

Graphical abstract
Keywords: Pentabrominated diphenyl ethers, Liver toxicity, Carcinogenic activity
Highlights
-
•
Pentabrominated diphenyl ether (PBDE) mixture was a multispecies carcinogen causing liver tumors in male and female rats and mice.
-
•
Hras or Ctnnb1 mutations characterized the PBDE-induced liver tumors.
-
•
PBDE-induced liver tumors increased with increasing PBDE exposure.
Abstract
Pentabrominated diphenyl ether (PBDE) flame retardants have been phased out in Europe and in the United States, but these lipid soluble chemicals persist in the environment and are found human and animal tissues. PBDEs have limited genotoxic activity. However, in a 2-year cancer study of a PBDE mixture (DE-71) (0, 3, 15, or 50 mg/kg (rats); 0, 3, 30, or 100 mg/kg (mice)) there were treatment-related liver tumors in male and female Wistar Han rats [Crl:WI(Han) after in utero/postnatal/adult exposure, and in male and female B6C3F1 mice, after adult exposure. In addition, there was evidence for a treatment-related carcinogenic effect in the thyroid and pituitary gland tumor in male rats, and in the uterus (stromal polyps/stromal sarcomas) in female rats. The treatment-related liver tumors in female rats were unrelated to the AhR genotype status, and occurred in animals with wild, mutant, or heterozygous Ah receptor. The liver tumors in rats and mice had treatment-related Hras and Ctnnb mutations, respectively. The PBDE carcinogenic activity could be related to oxidative damage, disruption of hormone homeostasis, and molecular and epigenetic changes in target tissue. Further work is needed to compare the PBDE toxic effects in rodents and humans.
1. Introduction
The pentabrominated diphenyl ethers (PBDEs) are used as flame retardants, and are found in human [1] and animal tissues (e.g. eagles [2], starlings [3], whales [4], fish [5]). The most prevalent PBDE congener in tissues is PBDE-47 (2,2′,4,4′-tetrabromodiphenyl ether) [[6], [7], [8]]. Environmental accumulation of PBDEs [[9], [10], [11]] occurs in densely populated areas as well as in remote locations [12]. Concentrations of PBDE in the atmosphere vary with temperature and humidity [13].
While PBDE use has been phased out in Europe [14] and in the United States [15], exposure to these chemicals continues [1], although some studies indicate that PBDE tissue levels are decreasing [16,17]. It is expected that PBDEs will continue to be released into the environment from disposal of TVs and computer parts [18]. Recent studies suggest that marine bacteria can synthesis PBDEs using flavin-dependent brominases [19]. Another source of PBDEs could be photodegradation of bromophenols [20]. Oral, dermal, and inhalation exposure are all possible routes of PBDE exposure [21].
PBDEs accumulate in tissues, in part, because these are fat soluble chemicals [[22], [23], [24]]. The whole-body half-life (representing primarily elimination from fat) increases with the number of bromine atoms (e.g. whole body half-life for commonly occurring PBDEs: PBDE-153 (six bromine atoms; 11.7 yrs.) > PBDE-99 (five bromine atoms; 5.4 yrs.) > PBDE-47 (four bromine atoms; 3 yrs.) [25]. The PBDE log Kow value, a measure of lipid solubility, also increases with increasing number of bromine atoms (e.g. log Kow 6.81, 7.32, 7.9 for PBDE-47, 99, 153, respectively) [26].
While PBDEs have limited genotoxic activity [26,27], PBDE exposure can disrupt liver and thyroid function. The toxicity of PBDE and its hydroxylated metabolites (e.g. 3-OH-BDE47, 6-OH-BDE47 [28]) may be related in part to the similarity of their structure to the thyroid hormone structure, and competition with T4 for the thyroid hormone receptor resulting in alteration of thyroid hormone transport and metabolism [29]. There is particular concern about early life PBDE exposure and the potential for disruption of thyroid hormone levels [30], because these chemicals occur in household dust [[31], [32], [33]], the hand-to-mouth behavior of young children [34], and the exposure to the infant from mother’s milk [35,36]. PBDEs in food, water, air, soil, sewage sludge, and at electronic waste sites allow for oral, dermal, and/or inhalation exposure [37,15]. PBDE exposure is linked to development toxicity in children [38].
This PBDE mixture (DE-71) oral gavage study examined the occurrence of treatment-related non-neoplastic and neoplastic lesions after an in utero/postnatal/adult exposure in rats, and after adult exposure in mice. The occurrence of treatment-related carcinogenic effects was studied in relationship to rat Ah receptor genotype, because activation of this receptor is involved in toxicity from other persistent organic pollutants [39,40]. We discuss how PBDE key characteristics could contribute to its toxic and carcinogenic activity.
2. Methods
2.1. Chemical and gavage formulation
The PBDE mixture (DE-71, lot 1550OK07A, Great Lakes Chemical Corp., El Dorado, AR) was identified using infrared (IR), proton and carbon-13 nuclear magnetic resonance (NMR) spectroscopy and gas chromatography (GC) with mass spectrometry (MS). The purity and composition were assessed using GC with flame ionization detection (FID). The DE-71 composition was: PBDE-99 (41.7%), PBDE-47 (35.7%), PBDE-100 (10.4%), PBDE-154 (3.6%), PBDE-153 (3.3%), and PBDE85 (2%); low levels of polybrominated dibenzodioxins and furans were also identified (approximately 7 × 10−6% by weight).
DE-71 formulations were prepared in corn oil to deliver by gavage to rats 0, 3, 15, or 50 mg/kg in a volume of 5 mL/kg body weight and to mice 0, 3, 30, 100 mg/kg at a volume of 10 mL/kg body weight. During the study, dose formulations were analyzed approximately every 2 months and were found to be within 10% of target concentration. Oral gavage dose formulation analysis and stability studies of DE-71 in corn oil was conducted using GC-FID. Stability of corn oil formulations were confirmed for at least for 46 days for formulations stored in amber glass containers sealed with Teflon lined lids at ambient temperature.
2.2. Experimental design
Wistar Han [Crl:WI(Han)] rat dams (referred to as Wistar Han rats) were obtained from Charles River Laboratories (Raleigh, NC) and B6C3F1/N mice from Taconic Farms, Inc. (Germantown, NY). Animals were stratified by weight then randomly assigned to dose groups to ensure equal initial mean weights across dose groups. Fifty B6C3F1/N mice (age 5–6 weeks of age) per dose group were dosed 5 days per week for 2-years. Twenty-five Wistar Han rat dams (12–13 weeks of age) per dose group were dosed (0, 3, 15 or 50 mg/kg/day) from gestation day (GD) 6 through postnatal day (PND) 21. At PND4 all litters were culled to 3 males and 3 females per litter; pups started on direct dosing (using the same dose as the dam) from PND12 – PND21. At PND22 the pups were assigned to the two-year study (generally 2 males and 2 females from 25 litters to each dose group), and received PBDE dosing 5 days/week for up to 2 years.
The top dose for the rat 2-year study was based on results from 3-month studies, which showed reduced body weights and liver lesions (marked liver hypertrophy; increased liver weight (∼ ↑1.7X) and liver enzyme levels) in rats at levels of 100 mg/kg and above. The top dose for mice was set at 100 mg/kg because of reduced survival and liver lesions (moderate to marked liver hypertrophy; increase in liver weight (∼↑3x) and enzyme levels) at 500 mg/kg) in the 13-week studies. The lower doses were selected to give a broader range of exposure [41].
Animals were housed by species and sex, two to three male rats per cage, five female rats per cage, one male mouse per cage, and five female mice per cage. Tap water and NTP-2000 diet (Zeigler Brothers, Inc. Gardners, PA) were made available ad libitum. The care of animals on this study was according to NIH procedures as described in the “The U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals, available from the Office of Laboratory Animal Welfare, National Institutes of Health, Department of Health and Human Services, RKLI, Suite 360, MSC 7982, 6705 Rockledge Drive, Bethesda, MD 20892-7982 or online at http://grants.nih.gov/grants/olaw/olaw.htm#pol. The use of the animals was approved by the Institute animal care and use committee.
2.3. Necropsy and pathology
Animals were euthanized with CO2 gas. All tissue collections, necropsies, tissue processing and histopathological examinations were performed according to NTP specifications (http://ntp.niehs.nih.gov/ntp/test_info/finalntp_toxcarspecsjan2011.pdf). NTP guidelines were used for all pathology nomenclature (http://ntp.niehs.nih.gov/nnl/). Histological examinations were performed by experienced board certified veterinary anatomic pathologists.
Complete necropsies were performed on all animals when moribund, when found dead, or at the end of the two-year exposure period. At necropsy, all organs and tissues were examined for grossly visible lesions. Tissues were preserved in 10% neutral buffered formalin (except eyes which were first fixed in Davidson’s solution, and testes and epididymis which were fixed in modified Davidson’s solution), embedded in paraffin, sectioned, and stained with hematoxylin and eosin. The following tissues were examined microscopically from male and/or female animals: gross lesions and tissue masses, adrenal gland, bone with marrow, brain, cervix, clitoral gland, esophagus, eyes, gallbladder (mice only), Harderian gland, heart, large intestine (cecum, colon, rectum), small intestine (duodenum, jejunum, ileum), kidney, liver, lungs, lymph nodes (mandibular and mesenteric), mammary gland, nose, ovary, pancreas, parathyroid gland, pituitary gland, preputial gland, prostate gland, salivary gland, skin, spleen, stomach (forestomach and glandular), testis with epididymis and seminal vesicle, thymus, thyroid gland, trachea, urinary bladder, uterus with cervix), and vagina. At terminal necropsy (at the 2-year exposure period) tissue samples were collected and stored frozen at −70 °C for use in molecular biology studies and for PBDE tissue level determinations.
Following completion of the studies, histopathology diagnoses were confirmed by NTP pathology peer reviews [42,43]. There were two pathology reviews of the female rat reproductive tissues termed the “original transverse review” and the “residual longitudinal review.” The original review involved a transverse section through each uterine horn, approximately 0.5 cm cranial to the cervix, and an evaluation of gross cervical and vaginal lesions. The residual longitudinal review involved collection and evaluation of remaining uteri, vaginas and cervices stored in formalin and sectioned in a longitudinal plane.
