

Abstract
It is well known that chemotherapy can cure only some cancers in advanced stage, mostly those with an intact p53 pathway. Hematological cancers such as lymphoma and certain forms of leukemia are paradigmatic examples of such scenario. Recent evidence indicates that the efficacy of many of the alkylating and intercalating agents, antimetabolites, topoisomerase, and kinase inhibitors used in cancer therapy is largely due to p53 stabilization and activation consequent to the inhibition of ribosome biogenesis. In this context, innovative drugs specifically hindering ribosome biogenesis showed preclinical activity and are currently in early clinical development in hematological malignancies. The mechanism of p53 stabilization after ribosome biogenesis inhibition is a multistep process, depending on specific factors that can be altered in tumor cells, which can affect the antitumor efficacy of ribosome biogenesis inhibitors (RiBi). In the present review, the basic mechanisms underlying the anticancer activity of RiBi are discussed based on the evidence deriving from available preclinical and clinical studies, with the purpose of defining when and why the treatment with drugs inhibiting ribosomal biogenesis could be highly effective in hematological malignancies.
Keywords: Ribosome biogenesis inhibitors, Chemotherapy, Lymphoma, Leukemia, Ribosomal proteins, MDM2, p53, pRb
Background
The ribosome biogenesis is defined as the process of building new ribosomes, the intracellular organelles where protein synthesis takes place.
In recent years, several studies on the relationship between cell growth and proliferation produced important data regarding the mechanisms linking ribosome biogenesis, which is at the basis of cell growth, to the progression through the cell cycle phases of the proliferating cell. There is now evidence that a perturbed ribosome biogenesis activates a pathway leading to the stabilization and activation of the tumor suppressor protein p53, which in turn induces cell cycle arrest and/or apoptotic cell death [1–4].
Current evidence indicates that inhibition of ribosome biogenesis represents a major mechanism by which many of the currently used chemotherapeutic drugs (alkylating and intercalating agents, antimetabolites, topoisomerase inhibitors) exert their cytotoxic activity on cancer cells [5, 6]. Importantly, a series of new drugs selectively hindering the transcription of ribosomal (r) RNA, thus inhibiting ribosome biogenesis without having genotoxic effects, have been proposed as a new therapeutic approach, based on p53 activation [7–12].
However, it is known since long time that chemotherapy can cure only some cancers once they reach advanced stages. In fact, despite initial responses, the majority of metastatic solid tumors ultimately progress under chemotherapy treatment. Hematological malignancies (such as lymphomas and acute leukemias) represent paradigmatic examples of the few cancers that can be cured by chemotherapeutic agents and will be the main topic of the present review [13]. The basic biological characteristic underlying the intrinsic curability of such cancers is that, in a significant fraction of cases, they retain a functional p53-mediated response to nucleolar stress arising from ribosomal biogenesis inhibition; on the other hand, as a matter of fact, the presence of genomic alterations of the TP53 gene is an established negative prognostic predictor in lymphoma, acute and chronic leukemias treated with chemotherapy regimens [14–17].
Since p53 stabilization and activation is a multistep and tightly regulated process, in principle, the prerequisite for the antitumor efficacy of drugs inhibiting ribosome biogenesis should be the presence in the tumor cells, other than a normally functioning p53, also of those factors necessary for the activation of p53 and the induction of a p53-mediated cell cycle arrest and/or the apoptosis. These factors, which control cell cycle progression in normally proliferating cells [18], are qualitatively and quantitatively altered in the large number of cancers [19, 20], thus influencing the sensitivity to ribosome biogenesis (RiBi) inhibitors.
Therefore, it seems timely to critically review the characteristics of cancer cells which affect their sensitivity to RiBi inhibitors, with the purpose of highlighting those parameters which render the treatment with these drugs appropriate or not in hematological malignancies. For the convenience of the reader, the normal process of ribosome biogenesis will be first briefly described.
Ribosome biogenesis
Ribosomes are ribonucleoprotein particles which are located in the cytoplasm where, either free or membrane-bound, are engaged in protein synthesis. Four types of ribosomal RNA (rRNA) molecules and about 80 different ribosomal proteins constitute the ribosome. Ribosome formation occurs mainly in the nucleolus, being later completed in the nucleoplasm and in the cytoplasm (see for reviews: [21–24]). In the nucleolus, ribosomal genes are transcribed by RNA polymerase I (Pol I) to generate the 47S rRNA precursor, which undergoes to site-specific methylation and pseudo uridylation, and processing to give rise to the mature 18S, 5.8S, and 28S rRNA. The fourth types of rRNA, the 5S rRNA, is synthesized in the nucleoplasm by RNA polymerase III (Pol III) and then imported in the nucleolus together with the ribosomal proteins (RPs), whose mRNA is transcribed by RNA polymerase II (Pol II). The assembling of rRNA molecules with the RPs constitutes the two subunits of the mature ribosome, the large 60S and the small 40S subunit. The large 60S subunit is constituted by one each of the 28S, 5.8S, and 5S RNA molecules, together with 47 ribosomal proteins (RPLs); the small 40S subunit contains only one 18S RNA molecule and 33 ribosomal proteins (RPSs) [25, 26]. Both subunits migrate from the nucleolus to the cytoplasm where they form the 80S ribosome particle. In the process of ribosome biogenesis, more than 150 non-ribosomal proteins and around 70 small nucleolar RNAs are involved [27–32].
For the transcription of the of 47S pre-rRNA, the assembly of a specific multiprotein complex at the rDNA promoter containing Pol I is required. In this complex, three basal factors, termed transcription initiation factor I (TIF-I) A, selectivity factor 1 (SL1), and upstream binding factor (UBF), are present [33]. For the transcription of the 5S rRNA by Pol III, the transcription factors TFIIIC and TFIIIB are necessary [34–36]. In proliferating cells, the rate of ribosome biogenesis is enhanced in order to assure an adequate ribosome complement for the daughter cells and inhibition of ribosome biogenesis arrests cell cycle progression [37]. Furthermore, the rate of ribosome biogenesis influences the length of the cell cycle: higher the level of ribosome biogenesis, more rapid the cell cycle progression [38]. Ribosome biogenesis rate in cancer shows high variability, depending on a multiplicity of factors including the activation of specific intracellular signaling pathways and deregulated activity of oncogenes and tumor suppressors. On the other hand, quantitative and qualitative changes in ribosome biogenesis have been shown to facilitate neoplastic transformation. For a detailed description of the relationship between ribosome biogenesis and cancer, the reader should refer to [39–44]. In hematological malignancies, such as aggressive lymphoproliferative neoplasms, it is worth mentioning the oncogenic cooperation between the MYC oncogene and the phosphatidyl-inositol-3-kinase (PI3K) signaling pathway [45], which converge in stimulating rRNA synthesis and ribosome biogenesis [46].
