B) Competing transition states for halolactonizations of acids 48 (n = 1,2; R2 = R3 = H). (C) Competing transition states for halolactonizations of acids 48 (n=1; R1 = R3 = H).
An inspection of models of these transition states reveals dominating steric interactions between the benzylic methyene group that is attached to the amidine substituent and either the olefinic R3 group (e.g., 52, Figure 4A) or the allylic methylene group in the chain linking the double bond with the carboxylic acid moiety (e.g., 55 and 57, Figure 4B, C) in the disfavored transition states. The transition states 53, 54, and 56 leading to the observed products do not suffer such unfavorable steric interactions. In a recent study of bromocycloetherifications of 5-aryl-4(E/Z)-pentenols, Denmark noted that the catalyst directed the electrophilic bromine ion to one face of the olefin, irrespective of olefin geometry.2d,f Similarly, we find that the electrophilic bromo or iodo species is preferentially delivered from the same face of the olefin in all cyclizations of unsaturated pentenoic and hexenoic acids that lack a substituent on the olefinic carbon atom proximal to the carboxyl group (i.e., 48, n = 1,2 and R3 = H) (see Tables 6–8 and Figure 4B,C). When the internal carbon atom of the olefin is geminally substituted (i.e., 48, n = 1,2 and R3 = alkyl, aryl) (see Tables 4,5), however, attack of the electrophilic halogen moiety is from the opposite face.
Applications to Methodological Problems
At the outset of our studies, we identified a number of synthetic challenges that needed to be addressed in the context of enantioselective halolactonizations. The first of these was to induce transformations that created new C–Br of C–I at stereogenic centers exo to a ring; we had now achieved that goal. However, other problems that needed to be solved involved desymmetrization of prochiral substrates and kinetic resolution of racemic substrates, although there have been several reports of alternative solutions since we completed our own work.16
The potential of 16 as a catalyst to induce desymmetrization reactions is exemplified by the bromolactonization of 1,4-dihydrobenzoic acid (58) in the presence of 16 to furnish lactone 59 in 73:27 er (Equation 5). Although the enantioselectivity in this reaction was modest, the enantiopurity of 59 was easily improved to 99:1 er after a single recrystallization. This process was then applied in the syntheses of several synthons of the F ring subunit of the natural product kibdelone C (vide infra).9a Iodolactonization of 58 could be effected using catalyst 20, but the resultant iodolactone was unstable. On the other hand, the utility of 20 as a catalyst to promote kinetic resolutions of racemic olefinic acids is illustrated by the iodolactonizations of 60 and 62 to deliver the corresponding bicyclic iodolactones 61 and 63 in 83:17 er and 78:22 er, respectively (Equations 6 and 7). Both 61 and 63 are important intermediates for the syntheses of several natural products,17 including sieboldine A17b and phyllanthocin.17d
 |
(5) |
 |
(6) |
 |
(7) |
Another important extension of our halolactonization methodology is to the synthesis of enantioenriched cis-1,2-disubstituted epoxides. The direct epoxidation of Z-disubstituted olefins without directing groups remains a significant challenge,18 but we discovered that 4(Z)-alkenoic acids can be readily converted into the corresponding epoxides by a facile two-step process. For example, iodolactone 33e, which was obtained from the cis-disubstituted olefinic acid 32e (Table 6, entry 9), was converted to cis-epoxide 64 with no loss of enantiopurity simply upon treatment with Cs2CO3 in MeOH (Equation 8). A similar iodolactonization-epoxidation sequence was utilized in our concise enantioselective synthesis of (+)-disparlure (vide infra).9b
 |
(8) |
Application to Natural Product Synthesis: F-Ring Fragments of Kibdelone C
First isolated in 2007 by Capon and coworkers, kibdelone C (65) is a member of a novel family of polycyclic xanthone natural products that is produced by Kibdelosporangium sp, a rare soil actinomycete.19 Collectively, compounds belonging to this class possess an array of potentially useful biological activities that include low nanomolar GI50’s against a panel of human cancer cell lines as well as potent antibacterial and nematocidal properties. The promising biological activity of 65 coupled with its complex polycyclic architecture sparked considerable interest in the synthetic community, culminating in two successful enantioselective syntheses, (+)-kibdelone C completed by Porco,20 and (−)-kibdelone C completed by Ready.21 A key challenge for both groups was the synthesis of the chiral polyhydroxylated F ring (Scheme 3). Toward this end, for his synthesis of (+)-kibdelone C, Porco utilized 66a, which was prepared in 13 steps and 15% overall yield.20 Hudlicky subsequently reported an enzymatic approach that provided the related F ring precursor 66b in three steps, albeit in only 2.2% overall yield.22 Ready’s approach to (−)-kibdelone C required the use of fragment 67, which was in prepared in seven steps and 30% overall yield.21
Scheme 3.

F-ring fragments of kibdelone C used by Porco and Ready
With the view of developing a short entry to cyclohexene derivatives related to 66a,b and 67, we envisioned that the lactone ent-59 might be a viable intermediate (Scheme 4). Drawing from earlier work directed toward developing modified catalysts (vide supra), we initially queried whether the simplified catalyst ent-23b might deliver ent-59 with selectivity comparable to that of ent-16 (see Equation 5). However, desymmetrization of 58 using ent-23b afforded ent-59 with slightly diminished enantioselectivity (63:37 er), so we were relegated to using ent-16. In our initial experiments to desymmetrize 58 via an enantioselective bromolactonization using ent-16, the reaction was performed on a relatively small scale (100 mg) at low concentrations (0.03 M). However, we found this reaction was readily scalable to multigram quantities (2.5 g) at higher concentrations (0.1 M). Moreover, we discovered that the less expensive brominating agent N-bromosuccinimide (NBS) gave ent-59 in comparable yield and enantioselectivity to TBCO.
Scheme 4.

Synthesis of F-ring fragments of kibdelone C
Our initial foray to induce dihydroxylation of ent-59 was problematic because ent-59 was unstable in the basic media typical of standard Sharpless or Upjohn reaction conditions,23 so 68 was isolated in <35% yield and only <4:1 dr. After screening a variety of conditions, we discovered that dihydroxylation of ent-59 in the presence of citric acid proceeded with high selectivity from the less hindered face to deliver 68 in 60% yield (>20:1 dr).24 Treating the lactone 68 with methanol and potassium carbonate initiated a multistep cascade of reactions involving sequential transesterification and formation of the epoxide 69 that underwent spontaneous β-elimination to deliver triol 70, which was used without further purification. Formation of the acetonide moiety of the vicinal diol in 70 then gave 71, and subsequent protection of the distal hydroxyl group furnished 72 in 71% yield over three steps. Although we did not pursue the possibility, it is notable that 71, which is available in only four steps and 19% overall yield from 1,4-dihydrobenzoic acid (58) is also a potential F-ring precursor of (+)-kibdelone C.
Our initial plan was to directly install a halogen atom at the β-position of the enoate 72 to provide 73 (Equation 9), but numerous attempts to induce this transformation were unavailing. For example, treatment of 72 with a variety of reagents such as Br2 and triethylamine,25 NBS, NIS, or I(coll)2PF6 gave no detectable amounts of the desired product 73. Another approach to prepare 73 was inspired by Bartlett’s synthesis of (−)-2-chloroshikimic acid,26 but preliminary efforts to apply this procedure to the problem at hand were unsuccessful. A change of plan was needed, and we turned our attention to the synthesis of 77, a putative F-ring fragment similar to 67.
 |
(9) |
In the event, the methyl ester moiety of 72 was saponified to deliver the carboxylic acid 74 in 87% yield (Scheme 4). The use of CH3CN as solvent in this reaction proved to be critical, because when aqueous solvent mixtures of THF or MeOH were used, unavoidable epimerization of the C6 stereocenter was observed. Carbodiimide mediated coupling 74 with 75 afforded the intermediate thiohydroxamate ester 76 that was immediately subjected to a Barton–Hunsdiecker27 halodecarboxylation by irradiation in bromotrichloromethane to provide vinyl bromide 77 in 55% overall yield from 74. We thus completed a short synthesis of 77, an alternate F-ring synthon of kibdelone C, in eight steps and 9% overall yield from 1,4-dihydrobenzoic acid (58).