2.4. Statistical analysis
Survival was compared among dose groups using Tarone’s life table test to examine for dose-related trends [44] and Cox’s proportional hazards method for pairwise comparisons of each dose group to the control group [45]. The statistical analysis methods for the rats and mice differed to account for the possibility that rat littermates may have correlated endpoints
For the mice, Jonckheer’s test [46] was applied to body weights to determine if a dose-related trend was present (at p < 0.01). If a dose-related trend was present, mean body weights were compared to the control group using Williams’ test [47,48]; otherwise, Dunnett’s test [49] was used. The poly-3 test, which takes survival into account, was used to assess neoplastic and nonneoplastic lesion incidences [[50], [51], [52]]. When applied to all exposure groups, this test evaluates the significance of a dose-related trend in lesions; when applied to the control group and one exposure group, the test evaluates the significance of the pairwise difference of lesion incidence in the exposed group compared to the control group without correction for multiple testing.
The rats were exposed prenatally to DE-71 and usually two pups per sex per litter continued in the study for up to two years. Therefore, potential litter effects were controlled for in the statistical analyses of the rat study. For body weights, Johnkheere’s test was applied to determine if a dose-related trend was present (p < 0.01). Mixed effects analysis of variance was then applied to body weights, with Williams’ test (if a trend was present) or Dunnett’s test (if a trend was not present) used to compare each dosed group to the control group. Mixed effects logistic regression with litter ID as the random effect and a poly-3-weight adjustment for survival, was used to assess the effects of DE-71 on neoplastic and nonneoplastic lesion incidences.
2.5. Ah receptor (AhR) genotyping
Formalin fixed (FFPE) liver sections from female rats were taken from 60 control liver and 58 female rat liver tumor samples from the 50 mg/kg group (total of 118 female rat liver samples analyzed for AhR genotype). Formalin fixed kidney samples (from the some of the same animals used for liver sample collection) were also analyzed for AhR genotype (64 control and 58 female rats at 50 mg/kg). Genomic DNA (gDNA) was isolated from liver and kidney using the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA) per manufacturer’s procedure.
The AhR genotypes [39,40] were determined using QPCR and Sanger sequencing methods. A nested PCR product was used for QPCR. The nested PCR product was prepared to amplify the exon 10 region of interest that contained the AhR SNP to determine the genotype (wild, mutant, or heterozygous) for the single nucleotide polymorphism – SNP (mutation) [39,40]. Real time PCR was conducted with a TaqMan® platform in which [VIC/FAM] fluorescence was used to determine the genotype for each sample (adapted from Ref. [53]). The nested PCR product of each sample was analyzed with TaqMan® Genotyping Master Mix (Life Technologies) according to manufacturer’s procedure with the following cycling conditions (95 °C for 10 min, followed by 40 cycles of 95 °C for 20 s, and 66 °C for 1 min) on the ViiA™7 Real Time PCR System (Life Technologies). Upon completion of the PCR run, an endpoint Allelic Discrimination read was performed to determine the genotype for each sample (60 °C for 30 s). The ViiA™7 Real Time PCR System (Life Technologies) software assigned genotypes based on the [VIC/FAM] fluorescence of each sample. A genotype call was deemed 'Undetermined' if the Ct value was >35 or reported by the software. Further details on the AhR genotyping methods are found in the Data in Brief article [54].
2.6. Tumor mutation analysis
Genetic mutation studies were conducted on PBDE-induced liver tumors from male and female rats and mice to characterize the molecular nature of the tumors. FFPE mouse and rat hepatocellular tumors arising either spontaneously or from chronic PBDE exposure as well as age-matched normal liver tissue were used for mutation analyses. DE-71 exposed and control liver tissue were obtained from rats and mice in this study. The hepatocellular tumor tissues chosen for molecular biology analysis were based on their overall size and viability (minimal to no necrosis or hemorrhage observed microscopically) in order to maximize the amount and quality of DNA obtained from FFPE sections. Further details on the mutation analysis methods are found in the Data in Brief article [54].
2.7. Quantitation of PBDE-47, 99, and 153 tissue levels
PBDE-47, 99, and 153 levels were determined in liver, fat, and plasma of male and female rats (stored at −80 °C until analyses) and in liver and fat of male (except for 30 mg/kg) and female mice using validated analytical methods. Lipid levels in liver and adipose in male and female rats was determined following extraction with chloroform:methanol (1:1, v/v), hydrolysis with acid, reaction with vanillin reagent followed by detection at 490 nm to allow reporting tissue levels as μg/g lipid in the tissue. There were 4–15 tissue samples available per dose level/sex/species. The methods for the PBDE tissue analysis are found in the Data in Brief Article [54].
3. Results
3.1. Body weights and survival
There were no treatment-related effects on littering parameters after in utero/postnatal PBDE exposure in rats (Table 1). Final mean body weights of 0, 3, 15, and 50 mg/kg male rats and 3 and 15 mg/kg female rats were similar to controls (Table 2). Final mean body weights of 50 mg/kg female rats were reduced. Mean body weights of 30 and 100 mg/kg male and female mice were reduced relative to controls, and this reduction in body weight was attributed to the development of treatment-related liver tumors. Early deaths in rats and mice were attributed to the carcinogenic effects of the PBDE exposure (Table 2). All high dose mice were sacrificed by 18 months because at this time the mice that had died early were all diagnosed with multiple hepatocellular tumors, and the remaining mice in this group were moribund.
Table 1.
Littering Parameters for Wistar Han rats after in utero/postnatal exposure.
| Dose (mg/kg) | 0 | 3 | 15 | 50 |
|---|---|---|---|---|
| Time-Mated Females (GD 6) | 62 | 52 | 52 | 62 |
| Females Pregnant (%) | 54 (87%) | 42 (81%) | 43 (83%) | 51 (82%) |
| Dams with Evidence of Pregnancy Not Delivering (%) | 2 (4%) | 1 (2%) | 4 (9%) | 2 (4%) |
| Dams with Litters on PND 0 (%) | 52 (96%) | 41 (98%) | 39 (91%) | 49 (96%) |
| Dams, Moribund or Natural Deaths | 0 | 0 | 0 | 0 |
| Litters Post-Standardization (PND 4) | 36 | 29 | 28 | 37 |
| Post-Weaning Allocation | ||||
| F1 Males – Core (litters) | 50 (29a) | 50 (25) | 50 (25) | 50 (29) |
| F1 Females – Core (litters) | 50 (30) | 50 (25) | 50 (25) | 50 (28) |
There were no significant differences among the groups for any of the litter parameters.
Number of litters from which pups were taken at PND22 to populated the 50 rats/sex/dose group that went on for 2 year studies.
Table 2.
Survival and final mean body weight for male and female rats and mice.
| Dose (mg/kg) | 0 | 3 | 15 | 50 |
|---|---|---|---|---|
| Male rats | ||||
| Survivala | 36 | 35 | 38 | 27* |
| Mean body wt. (g)b % control |
673 ± 14 – |
669 ± 12 99% |
695 ± 14 103% |
678 ± 18 101% |
| Female rats | ||||
| Survival | 38 | 40 | 34 | 38 |
| Mean body wt. (g) % control |
390 ± 9 – |
374 ± 11 96% |
358 ± 10 92% |
314 ± 12† 81% |
| Dose (mg/kg) | 0 | 3 | 30 | 100 |
|---|---|---|---|---|
| Male mice | ||||
| Survival | 30 | 33 | 31 | 18 month termination |
| Mean body wt. (g) % control |
45.7 ± 1.5 – |
46.4 ± 1.1 102% |
38.2 ± 1.1† 84% |
|
| Female mice | ||||
| Survival | 33 | 35 | 38 | 18 month termination |
| Mean body wt. (g) % control |
51.7 ± 1.4 – |
53.4 ± 1.4 103% |
48.5 ± 1.2 94% |
Survival is significantly reduced compared to the control group, by Cox’s life table pairwise comparison, p < 0.05.
Body weight is significantly reduced compared to the control group, by Dunnett’s test, p < 0.05.
Number of animals surviving to end of the study of 50 per group starting the study.
Mean ± standard error of the mean.
3.2. Treatment-related lesions
3.2.1. Rat liver lesions
There were treatment-related benign and malignant liver tumors in male and female rats and mice (Table 3). Treatment-related nonneoplastic lesions of the liver also occurred in male and female rats and mice (Table 4) including centrilobular hepatocellular hypertrophy and fatty change. The hypertrophy was characterized by enlarged hepatocytes with granular eosinophilic cytoplasm and enlarged nuclei. The hepatocytes with fatty change were characterized by vacuolization within the cytoplasm that displaced the nucleus peripherally.
Table 3.
Treatment-related carcinogenic effects in male and female Wistar Han rats and B6C3F1 mice.