Inhibition of ribosome biogenesis activates the RPs/MDM2/p53 pathway
Available data indicate that the levels of p53 expression and activity are mainly regulated by interactions with the tumor suppressor MDM2 (murine double minute 2, and HDM2 in humans). MDM2 is an E3 ubiquitin ligase which negatively controls p53 activity in two ways: by binding to the protein and inhibiting its transactivation activity, and by facilitating its proteasome degradation [47–49]. In normal proliferating cells, the level of p53 is maintained low because of the binding with MDM2 with consequent p53 ubiquitination and proteasome digestion [50]. When a perturbation in the ribosome biogenesis occurs (ribosome stress), it results in the binding of several ribosomal proteins, no longer used for ribosome building, to MDM2. This binding relieves the inhibitory activity of MDM2 toward p53 (see reviews [2–4, 51, 52]) (Fig. 1). Although there is evidence that RPL5, RPL11, and RPL23 play a major role in neutralization of MDM2 activity and in the induction of p53 stabilization [50, 53–58], the list of ribosomal proteins (of both large and small ribosomal subunit) able to inhibit MDM2 activity and to stabilize p53 upon “ribosomal stress” is rapidly expanding [52]. For a valid binding to MDM2 and its inactivation, the RPL11 and RPL5 must form a complex with the 5S rRNA and all the components of this complex are necessary for its inhibitory function [59, 60]. p53 stabilization always causes cell cycle arrest in proliferating cells and, depending on the quantitative level of stabilized p53, also apoptotic cell death [61–63]. p53 arrests cell cycle progression by inhibiting the phosphorylation of the tumor suppressor retinoblastoma protein, pRb. In its hypo-phosphorylated form, pRb binds to and inhibits the activity of E2F1, a transcription factor whose target genes are necessary for cell cycle progression. The inhibition of E2F1 activity by hypo-phosphorylated pRb reduces the expression of both cyclin E and A, necessary factors for cell cycle progression from G1 to S phase and from G2 to M phase respectively, with consequent cell accumulation in G1 and G2 phase [64]. The induction of apoptotic cell death by p53 is a consequence of induced expression of the pro-apoptotic members of the B cell lymphoma 2 (Bcl-2) gene family, PUMA, and BAX [63, 65–67] (Fig. 1). Finally, it should be noted that additional factors may interact with the RPs/MDM2/p53 axis, such as the ARF tumor suppressor and the activation of the PI3K pathway. In fact, ARF loss is a common genetic event in cancer and especially in aggressive lymphoid neoplasms, resulting in increased MDM2 activity and increased p53 degradation (reviewed in [68]). On the other hand, MDM2 is a downstream target of the PI3K-AKT axis, and AKT-induced MDM2 phosphorylation results in increased stability of MDM2 with consequent p53 degradation [69, 70]. As mentioned before, constitutive PI3K signaling is common in lymphoproliferative neoplasms, and PI3K inhibitors are in clinical development in lymphoid cancers. These notions could be relevant for designing therapeutic combination strategies aimed at increasing the p53-mediated response to the inhibition of ribosome biogenesis.
Fig. 1.
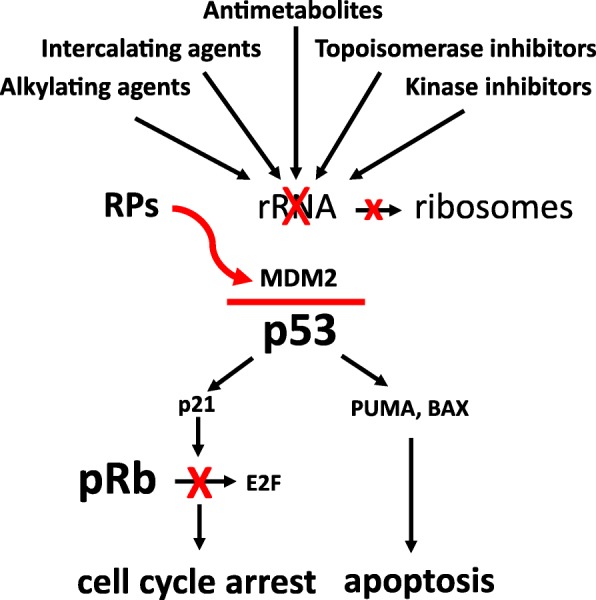
Schematic representation of the pathway activated by drug-induced perturbation of rRNA synthesis. Ribosomal proteins (RPs), no longer used for ribosome building, bind to MDM2, thus inhibiting its ubiquitin ligase activity toward p53 and the proteasome digestion of the tumor suppressor. As a consequence, p53 accumulates and induces transcription of p21, PUMA, and BAX. P21 is responsible for the cell cycle arrest by hindering pRb phosphorylation: in fact, hypo-phosphorylated pRb binds to and inhibits the activity of the transcription factor E2F1, whose target gene products are necessary for cell cycle progression. The induction of the pro-apoptotic factors PUMA and BAX activates the process of apoptotic cell death
Development of selective inhibitors of ribosome biogenesis
As briefly mentioned before, a strong contribution to p53 activation induced by chemotherapeutic agents is due to the inhibition of ribosomal biogenesis. As reported by Burger et al. [5], a series of drugs currently used for treating solid cancers and hematological malignancies inhibit ribosome biogenesis at the level of rRNA transcription and/or at the level of rRNA processing (Table 1). To this list, cyclophosphamide and mycophenolic acid should be added. Cyclophosphamide, a widely used anticancer drug, also inhibits rRNA transcription [71], after being converted to acrolein [72, 73], and the immunosuppressant mycophenolic acid has been demonstrated to inhibit the synthesis of rRNA [74].
Table 1.
Drugs used to treat hematological and solid malignancies which are effective or highly effective in the inhibition of rRNA transcription or processing (modified from Burger et al., 2010) [5]
| inibition of rRNA synthesis | ||
|---|---|---|
| transcription | processing | |
| Alkylating agents: | ||
| Melphalan* | + | - |
| Cisplatin* | + | - |
| Oxaliplatin* | + | - |
| Cyclophosphamide 1 * | + | - |
| Intercalating agents: | ||
| Doxorubicin * | + | - |
| Mitoxantrone * | + | - |
| Actinomycin D * | + | - |
| Mitomycin C | + | - |
| Antimetabolites: | ||
| Methotrexate * | + | - |
| 5-Fluorouracil | - | + |
| Topoisomerase inhibitors: | ||
| Camptothecin | - | + |
| Etoposide* | - | + |
| Kinase inhibitors: | ||
| Flavopiridol* | - | + |
| Roscovitine | - | + |
| Rapamycin | + | - |
| Proteasome inhibitors: | ||
| Bortezomib* | - | + |
| Translation inhibitors: | ||
| Homoharringtonine* | - | + |
| Mitosis inhibitors: | ||
| Vinblastine* | - | + |
| rRNA polymerase I inhibitors: | ||
| CX-5461 2 * | + | - |
* drugs currently used or in clinical development for the treatment of lymphomas and leukemia
1 Cyclophosphamide is metabolized to acrolein, which is responsible for the inhibition of rRNA transcription [60, 61]
2 CX-5461 is in phase I clinical trial in patients with haematological malignancies and in phase I/II trial in patients with breast cancer
In recent years, several efforts have been made to develop specific inhibitors of ribosomal biogenesis, in order to achieve a selective inhibition of rRNA synthesis without the genotoxic effects proper of chemotherapeutic drugs. In this light, it appears to be of particular relevance the CX-5461 molecule which selectively inhibits ribosome biogenesis, most likely by disrupting the SL-1/rDNA complex, promoting a cancer-specific activation of p53. Recent preclinical data indicate high activity of CX-5461 in MYC-driven lymphoma, providing the rationale for further clinical development of this compound [7, 75, 76]. CX-5361 is currently under phase I clinical trial for the treatment of patients with advanced hematologic malignancies, including acute myeloid leukemia.