Applications to Natural Product Synthesis: (+)-Disparlure (78)
The gypsy moth, Lymantria dispar, ranks as one of the most devastating forest pests because of the extensive deforestation it causes during outbreaks in North America, Europe, and Asia. Because the resultant defoliation has serious ecological and economic ramifications, the gypsy moth has become a target for eradication.28 Disparlure, (Z)-7,8-epoxy-2-methyloctadecane, was identified in 1970 by Bierl as the sex pheromone emitted by the female gypsy moth,29 and it was later shown that (+)-disparlure (78) is the major component.30 Since that time, (+)-disparlure has been widely used as an attractant in traps as a means of controlling and managing gypsy moth populations. However, owing to its scarcity from natural sources, it has been necessary to rely upon synthesis to ensure adequate quantities of synthetic (+)-disparlure are available. Accordingly, (+)-disparlure has been the target of innumerable chemical investigations, and more than 50 syntheses of racemic and enantiopure disparlure have been reported.31 Despite these many successes, there remains significant opportunity for improvements that may result from advances in enantioselective synthesis. It thus occurred to us that an enantioselective iodolactonization/epoxide forming sequence might be applied to a concise enantioselective synthesis of (+)-disparlure.
Based upon the chemistry that is summarized in Equation 8, we envisaged that (+)-disparlure (78) could be prepared from the cis-epoxide 79, which would be produced upon the reductive opening of the enantioenriched iodolactone 80 followed by epoxide formation (Scheme 5). This plan was predicated upon the ability of our BINOL-derived catalyst ent-16 to convert the unsaturated acid 81 to the corresponding iodolactone with a high degree of enantioselectivity. At the outset of our studies, however, it was unclear whether 81 would be a suitable substrate, because we had previously observed that unbranched, alkyl-substituted cis-alkenoic acids such as 31a cyclized to iodolactones with only moderate enantioselectivity (85:15 er) (Table 6, Entry 1). Accordingly, the key question that needed to be resolved whether a long, n-alkyl chain in 81 might behave differently from the mere ethyl group in 31a.
Scheme 5.

Approach to (+)-disparlure (78)
Our first generation approach to prepare (+)-disparlure via 81 commenced with the alkylation of the lithiated alkyne 82 with 1-iododecane to give 83 in 85% yield (Scheme 6).32 When 83 was subjected to the conditions of the Jones oxidation, the tetrahydropyran (THP) protecting group was removed, and the resulting alcohol underwent direct oxidation to the carboxylic acid 84 in 82% yield. Initial attempts to partially reduce the alkyne moiety of 84 with Lindlar’s catalyst gave inseparable mixtures of cis- and trans-isomers of 81 (~10:1 cis:trans). However, semi-hydrogenation of 84 with P2-Ni and dihydrogen cleanly gave the desired cis-olefinic acid 81 without any contamination by the trans-isomer. Gratifyingly, the enantioselective iodolactonization of 81 in the presence of ent-16 proceeded to give the iodolactone 80 in 81% yield and 90:10 er. Inclusion of I2 (10 mol %) further enhanced the enantioselectivity of the reaction, furnishing 80 with 95:5 er. Although these experiments established critical proof-of-principle for the highly enantioselective iodolactonization of 81, we viewed the sequence as too lengthy, so we then sought to streamline the synthesis of the key Z-olefinic acid 81.
Scheme 6.

Enantioselective iodolactonization of 81
A more appealing approach to 81 was inspired by a previously reported one-step procedure for the synthesis of (Z)-4-alkenoic acids by a carbocupration of acetylene followed by reaction of the intermediate vinylcuprate with β-propiolactone (Equation 10).33 However, despite extensive experimentation using the cuprate 86, we were not able to reliably prepare 81 in acceptable yield, so we abandoned this route.
 |
(10) |
A reliable, two-step route to 81 was then developed that commenced with alkylation of the dianion generated from 87 with 1-iododecane (Scheme 7). Because it was necessary to employ an excess of 1-iododecane to optimize C-alkylation, considerable amounts of the corresponding ester by-product were unavoidably formed. However, any ester was saponified in situ upon completion of the reaction to provide 84 (82% yield), which was elaborated to 80 as shown in Scheme 6. The transformation of 80 into the epoxide 88 was then achieved in 62% overall yield using a simple one-pot process that involved sequential reduction of the lactone moiety with DIBAL-H and a Wittig reaction of the intermediate cyclic hemiacetal. Consistent with literature precedent,34 we discovered that selective reduction of the double bond in 88 by catalytic hydrogenation was problematic because reductive opening of the epoxide was a significant side reaction. After some experimentation with different catalysts and conditions, however, we eventually solved the problem by hydrogenating 88 using PtO2 as a catalyst and hexanes as solvent to deliver 78 in 90% yield. Notably, this synthesis of (+)-disparlure proceeded in 33% overall yield in only five steps from commercially available 87, making this the shortest catalytic enantioselective synthesis of 78 to date.35
Scheme 7.

Synthesis of (+)-disparlure (78)
SUMMARY
In summary, we have discovered and developed 16 and 20 as novel bifunctional organic catalysts to promote highly enantioselective and regioselective bromo- and iodolactonizations of a diverse array of olefinic acids. The only other catalyst reported to promote enantioselective halolactonizations with different halogens is (DHQD)2PHAL (4).4a,5h Catalysts 16 and 20, which uniquely incorporate a phenolic moiety as a Brønsted acid (or base) in the bifunctional catalyst design, were the first to incorporate a binaphthyl moiety as a chiral scaffold, although several such catalysts have been reported subsequently.6h,7b Significantly, these organic catalysts were the first to induce enantioselective halolactonizations via exo cyclization modes to give halolactones having carbon–halogen bonds at newly created stereogenic centers exocyclic to the lactone. Notable applications of this halolactonization methodology include desymmetrization, kinetic resolution, and epoxidation of simple Z-alkenes. For example, we showed that the desymmetrization of 58 could be performed on multigram scale, leading to short enantioselective syntheses of two potential F-ring synthons of kibdelone C. The utility of 16 was also exemplified by its application to the enantioselective synthesis of (Z)-1,2-alkyl epoxides from iodolactones generated from 4(Z)-pentenoic acids, and this process was applied to the shortest catalytic enantioselective synthesis of the insect pheromone (+)-disparlure to date.
Experimental
General
Solvents were purified before use as follows unless otherwise noted. Dichloromethane (CH2Cl2) and benzene were distilled from calcium hydride immediately prior to use. Tetrahydrofuran and diethyl ether were dried by filtration through two columns of activated, neutral alumina according to the procedure described by Grubbs.36 Methanol (MeOH), acetonitrile (MeCN), and dimethylformamide (DMF) were dried by filtration through two columns of activated molecular sieves, and toluene was dried by filtration through one column of activated, neutral alumina followed by one column of Q5 reactant. These solvents were determined to have less than 50 ppm H2O by Karl Fischer coulometric moisture analysis. Chloroform and acetone were distilled from CaSO4 and stored over 4 A molecular sieves. Reagents were reagent grade and used without purification unless otherwise noted. Trifluoromethanesulfonic anhydride (Tf2O) was freshly distilled from P2O5. Alkyl halides were passed through a plug of silica and distilled. KCN was crushed and heated at 80 °C under vacuum for 3 h prior to use. Zinc powder was activated and stored under argon. Triethylamine (Et3N), ethylene diamine, diisopropylethylamine (Hünig’s base), and diisopropylamine were refluxed with, distilled from, and stored over KOH. In nickel(0), palladium(0), and copper(I)-catalyzed reactions, all solvents were freed from oxygen by three freeze-pump-thaw cycles prior to use. All reactions were performed in flame-dried glassware under nitrogen or argon; reaction temperatures refer to the temperature of the cooling/heating bath.