| Dose (mg/kg) | 0 | 3 | 15 | 50 |
|---|---|---|---|---|
|
Male rats liver, Number examined |
49 | 50 | 50 | 50 |
| Hepatocellular adenoma | 3* (6%) |
2 (4%) |
4 (8%) |
8 (16%) |
| Hepatocellular carcinoma | 0 | 0 | 0 | 2 (4%) |
| Hepatocellular adenoma or carcinoma | 3** (6%) |
2 (4%) |
4 (8%) |
9* (18%) |
| Hepatocholangioma | 0* | 0 | 0 | 2 |
| Hepatocholangioma, hepatocellular adenoma or hepatocellular carcinomaa | 3** (6%) |
2 (4%) |
4 (8%) |
11* (22%) |
| Male rats thyroid, Number examined | 45 | 45 | 48 | 46 |
| Thyroid gland: follicular cell adenoma |
1* (2%) |
3 (7%) |
2 (4%) |
6* (13%) |
| Thyroid gland: follicular cell carcinoma |
0 (0%) |
2 (4%) |
1 (2%) |
0 (0%) |
| Thyroid gland follicular cell adenoma or carcinomab |
1 (2%) |
5 (11%) |
3 (6%) |
6* (13%) |
|
Male rats pituitary, Number examined |
49 | 49 | 50 | 50 |
| Pituitary gland: pars distalis or unspecified site adenomac | 19** (39%) |
12 (24%) |
22 (44%) |
35** (70%) |
|
Female rats liver, Number examined |
50 | 49 | 50 | 47 |
| Hepatocellular adenoma | 3** (6%) |
2 (4%) |
8 (16%) |
16** (34%) |
| Hepatocellular carcinoma | 0** | 0 | 1 (2%) |
6** (13%) |
| Hepatocellular adenoma or carcinoma | 3** (6%) |
2 (4%) |
8 (16%) |
17** (36%) |
| Cholangiocarcinoma | 0* | 0 | 0 | 2 (4%) |
| Hepatocholangioma | 0** | 0 | 0 | 8** (17%) |
| Hepatocholangioma, hepatocellular adenoma, or hepatocellular carcinomad | 3** (6%) |
2 (4%) |
8 (16%) |
21** (45%) |
|
Female rats uterus, Number examined |
50 | 50 | 50 | 49 |
| Uterus, metaplasia, squamous | 0 | 2 (4%) |
5* (10%) |
6* (12%) |
| Cervix, squamous hyperplasia | 2** (4%) |
3 (6%) |
4 (8%) |
8* (16%) |
| Uterus polyp, stromal | 4 (8%) |
12* (24%) |
11* (22%) |
9 (18%) |
| Uterus, stromal sarcoma | 0 | 0 | 1 (2%) |
0 |
| Uterus stromal polyp or stromal sarcomae | 4 (8%) |
12* (24%) |
12* (24%) |
9 (18%) |
| Vaginal polyp | 0* | 0 | 0 | 2 (4%) |
| Dose (mg/kg) | 0 | 3 | 30 | 100 |
|---|---|---|---|---|
|
Male mice liver, Number examined |
50 | 50 | 50 | 50 |
| Hepatocellular adenoma | 23** (46%) |
35* (70%) |
49** (98%) |
40** (80%) |
| Hepatocellular carcinoma | 18** (36%) |
15 (30%) |
30* (60%) |
45** (90%) |
| Hepatoblastoma | 1** (2%) |
1 (2%) |
16** (32%) |
5* (10%) |
| Hepatocellular adenoma, carcinoma, or hepatoblastomaf |
31** (62%) |
40 (80%) |
49** (98%) |
47** (94%) |
|
Female mice liver, Number examined |
50 | 49 | 50 | 49 |
| Hepatocellular adenoma | 5** (10%) |
7 (14%) |
32** (64%) |
46** (94%) |
| Hepatocellular carcinoma | 4** (8%) |
2 (4%) |
6 (12%) |
27** (55%) |
| Hepatocellular adenoma or carcinomag |
8** (16%) |
8 (16%) |
33** (66%) |
47** (96%) |
*P ≤ 0.05; **P ≤ 0.01.
Historical Data:
Male rats – liver tumors
aHistorical controls, gavage corn oil:a3/99 (3.1% ± 4.3%), range.0%–6%.
aHistorical controls, all routes:a4/299 (1.4% ± 2.5%), range.0%–6%.
Male rats – Thyroid tumors
bHistorical controls, gavage corn oil: 4/95 (4.1% ± 2.7%), range.2%–6%.
bHistorical controls, all routes: 5/295 (1.7% ± 2.4%), range.0%–6%.
Male rats – Pituitary tumors
cHistorical controls, gavage corn oil: 40/99 (40.4% ± 2.3%),range 39%-42%.
cHistorical controls, all routes:101/298 (33.9% ± 5.7%), range.28%–42%.
Female rats – liver tumors
dHistorical controls, gavage corn oil:4/100 (4.0% ± 2.8%), range.2%–6%.
dHistorical controls, all routes:6/300 (2.0% ± 2.2%), range.0%–6%.
Female rats - uterus
Historical controls, all routes.
e29/194 (15.1% ± 6.3%), range.8%–22%.
Male mice – liver tumors
fHistorical controls, gavage corn oil: 221/300 (73.7% ± 6.1%), range.62%–78%.
fHistorical controls, all routes: 545/700 (77.3% ± 8.3%), range.62%–90%.
Female mice – liver tumors
gHistorical controls, gavage corn oil: 85/300 (28.3% ± 10.2%), range.16%–40%.
gHistorical controls, all routes: 320/698 (45.9% ± 21.9%), range.16%–82%.
Table 4.
Selected treatment-related nonneoplastic lesions of liver and thyroid in male and female rats and mice.
| Dose (mg/kg) | 0 | 3 | 15 | 50 |
|---|---|---|---|---|
| Male rat | ||||
| Liver Numbered examined |
49 | 50 | 50 | 50 |
| Hepatocyte hypertrophy | 1 (2%)** [1.0]a |
44 (88%)** [2.1] |
50 (100%)** [3.0] |
50(100 %)** [3.8] |
| Basophilic focus | 16(33%) | 21(42%) | 11(22% | 11(22%) |
| Clear cell focus | 39 (80%)** | 37 (74%) | 35 (70%) | 29 (58%)* |
| Eosinophilic focus | 3 (6%)** | 3 (6%) | 12 (24%)* | 15 (30%)** |
| Hyperplasia, nodular | 0 | 0 | 3 | 0 |
| Fatty change | 32 (65%)** | 37 (74%) | 48 (96%)** | 48 (96%)** |
| Cholangiofibrosis | 0 | 0 | 1 (2%) | 1 (2%) |
| Inflammation chronic | 1 (2%)* | 2 (4%) | 0 | 5 (10%) |
| Pigmentation | 0 ** | 0 | 1 (2%) | 6 (12%)* |
| Oval cell hyperplasia | 0 * | 0 | 2 (4%) | 3 (6%) |
| Thyroid, Number examined |
45 | 45 | 48 | 46 |
| Thyroid gland follicle hypertrophy | 1**(2%) [1.2] |
26**(58%) [1.4] |
34**(70%) [2.0] |
23**(50%) [2.4] |
| Female Rat | ||||
| Liver Number examined |
50 | 49 | 50 | 47 |
| Hepatocyte hypertrophy |
0 ** | 48 (98%)** [1.9] |
49 (100%)** [3.0] |
45 (96%)** [3.9] |
| Basophilic focus | 44 (88%) | 43 (88%) | 40 (80%) | 33 (70%)* |
| Clear cell focus | 35 (70%) | 21 (43%) | 26 (52%) | 31 (66%) |
| Eosinophilic focus | 5 (10%)** | 7 (14%) | 21 (42%)** | 31 (66%)** |
| Fatty change | 15 (30%)** | 12 (24%) | 28 (56%)** | 39 (83%)** |
| Cholangiofibrosis | 0 ** | 0 | 0 | 3 (6%) |
| Hyperplasia, nodular | 0 ** | 0 | 2 (4%) | 7 (15%)** |
| Hepatocyte Necrosis | 4 (8%)* | 2 (4%) | 1 (2%) | 8 (17%) |
| Bile duct cyst | 2 (4%)* | 2 (4%) | 5 (10%) | 7 (15%)* |
| Oval cell hyperplasia | 1 (2%) | 3 (6%) | 3 (6%) | 10 (21%)** |
| Uterus Numbered examined |
50 | 50 | 50 | 49 |
| Vaginal polyp | 0 | 0 | 0 | 1(2%) |
| Uterus histiocytic sarcoma | 0* | 0 | 0 | 2(4%) |
| Uterus metaplasia | 0** | 0 | 1(2%) | 4(8%)* |
| Cervix Hyperplasia | 0* | 0 | 0 | 2 (4%) |
| Thyroid, Number examined | 45 | 49 | 47 | 42 |
| Thyroid gland Follicle hypertrophy |
8(16%)** [1.1 |
17 (34%) [1.3] |
22**(47%) [1,4] |
35**(83%) [1.9] |
| Dose (mg/kg) | 0 | 3 | 30 | 100 |
|---|---|---|---|---|
| Male mouse | ||||
| Liver Number examined |
50 | 49 | 50 | 49 |
| Centrilobular, hepatocyte, hypertrophy | 0** | 28(56%)** [1.6] |
46(92%)** [3.8] |
48(96%)** [3.9] |
| Fatty change | 17(34%) | 25(50%) | 17(34%) | 5(10%) |
| Necrosis, focal | 2(4%)* | 2(4%) | 16(32%)** | 2(4%) |
| Kupffer cell pigmentation | 5(10%)** | 15(305)* | 33(66%)** | 25(50%)** |
| Clear cell focus | 10 | 13 | 20* | 7 |
| Thyroid Number examined |
||||
| Thyroid gland Follicle, hypertrophy | 25** [1.3] |
35* [1.5] |
41* [1.5] |
45* [2.4] |
| Female mouse | ||||
| Liver Number examined |
50 | 49 | 50 | 49 |
| Centrilobular hypertrophy | 0** | 7(14%)** [1.0] |
45(90%)** [2.0] |
47(96%)** [3.6] |
| Eosinophilic focus | 3(6%)** | 2(4%) | 16(325)** | 15(31%)** |
| Fatty change | 18(36%)** | 18(37%) | 39(78%)** | 20(41%)* |
| Hemorrhage | 4(8%)* | 1(2%) | 4(8%) | 5(10%) |
| Necrosis, focal | 1(2%)* | 1(2%) | 4(8%) | 3(6%) |
| Kupffer cell pigmentation | 3(6%)** | 10(20%)* | 24(48%)** | 27(55%)** |
| Thyroid gland Number examined |
50 | 49 | 48 | 47 |
| Thyroid gland Follicle, hypertrophy |
24 [1.3] |
31 [1.5] |
37** [1.5] |
42** [2.4] |
Trend statistic under control column; pairwise statistic under dose columns: *p ≤ 0.05 **p ≤ 0.01.
Average severity greade of lesions in affected animals: 1 = minimal, 2-mild, 3 = moderate, 4 = marked.
In rats, the incidences of hepatocellular neoplasms (hepatocellular adenomas and hepatocellular carcinomas) were significantly increased in both male and female rats exposed to 50 mg/kg. Additionally, the incidence of hepatocholangiomas was significantly increased in females exposed to 50 mg/kg. Hepatocellular adenomas typically consisted of well-circumscribed masses that caused compression of the surrounding hepatic parenchyma. These neoplasms were composed of a uniform population of hepatocytes and lacked the normal lobular architecture. Some adenomas displayed a little cellular atypia, but it was less common and less pronounced than that seen in the hepatocellular carcinomas. Hepatocellular carcinomas were also invasive and less well-demarcated than adenomas, and frequently contained areas of necrosis and blood-filled spaces. Their growth pattern was characterized by thickened hepatic trabeculae, composed of at least three cell layers wide compared with single-cell wide hepatic cords found in normal liver.