Finally, there is experimental evidence that a small molecular compound, BMH-21, and a small-molecule peptide (22mer) also selectively inhibit rDNA transcription. BMH21 binds to GC-rich sequences and inhibits RNA Pol I activity [9]. It also induces the proteasome-dependent destruction of the large catalytic subunit in the Pol I complex, as do three other small molecular compounds, BMH-9, BMH-22, and BMH-23 [10]. The 22mer targets the interface between RNA polymerase I and Rrn3, thus selectively inhibiting the synthesis of rRNA [11].
Factors determining cancer cell sensitivity to drugs inhibiting ribosome biogenesis
The p53 status
Since a major effect of ribosome biogenesis inhibition is the activation of p53, the cytostatic and cytotoxic effects of chemotherapeutic agents inhibiting ribosome biogenesis should be obviously affected by the status of p53 [64, 77, 78]. Several lines of preclinical and clinical evidence support this notion. Indeed, actinomycin D, at a dose that exclusively hinders rDNA transcription, induced a cell cycle arrest with cell accumulation in G1 and, to a lesser extent, in G2 phase in p53 proficient cell lines [38, 64] whereas these changes in cell cycle distribution appeared to be reduced if cells were previously silenced for p53 expression [64]. The same occurs in cells with inactivated p53 in which the synthesis of rRNA was hindered by polymerase I silencing [79]. Also, p53 silencing significantly reduced the antiproliferative effects of 5-fluorouracil and methotrexate or doxorubicin, in human cancer cell lines harboring wild type (wt) p53 [78] and treatment of human leukemia and lymphoma cell lines with CX-5461, a selective inhibitor of Pol I transcription [7], was much more effective in cells with wt p53 in comparison with those with mutated p53 [75, 79].
On the other hand, it is worth noting that although p53 stabilization appears to be the main mechanism by which inhibitors of ribosomal biogenesis exert their cytostatic and cytotoxic action, there is evidence that these effects can be also caused in a p53-independent way. Depletion of the catalytic subunit of RNA polymerase I inhibited the synthesis of rRNA and hindered cell cycle progression in cells with inactivated p53, as a consequence of downregulation of the transcription factor E2F-1. Downregulation of E2F-1 was due to release of the ribosomal protein L11, which inactivated the E2F-1-stabilizing function of the E3 ubiquitin protein ligase MDM2 [79]. Furthermore, CX-5461 can induce p53-independent G2 checkpoint and apoptosis through activation of the ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) kinase pathway, in the absence of DNA damage [80, 81].
Regarding hematological malignancies, there is evidence that p53 status is an important factor determining the response to currently used chemotherapy regimens for lymphoma and leukemia treatment, which are based on drugs hindering ribosome biogenesis [5]. Anthracycline-based polychemotherapy represents the standard therapeutic approach for pediatric acute lymphoblastic leukemia (ALL) and multiple lymphoma subtypes of the adult [including Hodgkin lymphoma (HL), diffuse large B cell lymphoma (DLBCL), and anaplastic large T cell lymphoma (ALCL)]. More in detail, the ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) and the CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) regimens represent the treatments of choice in HL, DLBCL, and ALCL respectively. In general, the cure rates of antracycline-based regimens have been proved to be variable, being high for pediatric ALL and Hodgkin lymphoma [82, 83], intermediate for DLBCL [84–86] and ALCL [87, 88], and low for in indolent B cell lymphoma [89]. Similar considerations apply for myeloid disorders where anthracycline-based polychemotherapy has been shown to be effective certain forms of acute myeloid leukemia (reviewed in [90–92]), whereas chronic myeloid neoplasms are considered virtually incurable with standard polychemotherapy (reviewed in [93]). The intrinsic curability of the aforementioned hematologic cancers relies on precise biological characteristics of cancer cells, and the p53 status has been demonstrated to represent an important prognostic factor.
In line with this concept, the presence of TP53 genomic alterations in DLBCL and chronic lymphoid leukemia is a well-established negative prognostic predictor [14, 16, 94, 95]. DLBCL harboring alterations of the p53 pathway are often nonresponsive to CHOP plus rituximab (R) chemoimmunotherapy and are characterized by shorter overall survival. In CLL, patients harboring 17p deletions or TP53 mutations are refractory to standard chemotherapy and are currently treated with chemo-free treatments including inhibitors of B cell receptor signaling or bcl-2 inhibitors [96]. In acute myeloid leukemia, the presence of TP53 mutations is a powerful negative prognostic predictor, being associated with refractoriness to current anthracycline-based induction therapies [92, 97]. Finally, the presence of TP53 gene mutations predicts the outcome after induction and reinduction chemotherapy in acute lymphoid leukemia [98].
The prognostic value of genomic alterations of TP53 has been recently evaluated across a wide variety of hematological malignancies confirming the role of the p53 axis in determining the efficacy of chemotherapy in this setting [15].
The pRb status
These experimental and clinical data indicate that wild-type TP53 is a necessary requisite for the activation of the mechanisms leading to cell cycle arrest and/or apoptotic cell death in cancer cells treated with drugs inhibiting ribosome biogenesis.
There is evidence that this could be mostly true in the case of a normally functioning pRb pathway. Indeed, the absence of pRb could be a major factor conditioning the sensitivity of cancer cells to the exposure of RiBi inhibitors, also when the p53 pathway is dysfunctional [77]. Preliminary studies on this topic were conducted on solid tumor models, such as breast cancer. In fact, the contemporary absence of pRb and functional p53 has been shown to be responsible for a marked reduction of the cell population growth after the inhibition of ribosome biogenesis by actinomycin D, 5-fluorouracyl, methotrexate, and doxorubicin, which was even greater than that observed in p53 proficient cells [64, 78]. The cause of this increased sensitivity lies in the complete abrogation of the two cell cycle checkpoints in the absence of RB [19, 99, 100]: in cells lacking RB, the inhibition of ribosome biogenesis does not hinder the cell cycle progression, thus leading the cells to divide without having reached an appropriate ribosome complement. Very rapidly, the reduction of ribosome complement becomes incompatible with cell survival and a progressive increase of apoptotic cell death occurs [64]. These experimental data are consistent with studies investigating the relationship between the p53 and RB status and its implications on the clinical outcome after treatment with drugs inhibiting ribosome biogenesis. In a series of breast cancers treated with an adjuvant chemotherapeutic protocol including cyclophosphamide, methotrexate, and 5-fluorouracil, the presence of a wild-type or mutated p53, considered independently of the RB status, proved to have a null prognostic value. However, by excluding the cases with no pRb expression or inactivated-hyper-phosphorylated pRb, the p53 status resulted the only factor predicting the patient clinical outcome with patients with wt TP53 having a much better prognosis compared to those with mutated TP53. Worth of noting, the lack of pRb expression was the only independent factor predicting a good clinical outcome in patients treated with adjuvant chemotherapy [101, 102]. Moreover, an RB loss gene expression signature was demonstrated to be associated with increased pathological complete response to neoadjuvant chemotherapy in both estrogen-receptor positive and negative breast cancers [102]. Although the role of pRb pathway has not been evaluated as extensively as p53, similar observations were reported in hematological malignancies. In anaplastic large cell lymphoma, absence of pRb expression was observed in 40% of cases and hyperphosphorylation of pRb was detected in a significant fraction of RB positive patients, consistent with RB inactivation. Notably, these alterations correlated with a favorable clinical outcome [103]. In chronic lymphoid leukemia, 13q14 deletion is a frequent genomic alteration, and although the specific pathogenetic role of RB1 loss in the context of 13q14 deletion is yet to be determined, this cytogenetic abnormality predicts good clinical outcome following therapy with the FCR (fludarabine, cyclophosphamide, rituximab) regimen [104].