Analytical HPLC separations were performed using a Pirkel Covalent (S,S) Whelk-O1 (Regis Technologies, Inc.), Chiralcel OD-H (Daicel Chemical Industries, Ltd.), Chiralcel OB (Daicel Chemical Industries, Ltd.) column, as indicated. Infrared (IR) spectra were obtained either neat on sodium chloride or as solutions in the solvent indicated and reported as wavenumbers (cm−1). Proton nuclear magnetic resonance (1H NMR) and proton-decoupled, carbon nuclear magnetic resonance (13C NMR) spectra were obtained at the indicated field as solutions in CDCl3 unless otherwise indicated. Chemical shifts are referenced to the deuterated solvent (e.g., for CDCl3, δ = 7.26 ppm and 77.0 ppm for 1H and 13C NMR, respectively) and are reported in parts per million (ppm, δ) relative to tetramethylsilane (TMS, δ = 0.00 ppm). Coupling constants (J) are reported in Hz and the splitting abbreviations used are: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; comp, overlapping multiplets of magnetically nonequivalent protons; br, broad; app, apparent.
The catalysts 16 and 20, and the bromolactones 22, 26a,b, 29c, 32a–e, 19a, 41a,b, 45 (X=Br), and 47 (X=Br) were prepared according to our previously reported procedures8a as were the iodolactones 33b–i, 39a–d, 42a+43a, 42c, 43d, and 57.8b The kibdelone fragments 67 and 739a and (+)-disparlure (74)9b were synthesized according to our previously reported procedures.
General Procedure for Enantioselective Bromolactonization
(S)-5-((S)-1-Bromo-2-methylpropyl)dihydrofuran-2(3H)-one (32b)
A solution of TBCO (0.197 g, 0.480 mmol) in CH2Cl2 (4 mL) was added dropwise to a solution of (Z)-6-methylhept-4-enoic acid 31b (0.057 g, 0.400 mmol) and catalyst 16 (0.018 g, 0.040 mmol) in toluene (8 mL) at −50 °C, and the solution was stirred for 14 h. The reaction was quenched with saturated aqueous Na2SO3 (10 mL), and the mixture was warmed to room temperature with vigorous stirring. The mixture was diluted with Et2O (40 mL) and water (10 mL), and the organic fraction was washed with 5% aqueous Na2CO3 (2 × 20 mL), dried (MgSO4), filtered and concentrated under reduced pressure. The crude residue was purified by column chromatography, eluting with CH2Cl2/toluene (2:1) to give 0.083 g (94%) of 32b as a clear, colorless oil: 1H NMR (CDCl3, 400 MHz) δ 4.76–4.71 (m, 1 H), 3.87 (dd, J = 6.0, 3.6 Hz, 1 H), 2.76–2.66 (m, 1 H), 2.59–1.48 (m, 1 H), 2.44–2.33 (m, 1 H), 2.22–2.06 (comp, 2 H), 1.10 (d, J = 6.4 Hz, 6 H); 13C NMR (CDCl3, 100 MHz)δ 176.4, 79.9, 66.8, 32.8, 28.3, 26.7, 21.2, 20.3; IR (neat) 2966, 2933, 2876, 1769, 1176, 1022, 908 cm−1; HRMS (ESI/Q-TOF) m/z [M + Na]+ Calcd for C8H13BrNaO2 242.9991; found 242.9992; [α]25 D +53.0 (c = 1.0, CHCl3); HPLC (210 nm): Whelk-O1 (20% i-PrOH/hexanes, 1.2 mL/min) 17.4 min (minor), 20.3 min (major); 97:3 er.
General Procedure for Enantioselective Iodolactonization
(S)-5-(Iodomethyl)-5-phenyldihydrofuran-2(3H)-one (24)
NIS (0.027 g, 0.12 mmol) (and I2 (0.0026 g, 0.010 mmol) for reactions with addition of 10 mol % iodine) was added to a solution of 4-phenyl-4-pentenoic acid 21 (0.018 g, 0.100 mmol) and catalyst 20 (0.005 g, 0.010 mmol) in toluene (2 mL) and CH2Cl2 (1 mL) at −20 °C, and the solution was stirred for 14 h. The reaction was quenched with saturated aqueous Na2S2O3 (1 mL), and the mixture was warmed to room temperature with vigorous stirring. The two layers were separated, and the aqueous layers were extracted with CH2Cl2 (2 × 5 mL), and the combined organic layers were dried (Na2SO4), filtered and concentrated under reduced pressure. The crude residue was purified by column chromatography, eluting with hexanes/EtOAc (5:1, v/v) to give 0.027 g (89%) of 24 as a clear, colorless oil: 1H NMR (CDCl3, 300 MHz) δ 7.42–7.33 (comp, 5 H), 3.65 (d, J = 11.1 Hz, 1 H), 3.61 (d, J = 11.1 Hz, 1 H), 2.83–2.68 (comp, 2 H), 2.66–2.45 (comp, 2 H),; 13C NMR (CDCl3, 75 MHz) δ 175.3, 140.6, 128.8, 128.6, 124.8, 86.0, 33.9, 29.2, 16.2; IR (neat) 2924, 2853, 1779, 1448, 1151, 700 cm−1; HRMS (CI/Magnetic Sector) m/z [M + H]+ Calcd for C11H12IO2 302.9882; Found 302.9881; [α]24D −15.3 (c = 1.0, CHCl3); HPLC (214 nm): OD-H (5% i-PrOH/hexanes, 1.0 mL/min) 19.0 min (minor), 23.8 min (major); 93:7 er.
(S)-4-(2-(Iodomethyl)-5-oxotetrahydrofuran-2-yl)benzonitrile (27a)
Isolated 0.024 g (73%) of 27a as a white solid: mp 154 °C (decomposition); 1H NMR (CDCl3, 300 MHz) δ 7.72 (d, J = 8.4 Hz, 2 H), 7.54 (d, J = 8.4 Hz, 2 H), 3.60 (s, 2 H), 2.85–2.74 (comp, 2 H), 2.64–2.51 (comp, 2 H); 13C NMR (CDCl3, 75 MHz) δ 174.4, 145.9, 132.6, 125.8, 118.1, 112.7, 85.3, 33.8, 29.0, 14.6; IR (neat) 2921, 2230, 1783, 1413, 1164, 1028, 841 cm−1; HRMS (CI/Magnetic Sector) m/z [M + H]+ Calcd for C12H11NO2I 327.9835; Found 327.9835; [α]25D −10.6 (c = 1.0, CHCl3); HPLC (231 nm): Whelk-O1 (3% CH3CN/20% i-PrOH/hexanes, 1.2 mL/min) 17.5 min (minor), 20.2 min (major); 90:10 er.
(S)-5-(Iodomethyl)-5-(4-methoxyphenyl)dihydrofuran-2(3H)-one (27c)
Isolated 0.030 g (90%) of 27c as a colorless oil; 1H NMR (CDCl3, 300 MHz) δ 7.34-7.31 (m, 2 H), 6.93-6.90 (m, 2 H), 3.83 (s, 3 H), 3.62 (d, J = 11.1 Hz, 1 H), 3.58 (d, J = 11.1 Hz, 1 H), 2.77–2.48 (comp, 4 H); 13C NMR (CDCl3, 75 MHz) δ 175.4, 159.6, 132.3, 126.2, 114.1, 86.0, 55.3, 33.7, 29.3, 16.6; IR (neat) 2957, 2837, 1779, 1514, 1253, 1178, 1029, 928, 834 cm−1; HRMS (CI/Magnetic Sector) m/z: [M + H]+ Calcd for C12H14O3I, 332.9988; Found 332.9991; [α] 25D −8.3 (c = 1.0, CHCl3); HPLC (230 nm): Whelk-O1 (3% CH3CN/20% i-PrOH/hexanes, 1.2 mL/min) 10.3 min (minor), 12.7 min (major); 74:26 er.