Hepatocholangiomas are thought to arise from cells that can differentiate into either hepatocytes or biliary cells. Hepatocholangiomas were distinguished from hepatocellular adenomas by the presence of dilated nonneoplastic bile ducts, by the increased number of bile ducts within hepatocholangiomas, and by the fact that hepatocellular adenomas typically lack bile ducts. The biliary epithelial component of the hepatocholangiomas was cuboidal, in contrast to the typically flattened epithelium found in biliary cysts.
Cholangiocarcinoma occurred in two 50 mg/kg females and cholangiofibrosis occurred in three other 50 mg/kg female rat. Cholangiofibrosis is believed to be a precursor lesion to cholangiocarcinoma [55]. Cholangiocarcinoma consisted of an irregular, relatively large, noncircumscribed lesion that effaced and invaded normal liver parenchyma. The lesion consisted of fibrous connective tissue stroma containing numerous atypical bile ducts, which frequently contained mucinous material and cellular debris. The epithelium forming the atypical bile ducts was often discontinuous, consisted of large atypical cells and intestinal goblet cells, and displayed degenerative changes. The distinction between cholangiofibrosis and cholangiocarcinoma was primarily based upon liver invasion and size. Cholangiocarcinoma and cholangiofibrosis are uncommon in control rats, but have been observed in previous NTP studies of rats exposed to hepatic carcinogens. Consequently, the observations of these neoplasms in the livers of rats exposed to the PBDE mixture were considered related to exposure.
3.2.2. Mouse liver lesions
Hepatocellular adenomas were significantly increased in incidence in all dosed groups of male mice, and in the 30 and 100 mg/kg groups of female mice. In addition, there were significantly increased incidences of hepatocellular carcinoma in 30 and 100 mg/kg males and 100 mg/kg females; and increased incidences of hepatoblastomas in 30 and 100 mg/kg males.
Hepatocellular adenomas were discrete, well-circumscribed lesions that compressed surrounding parenchyma. They were composed of irregular plates of hepatocytes, which were most commonly eosinophilic, but also basophilic or vacuolated. Central veins and portal areas were generally absent. Hepatocellular carcinomas were large lesions, frequently with areas of necrosis, which caused compression of, and invasion into, surrounding parenchyma. Typically, hepatocellular carcinomas were characterized by hepatocytes forming trabeculae that were at least three cells thick, although some of the areas of carcinomas were of a solid pattern of growth. Cells within the hepatocellular carcinomas ranged from eosinophilic to basophilic in staining, and displayed marked pleomorphism and an increased mitotic rate.
Hepatoblastomas were composed of small cells with scant cytoplasm and hyperchromatic, oval nuclei. Cells were often arranged in rows around variably sized vascular spaces. Most often, hepatoblastomas arose from within a hepatocellular adenoma or carcinoma, and when this occurred only the hepatoblastoma was recorded.
3.2.3. Treatment-related findings in other tissues
When combined, the incidence of thyroid follicular cell adenoma and follicular cell carcinoma were significantly increased in 50 mg/kg male rats. The thyroid follicular cell adenoma was a discrete, compressive mass composed of proliferation of follicular cells forming complex papillary infoldings and irregular follicular structures. The cells were slightly pleomorphic and larger than normal follicular cells. Follicular cell carcinoma displayed more disorganized growth patterns and cellular pleomorphism and invaded the thyroid gland capsule. Treatment-related thyroid gland follicle hypertrophy was also present in male rats (Table 4), and progression from thyroid gland follicle hypertrophy, to follicular cell adenoma and follicular cell carcinoma occurs in rodents. These two neoplasms (thyroid gland follicular cell adenoma and carcinoma) are frequently combined for statistical purposes. There were no treatment-related thyroid tumors in mice or female rats, although the incidence and severity of thyroid gland follicle hypertrophy was increased in the treated groups (Table 4).
In male rats, there was a statistically significant increase in the incidence of adenomas of the pars distalis of the pituitary gland. Pituitary pars distalis adenomas were typically composed of sheets of chromophobes, although scattered acidophils and basophils could be found in some neoplasms. Variable-sized blood vessels, some angiectatic, as well as hemorrhage, were present in many of the neoplasms. The adenomas were usually well-demarcated masses that caused compression of the surrounding parenchyma, with larger neoplasms causing dorsal compression of the hypothalamic region of the brain. There were no increases in pituitary gland tumors in female rats or in male and female mice.
In female rats, there were significantly increased incidences of uterine stromal polyps in the 3 and 15 mg/kg groups based upon examination of the longitudinal cuts and the cross sections of the uterus. In addition, two animals in the 50 mg/kg group had vaginal polyps. The uterine stromal polyps were solitary exophytic nodules that projected into the uterine lumen. They were covered by normal-appearing endometrial surface epithelium, and supported by a broad stalk of endometrial stroma, blood vessels, and a few entrapped glands. Polyps in the vagina were similar to those found in the uterus. Stromal sarcomas were composed of spindle-shaped cells with indistinct cytoplasmic borders that invaded into the uterine wall. Squamous metaplasia was recorded in the uterus when the normal cuboidal to columnar epithelium lining the uterus or endometrial glands was replaced by stratified squamous epithelium. In the cervix, squamous hyperplasia was characterized by increased layers of the normally present squamous epithelium. There were no increases in uterine tumors in female mice.
3.3. Mutations in rat and mouse liver tumors
Mutational analysis of PBDE-induced rat and mouse liver tumors for Hras and/or Ctnnb1 mutations was conducted to better characterize PBDE effects (Table 5, Table 6). In this study, the rat hepatocellular tumors resulting from PBDE mixture exposure (DE-71) demonstrated mutations exclusively within Hras codon 60 [20% (7/35)], rather than at the common target Hras codon 61 found in spontaneous mouse and human liver tumors. All the mutations were the same G to A transition (Gly to Asp). In the rat, Ctnnb1 mutations were fewer [11% (4/35)], more diverse, identified between codons 33 to 40, and consisted of transitions and transversions. No Hras or Ctnnb1 mutations were noted in the spontaneous hepatocellular adenomas in rats. There were no significant differences in the incidences of mutations between male and female rats (data not presented) and hence the combined data from both male and female rats are presented in Table 5.
Table 5.
Hras and Ctnnb1 Mutations in liver tumors in male and female rats.
| DE-71 | Mutation frequency |
Hras Cdn60 | Ctnnb1 | |
|---|---|---|---|---|
| Hras (%) | Ctnnb1 (%) | GGT to GAT | Cdn 33-40 | |
| Control – Non-tumor Liver | 0/10 (0%) | 0/10 (0%) | 0 | 0 |
| Control – Spontaneous Liver Tumors | 0/5 (0%) | 0/5 (0%) | 0 | 0 |
| 3 mg/kg | 1/3 (33%) | 0/3 (0%) | 1 | 0 |
| 15 mg/kg | 1/12 (8%) | 1/12 (8%) | 1 | 1 |
| 30 mg/kg | 5/20 (25%) | 3/20 (15%) | 5 | 3 |
| All DE-71 treated groups (Combined) | 7/35 (20%) | 4/35 (11%) | 7 | 4 |
Male and female Wistar Han rats were exposed to 0, 3, 15, or 50 mg/kg PBDE mixture for 2 years. Silent mutations not included; Compared to mice, the hepatocellular carcinoma (HCC) incidence was lower in the rats and hence, hepatocellular adenomas (HCA) were also included in the mutation analysis. The rat HCA and HCC included in this study included: controls (5 HCA); 3 mg/kg (3 HCA); 15 mg/kg (11 HCA and 1 HCC); 50 mg/kg (14 HCA, 6 HCC (3 HCC had Hras mutations, 1 HCC had Ctnnb1 mutation)).
Table 6.
Hras and Ctnnb1 mutations in hepatocellular carcinomas in mice.
| DE-71 | Mutation frequency |
Hras Cdn61 (CAA) |
Ctnnb1 | |||
|---|---|---|---|---|---|---|
| Hras (%) | Ctnnb1 (%) | AAA | CGA | CTA | Cdn 15-46 | |
| Control – Non-tumor liver | 0/8 (0%) | 0/8 (0%) | 0 | 0 | 0 | 0 |
| Control – Spontaneous hepatocellular tumors | 2/17(12%) | 0/17 (05)## | 2 | 0 | 0 | 0 |
| 3 mg/kg | 2/14 (14%) | 3/14 (21%) | 1 | 1 | 0 | 3 |
| 30 mg/kg | 3/19 (16%) | 1/19 (5%) | 2 | 0 | 1 | 1 |
| 100 mg/kg | 1/29 (3%) | 9/29 (31%)** | 1 | 0 | 0 | 9 |
| All DE-71 treated groups (Combined) |
6/62 (105) | 13/62 (21%)* | 4 | 1 | 1 | 13 |
Male and female mice were dosed with 0, 3, 30, or 100 mg/kg PBDE mixture by oral gavage for 2 years. Silent mutations are not included. Non-tumor Liver – 0 mg/kg (3 males +5 females); Liver Tumors- 0 mg/kg (14 males +3 females); 3 mg/kg (12 males +2 females); 30 mg/kg (13 males +6 females); 100 mg/kg (15 males +14 females).
Silent mutations not included.
* p < 0.05, **p < 0.01 by Fisher’s exact test to compare each dose group to the controls ## p < 0.01 for trend by the Cochran–Armitage trend test.
In the mouse hepatocellular carcinomas, the incidences of Hras mutations were low [10% (6/62)] and were located within codon 61 mainly C to A or A to T transversions (Table 6). However, there were no significant differences in the incidences of Hras mutations or the mutation spectra between hepatocellular carcinomas occurring spontaneously or resulting from PBDE exposures. Conversely, statistically significant increased incidences of Ctnnb1 mutations were noted in mouse hepatocellular carcinomas resulting from administration of the PBDE mixture compare to controls. None of the hepatocellular carcinomas arising spontaneously harbored Ctnnb1 mutations. These mutations were present within codons 15 to 46 and contained a mixture of transitions and transversions. In addition, there was a deletion of codons 15 to 46 in one carcinoma. There were no significant differences in the incidences or types of mutations between male and female mice (data not presented), and, thus, the combined data from both male and female mice are presented in Table 6.