Similarly, trisomy 12 (resulting in copy number gain of CDK4 with consequent hyperphosphorylation and inactivation of pRb) is associated with excellent outcomes following chemoimmunotherapy [104]. Of note, the contemporary presence of 13q14 deletion seems to attenuate the adverse outcome related to the presence of TP53 deletions in CLL [105]. Since the RB1 locus is affected in less than 50% of CLL cases harboring 13q14 deletions [106], it would be interesting to investigate whether specific loss of RB1 attenuates the poor prognosis related to TP53 alterations. In conclusion, these data taken together indicate that (1) the presence of wt p53 associated with a normal downstream pRB pathway is an important characteristic which render cancer cells very sensitive to drugs inhibiting ribosome biogenesis and (2) cancer cells with RB1 loss could be sensitive to ribosome biogenesis inhibitors irrespective of the p53 status.
However, the integrity of the p53/pRb pathway might not be the only factor affecting response to ribosomal biogenesis inhibition, as described below.
The rate of ribosome biogenesis of the cell
Other than arresting cell cycle progression, stabilized p53 may cause programmed cell death by inducing transcription of pro-apoptotic factors [63, 65, 66]. Induction of apoptosis by inhibitors of ribosome biogenesis depends on the level of p53 stabilization, apoptosis being activated only by high amount of stabilized p53. In turn, the amount of stabilized p53 was shown to be directly related to the ribosome biogenesis rate of the cell. This was demonstrated by using four drugs, which inhibit rRNA synthesis at different steps: actinomycin D, doxorubicin, 5-fluorouracyl, and CX-5461 [63]. In cells characterized by a high rate of rRNA transcription, the inhibition of ribosome biogenesis caused a significantly greater degree of p53 stabilization and consequent greater expression of the pro-apoptotic members of the Bcl-2 gene family, PUMA, and BAX, compared to those characterized by a lower baseline rRNA synthesis. Accordingly, apoptotic cell death occurred in cells with a high rRNA synthesis and not in cells with a low ribosome biogenesis rate, the latter showing only cell cycle arrest. The tight relationship between the level of p53 stabilization and the rRNA synthesis rate was due to the fact that, upon ribosome biogenesis inhibition, different amounts of RPs, no longer used for ribosome building, bind to MDM2, thus hindering with higher efficiency the proteasomal degradation of p53 [63]. Interestingly, in cells with low rRNA synthesis (in which the inhibition of ribosome biogenesis stabilized p53 in a level that was not sufficient for apoptosis induction), the combined treatment with hydroxyurea which activates p53 with a different mechanism allowed to increase the total amount of stabilized p53 inducing apoptotic cell death [55].
Since the induction of cell death, and not cell cycle arrest, is the main goal of cancer chemotherapy, these observations might be relevant for establishing more effective and appropriate therapeutic protocols. In fact, this model implies that ribosome biogenesis inhibitors as single agents could be highly effective in p53 wild-type cancers with a high ribosome biogenesis rate, by inducing apoptotic cell death, whereas for treating cancers with a low ribosome biogenesis rate, they should be combined with drugs capable of stabilizing p53 or inducing apoptosis through different mechanisms. This model applies well in the setting of TP53 wild-type lymphoproliferative neoplasms, where aggressive lymphomas such as DLBCLs, characterized by high ribosomal biogenesis rates [107], can be cured with standard R-CHOP polychemotherapy [84–86], whereas indolent B cell non-Hodgkin lymphomas (such as small lymphocytic lymphoma/chronic lymphoid leukemia, marginal zone lymphoma, and follicular lymphomas), characterized by low ribosomal biogenesis rates [107], are virtually incurable with the same type of polychemotherapy [89].
Ribosomal protein deletions and mutations
Since the main mechanism involved in p53 stabilization upon ribosome biogenesis inhibition is represented by the binding of RPs to MDM2, mutations of ribosomal proteins may constitute another factor influencing the response of cancer cells to ribosome biogenesis inhibitors. As reported above, RPL5 and RPL11 play a major role in MDM2 inactivation. However, many other RPs, including RPL3, RPL6, RPL23, RPL26, RPL37, RPS7, RPS14, RPS15, RPS19, RPS20, RPS25, RPS26, and RPS27, have been shown to bind to MDM2, thus stabilizing p53 after induction of ribosomal stress (see for a recent and comprehensive review: [52]). There is increasing evidence for the presence of ribosomal protein copy number changes and mutations in many types of cancer. Regarding the RPs of the large ribosome subunit, exome sequencing demonstrated the presence of mutations of RPL5 in T cell acute lymphoblastic leukemia (T-ALL) [108] and in glioblastoma [109], and loss of the 1p22.1 region encompassing the RPL5 gene was found in 20% of multiple myeloma cases (MM) [110]. Furthermore, RPL5 and RPL10 mutations were recently observed, even though at low frequency, in MM [111]. The frequency of inactivating RPL5 mutations and deletions was found to be 11% in glioblastoma, 28% in melanoma, and 34% in breast cancer patients [112]. In T-ALL, RPL10 and RPL11 mutations have been also described [108, 113] and RPL22 was found to be deleted in about 10% patients [114]. RPL22 mutations were observed to occur with high frequency in endometrial [115, 116] and colorectal cancer [117] with microsatellite instability. Regarding the proteins constituting the small ribosome subunit, whole exome sequencing of chronic lymphocytic leukemia showed recurrent mutations of RPS15 [117, 118] while mutations of RPS20 are associated with colorectal carcinoma [119]. There are still few data on the effect of ribosomal protein deletion or mutations on the response to chemotherapeutic treatments. Experiments conducted using cancer cell lines demonstrated that silencing the expression of RPL5 and RPL11 strongly reduced the stabilization and activation of p53 caused by selective rRNA transcription inhibitors [120, 121], suggesting that cancers carrying these genetic changes should be resistant to chemotherapy based on inhibitors of ribosome biogenesis.