(R)-5-(Iodomethyl)-5-methyldihydrofuran-2(3H)-one (27d)
Isolated 0.023 g (96%) of 27d as a colorless oil; spectra was consistent with previsouly reported data for racemic 27d.37 1H NMR (CDCl3, 300 MHz) δ 3.42 (d, J = 10.5 Hz, 1 H), 3.37 (d, J = 10.5 Hz, 1 H), 2.71–2.64 (comp, 2 H), 2.38–2.78 (m, 1 H), 2.19–2.09 (m, 1 H); 13C NMR (CDCl3, 75 MHz) δ 175.7, 83.8, 32.9, 29.4, 25.9, 14.1; IR (neat) 2976, 1769, 1158 cm−1; HRMS (ESI) m/z calcd for [C6H9O2INa]+ (M+Na), 262.9539; found 262.9537; [α] 25D −21 (c = 1.0, CHCl3); HPLC (254 nm): Whelk-O1 (20% i-PrOH/hexanes, 1.2 mL/min) 17.8 min (minor), 19.2 min (major); 65:35 er.
(S)-5-(tert-Butyl)-5-(iodomethyl)dihydrofuran-2(3H)-one (27e)
Reaction executed on 0.128 mmol scale. Isolated 0.033 g (91%) of 27e as a colorless oil; 1H NMR (CDCl3, 400 MHz) δ 3.83 (d, J = 11.2 Hz, 1 H), 3.47 (d, J = 10.8 Hz, 1 H), 2.85–2.76 (m, 1 H), 2.55–2.36 (comp, 2 H), 2.16–2.08 (m, 1 H); 13C NMR (CDCl3, 100 MHz) δ 176.5, 89.9, 38.2, 30.2, 28.4, 25.3, 16.4; IR (neat) 2969, 1762, 1469, 1166, 986 cm−1; HRMS (ESI/QTOF) m/z [M + Na]+ Calcd for C9H15O2INa 305.0009; Found 305.0008; [α]25D −35 (c = 1.0, CHCl3 ); HPLC (256 nm): Whelk-O1 (20% i-PrOH/hexanes, 1.2 mL/min) 11.0 min (minor), 12.0 min (major); 83:17 er.
(S)-6-(Bromomethyl)-6-phenyltetrahydro-2H-pyran-2-one (29a)
Reaction executed on a 0.2 mmol scale. Isolated 0.042 g (75%) of 29a as a colorless oil, spectra consistent with the previously reported data.5e HPLC (217 nm) OD-H (1% i-PrOH/hexanes, 1 mL/min) 53.3 min (major), 71.5 min (minor); 62:38 er.
(R)-6-(Bromomethyl)-6-methyltetrahydro-2H-pyran-2-one (29b)
Reaction executed on a 0.2 mmol scale. Isolated 0.037 g of 29b as a colorless oil, spectra consistent with the previously reported data.5e HPLC (217 nm) OD-H (1% i-PrOH/hexanes, 1 mL/min) 62.28 min (minor), 64.89 min (major); 82:18 er.
(S)-6-(Iodomethyl)-6-phenyltetrahydro-2H-pyran-2-one (30a)
Isolated 0.031 g (98%) of 30a as a clear, colorless oil: 1H NMR (CDCl3, 300 MHz) δ 7.41–7.35 (m, 5 H), 3.58 (s, 2 H), 2.50–2.44 (comp, 2 H), 2.40-2.34 (comp, 2 H), 1.87–1.79 (comp, 1 H), 1.64–1.55 (m, 1 H); 13C NMR (CDCl3, 75 MHz) δ 170.4, 140.2, 129.0, 128.4, 125.2, 84.4, 32.0, 29.0, 17.6, 16.5; IR (neat) 2958, 1738, 1257, 1179, 1037, 701 cm−1; HRMS (ESI/Q-TOF) m/z [M + Na]+ Calcd for C12H13NaIO2, 338.9852; Found 338.9853; [α]26D +12.3 (c = 1.0, CHCl3); HPLC (214 nm): Whelk-O1 (20% i-PrOH/hexanes, 1.2 mL/min) 20.4 min (minor), 27.2 min (major); 76:24 er.
(R)-6-(Iodomethyl)-6-methyltetrahydro-2H-pyran-2-one (30b)
Isolated 0.024 (89%) of 30b as clear, slightly yellow oil. 1H NMR (CDCl3, 300 MHz) δ 3.4 (dd, J = 10.5, 14.5 Hz, 2 H), 2.62–2.42 (comp, 2 H), 2.16-2.02 (m, 1 H), 1.95–1.82 (comp, 3 H), 1.59 (s, 3 H); 13C NMR (75 MHz; CDCl3) δ 170.4, 81.9, 31.9, 29.4, 26.5, 16.9, 15.3; IR (neat) 2955, 1730, 1275, 1215, 1183, 1050 cm−1; HRMS (CI/Magnetic Sector) m/z [M + H]+ Calcd for C7H11IO2 (M+H)+, 254.9882; Found 254.9872; [α]25D −32 (c = 1.0, CHCl3); HPLC (259 nm): Whelk-O1 (20% i-PrOH/hexanes, 1.2 mL/min) 22.5 min (minor), 27.3 min (major); 79:21 er.
(R)-6-(Iodomethyl)-6-phenyl-1,4-dioxan-2-one (30c)
Isolated 0.029 g (91%) of 30c. 1H NMR (400 MHz, CDCl3) δ 7.50–7.35 (comp, 5 H), 4.43 (d, J = 17.7 Hz, 1 H), 4.32 (d, J = 12.9 Hz, 1 H), 4.28 (d, J = 17.7 Hz, 1 H), 4.19 (d, J = 12.9 Hz, 1 H), 3.70 (d, J = 11.2 Hz, 1 H), 3.66 (d, J = 11.2 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 166.0, 138.0, 129.0, 128.9, 125.1, 83.0, 69.6, 65.4, 10.7; HRMS (CI/Magnetic Sector) m/z [M + H]+ Calcd for C11H11IO3 317.9753; Found 317.9753; HPLC (210 nm): Whelk-O1 (20% i-PrOH/hexanes, 1.2 mL/min) 19.5 min (minor), 24.3 min (major); 84:16 er (without I2), 90:10 er (with 10 mol % I2)
(R)-5-((S)-1-Iodoethyl)-5-methyldihydrofuran-2(3H)-one (45, X = I)
Isolated 0.020 g (80%) of 45-I as a clear, colorless oil: 1H NMR (CDCl3, 300 MHz) δ 4.29 (q, J = 7.0 Hz, 1 H), 2.70-2.64 (comp, 2 H), 2.35-2.13 (comp, 2 H), 1.97 (d, J = 7.0 Hz, 3 H), 1.59 (s, 3 H). 13C NMR (75 MHz; CDCl3): δ 176.1, 88.7, 34.8, 34.6, 29.7, 23.6, 21.7; IR (neat) 2978, 2931, 1777, 1240, 1176, 1073 cm−1; HRMS (ESI/Q-TOF) m/z [M + Na]+ Calcd for C7H11INaO2276.9696, Found 276.9695; [α]25D −35.3 (c = 0.5, CHCl3); HPLC (259 nm): Whelk-O1 (20% i-PrOH/hexanes, 1.2 mL/min) 14.4 min (minor), 17.4 min (major); 68:32 er.