3.4. Ah receptor genotyping in female rats
Because the toxicity of some other persistent organic pollutants depends on a functional AhR receptor, the status of this receptor was analyzed in female rats. The liver tumors in female rats developed independently of the AhR status of the outbred Wistar Han rats (Table 7; Supplement 3), as determined by PCR SNP analysis of the AhR type (whether wild, heterozygous, or homozygous mutant). Of the 118 liver FFPE samples analyzed for AhR genotype, 26 (22.0%) were homozygous wild type G/G; 51 (43.2%) were heterozygous G/A; and 39 (33.1%) were homozygous mutant A/A; and 2 (1.7%) undetermined. The 122 kidney FFPE samples yielded the following genotype totals: 21 (17.2%) homozygous wild type G/G, 51 (41.8%) heterozygous G/A, 38 (31.1%) homozygous mutant A/A, and 12 (9.8%) undetermined. Of the 21 female rats with liver tumors in the 50 mg/kg PBDE exposure group, 6 occurred in female rats with wild AhR (G/G); 4 in female rats with mutant AhR (A/A); 9 with heterozygous AhR (G/A); and 2 in rats with undetermined AhR status.
Table 7.
AhR genotyping of female rats.
| AhR genotype at exon 10 | Wild G/G |
Mutant A/A |
Heterozygous G/A |
Undetermined | Total |
|---|---|---|---|---|---|
| Control – no liver tumors | 13 (23%) |
16 (29%) |
27 (48%) |
0 (0%) |
56 |
| 50 mg/kg – no liver tumors | 5 (13%) |
18 (49%) |
13 (35%) |
1 (3%) |
37 |
| Control – with liver tumors | 1 (25%) |
2 (50%) |
1 (25%) |
0 (0%) |
4 |
| 50 mg/kg – with liver tumors | 7 (33%) |
3 (14%) |
10 (48%) |
1 (5%) |
21 |
3.5. PBDE-47, 99, and 153 tissue concentrations
The tissue levels (taken at the terminal necropsy at 2-years) of PBDE-47, 99, and 153 for a given matrix (liver, adipose, or plasma (rat only)) generally occurred at similar tissue concentrations (μg PBDE/g tissue) in both rats and mice [54]. PBDE-153 accumulated in tissues to a greater extent in tissues than predited based on its content in the mixture (3% PBDE-153, 36% PBDE-47, and 42%, PBDE-99). The levels of PBDEs in adipose tissue were generally 30–60 times the PBDE level in liver tissue in rats and mice. PBDE-47, 99, or 153 concentrations in adipose or liver in female rats or female mice were greater than the corresponding matrix level in male rats or male mice. The total plasma concentration of PBDE-47, 99, and 153 (combined) in male and female rats at 50 mg/kg was 15.09 and 24.68 μg PBDE/g plasma, respectively (6560 and 10,730 μg PBDE/g lipid, using plasma lipid concentration of 0.0023 per fraction of plasma weight [56]).
The PBDE tissue levels were linearly related to the administered doses, and the incidence of liver tumors increased with increasing PBDE dose. Female rats had a higher incidence of hepatocellular tumors per dose group than male rats, and this may have been related to the higher tissue levels of PBDE-47, 99, or 153 than in male rats.
4. Discussion
In these studies, PBDE mixture (DE-71) induced treatment-related carcinogenic effects in the liver of male and female rats and mice, in the thyroid and pituitary of male rats, and the uterus of female rats. This liver carcinogenic response in both sexes and species is an unusual finding, occurring in 11/594 NTP chemical carcinogenic studies (1-amino-2,4-dibromoantraquinone (TR 383); 3-amino-9-ethylcarbazole HCl (TR093); chlodecone (kepone) (TR 000); chlorinated paraffins (TR 308); di(2-ethylhexyl)phthalate (TR 217); furan (TR 402); methyleugenol (TR 491); 4,4′-oxydianiline (TR 205); polybrominated biphenyl mixture (Firemaster FF-1) (TR 244); tetrafluoroethylene (TR 450); N,N-dimethyl-p-toluidine (TR (579); pentabromodiphenyl ether mixture (TR 598) [57]).
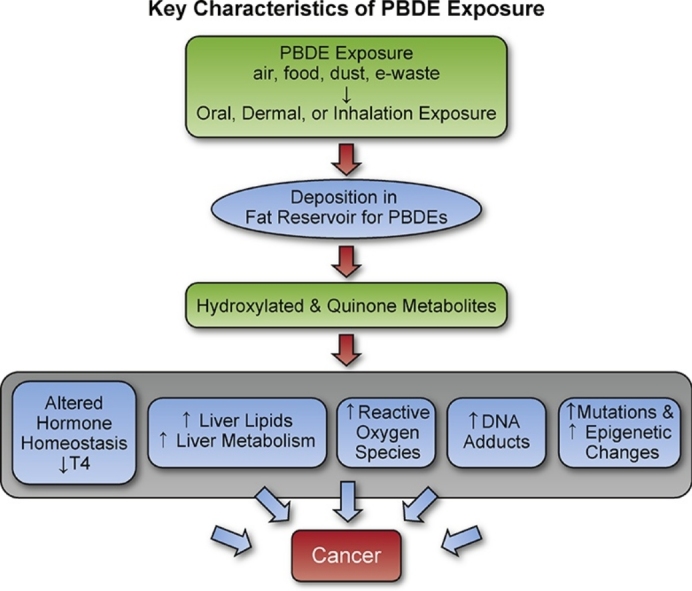
Hras mutations, commonly occurring in rodent liver tumors at codon 61, occurred at codon 60 (G to A transition, Gly to Asp) in the PBDE rat liver tumors. In contrast, Hras mutations were not statistically increased in the PBDE mouse liver tumors (vs. spontaneous liver tumors). In mice, Ctnnb1 mutations occurred in PBDE hepatocellular tumors, a mutation also found in other chemical-induced mouse liver tumors [58], and in human hepatocellular cancers [59,60]. Other studies examined the PBDE mouse liver tumors from this study, and found hypomethylation within the Tbx3 locus, and upregulation of Tbx3 mRNA and protein [61]. Tbx3 is a downstream target of the Wnt/Ctnnb1 pathway, and a transcription factor important in liver tumorigenesis and in proliferation of progenitor cells, such as embryonic and cancer stem cells [62,63].
These molecular and epigenetic events in the PBDE liver tumors may be related to oxidative damage. Other in vitro and in vivo studies reported in the literature found that PBDE exposure caused oxidative damage and increased reactive oxygen species [[64], [65], [66], [67], [68], [69]]. In addition, Br-C bonds once broken release free radicals [70]. Oxidative damage can eventually lead to DNA damage and mutations [71]. With the PBDEs, with a long tissue half-life, there is potential for continued exposure, as demonstrated by the high levels of PBDE found in the adipose tissue in these studies. As animals age the ability to mount an antioxidant defense decreases [71,72], and, thus, oxidative damage could become more pronounced in older animals.
PBDE metabolic alterations (e.g. upregulation of cytochrome P450s [73,74]) may also have contributed to PBDE carcinogenic effects. Upregulation of cytochrome P450 systems after PBDE exposure can activate nuclear hormone receptors including the peroxisome proliferation-activated receptor (PPAR) and/or constitutive activated/androstane receptor (CAR) receptor [75,76], both of which regulate lipid metabolism [77]. Metabolism by cyp450 enzymes can generate free reactive oxygen species [[78], [79], [80]], and oxidative damage is a key characteristic for increased risk for liver cancer [81].
Studies in the literature, report that PBDE exposure may lead to DNA adduct formation. PBDEs undergo metabolism to hydroxylated metabolites [82] and to PBDE-quinone metabolites that interact with DNA forming DNA adducts [83]. In vitro studies show that various types of DNA adducts may be generated from PBDE quinone (2-(2′,4′-bromophenoxyl)-benzoquinone (2′4′BrPhO-BQ)) including deoxyguanosine (dG), 2′-deoxyadenosine (dA), 2′-deoxycytidine (dC), or thymidine [84].
Fatty change and cytoplasmic vacuolization in the liver (an indicator for liver fat accumulation [85]) were found after PBDE exposure. Fat accumulation may be a factor that increases the risk for liver cancer development [81]. Other contributing factors to the PBDE-induced liver toxic and carcinogenic effects were the occurrence of hepatocyte hypertrophy; however, not all chemical exposures that cause this effect go on to be liver carcinogens [86,87].
PBDE exposures have been found to cause alterations in thyroid hormone levels [88]. Hydroxylated PBDE metabolites are more toxic [89] than the parent compound, and have higher binding affinity to thyroid receptors [90]. Changes in thyroid hormone levels may not only affect carcinogenic processes in the thyroid, but also in the liver as these hormones are critical regulators of hepatic lipid metabolism [91]. PBDE-induced changes in thyroid hormone levels can occur in both rodents and humans. For example, a human PBDE serum level of 130 pg/g was associated with a decrease in T4 serum level [92]. In contrast, in rodents, oral PBDE exposure in the mg/kg range (∼PBDE plasma level in μg/g range) is usually needed to lower T4 serum levels [88].
PBDE exposures have the potential to also alter estrogen levels. Binding of hydroxylated PBDE metabolites to sulfotransferase can interfere with sulfonation of estrogen and its excretion, ultimately leading to increased estrogen levels [93]. Such effects on hormone levels could contribute to PBDE carcinogenic effects in the uterus [94] or pituitary gland [95] as occurred in these studies.
The PBDE mixture (DE-71) used in this study contained very low levels of brominated dioxins and furans, and these brominated dioxins and furans have a toxic equivalence factor of 10% that of TCDD [96]. However, to fully investigate whether PBDE-induced liver tumors in the outbred Wistar rat were related to activation of AhR, we characterized AhR status in female Wistar rats. In this outbred rat, the AhR genotype at exon 10 may have a wild, mutant, or heterozygous sequence ([39,40]; Yao et al., 2012). We found that the occurrence of treatment-related liver tumors in female rats was not correlated with AhR type, and the PBDE-induced female rat liver tumors occurred in animals with wild, mutant, or heterozygous AhR. Thus, the occurrence of PBDE-induced liver tumors did not require a functional Ah receptor, and PBDEs themselves are generally not thought to be activators of the AhR [96].