Up to now, the only clinical evidence of the impact of RP genetic changes on chemotherapy resistance based on a reduced activation of the RP-MDM2-p53 pathway comes from the study by Ljungström et al. [118] on the relationship between RPS15 mutations and clinical outcome of patients with chronic lymphocytic leukemia. The authors found that patients with RPS15 mutations, but carrying wild-type TP53, treated with standard chemoimmunotherapy (combination of fludarabine, cyclophosphamide, and rituximab), had a shorter 10-year survival compared with patients without mutated RPS15, and an overall survival similar to patients characterized by other adverse-prognostic markers. In the same study, the authors, using a human tumor cell line, demonstrated that transiently expressed mutant RPS15 reduced the expression of p53 due to an increased ubiquitin-mediated p53 degradation in comparison with cells carrying wild-type RPS15. It could be possible that mutated RPS15 is not capable of neutralizing the MDM2-mediated p53 digestion [122], thus reducing the induction of stabilized p53 upon chemotherapy treatment. In line with these data, our group recently found non-recurrent mutations of multiple RP genes in a significant fraction of DLBCL cases (> 10%) and RPS12 and RPL22 deletions in up to 20% of cases. Furthermore, our preliminary data indicate that these alterations are mutually exclusive with TP53 mutations and that RP mutations could be associated with adverse outcome in TP53 wild-type patients (manuscript submitted).
In conclusion, although preliminary evidence suggests that RP mutations could provide cancer cells with alternative mechanisms to inactivate p53-mediated responses to nucleolar stress, more studies are needed on the occurrence of RP gene deletions and mutations in cancer cells and their influence on p53 stabilization and therapeutic response after treatment with ribosome biogenesis inhibitors.
Mutated nucleophosmin
Nucleophosmin (NPM1), also called protein B23, numatrin, and NO38, is a non-ribosomal phosphoprotein, primary located in the nucleolus [123, 124]. NPM1 shuttles between the nucleolus and the cytoplasm [125] and exerts a series of different biochemical functions, some of them being independent of ribosome biogenesis (see for review [126–129]). Regarding the relationship between NPM1 and ribosome biogenesis, there is evidence that NPM1 plays a role in rRNA maturation [130] and its chaperone activity may facilitate the process of ribosome assembly [131]. Furthermore, NPM1 has been shown to be an important mediator, connecting the BCR-ABL network to ribosome biogenesis and, hence, protein synthesis and cell growth in chronic myelogenous leukemia [132]. Lastly, in proliferating cells, the amount of NPM1 is directly related to the rRNA transcription rate [133] and in human cancer cell lines to the nucleolar size and to the rate of cell proliferation [134].
Quantitative and qualitative changes of NPM1 have been reported to occur in many human malignancies (see for review [126]). Heterozygous NPM1 mutations were observed to occur in about 30% of patients with acute myeloid leukemia (AML) and, with very few exceptions, were restricted to exon 12 [135, 136]. Mutant NPM1 is delocalized to the cytoplasm (NPM1c+) while the amount of wild-type NPM1 located in the nucleolus is reduced as a consequence of haploinsufficiency and formation of heterodimers with mutated NPM1 in the cytoplasm [136]. Importantly, NPM1 mutations are mutually exclusive with TP53 mutations [137] and consistent with this observation the presence of NPM1c+ inhibits p53-mediated responses: in fact cytoplasmic NPM1 localization determines sequestration of ARF tumor suppressor in the cytoplasm, therefore limiting the interaction of ARF with MDM2 with consequent increased p53 degradation [138–140]. It is noteworthy that from the clinical point of view acute myeloid leukemia with mutated NPM1 is characterized by a better prognosis due to a higher remission rate after chemotherapy containing anthracyclines and cytarabine [91, 141]. This is probably due to the fact that leukemic cells with mutated NPM1 maintain a functional wild-type p53 [137]. In line with this data, a recent study reported that patients with AML with mutated NPM1, not eligible for intensive chemotherapy or with refractory or relapsed disease, may be successfully treated with actinomycin D, at the same dose as that used for low-risk gestational trophoblastic tumors [142]. The rationale at the basis of this therapeutic strategy is that leukemic cells with mutated NPM1 may have a more vulnerable nucleolus to the stress induced by the inhibition of ribosome biogenesis, resulting in a very strong p53-mediated response. NPM1 is also a frequent target of chromosomal translocations. The NPM1-ALK (anaplastic lymphoma kinase) fusion protein is the hallmark of ALK-positive anaplastic large cell lymphoma (reviewed in [143]). The NPM1-ALK fusion protein activates a series of cellular signaling pathways boosting lymphomagenesis while inhibiting p53 activity with MDM2 and JNK (c-Jun N-terminal kinase) dependent mechanisms [144]. Therefore, ALK-positive ALCL often retain a functional p53-mediated response to nucleolar stress, and accordingly TP53 mutations are rare in NPM1-ALK-positive ALCL. In line with these findings, NPM1-ALK-positive ALCL are characterized by a better prognosis following conventional CHOP compared to their ALK negative counterparts. Further investigations on the relationship between the functional state of the nucleolus and the response to ribosome biogenesis inhibitors should be conducted with the aim of establishing therapeutic protocols based on selective inhibition of ribosome biogenesis.
Conclusions
Despite the advent of personalized medicine, current treatment algorithms do not take into account important biological parameters which have been demonstrated to affect the cancer response to chemotherapeutic agents (these factors are summarized in Table 2) [14–16, 91, 92, 94, 103, 108, 110, 111, 118, 146–161]. There is now evidence that the efficacy of many of the chemotherapeutic drugs used for cancer treatment is related to p53 stabilization consequent to ribosome biogenesis inhibition (Fig. 1), and efforts are ongoing to develop new drugs that can selectively target ribosome biogenesis, without having the genotoxic effects proper of standard chemotherapeutic agents. In this context, it is worth mentioning the selective inhibitor of rRNA transcription, the CX-5461 molecule [7, 75], which may represent a new, very interesting strategy for cancer therapy [12, 162–164]. In this research field, other molecular compounds specifically hindering rDNA transcription have been proposed, demonstrating the increasing interest in this new therapeutic approach [9–11, 165]. On the other hand, as reported in the present review, a series of experimental and clinical data indicate that human tumors are characterized by several genomic alterations determining a highly variable response to the treatment with ribosome biogenesis inhibitors. In fact, several mechanisms converge in attenuating the anticancer activity of ribosome biogenesis inhibitors, mostly by reducing the amount of stabilized p53 and/or the extent of apoptotic responses to RIBi inhibitor-dependent nucleolar stress (Table 2). Accurate knowledge of these mechanisms could provide the rationale for treatment strategies able to by-pass resistance to RIBi inhibitors, such as combinations with MDM2 inhibitors or small molecule inhibitors of phosphatidyl-inositol-3-kinase (PI3K) pathway or antiapoptotic proteins such as bcl-2. The main characteristics influencing the response of hematologic malignancies to drugs inhibiting ribosome biogenesis are summarized in Fig. 2. These characteristics should be considered and evaluated in advance, in order to predict the degree of therapeutic response, especially when using selective inhibitors of ribosome biogenesis.