(R)-6-((S)-1-Iodoethyl)-6-methyl-1,4-dioxan-2-one (47, X = I)
Isolated 0.026 g (81%) of 47-I as a clear, colorless oil: 1H NMR (CDCl3, 300 MHz) δ 4.41 (q, J = 7.1 Hz, 1 H), 4.31 (s, 2 H), 4.05 (d, J = 12.6 Hz, 1 H), 3.96 (dd, J = 56.7, 12.6 Hz, 2 H), 3.87 (d, J = 12.6 Hz, 1 H), 2.02 (d, J = 7.1 Hz, 3 H), 1.61 (s, 3 H); 13C NMR (75 MHz; CDCl3): δ 166.6, 84.1, 71.8, 65.5, 28.2, 22.5, 19.0; IR (neat) 2985, 2932, 2872, 1749, 1273, 1102 cm−1; HRMS (ESI/Q-TOF) m/z [M + Na]+ Calcd for C7H11INaO3 292.9645; Found 292.9644; [α]24D +5.3 (c = 0.5, CHCl3); HPLC (259 nm): Whelk-O1 (20% i-PrOH/hexanes, 1.2 mL/min) 10.7 min (minor), 12.8 min (major); 79:21 er.
(1S,4S,5S)-4-Iodo-6-oxabicyclo[3.2.1]octan-7-one (63)
Reaction executed according to the general iodolactonization procedure with 0.5 equiv NIS. Isolated 0.011 g (43%) of 63 as a clear, colorless oil. Spectra consistent with the previously reported data.38 HPLC (210 nm) Chiracel OB (0.5% i-PrOH/hexanes, 1.05 mL/min) 12.0 min (major), 13.0 min (minor); 78:22 er.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM31077) and the Robert A. Welch Foundation (F-0652) for generous support of this research. DHP thanks the NIH for a postdoctoral fellowship (GM 96557). CRS thanks the NSF for support via an NSF-REU grant (CHE-1003947). We also thank Jonathan Kim (The University of Texas) for assistance with catalyst synthesis and Dr. Vincent Lynch (The University of Texas) for X-ray crystallography.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. characterization of new compounds, and X-ray crystal data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews, see: Chen G, Ma S. Enantioselective Halocyclization Reactions for the Synthesis of Chiral Cyclic Compounds. Angew Chem Int Ed. 2010;49:8306–8308. doi: 10.1002/anie.201003114.Tan CK, Zhou L, Yeung YY. Organocatalytic Enantioselective Halolactonizations: Strategies of Halogen Activation. Synlett. 2011:1335–1339.Snyder SA, Treitler DS, Brucks AP. Halonium-Induced Cyclization Reactions. 2011;44:27–40.Denmark SE, Kuester WE, Burk MT. Catalytic, Asymmetric Halofunctionalization of Alkenes-a Critical Perspective. Angew Chem Int Ed. 2012;51:10938–10953. doi: 10.1002/anie.201204347.Murai K, Fujioka H. Recent Progress in Organocatalytic Asymmetric Halocyclization. Heterocycles. 2013;87:763–805.Nolsøe JMJ, Hansen TV. Asymmetric Iodolactonization: An Evolutionary Account. Eur J Org Chem. 2014;2014:3051–3065.Kalyani D, Kornfilt DJ-P, Burk MT, Denmark SE. Lewis Base Catalysis in Organic Synthesis. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2016. Lewis Base Catalysis: A Platform for Enantioselective Addition to Alkenes Using Group 16 and 17 Lewis Acids (N → Σ*) pp. 1153–1212.Gieuw MH, Ke Z, Yeung YY. Lewis Base Catalyzed Stereo- and Regioselective Bromocyclization. Chem Rec. 2017;17:287–311. doi: 10.1002/tcr.201600088.
- 2.(a) Kang SH, Lee SB, Park CM. Catalytic Enantioselective Iodocyclization of γ-Hydroxy-Cis-Alkenes. J Am Chem Soc. 2003;125:15748–15749. doi: 10.1021/ja0369921. [DOI] [PubMed] [Google Scholar]; (b) Huang D, Wang H, Xue F, Guan H, Li L, Peng X, Shi Y. Enantioselective Bromocyclization of Olefins Catalyzed by Chiral Phosphoric Acid. Org Lett. 2011;13:6350–6353. doi: 10.1021/ol202527g. [DOI] [PubMed] [Google Scholar]; (c) Hennecke U, Müller CH, Fröhlich R. Enantioselective Haloetherification by Asymmetric Opening of Meso-Halonium Ions. Org Lett. 2011;13:860–863. doi: 10.1021/ol1028805. [DOI] [PubMed] [Google Scholar]; (d) Denmark SE, Burk MT. Enantioselective Bromocycloetherification by Lewis Base/Chiral Brønsted Acid Cooperative Catalysis. Org Lett. 2012;14:256–259. doi: 10.1021/ol203033k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhao Y, Jiang X, Yeung YY. Catalytic, Enantioselective, and Highly Chemoselective Bromocyclization of Olefinic Dicarbonyl Compounds. Angew Chem Int Ed. 2013;52:8597–8601. doi: 10.1002/anie.201304107. [DOI] [PubMed] [Google Scholar]; (f) Denmark SE, Burk MT. Development and Mechanism of an Enantioselective Bromocycloetherification Reaction via Lewis Base/Chiral Brønsted Acid Cooperative Catalysis. Chirality. 2014;26:344–355. doi: 10.1002/chir.22259. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ke Z, Tan CK, Chen F, Yeung YY. Catalytic Asymmetric Bromoetherification and Desymmetrization of Olefinic 1,3-Diols with C2-Symmetric Sulfides. J Am Chem Soc. 2014;136:5627–5630. doi: 10.1021/ja5029155. [DOI] [PubMed] [Google Scholar]; (i) Ke Z, Tan CK, Liu Y, Lee KGZ, Yeung YY. Catalytic and Enantioselective Bromoetherification of Olefinic 1,3-Diols: Mechanistic Insight. Tetrahedron. 2016;72:2683–2689. [Google Scholar]; (j) Böse D, Denmark SE. Investigating the Enantiodetermining Step of a Chiral Lewis Base Catalyzed Bromocycloetherification of Privileged Alkenes. Synlett. 2018;29:433–439. doi: 10.1055/s-0036-1590951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Zhou L, Chen J, Tan CK, Yeung YY. Enantioselective Bromoaminocyclization Using Amino-Thiocarbamate Catalysts. J Am Chem Soc. 2011;133:9164–9167. doi: 10.1021/ja201627h. [DOI] [PubMed] [Google Scholar]; (b) Chen F, Tan CK, Yeung YY. C2-Symmetric Cyclic Selenium-Catalyzed Enantioselective Bromoaminocyclization. J Am Chem Soc. 2013;135:1232–1235. doi: 10.1021/ja311202e. [DOI] [PubMed] [Google Scholar]; (c) Egart B, Lentz D, Czekelius C. Diastereoselective Bromocyclization of O-Allyl-NTosyl-Hydroxylamines. J Org Chem. 2013;78:2490–2499. doi: 10.1021/jo3026725. [DOI] [PubMed] [Google Scholar]; (d) Zhou L, Tay DW, Chen J, Leung GYC, Yeung YY. Enantioselective Synthesis of 2-Substituted and 3-Substituted Piperidines through a Bromoaminocyclization Process. Chem Commun. 2013;6:4412–4414. doi: 10.1039/c2cc36578b. [DOI] [PubMed] [Google Scholar]; (e) Mizar P, Burrelli A, Günther E, Söftje M, Farooq U, Wirth T. Organocatalytic Stereoselective Iodoamination of Alkenes. Chem Eur J. 2014;20:13113–13116. doi: 10.1002/chem.201404762. [DOI] [PubMed] [Google Scholar]; (f) Tripathi CB, Mukherjee S. Catalytic Enantioselective Halocyclizations Beyond Lactones: Emerging Routes to Enantioenriched Nitrogenous Heterocycles. Synlett. 