5. Conclusion
In conclusion, PBDE mixture exposure (DE-71) caused toxic and carcinogenic effects in rats and mice. Several key carcinogenic characteristics reported in the literature may contribute to the PBDE carcinogenic activity observed in this study including oxidative damage, alteration in hormone homeostasis, and molecular and epigenetic changes in target tissue. Further work is needed to compare the sensitivity PBDE toxic effects in rodents and humans.
Financial support
The intramural program of the National Institute of Environmental Health Sciences (NIEHS), Research Triangle Park, NC, supported this work. However, the statements, opinions or conclusions contained therein do not necessarily represent the statements, opinions or conclusions of NIEHS or NIH. The in-life phase of the study was conducted under NIEHS Contract N0T1-ES-45516; AhR work under NIEHS Contract N01-ES-00005; and PBDE tissue level measurements under Contract HHSN273201000016C.
Conflict of interest
None of the authors have any conflict of interest to declare.
Transparency document
Acknowledgements
We thank Dr. A. Harrill and R. Kovi, NIEHS, for their review of this manuscript. All persons gave their informed consent prior to their inclusion as a paper author. This article does not contain clinical studies or patient data.
References
- 1.Sjodin A., Jones R.S., Caudill S.P., Wong L.Y., Turner W.E., Calafat A.M. Polybrominated diphenyl ethers, polychlorinated biphenyls, and persistent pesticides in serum from the national health and nutrition examination survey: 2003–2008. Environ. Sci. Technol. 2014;48:753–760. doi: 10.1021/es4037836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spears B.L., Isanhart J. Polybrominated diphenyl ethers in bald (Haliaeetus leucocephalus) and golden (Aquila chrysaetos) eagles from Washington and Idaho, USA. Environ. Toxicol. Chem. 2014;33:2795–2801. doi: 10.1002/etc.2742. [DOI] [PubMed] [Google Scholar]
- 3.Erratico C., Currier H., Szeitz A., Bandiera S., Covaci A., Elliott J. Levels of PBDEs in plasma of juvenile starlings (Sturnus vulgaris) from British Columbia, Canada and assessment of PBDE metabolism by avian liver microsomes. Sci. Total Environ. 2015;518–519:31–37. doi: 10.1016/j.scitotenv.2014.12.102. [DOI] [PubMed] [Google Scholar]
- 4.Dorneles P.R., Lailson-Brito J., Secchi E.R., Dirtu A.C., Weijs L., Dalla Rosa L., Bassoi M., Cunha H.A., Azevedo A.F., Covaci A. Levels and profiles of chlorinated and brominated contaminants in Southern Hemisphere humpback whales, Megaptera novaeangliae. Environ. Res. 2015;138:49–57. doi: 10.1016/j.envres.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Airaksinen R., Hallikainen A., Rantakokko P., Ruokojarvi P., Vuorinen P.J., Mannio J., Kiviranta H. Levels and congener profiles of PBDEs in edible Baltic, freshwater, and farmed fish in Finland. Environ. Sci. Technol. 2015;49:3851–3859. doi: 10.1021/es505266p. [DOI] [PubMed] [Google Scholar]
- 6.Petreas M., She J., Brown F.R., Winkler J., Windham G., Rogers E., Zhao G., Bhatia R., Charles M.J. High body burdens of 2,2’,4,4’-tetrabromodiphenyl ether (BDE-47) in California women. Environ. Health Perspect. 2003;111:1175–1179. doi: 10.1289/ehp.6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schecter A., Haffner D., Colacino J., Patel K., Papke O., Opel M., Birnbaum L. Polybrominated diphenyl ethers (PBDEs) and hexabromocyclodecane (HBCD) in composite U.S. food samples. Environ. Health Perspect. 2010;118:357–362. doi: 10.1289/ehp.0901345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schecter A., Smith S., Haffner D., Colacino J., Malik N., Patel K., Harris T.R., Opel M., Paepke O. Does flying present a threat of polybrominated diphenyl ether exposure? J. Occup. Environ. Med. 2010;52:1230–1235. doi: 10.1097/JOM.0b013e3181fe0a8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eskenazi B., Fenster L., Castorina R., Marks A.R., Sjodin A., Rosas L.G., Holland N., Guerra A.G., Lopez-Carillo L., Bradman A. A comparison of PBDE serum concentrations in Mexican and Mexican-American children living in California. Environ. Health Perspect. 2011;119:1442–1448. doi: 10.1289/ehp.1002874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hurley S., Reynolds P., Goldberg D., Nelson D.O., Jeffrey S.S., Petreas M. Adipose levels of polybrominated diphenyl ethers and risk of breast cancer. Breast Cancer Res. Treat. 2011;129:505–511. doi: 10.1007/s10549-011-1481-7. [DOI] [PubMed] [Google Scholar]
- 11.Park J.S., She J., Holden A., Sharp M., Gephart R., Souders-Mason G., Zhang V., Chow J., Leslie B., Hooper K. High postnatal exposures to polybrominated diphenyl ethers (PBDEs) and polychlorinated biphenyls (PCBs) via breast milk in California: does BDE-209 transfer to breast milk? Environ. Sci. Technol. 2011;45:4579–4585. doi: 10.1021/es103881n. [DOI] [PubMed] [Google Scholar]
- 12.Dallaire R., Ayotte P., Pereg D., Dery S., Dumas P., Langlois E., Dewailly E. Determinants of plasma concentrations of perfluorooctanesulfonate and brominated organic compounds in Nunavik Inuit adults (Canada) Environ. Sci. Technol. 2009;43:5130–5136. doi: 10.1021/es9001604. [DOI] [PubMed] [Google Scholar]
- 13.Dien N.T., Hirai Y., Miyazaki T., Sakai S.I. Factors influencing atmospheric concentrations of polybrominated diphenyl ethers in Japan. Chemosphere. 2015;144:2073–2080. doi: 10.1016/j.chemosphere.2015.10.119. [DOI] [PubMed] [Google Scholar]
- 14.European Parliament and the Council of the European Union (EPCEU) 2003. Directive 2003/11/EC of the European Parliament and of the Counceil of 6 February 2003 Amending for the 24th Time Council Directive 76/769/EEC Relating to Restrictions on the Marketing and Use of Certain Dangerous Substances and Preparations (Pentabromodiphenyl Ether, Octabromodiphenyl Ether). Off J Eur Union 15.2.2003, L42/45-L42/46. [Google Scholar]
- 15.U.S. Environmental Protection Agency, D.f.t.E . 2014. Flame Retardants Used in Flexible Polyurethane Foam: Al Aternatives Asessement Update.http://www.epa.gov/dfe/alternative_assessments.html [Google Scholar]
- 16.Darnerud P.O., Lignell S., Aune M., Isaksson M., Cantillana T., Redeby J., Glynn A. Time trends of polybrominated diphenylether (PBDE) congeners in serum of Swedish mothers and comparisons to breast milk data. Environ. Res. 2015;138:352–360. doi: 10.1016/j.envres.2015.02.031. [DOI] [PubMed] [Google Scholar]
- 17.Ma W.L., Yun S., Bell E.M., Druschel C.M., Caggana M., Aldous K.M., Buck Louis G.M., Kannan K. Temporal trends of polybrominated diphenyl ethers (PBDEs) in the blood of newborns from New York State during 1997 through 2011: analysis of dried blood spots from the newborn screening program. Environ. Sci. Technol. 2013;47:8015–8021. doi: 10.1021/es401857v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S., Jang Y.C., Kim J.G., Park J.E., Kang Y.Y., Kim W.I., Shin S.K. Static and dynamic flow analysis of PBDEs in plastics from used and end-of-life TVs and computer monitors by life cycle in Korea. Sci. Total Environ. 2015;506–507:76–85. doi: 10.1016/j.scitotenv.2014.10.116. [DOI] [PubMed] [Google Scholar]
- 19.Agarwal V., El Gamal A.A., Yamanaka K., Poth D., Kersten R.D., Schorn M., Allen E.E., Moore B.S. Biosynthesis of polybrominated aromatic organic compounds by marine bacteria. Nat. Chem. Biol. 2014;10:640–647. doi: 10.1038/nchembio.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao H., Jiang J., Wang Y., Lehmler H.J., Buettner G.R., Quan X., Chen J. Monohydroxylated polybrominated diphenyl ethers (OH-PBDEs) and dihydroxylated polybrominated biphenyls (Di-OH-PBBs): novel photoproducts of 2,6-dibromophenol. Environ. Sci. Technol. 2015;49:14120–14128. doi: 10.1021/acs.est.5b03637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones D.P. Sequencing the exposome: a call to action. Toxicol. Rep. 2016;3:29–45. doi: 10.1016/j.toxrep.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.U.S. Environmental Protection Agency . 2008. Toxicological Review of 2,2’-tetrabromodiphenyl Ether (BDE-47)http://www.epa.gov/NCEA/iris/ [Google Scholar]
- 23.U.S. Environmental Protection Agency . 2008. Toxicological Review of 2,2’,4,4’,5,5’-hexabromodiphenyl Ether (BDE-153)http://www.epa.gov/NCEA/iris/ [Google Scholar]
- 24.U.S. Environmental Protection Agency . 2008. Toxicological Review of 2,2’.4,4’,5-pentabromodiphenyl Ether (BDE-99)http://www.epa.gov/NCEA/iris/ [Google Scholar]
- 25.U.S. Environmental Protection Agency . 2010. An Exposure Assessment of Polybrominated Diphenyl Ethers.www.epa.gov/dfe [Google Scholar]
- 26.Braekevelt E., Tittlemeir S.A., Tomy G.T. Direct measurement of octanol-water partition coefficients of some environmentally relevant brominated diphenyl ether congeners. Chenmosphere. 2003;51:563–567. doi: 10.1016/S0045-6535(02)00841-X. [DOI] [PubMed] [Google Scholar]
- 27.Witt K.L., Livanos E., Kissling G.E., Torous D.K., Caspary W., Tice R.R., Recio L. Comparison of flow cytometry- and microscopy-based methods for measuring micronucleated reticulocyte frequencies in rodents treated with nongenotoxic and genotoxic chemicals. Mutat. Res. 2008;649:101–113. doi: 10.1016/j.mrgentox.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeiger E., Anderson g., Haworth S., Lawlor T., Mortelmans K., Speck W. Salmonella mutagenicity tests; III. Results from the testing of 255 chemicals. Environ. Mutagen. 1987;9(Suppl):1–110. [PubMed] [Google Scholar]