Table 2.
Overview of genomic alterations involved in the regulation of the RP/MDM2/p53 axis in hematologic malignancies
| Genomic alteration | Disease type | Incidence of the alteration | Prognostic impact | Proposed Mechanism | Reference |
|---|---|---|---|---|---|
| TP53 mutation | DLBCL | 22%-24% | Poor | Impaired p53 mediated response to nucleolar stress | [14, 146] |
| CLL | 7-9% | Poor | [94, 147–149] | ||
| ALCL | 8% | Poor | [145] | ||
| ALL | 14-15% | Poor | [15, 150] | ||
| AML | 5%-9% | Poor | [92, 151] | ||
| MM | <5% | Poor | [152] | ||
| TP53 deletion | DLBCL | 12% | Poor | [16] | |
| CLL | 5-12% | Poor | [147, 148] | ||
| ALL | 11% | Poor | [15] | ||
| MM | 9.5% | Poor | [152] | ||
| ARF deletion | DLBCL | 35% | Poor | Increased MDM2-dependent p53 degradation | [153] |
| FL | 8% | Poor | [154] | ||
| ALL | 14-15% | Poor | [15, 150, 155] | ||
| RB1 loss | DLBCL | 11% | Neutral | Loss of G1/S checkpoint | [156] |
| CLL | 20% | Neutral | [157] | ||
| ALCL | 40% | Good | [103] | ||
| ALL | 9% | Neutral | [158, 159] | ||
| RPS15 mutation | CLL | 19% (RELAPSE) | Poor | Impaired p53 mediated response to nucleolar stress | [118] |
| RPL5 mutation | MM | Sporadic | NE | [111] | |
| T-ALL | <5% | NE | [108] | ||
| RPL5 deletion | MM | 20% | Poor | [110] | |
| RPL10 mutation | T-ALL | 5% | NE | [108] | |
| RPL22 deletion | T-ALL | 10% | NE | [160] | |
| NPM1 mutation | AML | 53% | Good* | Increased sensitivity to nucleolar stress | [91] |
| NPM1-ALK | ALCL | 55% | Good | [161] |
Abbreviations: NE (not evaluated), DLBCL (diffuse large B-cell lymphoma), FL (Follicular lymphoma), CLL (chronic lymphoid leukemia), ALCL (anaplastic large T-cell lymphoma), ALL (acute lymphoid leukemia), T-ALL (T-cell acute lymphoid leukemia), MM (Multiple Myeloma), AML (acute myeloid leukemia)
*Associated with good prognosis in the absence of FLT3 genomic alterations
Fig. 2.
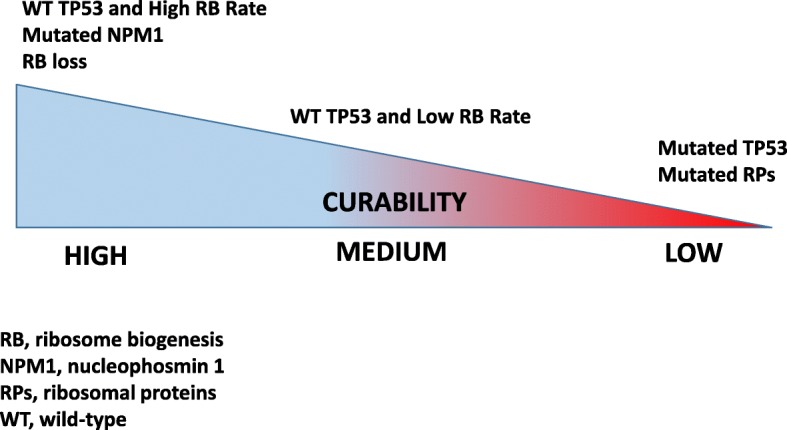
Schematic model representing the relationship between certain intrinsic cancer cell characteristics and curability of hematologic malignancies following chemotherapy based on drugs inhibiting ribosome biogenesis. Cancers with wild-type TP53, high ribosome biogenesis rate, loss of retinoblastoma protein, mutated NPM1 are characterized by good prognosis following chemotherapy (this is the case of TP53 wild-type HL, ALCL, DLBCL, NPM1c+ AML). At the opposite side of the spectrum, cancers characterized by mutant TP53 or mutant ribosomal proteins genes are associated with a low cure rate (certain forms of DLBCL, MM, T-ALL, CLL, AML). In the middle, cancers with low ribosomal biogenesis rate and wild-type TP53 harbor an intermediate cure rate (FL, other indolent B cell lymphoma subtypes)
Acknowledgments
Funding
This work was supported by the Roberto and Cornelia Pallotti Legacy for Cancer Research.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- ABVD
Doxorubicin, bleomycin, vinblastine, and dacarbazine
- ALCL
Anaplastic large cell lymphoma
- ALK
Anaplastic lymphoma kinase
- ALL
Acute lymphoblastic leukemia
- BAX
Bcl-2-associated X protein
- Bcl-2
B cell lymphoma 2
- CHOP
Cyclophosphamide, doxorubicin, vincristine, and prednisone
- DLBCL
Diffuse large B cell lymphoma
- DRB
Dichloro-ribofuranosylbenzimidazole
- FCR
Fludarabine, cyclophosphamide, rituximab
- HL
Hodgkin lymphoma
- JNK
c-Jun N-terminal kinase
- MDM2
Murine double minute 2
- NPM1
Nucleophosmin
- Pol I
RNA polymerase I
- Pol II
RNA polymerase II
- Pol III
RNA polymerase III
- pRb
Retinoblastoma protein
- PUMA
P53 upregulated modulator of apoptosis
- R
Rituximab
- RiBi
Ribosome biogenesis
- RPLs
Ribosomal proteins of the large subunit
- RPs
Ribosomal proteins
- RPSs
Ribosomal proteins of the small subunit
- SL1
Selectivity factor 1
- TIF-I
Transcription initiation factor I
- TP53
Tumor protein 53
- UBF
Upstream binding factor
Authors’ contributions
ED conceived the structure of the review and wrote the manuscript; AL helped with the manuscript writing; DT conceived the structure of the review and wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Enrico Derenzini, Phone: +39 02 57489.538, Email: enrico.derenzini@ieo.it.
Alessandra Rossi, Email: alessandra.rossi2@ieo.it.
Davide Treré, Phone: +39 051 208 3040, Email: davide.trere@unibo.it.