2014;25:163–169. [Google Scholar]; (g) Cai Y, Zhou P, Liu X, Zhao J, Lin L, Feng X. Diastereoselectively Switchable Asymmetric Haloaminocyclization for the Synthesis of Cyclic Sulfamates. Chem Eur J. 2015;21:6386–6389. doi: 10.1002/chem.201500454. [DOI] [PubMed] [Google Scholar]; (h) Liu W, Pan H, Tian H, Shi Y. Enantioselective 6-exo-Bromoaminocyclization of Homoallylic N-Tosylcarbamates Catalyzed by a Novel Monophosphine-Sc(OTf)3 Complex. Org Lett. 2015;17:3956–3959. doi: 10.1021/acs.orglett.5b01779. [DOI] [PubMed] [Google Scholar]; (i) Jiang HJ, Liu K, Yu J, Zhang L, Gong LZ. Switchable Stereoselectivity in Bromoaminocyclization of Olefins: Using Brønsted Acids of Anionic Chiral Cobalt(III) Complexes. Angew Chem Int Ed. 2017;56:11931–11935. doi: 10.1002/anie.201705066. [DOI] [PubMed] [Google Scholar]
- 4.For chlorolactonizations, see: Whitehead DC, Yousefi R, Jaganathan A, Borhan B. An Organocatalytic Asymmetric Chlorolactonization. J Am Chem Soc. 2010;132:3298–3300. doi: 10.1021/ja100502f.Zhang W, Liu N, Schienebeck CM, Decloux K, Zheng S, Werness JB, Tang W. Catalytic Enantioselective Halolactonization of Enynes and Alkenes. Chem Eur J. 2012;18:7296–7305. doi: 10.1002/chem.201103809.Yousefi R, Ashtekar KD, Whitehead DC, Jackson JE, Borhan B. Dissecting the Stereocontrol Elements of a Catalytic Asymmetric Chlorolactonization: Syn Addition Obviates Bridging Chloronium. J Am Chem Soc. 2013;135:14524–14527. doi: 10.1021/ja4072145.Han X, Dong C, Zhou HB. C3-Symmetric Cinchonine-Squaramide-Catalyzed Asymmetric Chlorolactonization of Styrene-Type Carboxylic Acids with 1,3-Dichloro-5,5-Dimethylhydantoin: An Efficient Method to Chiral Isochroman-1-Ones. Adv Synth Catal. 2014;356:1275–1280.Denmark SE, Ryabchuk P, Burk MT, Gilbert BB. Toward Catalytic, Enantioselective Chlorolactonization of 1,2-Disubstituted Styrenyl Carboxylic Acids. J Org Chem. 2016;81:10411–10423. doi: 10.1021/acs.joc.6b01455.Ashtekar KD, Vetticatt M, Yousefi R, Jackson JE, Borhan B. Nucleophile-Assisted Alkene Activation: Olefins Alone Are Often Incompetent. J Am Chem Soc. 2016;138:8114–8119. doi: 10.1021/jacs.6b02877.Salehi Marzijarani N, Yousefi R, Jaganathan A, Ashtekar KD, Jackson JE, Borhan B. Absolute and Relative Facial Selectivities in Organocatalytic Asymmetric Chlorocyclization Reactions. Chem Sci. 2018;9:2898–2908. doi: 10.1039/c7sc04430e.
- 5.For bromolactonizations, see: Zhang W, Zheng S, Liu N, Werness JB, Guzei IA, Tang W. Enantioselective Bromolactonization of Conjugated (Z)-Enynes. J Am Chem Soc. 2010;132:3664–3665. doi: 10.1021/ja100173w.Zhou L, Tan CK, Jiang X, Chen F, Yeung YY. Asymmetric Bromolactonization Using Amino-Thiocarbamate Catalyst. J Am Chem Soc. 2010;132:15474–15476. doi: 10.1021/ja1048972.Tan CK, Zhou L, Yeung Y. Amino-Thiocarbamate Catalyzed Asymmetric Bromolactonization of 1,2-Disubstituted Olefinic Acids. Org Lett. 2011;13:2738–2741. doi: 10.1021/ol200840e.Tan CK, Le C, Yeung YY. Enantioselective Bromolactonization of Cis-1,2-Disubstituted Olefinic Acids Using an Amino-Thiocarbamate Catalyst. Chem Commun. 2012;48:5793–5795. doi: 10.1039/c2cc31148h.Jiang X, Tan CK, Zhou L, Yeung YY. Enantioselective Bromolactonization Using an S-Alkyl Thiocarbamate Catalyst. Angew Chem Int Ed. 2012;51:7771–7775. doi: 10.1002/anie.201202079.Murai K, Matsushita T, Nakamura A, Fukushima S, Shimura M, Fujioka H. Asymmetric Bromolactonization Catalyzed by a C3-Symmetric Chiral Trisimidazoline. Angew Chem Int Ed. 2010;49:9174–9177. doi: 10.1002/anie.201005409.Murai K, Nakamura A, Matsushita T, Shimura M, Fujioka H. C3-Symmetric Trisimidazoline-Catalyzed Enantioselective Bromolactonization of Internal Alkenoic Acids. Chem Eur J. 2012;18:8448–8453. doi: 10.1002/chem.201200647.Armstrong A, Braddock CD, Jones AX, Clark S, Braddock DC, Jones AX, Clark S. Catalytic Asymmetric Bromolactonization Reactions Using (DHQD)2PHAL-Benzoic Acid Combinations. Tetrahedron Lett. 2013;54:7004–7008.Aursnes M, Tungen JE, Hansen TV. Enantioselective Organocatalyzed Bromolactonizations: Applications in Natural Product Synthesis. J Org Chem. 2016;81:8287–8295. doi: 10.1021/acs.joc.6b01375.
- 6.For iodolactonizaions, see: Veitch GE, Jacobsen EN. Tertiary Aminourea-Catalyzed Enantioselective Iodolactonization. Angew Chem Int Ed. 2010;49:7332–7335. doi: 10.1002/anie.201003681.Tungen JE, Nolsøe JMJ, Hansen TV. Asymmetric Iodolactonization Utilizing Chiral Squaramides. Org Lett. 2012;14:5884–5887. doi: 10.1021/ol302798g.Dobish MC, Johnston JN. Achiral Counterion Control of Enantioselectivity in a Bronsted Acid-Catalyzed Iodolactonization. J Am Chem Soc. 2012;134:6068–6071. doi: 10.1021/ja301858r.Oderinde MS, Hunter HN, Bremner SW, Organ MG. Iodolactonization: Synthesis, Stereocontrol, and Compatibility Studies. Eur J Org Chem. 2012:175–182.Arai T, Kajikawa S, Matsumura E. The Role of Ni-Carboxylate during Catalytic Asymmetric Iodolactonization Using PyBidine-Ni(OAc)2. Synlett. 2013;24:2045–2048.Arai T, Sugiyama N, Masu H, Kado S, Yabe S, Yamanaka M. A Trinuclear Zn3(OAc)4-3,3′-bis(aminoimino)binaphthoxide Complex for Highly Efficient Catalytic Asymmetric Iodolactonization. Chem Commun. 2014;50:8287–8290. doi: 10.1039/c4cc02415j.Filippova L, Stenstrøm Y, Hansen TV. An Asymmetric Iodolactonization Reaction Catalyzed by a Zinc Bis-Proline-Phenol Complex. Tetrahedron Lett. 2014;55:419–422.Nakatsuji H, Sawamura Y, Sakakura A, Ishihara K. Cooperative Activation with Chiral Nucleophilic Catalysts and N-Haloimides: Enantioselective Iodolactonization of 4-Arylmethyl-4-Pentenoic Acids. Angew Chem Int Ed. 2014;53:6974–6977. doi: 10.1002/anie.201400946.Kristianslund R, Aursnes M, Tungen JE, Hansen TV. Squaramide Catalyzed Enantioselective Iodolactonization of Allenoic Acids. Tetrahedron Lett. 2016;57:5232–5236.