- 29.Butt C.M., Miranda M.L., Stapleton H.M. Development of an analytical method to quantify PBDEs, OH-BDEs, HBCDs, 2,4,6-TBP, EH-TBB, and BEH-TEBP in human serum. Anal. Bioanal. Chem. 2016;408:2449–2459. doi: 10.1007/s00216-016-9340-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDonald T.A. A perspective on the potential health risks of PBDEs. Chemosphere. 2002;46:745–755. doi: 10.1016/s0045-6535(01)00239-9. [DOI] [PubMed] [Google Scholar]
- 31.Bradman A., Castorina R., Gaspar F., Nishioka M., Colon M., Weathers W., Egeghy P.P., Maddalena R., Williams J., Jenkins P.L., McKone T.E. Flame retardant exposures in California early childhood education environments. Chemosphere. 2014;116:61–66. doi: 10.1016/j.chemosphere.2014.02.072. [DOI] [PubMed] [Google Scholar]
- 32.Frederiksen M., Thomsen C., Froshaug M., Vorkamp K., Thomsen M., Becher G., Knudsen L.E. Polybrominated diphenyl ethers in paired samples of maternal and umbilical cord blood plasma and associations with house dust in a Danish cohort. Int. J. Hyg. Environ. Health. 2011;213:233–242. doi: 10.1016/j.ijheh.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Harrad S., Goosey E., Desborough J., Abdallah M.A., Roosens L., Covaci A. Dust from U.K. primary school classrooms and daycare centers: the significance of dust as a pathway of exposure of Young U.K. Children to brominated flame retardants and polychlorinated biphenyls. Environ. Sci. Technol. 2010;44:4198–4202. doi: 10.1021/es100750s. [DOI] [PubMed] [Google Scholar]
- 34.Johnson P.I., Stapleton H.M., Sjodin A., Meeker J.D. Relationships between polybrominated diphenyl ether concentrations in house dust and serum. Environ. Sci. Technol. 2010;44:5627–5632. doi: 10.1021/es100697q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stapleton H.M., Kelly S.M., Allen J.G., McClean M.D., Webster T.F. Measurement of polybrominated diphenyl ethers on hand wipes: estimating exposure from hand-to-mouth contact. Environ. Sci. Technol. 2008;42:3329–3334. doi: 10.1021/es7029625. [DOI] [PubMed] [Google Scholar]
- 36.Schecter A., Colacino J., Sjodin A., Needham L., Birnbaum L. Partitioning of polybrominated diphenyl ethers (PBDEs) in serum and milk from the same mothers. Chemosphere. 2010;78:1279–1284. doi: 10.1016/j.chemosphere.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 37.Schecter A., Papke O., Harris T.R., Tung K.C., Musumba A., Olson J., Birnbaum L. Polybrominated diphenyl ether (PBDE) levels in an expanded market basket survey of U.S. food and estimated PBDE dietary intake by age and sex. Environ. Health Perspect. 2006;114:1515–1520. doi: 10.1289/ehp.9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lam J., Lanphear B.P., Bellinger D., Axelrad D.A., McPartland J., Sutton P., Davidson L., Daniels N., Sen S., Woodruff T.J. Developmental PBDE exposure and IQ/ADHD in childhood: a systematic review and meta-analysis. Environ. Health Perspect. 2017;125:086001. doi: 10.1289/EHP1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pohjanvirta R., Viluksela M., Tuomisto J.T., Unkila M., Karasinska J., Franc M.A., Holowenko M., Giannone J.V., Harper P.A., Tuomisto J., Okey A.B. Physicochemical differences in the AH receptors of the most TCDD-susceptible and the most TCDD-resistant rat strains. Toxicol. Appl. Pharmacol. 1999;155:82–95. doi: 10.1006/taap.1998.8565. [DOI] [PubMed] [Google Scholar]
- 40.Pohjanvirta R., Wong J.M., Li W., Harper P.A., Tuomisto J., Okey A.B. Point mutation in intron sequence causes altered carboxyl-terminal structure in the aryl hydrocarbon receptor of the most 2,3,7,8-tetrachlorodibenzo-p-dioxin-resistant rat strain. Mol. Pharmacol. 1998;54:86–93. doi: 10.1124/mol.54.1.86. [DOI] [PubMed] [Google Scholar]
- 41.Dunnick J.K., Nyska A. Characterization of liver toxicity in F344/N rats and B6C3F1 mice after exposure to a flame retardant containing lower molecular weight polybrominated diphenyl ethers. Exp. Toxicol. Pathol. 2009;61:1–12. doi: 10.1016/j.etp.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boorman G.A., Eustis S.L. The pathology working roup as a means for assuring pathology quality in toxicologic studies. In: Whitmire C., Davis C.L., Bristol D.W., editors. Managing Conduct and Data Quality of Toxicologic Studies. Princeton Scientific Publishing; Princeton: 1986. pp. 271–275. [Google Scholar]
- 43.Hardisty J.G., Boorman G.A. National toxicology program pathology quality assurrance procedures. In: Whitmire C., editor. Davis, CL, and Bristol, DW (Ed), Managing Conduct and Data of toxicologic studies. Princeton Publishing Company; Princeton: 1986. pp. 263–269. [Google Scholar]
- 44.Tarone R.E. Tests for trend in life table analysis. Biometrika. 1975;62:679–682. [Google Scholar]
- 45.Cox D.R. Regression models and life-tables. J. R. Stat. Soc. 1972;B34:187–220. [Google Scholar]
- 46.Jonckheere A.R. A distribution-free k-sample test against ordered alternatives. Biometrika. 1954;41:133–145. [Google Scholar]
- 47.Williams D.A. A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics. 1971;27:103–117. [PubMed] [Google Scholar]
- 48.Williams D.A. The comparison of several dose levels with a zero dose control. Biometrics. 1972;28:519–531. [PubMed] [Google Scholar]
- 49.Dunnett C.W. A multiple comparison procedure for comparting several treatments with a control. J. Am. Stat. Assoc. 1955;50:1096–1121. [Google Scholar]
- 50.Bailer A.J., Portier C.J. Effects of treatment-induced mortality and tumor-induced mortality on tests for carcinogenicity in small samples. Biometrics. 1988;44:417–431. [PubMed] [Google Scholar]
- 51.Piegorsch W.W., Bailer A.J. In: Statistics for Environmental Biology and Toxicology. Hall C.A., editor. Chapman and Hall; London: 1997. [Google Scholar]
- 52.Portier C.J., Bailer A.J. Testing for increased carcinogenicity using a survival-adjusted quantal response test. Fundam. Appl. Toxicol. 1989;12:731–737. doi: 10.1016/0272-0590(89)90004-3. [DOI] [PubMed] [Google Scholar]
- 53.Jiang T., Bell D.R., Clode S., Fan M.Q., Fernandes A., Foster P.M., Loizou G., MacNicoll A., Miller B.G., Rose M., Tran L., White S. A truncation in the aryl hydrocarbon receptor of the CRL:WI(Han) rat does not affect the developmental toxicity of TCDD. Toxicol. Sci. 2009;107:512–521. doi: 10.1093/toxsci/kfn252. [DOI] [PubMed] [Google Scholar]
- 54.Dunnick J.K., Pandiri A.R., Merrick B.A., Mutlu E., Waidyanatha S. Mutational analysis of pentabrominated dihenyl—induced tumors, AhR genotyping of Wistar Han rats, and PBDE tissue levels in rats. Data In Brief. 2018 doi: 10.1016/j.dib.2018.10.104. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thoolen B., Maronpot R.R., Harada T., Nyska A., Rousseaux C., Nolte T., Malarkey D.E., Kaufmann W., Kuttler K., Deschl U., Nakae D., Gregson R., Vinlove M.P., Brix A.E., Singh B., Belpoggi F., Ward J.M. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol. Pathol. 2010;38:5s–81s. doi: 10.1177/0192623310386499. [DOI] [PubMed] [Google Scholar]
- 56.El-Masri H.A., Portier C.J. Physiologically based pharmacokinetics model of primidone and its metabolites phenobarbital and phenylethylmalonamide in humans, rats, and mice. Drug Metab. Dispos. 1998;26:585–594. [PubMed] [Google Scholar]
- 57.National Toxicology Program accessed . 2017. NTP Technical Reports.https://ntp.niehs.nih.gov/results/pubs/longterm/reports/longterm/index.html [Google Scholar]
- 58.Hoenerhoff M.J., Pandiri A.R., Snyder S.A., Hong H.H., Ton T.V., Peddada S., Shockley K., Witt K., Chan P., Rider C., Kooistra L., Nyska A., Sills R.C. Hepatocellular carcinomas in B6C3F1 mice treated with Ginkgo biloba extract for two years differ from spontaneous liver tumors in cancer gene mutations and genomic pathways. Toxicol. Pathol. 2013;41:826–841. doi: 10.1177/0192623312467520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de La Coste A., Romagnolo B., Billuart P., Renard C.A., Buendia M.A., Soubrane O., Fabre M., Chelly J., Beldjord C., Kahn A., Perret C. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. U. S. A. 1998;95:8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dong B., Lee J.S., Park Y.Y., Yang F., Xu G., Huang W., Finegold M.J., Moore D.D. Activating CAR and beta-catenin induces uncontrolled liver growth and tumorigenesis. Nat. Commun. 2015;6:5944. doi: 10.1038/ncomms6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shimbo T., Dunnick J.K., Brix A., Mav D., Shah R., Roberts J.D., Wade P.A. DNA methylation changes in Tbx3 in a mouse model exposed to polybrominated diphenyl ethers. Int. J. Toxicol. 2017;36:229–238. doi: 10.1177/1091581817706676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Renard C.A., Labalette C., Armengol C., Cougot D., Wei Y., Cairo S., Pineau P., Neuveut C., de Reynies A., Dejean A., Perret C., Buendia M.A. Tbx3 is a downstream target of the Wnt/beta-catenin pathway and a critical mediator of beta-catenin survival functions in liver cancer. Cancer Res. 2007;67:901–910. doi: 10.1158/0008-5472.CAN-06-2344. [DOI] [PubMed] [Google Scholar]