References
- 1.Mayer C, Grummt I. Cellular stress and nucleolar function. Cell Cycle. 2005;4:1036–1038. doi: 10.4161/cc.4.8.1925. [DOI] [PubMed] [Google Scholar]
- 2.Deisenroth C, Zhang Y. Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene. 2010;29:4253–4260. doi: 10.1038/onc.2010.189. [DOI] [PubMed] [Google Scholar]
- 3.Bursac S, Brdovcak MC, Donati G, Volarevic S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim Biophys Acta. 2014;1842(6):817–830. doi: 10.1016/j.bbadis.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 4.Golomb L, Volarevic S, Oren M. p53 and ribosome biogenesis stress: the essentials. FEBS Lett. 2014;588(16):2571–2579. doi: 10.1016/j.febslet.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 5.Burger K, Mühl B, Harasim T, Rohrmoser M, Malamoussi A, Orban M, et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem. 2010;285(16):12416–12425. doi: 10.1074/jbc.M109.074211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burger K, Eick D. Functional ribosome biogenesis is a prerequisite for p53 destabilization: impact of chemotherapy on nucleolar functions and RNA metabolism. Biol Chem. 2013;394(9):1133–1143. doi: 10.1515/hsz-2013-0153. [DOI] [PubMed] [Google Scholar]
- 7.Drygin D, Lin A, Bliesath J, Ho CB, O'Brien SE, Proffitt C, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71(4):1418–1430. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- 8.Hannan RD, Drygin D, Pearson RB. Targeting RNA polymerase I transcription and the nucleolus for cancer therapy. Expert Opin Ther Targets. 2013;17(8):873–878. doi: 10.1517/14728222.2013.818658. [DOI] [PubMed] [Google Scholar]
- 9.Peltonen K, Colis L, Liu H, Jäämaa S, Zhang Z, Af Hällström T, et al. Small Molecule BMH-Compounds That Inhibit RNA Polymerase I and Cause Nucleolar Stress. Mol Cancer Ther. 2014;13:2537–2546. doi: 10.1158/1535-7163.MCT-14-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peltonen K, Colis L, Liu H, Trivedi R, Moubarek MS, Moore HM, et al. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell. 2014;25:77–90. doi: 10.1016/j.ccr.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rothblum K, Hu Q, Penrod Y, Rothblum LI. Selective inhibition of rDNA transcription by a small-molecule peptide that targets the interface between RNA polymerase I and Rrn3. Mol Cancer Res. 2014;12:1586–1596. doi: 10.1158/1541-7786.MCR-14-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woods SJ, Hannan KM, Pearson RB, Hannan RD. The nucleolus as a fundamental regulator of the p53 response and a new target for cancer therapy. Biochim Biophys Acta. 2015;1849(7):821–829. doi: 10.1016/j.bbagrm.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Savage P, Stebbing J, Bower M, Crook T. Why does cytotoxic chemotherapy cure only some cancers? Nat Clin Pract Oncol. 2009;6(1):43–52. doi: 10.1038/ncponc1260. [DOI] [PubMed] [Google Scholar]
- 14.Zenz T, Kreuz M, Fuge M, Klapper W, Horn H, Staiger AM, German High-Grade Non-Hodgkin Lymphoma Study Group (DSHNHL) et al. TP53 mutation and survival in aggressive B cell lymphoma. Int J Cancer. 2017;141(7):1381–1388. doi: 10.1002/ijc.30838. [DOI] [PubMed] [Google Scholar]
- 15.Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia. 2017;31(3):705–711. doi: 10.1038/leu.2016.263. [DOI] [PubMed] [Google Scholar]
- 16.Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood. 2012;120(19):3986–3996. doi: 10.1182/blood-2012-05-433334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eskelund CW, Dahl C, Hansen JW, Westman M, Kolstad A, Pedersen LB, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017;130(17):1903–1910. doi: 10.1182/blood-2017-04-779736. [DOI] [PubMed] [Google Scholar]
- 18.David-Pfeuty T. The flexible evolutionary anchorage-dependent Pardee’s restriction point of mammalian cells: how its deregulation may lead to cancer. Biochim Biophys Acta. 2006;1765:38–66. doi: 10.1016/j.bbcan.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 20.Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60:3689–3695. [PubMed] [Google Scholar]
- 21.Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006;25(48):6384–6391. doi: 10.1038/sj.onc.1209883. [DOI] [PubMed] [Google Scholar]
- 22.Kopp K, Gasiorowski JZ, Chen D, Gilmore R, Norton JT, Wang C, et al. Pol I transcription and pre-rRNA processing are coordinated in a transcription-dependent manner in mammalian cells. Mol Biol Cell. 2007;18(2):394–403. doi: 10.1091/mbc.e06-03-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lempiäinen H, Shore D. Growth control and ribosome biogenesis. Curr Opin Cell Biol. 2009;21(6):855–863. doi: 10.1016/j.ceb.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Grummt I. Wisely chosen paths--regulation of rRNA synthesis. FEBS J. 2010;277(22):4626–4639. doi: 10.1111/j.1742-4658.2010.07892.x. [DOI] [PubMed] [Google Scholar]
- 25.Vladimirov SN, Ivanov AV, Karpova GG, Musolyamov AK, Egorov TA, Thiede B, et al. Characterization of the human small-ribosomal-subunit proteins by N-terminal and internal sequencing, and mass spectrometry. Eur J Biochem. 1996;239(1):144–149. doi: 10.1111/j.1432-1033.1996.0144u.x. [DOI] [PubMed] [Google Scholar]
- 26.Odintsova TI, Müller EC, Ivanov AV, Egorov TA, Bienert R, Vladimirov SN, et al. Characterization and analysis of posttranslational modifications of the human large cytoplasmic ribosomal subunit proteins by mass spectrometry and Edman sequencing. J Protein Chem. 2003;22(3):249–258. doi: 10.1023/A:1025068419698. [DOI] [PubMed] [Google Scholar]
- 27.Fatica A, Tollervey D. Making ribosomes. Curr Opin Cell Biol. 2002;14(3):313–318. doi: 10.1016/S0955-0674(02)00336-8. [DOI] [PubMed] [Google Scholar]
- 28.Fromont-Racine M, Senger B, Saveanu C, Fasiolo F. Ribosome assembly in eukaryotes. Gene. 2003;313:17–42. doi: 10.1016/S0378-1119(03)00629-2. [DOI] [PubMed] [Google Scholar]
- 29.Tschochner H, Hurt E. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol. 2003;13(5):255–263. doi: 10.1016/S0962-8924(03)00054-0. [DOI] [PubMed] [Google Scholar]
- 30.Chédin S, Laferté A, Hoang T, Lafontaine DL, Riva M, Carles CI. ribosome synthesis controlled by pol I transcription? Cell Cycle. 2007;6(1):11–15. doi: 10.4161/cc.6.1.3649. [DOI] [PubMed] [Google Scholar]
- 31.Lam YW, Lamond AI, Mann M, Andersen JS. Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr Biol. 2007;17(9):749–760. doi: 10.1016/j.cub.2007.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kressler D, Hurt E, Bassler J. Driving ribosome assembly. Biochim Biophys Acta. 2010;1803(6):673–683. doi: 10.1016/j.bbamcr.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 33.Grummt I. Life on a planet of its own: regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003;17(14):1691–1702. doi: 10.1101/gad.1098503R. [DOI] [PubMed] [Google Scholar]
- 34.White RJ. RNA polymerase III transcription and cancer. Oncogene. 2004;23(18):3208–3216. doi: 10.1038/sj.onc.1207547. [DOI] [PubMed] [Google Scholar]
- 35.White RJ. RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol. 2005;6(1):69–78. doi: 10.1038/nrm1551. [DOI] [PubMed] [Google Scholar]