- 7.For fluorolactonizations, see: Parmar D, Maji MS, Rueping M. Catalytic and Asymmetric Fluorolactonisations of Carboxylic Acids through Anion Phase Transfer. Chem Eur J. 2014;20:83–86. doi: 10.1002/chem.201303385.Egami H, Asada J, Sato K, Hashizume D, Kawato Y, Hamashima Y. Asymmetric Fluorolactonization with a Bifunctional Hydroxyl Carboxylate Catalyst. J Am Chem Soc. 2015;137:10132–10135. doi: 10.1021/jacs.5b06546.Woerly EM, Banik SM, Jacobsen EN. Enantioselective, Catalytic Fluorolactonization Reactions with a Nucleophilic Fluoride Source. J Am Chem Soc. 2016;138:13858–13861. doi: 10.1021/jacs.6b09499.
- 8.(a) Paull DH, Fang C, Donald JR, Pansick AD, Martin SF. Bifunctional Catalyst Promotes Highly Enantioselective Bromolactonizations to Generate Stereogenic C-Br Bonds. J Am Chem Soc. 2012;134:11128–11131. doi: 10.1021/ja305117m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fang C, Paull DH, Hethcox JC, Shugrue CR, Martin SF. Enantioselective Iodolactonization of Disubstituted Olefinic Acids Using a Bifunctional Catalyst. Org Lett. 2012;14:6290–6293. doi: 10.1021/ol3030555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Klosowski DW, Martin SF. Enantioselective Synthesis of F-Ring Fragments of Kibdelone C via Desymmetrizing Bromolactonization of 1,4-Dihydrobenzoic Acid. Synlett. 2018;29:430–432. [Google Scholar]; (b) Klosowski DW, Martin SF. Synthesis of (+)-Disparlure via Enantioselective Iodolactonization. Org Lett. 2018;20:1269–1271. doi: 10.1021/acs.orglett.7b03911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Brown RS. Investigation of the Early Steps in Electrophilic Bromination through the Study of the Reaction with Sterically Encumbered Olefins. Acc Chem Res. 1997;30:131–137. [Google Scholar]; (b) Denmark SE, Burk MT, Hoover AJ. On the Absolute Configurational Stability of Bromonium and Chloronium Ions. J Am Chem Soc. 2010;132:1232–1233. doi: 10.1021/ja909965h. [DOI] [PubMed] [Google Scholar]; (c) Denmark SE, Burk MT. Lewis Base Catalysis of Bromo- and Iodolactonization, and Cycloetherification. Proc Natl Acad Sci. 2010;107:20655–20660. doi: 10.1073/pnas.1005296107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee DG, Srinivasan R. The Oxidation of Benzyldimethylamine by Bromine. Can J Chem. 1973;51:2546–2554. [Google Scholar]
- 12.Volla CMR, Atodiresei I, Rueping M. Catalytic C-C Bond-Forming Multi-Component Cascade or Domino Reactions: Pushing the Boundaries of Complexity in Asymmetric Organocatalysis. Chem Rev. 2014;114:2390–2431. doi: 10.1021/cr400215u. [DOI] [PubMed] [Google Scholar]
- 13.Shi M, Chen LH, Li CQ. Chiral Phosphine Lewis Bases Catalyzed Asymmetric Aza-Baylis-Hillman Reaction of N-Sulfonated Imines with Activated Olefins. J Am Chem Soc. 2005;127:3790–3800. doi: 10.1021/ja0447255. [DOI] [PubMed] [Google Scholar]
- 14.Amidines are known to promote the transfer of bromine from NBS to alkenes: Ahmad SM, Braddock DC, Cansell G, Hermitage SA, Redmond JM, White AJP. Amidines as Potent Nucleophilic Organocatalysts for the Transfer of Electrophilic Bromine from N-Bromosuccinimide to Alkenes. Tetrahedron Lett. 2007;48:5948–5952.Ahmad SM, Braddock DC, Cansell G, Hermitage SA. Dimethylformamide, Dimethylacetamide and Tetramethylguanidine as Nucleophilic Organocatalysts for the Transfer of Electrophilic Bromine From N-Bromosuccinimide to Alkenes. Tetrahedron Lett. 2007;48:915–918.
- 15.(a) Nicolaou KC, Prasad CVC, Somers PK, Hwang C-K. Activation of 6-Endo over 5-Exo Hydroxy Epoxide Openings. Stereoselective and Ring Selective Synthesis of Tetrahydrofuran and Tetrahydropyran Systems. J Am Chem Soc. 1989;111:5330–5334. [Google Scholar]; (b) Nicolaou KC, Prasad CVC, Somers PK, Hwang C-K. Activation of 7-Endo over 6-Exo Epoxide Openings. Synthesis of Oxepane and Tetrahydropyran Systems. J Am Chem Soc. 1989;111:5335–5340. [Google Scholar]
- 16.(a) Ikeuchi K, Ido S, Yoshimura S, Asakawa T, Inai M, Hamashima Y, Kan T. Catalytic Desymmetrization of Cyclohexadienes by Asymmetric Bromolactonization. Org Lett. 2012;14:6016–6019. doi: 10.1021/ol302908a. [DOI] [PubMed] [Google Scholar]; (b) Murai K, Matsushita T, Nakamura A, Hyogo N, Nakajima J, Fujioka H. Kinetic Resolution of β-Substituted Olefinic Carboxylic Acids by Asymmetric Bromolactonization. Org Lett. 2013;15:2526–2529. doi: 10.1021/ol401007u. [DOI] [PubMed] [Google Scholar]; (c) Wilking M, Mück-Lichtenfeld C, Daniliuc CG, Hennecke U. Enantioselective, Desymmetrizing Bromolactonization of Alkynes. J Am Chem Soc. 2013;135:8133–8136. doi: 10.1021/ja402910d. [DOI] [PubMed] [Google Scholar]; (d) Hennecke U, Wilking M. Desymmetrization as a Strategy in Asymmetric Halocyclization Reactions. Synlett. 2014;25:1633–1637. [Google Scholar]; (e) Wilking M, Daniliuc CG, Hennecke U. Monomeric Cinchona Alkaloid-Based Catalysts for Highly Enantioselective Bromolactonisation of Alkynes. Chem Eur J. 2016;22:18601–18607. doi: 10.1002/chem.201604003. [DOI] [PubMed] [Google Scholar]; (f) Knowe MT, Danneman MW, Sun S, Pink M, Johnston JN. Biomimetic Desymmetrization of a Carboxylic Acid. J Am Chem Soc. 2018;140:1998–2001. doi: 10.1021/jacs.7b12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Rohrig S, Hennig L, Findeisen M, Welzel P, Muller D. Use of Winterfeldt’s Template to Control the C-2′ Configuration in the Synthesis of Strigol-Type Compounds. Tetrahedron. 1998;54:3439–3456. [Google Scholar]; (b) Canham SM, France DJ, Overman LE. Total Synthesis of (+)-Sieboldine A. J Am Chem Soc. 2010;132:7876–7877. doi: 10.1021/ja103666n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trost BM, Kondo Y. An Asymmetric Synthesis of (+)-Phyllanthocin. Tetrahedron Lett. 1991;32:1613–1616. [Google Scholar]; (d) Martin SF, Colapret JA, Dappen MS, Dupre B, Murphy CJ. Application of Nitrile Oxide Cycloadditions to a Convergent, Asymmetric Synthesis of (+)-Phyllanthocin. J Org Chem. 1989;54:2209–2216. [Google Scholar]
- 18.Shi reported a modified fructose-derived ketone to promote enantioselective epoxidation of limited scope of cis-olefins with moderate enantioselectivity: Burke CP, Shi Y. Enantioselective Epoxidation of Nonconjugated. Cis-Olefins by Chiral Dioxirane. 2009;11:2007–2010. doi: 10.1021/ol901724v.Zhu Y, Wang Q, Cornwall RG, Shi Y. Organocatalytic Asymmetric Epoxidation and Aziridination of Olefins and Their Synthetic Applications. Chem Rev. 2014;114:8199–8256. doi: 10.1021/cr500064w.