- 63.Willmer T., Hare S., Peres J., Prince S. The T-box transcription factor TBX3 drives proliferation by direct repression of the p21(WAF1) cyclin-dependent kinase inhibitor. Cell Div. 2016;11:6. doi: 10.1186/s13008-016-0019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.An J., Li S., Zhong Y., Wang Y., Zhen K., Zhang X., Wang Y., Wu M., Yu Z., Sheng G., Fu J., Huang Y. The cytotoxic effects of synthetic 6-hydroxylated and 6-methoxylated polybrominated diphenyl ether 47 (BDE47) Environ. Toxicol. 2011;26:591–599. doi: 10.1002/tox.20582. [DOI] [PubMed] [Google Scholar]
- 65.Blanco J., Mulero M., Domingo J.L., Sanchez D.J. Gestational exposure to BDE-99 produces toxicity through upregulation of CYP isoforms and ROS production in the fetal rat liver. Toxicol. Sci. 2012;127:296–302. doi: 10.1093/toxsci/kfs082. [DOI] [PubMed] [Google Scholar]
- 66.Costa L.G., Pellacani C., Dao K., Kavanagh T.J., Roque P.J. The brominated flame retardant BDE-47 causes oxidative stress and apoptotic cell death in vitro and in vivo in mice. Neurotoxicology. 2015;48:68–76. doi: 10.1016/j.neuro.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumar J., Monica Lind P., Salihovic S., van Bavel B., Lind L., Ingelsson E. Influence of persistent organic pollutants on oxidative stress in population-based samples. Chemosphere. 2014;114:303–309. doi: 10.1016/j.chemosphere.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 68.Park H.R., Kamau P.W., Loch-Caruso R. Involvement of reactive oxygen species in brominated diphenyl ether-47-induced inflammatory cytokine release from human extravillous trophoblasts in vitro. Toxicol. Appl. Pharmacol. 2014;274:283–292. doi: 10.1016/j.taap.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Usenko C.Y., Abel E.L., Kudela M., Janise A., Bruce E.D. Comparison of PBDE congeners as inducers of oxidative stress in zebrafish. Environ. Toxicol. Chem. 2015;34:1154–1160. doi: 10.1002/etc.2922. [DOI] [PubMed] [Google Scholar]
- 70.Rawat S., Bruce E.D. Designing quantitative structure activity relationships to predict specific toxic endpoints for polybrominated diphenyl ethers in mammalian cells. SAR QSAR Environ. Res. 2014;25:527–549. doi: 10.1080/1062936X.2014.899512. [DOI] [PubMed] [Google Scholar]
- 71.Finkel T., Holbrook N.J. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 72.Nobrega-Pereira S., Fernandez-Marcos P.J., Brioche T., Gomez-Cabrera M.C., Salvador-Pascual A., Flores J.M., Vina J., Serrano M. G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 2016;7:10894. doi: 10.1038/ncomms10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dunnick J.K., Brix A., Cunny H., Vallant M., Shockley K.R. Characterization of polybrominated diphenyl ether toxicity in Wistar Han rats and use of liver microarray data for predicting disease susceptibilities. Toxicol. Pathol. 2012;40:93–106. doi: 10.1177/0192623311429973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sanders J.M., Burka L.T., Smith C.S., Black W., James R., Cunningham M.L. Differential expression of CYP1A, 2B, and 3A genes in the F344 rat following exposure to a polybrominated diphenyl ether mixture or individual components. Toxicol. Sci. 2005;88:127–133. doi: 10.1093/toxsci/kfi288. [DOI] [PubMed] [Google Scholar]
- 75.Mullur R., Liu Y.Y., Brent G.A. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94:355–382. doi: 10.1152/physrev.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sueyoshi T., Li L., Wang H., Moore R., Kodavanti P.R., Lehmler H.J., Negishi M., Birnbaum L.S. Flame retardant BDE-47 effectively activates nuclear receptor CAR in human primary hepatocytes. Toxicol. Sci. 2014;137:292–302. doi: 10.1093/toxsci/kft243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Y.Y., Brent G.A. Thyroid hormone crosstalk with nuclear receptor signaling in metabolic regulation. Trends Endocrinol. Metab. 2010;21:166–173. doi: 10.1016/j.tem.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bondy S.H., Naderi S. Contribution of hepatic cytochrome p450 systems to the generation of reactive oxygen species. Biochem. Pharmacol. 1994;45:155–159. doi: 10.1016/0006-2952(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 79.Khan W.A., Ali Khan M.W. Cytochrome P450-mediated estrogen metabolites and autoimmunity: relationship and link to free radicals. Curr. Drug Metab. 2015;17:65–74. doi: 10.2174/1389200216666151103115210. [DOI] [PubMed] [Google Scholar]
- 80.Zangar R.C., Davydov D.R., Verma S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol. 2004;199:316–331. doi: 10.1016/j.taap.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 81.Michelotti G.A., Machado M.V., Diehl A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013;10:656–665. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 82.Lai Y., Chen X., Lam M.H., Cai Z. Analysis of hydroxylated polybrominated diphenyl ethers in rat plasma by using ultra performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2011;879:1086–1090. doi: 10.1016/j.jchromb.2011.03.024. [DOI] [PubMed] [Google Scholar]
- 83.Huang L., Lai Y., Li C., Qiu B., Cai Z. Formation and characterization of glutathione adducts derived from polybrominated diphenyl ethers. Chemosphere. 2015;120:365–370. doi: 10.1016/j.chemosphere.2014.07.091. [DOI] [PubMed] [Google Scholar]
- 84.Huang L., Li C., Lai Y., Qiu B., Cai Z. Interaction of 2-(2’,4’-bromophenoxyl)-benzoquinone with deoxynucleosides and DNA in vitro. Chemosphere. 2015;118:29–34. doi: 10.1016/j.chemosphere.2014.04.108. [DOI] [PubMed] [Google Scholar]
- 85.Eustis S.L., Boorman G.A., Harada T., Popp J.A. Liver. In: Boorman G.A., Elwell M.R., Montgomery J.A.W.F.M.C.A., editors. Pathology of the Fischer Rat, 2018 Pathology of the Fischer Rat. Academic Press; 1990. pp. 78–81. S.L.E. [Google Scholar]
- 86.Allen D.G., Pearse G., Haseman J.K., Maronpot R.R. Prediction of rodent carcinogenesis: an evaluation of prechronic liver lesions as forecasters of liver tumors in NTP carcinogenicity studies. Toxicol. Pathol. 2004;32:393–401. doi: 10.1080/01926230490440934. [DOI] [PubMed] [Google Scholar]
- 87.Maronpot R.R., Yoshizawa K., Nyska A., Harada T., Flake G., Mueller G., Singh B., Ward J.M. Hepatic enzyme induction: histopathology. Toxicol. Pathol. 2010;38:776–795. doi: 10.1177/0192623310373778. [DOI] [PubMed] [Google Scholar]
- 88.National Toxicology Program . 2016. NTP Technical Report on the Toxicology of a Pentabromodiphenyl Oxide Mixture (DE-71) (Cas No. 32534-81-9) in F344/N Rats and B6C3F1/N Mice and Toxicology and Carcinogenesis Studies of a Pentabromodiphenyl Oxide Mixture (DE-71) in Wistar Han [Crl:WI(Han)] Rats and B6C3F1/N Mice (Gavage and Perinatal and Postnatal Gavage Studies) NTP TR 589.http://ntp.niehs.nih.gov/results/pubs/longterm/reports/longterm/index.html [Google Scholar]
- 89.Li T., Wang W., Pan Y.W., Xu L., Xia Z. A hydroxylated metabolite of flame-retardant PBDE-47 decreases the survival, proliferation, and neuronal differentiation of primary cultured adult neural stem cells and interferes with signaling of ERK5 MAP kinase and neurotrophin 3. Toxicol. Sci. 2013;134:111–124. doi: 10.1093/toxsci/kft083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Meerts I.A., van Zanden J.J., Luijks E.A., van Leeuwen-Bol I., Marsh G., Jakobsson E., Bergman A., Brouwer A. Potent competitive interactions of some brominated flame retardants and related compounds with human transthyretin in vitro. Toxicol. Sci. 2000;56:95–104. doi: 10.1093/toxsci/56.1.95. [DOI] [PubMed] [Google Scholar]
- 91.Sinha R.A., Singh B.K., Yen P.M. Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends Endocrinol. Metab. 2014;25:538–545. doi: 10.1016/j.tem.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 92.Makey C.M., McClean M.D., Braverman L.E., Pearce E.N., Sjodin A., Weinberg J., Webster T.F. Polybrominated diphenyl ether exposure and reproductive hormones in North American men. Reprod. Toxicol. 2016;62:46–52. doi: 10.1016/j.reprotox.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gosavi R.A., Knudsen G.A., Birnbaum L.S., Pedersen L.C. Mimicking of estradiol binding by flame retardants and their metabolites: a crystallographic analysis. Environ. Health Perspect. 2013;121:1194–1199. doi: 10.1289/ehp.1306902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Busch E.L., Crous-Bou M., Prescott J., Chen M.M., Downing M.J., Rosner B.A., Mutter G.L., De Vivo I. Endometrial cancer risk factors, hormone receptors, and mortality prediction. Cancer Epidemiol. Biomark. Prev. 2017;26:727–735. doi: 10.1158/1055-9965.EPI-16-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heaney A.P., Fernando M., Melmed S. Functional role of estrogen in pituitary tumor pathogenesis. J. Clin. Invest. 2002;109:277–283. doi: 10.1172/JCI14264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van den Berg M., Denison M.S., Birnbaum L.S., Devito M.J., Fiedler H., Falandysz J., Rose M., Schrenk D., Safe S., Tohyama C., Tritscher A., Tysklind M., Peterson R.E. Polybrominated dibenzo-p-dioxins, dibenzofurans, and biphenyls: inclusion in the toxicity equivalency factor concept for dioxin-like compounds. Toxicol. Sci. 2013;133:197–208. doi: 10.1093/toxsci/kft070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.