- 19.Ratnayake R, Lacey E, Tennant S, Gill JH, Capon RJ. Kibdelones: Novel Anticancer Polyketides from a Rare Australian Actinomycete. Chem Eur J. 2007;13:1610–1619. doi: 10.1002/chem.200601236. [DOI] [PubMed] [Google Scholar]
- 20.(a) Sloman DL, Bacon JW, Porco JA. Total Synthesis and Absolute Stereochemical Assignment of Kibdelone C. J Am Chem Soc. 2011;133:9952–9955. doi: 10.1021/ja203642n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Winter DK, Sloman DL, Porco JA., Jr Polycyclic Xanthone Natural Products: Structure, Biological Activity and Chemical Synthesis. Nat Prod Rep. 2013;30:382. doi: 10.1039/c3np20122h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Butler JR, Wang C, Bian J, Ready JM. Enantioselective Total Synthesis of (−)-Kibdelone C. J Am Chem Soc. 2011;133:9956–9959. doi: 10.1021/ja204040k. [DOI] [PubMed] [Google Scholar]; (b) Rujirawanich J, Kim S, Ma AJ, Butler JR, Wang Y, Wang C, Rosen M, Posner B, Nijhawanc D, Ready JM. Synthesis and Biological Evaluation of Kibdelone C and Its Simplified Derivatives. J Am Chem Soc. 2016;138:10561–10570. doi: 10.1021/jacs.6b05484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Endoma-Arias MAA, Hudlicky T. A Short Synthesis of Nonracemic Iodocyclohexene Carboxylate Fragment for Kibdelone and Congeners. Tetrahedron Lett. 2011;52:6632–6634. [Google Scholar]; (b) Froese J, Endoma-Arias MAa, Hudlicky T. Processing of O-Halobenzoates by Toluene Dioxygenase. The Role of the Alkoxy Functionality in the Regioselectivity of the Enzymatic Dihydroxylation Reaction. Org Process Res Dev. 2014;18:801–809. [Google Scholar]
- 23.Kolb HC, VanNieuwenhze MS, Sharpless KB. Catalytic Asymmetric Dihydroxylation. Chem Rev. 1994;94:2483–2547. [Google Scholar]
- 24.Dupau P, Epple R, Thomas AA, Fokin VV, Sharpless KB. Osmium-Catalyzed Dihydroxylation of Olefins in Acidic Media: Old Process, New Tricks. Adv Synth Catal. 2002;344:421–433. [Google Scholar]
- 25.Nicolaou KC, Li A. Total Syntheses and Structural Revision of α- and β-Diversonolic Esters and Total Syntheses of Diversonol and Blennolide C. Angew Chem Int Ed. 2008;47:6579–6582. doi: 10.1002/anie.200802632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rich RH, Lawrence BM, Bartlett PA. Synthesis of (−)-2-Chloroshikimic Acid. J Org Chem. 1994;59:693–694. [Google Scholar]
- 27.(a) Barton D, Crich D, Motherwell W. The Invention of New Radical Chain Reactions .8. Radical Chemistry of Thiohydroxamic Esters - a New Method for the Generation of Carbon Radicals From Carboxylic-Acids. Tetrahedron. 1985;41:3901–3924. [Google Scholar]; (b) Carbain B, Hitchcock PB, Streicher H. New Aspects of the Hunsdiecker-Barton Halodecarboxylation-Syntheses of Phospha-Shikimic Acid and Derivatives. Tetrahedron Lett. 2010;51:2717–2719. [Google Scholar]
- 28.(a) Journal S, Sep N, Liebhold AM, Halverson JA, Northeasternforest GAE. Gypsy Moth Invasion in North America: A Quantitative Analysis. J Biogeogr. 1992;19:513–520. [Google Scholar]; (b) Liebhold AM, Tobin PC. Population Ecology of Insect Invasions and Their Management. Annu Rev Entomol. 2008;53:387–408. doi: 10.1146/annurev.ento.52.110405.091401. [DOI] [PubMed] [Google Scholar]
- 29.Bierl BA, Beroza M, Collier CW. Potent Sex Attractant of the Gypsy Moth: Its Isolation, Identification, and Synthesis. Science. 1970;170:87–89. doi: 10.1126/science.170.3953.87. [DOI] [PubMed] [Google Scholar]
- 30.(a) Iwaki S, Marumo S, Saito T, Yamada M, Kazumasa K. Synthesis and Activity of Optically Active Disparlure. J Am Chem Soc. 1974;96:7842–7844. [Google Scholar]; (b) Plettner E, Lazar J, Prestwich EG, Prestwich GD. Discrimination of Pheromone Enantiomers by Two Pheromone Binding Proteins from the Gypsy Moth Lymantria Dispar. Biochemistry. 2000;39:8953–8962. doi: 10.1021/bi000461x. [DOI] [PubMed] [Google Scholar]
- 31.For recent syntheses, see: Garg Y, Kumar Tiwari A, Kumar Pandey S. Enantioselective Total Synthesis of Cis-(+)- and Trans-(+)-Disparlure. Tetrahedron Lett. 2017;58:3344–3346.Bethi V, Kattanguru P, Fernandes RA. Domino Recombinant γ-Isomerization and Reverse Wacker Oxidation of γ-Vinyl-γ-Butyrolactone: Synthesis of (+)-trans-(–)- and (+)-Disparlures. Eur J Org Chem. 2014;2014:3249–3255.Herbert MB, Marx VM, Pederson RL, Grubbs RH. Concise Syntheses of Insect Pheromones Using Z-Selective Cross Metathesis. Angew Chem Int Ed. 2013;52:310–314. doi: 10.1002/anie.201206079.Kim SGG. Synthesis. 2009. Concise Total Synthesis of (+)-Disparlure and Its trans-Isomer Using Asymmetric Organocatalysis; pp. 2418–2422.Prasad KRR, Anbarasan P. Enantiodivergent Synthesis of Both Enantiomers of Gypsy Moth Pheromone Disparlure. J Org Chem. 2007;72:3155–3157. doi: 10.1021/jo070060o.
- 32.Avedissian H, Sinha SC, Yazbak A, Sinha A, Neogi P, Sinha SC, Keinan E. Total Synthesis of Asimicin and Bullatacin. J Org Chem. 2000;65:6035–6051. doi: 10.1021/jo000500a. [DOI] [PubMed] [Google Scholar]
- 33.Fujisawa T, Sato T, Kawara T, Naruse K. One-Pot Synthesis of (Z)-Alkenoic Acids. Chem Lett. 1980:1123–1124. [Google Scholar]
- 34.Sajiki H, Hattori K, Hirota K. Pd/C(en)-Catalyzed Regioselective Hydrogenolysis of Terminal Epoxides to Secondary Alcohols. Chem Commun. 1999:1041–1042. [Google Scholar]
- 35.To the best of our knowledge, the shortest enantioselective synthesis of (+)-disparlure was achieved in five steps through the use of a chiral sulfoxide auxiliary Satoh T, Oohara T, Ueda Y, Yamakawa K. A Novel Approach to the Asymmetric Synthesis of Epoxides, Allylic Alcohols, α-Amino Ketones, and α-Amino Aldehydes from Carbonyl Compounds through α,β-Epoxy Sulfoxides Using the Optically Active p-Tolylsulfinyl Group To Induce Chirality. J Org Chem. 1989;54:3130–3136.
- 36.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Safe and Convenient Procedure for Solvent Purification. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 37.Meng C, Liu Z, Liu Y, Wang Q. 4-(Dimethylamino)pyridine-Catalysed Iodolactonisation of γ,δ-Unsaturated Carboxylic Acids. Org Biomol Chem. 2015;13:6766–6772. doi: 10.1039/c5ob00806a. [DOI] [PubMed] [Google Scholar]
- 38.Raghavan S, Babu VS. Enantioselective Synthesis of Oseltamivir Phosphate. Tetrahedron. 2011;67:2044–2050. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.