- 36.Goodfellow SJ, White RJ. Regulation of RNA polymerase III transcription during mammalian cell growth. Cell Cycle. 2007;6(19):2323–2326. doi: 10.4161/cc.6.19.4767. [DOI] [PubMed] [Google Scholar]
- 37.Volarevic S, Stewart MJ, Ledermann B, Zilberman F, Terracciano L, Montini E, et al. Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science. 2000;288:2045–2047. doi: 10.1126/science.288.5473.2045. [DOI] [PubMed] [Google Scholar]
- 38.Derenzini M, Montanaro L, Chillà A, Tosti E, Vici M, Barbieri S, et al. Key role of the achievement of an appropriate ribosomal RNA complement for G1-S phase transition in H4-II-E-C3 rat hepatoma cells. J Cell Physiol. 2005;202(2):483–491. doi: 10.1002/jcp.20144. [DOI] [PubMed] [Google Scholar]
- 39.Montanaro L, Treré D, Derenzini M. Changes in ribosome biogenesis may induce cancer by down-regulating the cell tumor suppressor potential. Biochim Biophys Acta. 2012;1825(1):101–110. doi: 10.1016/j.bbcan.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Goudarzi KM, Lindström MS. Role of ribosomal protein mutations in tumor development (Review) Int J Oncol. 2016;48(4):1313–1324. doi: 10.3892/ijo.2016.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orsolic I, Jurada D, Pullen N, Oren M, Eliopoulos AG, Volarevic S. The relationship between the nucleolus and cancer: Current evidence and emerging paradigms. Semin Cancer Biol. 2016;37-38:36–50. doi: 10.1016/j.semcancer.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 42.Derenzini M, Montanaro L, Trerè D. Ribosome biogenesis and cancer. Acta Histochem. 2017;119(3):190–197. doi: 10.1016/j.acthis.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 43.Pelletier J, Thomas G, Volarević S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer. 2018;18(1):51–63. doi: 10.1038/nrc.2017.104. [DOI] [PubMed] [Google Scholar]
- 44.Bustelo XR, Dosil M. Ribosome biogenesis and cancer: basic and translational challenges. Curr Opin Genet Dev. 2018;48:22–29. doi: 10.1016/j.gde.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 45.Sander S, Calado DP, Srinivasan L, et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012;22(2):167–179. doi: 10.1016/j.ccr.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan JC, Hannan KM, Riddell K, Ng PY, Peck A, Lee RS, Hung S. AKT promotes rRNA synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci Signal. 2011;4(188):ra56. doi: 10.1126/scisignal.2001754. [DOI] [PubMed] [Google Scholar]
- 47.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-R. [DOI] [PubMed] [Google Scholar]
- 48.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 49.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 50.Ljungman M. Dial 9-1-1 for p53: mechanisms of p53 activation by cellular stress. Neoplasia. 2000;2(3):208–225. doi: 10.1038/sj.neo.7900073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16:369–377. doi: 10.1016/j.ccr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stępiński D. Nucleolus-derived mediators in oncogenic stress response and activation of p53-dependent pathways. Histochem Cell Biol. 2016;146(2):119–139. doi: 10.1007/s00418-016-1443-6. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23(23):8902–8912. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3(6):577–587. doi: 10.1016/S1535-6108(03)00134-X. [DOI] [PubMed] [Google Scholar]
- 55.Bhat KP, Itahana K, Jin A, Zhang Y. Essential role of ribosomal protein L11 in mediating growth inhibition-induced p53 activation. EMBO J. 2004;23(12):2402–12. doi: 10.1038/sj.emboj.7600247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dai MS, Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem. 2004;279(43):44475–44482. doi: 10.1074/jbc.M403722200. [DOI] [PubMed] [Google Scholar]
- 57.Dai MS, Zeng SX, Jin Y, Sun XX, David L, Lu H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol. 2004;24(17):7654–7668. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jin A, Itahana K, O'Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol. 2004;24(17):7669–7680. doi: 10.1128/MCB.24.17.7669-7680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Donati G, Peddigari S, Mercer CA, Thomas G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 2013;4:87–98. doi: 10.1016/j.celrep.2013.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sloan KE, Bohnsack MT, Watkins NJ. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013;5:237–247. doi: 10.1016/j.celrep.2013.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pestov DG, Strezoska Z, Lau LF. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: effects of nucleolar protein Bop1 on G(1)/S transition. Mol Cell Biol. 2001;21(13):4246–4255. doi: 10.1128/MCB.21.13.4246-4255.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan X, Zhou Y, Casanova E, Chai M, Kiss E, Gröne HJ, et al. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol Cell. 2005;19(1):77–87. doi: 10.1016/j.molcel.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 63.Scala F, Brighenti E, Govoni M, Imbrogno E, Fornari F, Treré D, et al. Direct relationship between the level of p53 stabilization induced by rRNA synthesis-inhibiting drugs and the cell ribosome biogenesis rate. Oncogene. 2016;35(8):977–989. doi: 10.1038/onc.2015.147. [DOI] [PubMed] [Google Scholar]
- 64.Montanaro L, Mazzini G, Barbieri S, Vici M, Nardi-Pantoli A, Govoni M, et al. Different effects of ribosome biogenesis inhibition on cell proliferation in retinoblastoma protein- and p53-deficient and proficient human osteosarcoma cell lines. Cell Prolif. 2007;40(4):532–549. doi: 10.1111/j.1365-2184.2007.00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. J Cell Sci. 2003;116(Pt 20):4077–4085. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- 66.Jin S, Levine AJ. The p53 functional circuit. J Cell Sci. 2001;114(Pt 23):4139–4140. doi: 10.1242/jcs.114.23.4139. [DOI] [PubMed] [Google Scholar]
- 67.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137(3):413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 68.Tessoulin B, Eveillard M, Lok A, Chiron D, Moreau P, Amiot M, et al. p53 dysregulation in B-cell malignancies: More than a single gene in the pathway to hell. Blood Rev. 2017;31(4):251–259. doi: 10.1016/j.blre.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 69.Ogawara Y, Kishishita S, Obata T, Isazawa Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277(24):21843–21850. doi: 10.1074/jbc.M109745200. [DOI] [PubMed] [Google Scholar]
- 70.Feng J, Tamaskovic R, Yang Z, Brazil DP, Merlo A, Hess D, Hemmings BA. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J Biol Chem. 2004;279(34):35510–35517. doi: 10.1074/jbc.M404936200. [DOI] [PubMed] [Google Scholar]
- 71.Wang HT, Chen TY, Weng CW, Yang CH, Tang MS. Acrolein preferentially damages nucleolus eliciting ribosomal stress and apoptosis in human cancer cells. Oncotarget. 2016;7(49):80450–80464. doi: 10.18632/oncotarget.12608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fraiser LH, Kanekal S, Kehrer JP. Cyclophosphamide toxicity. Characterising and avoiding the problem. Drugs. 1991;42:781–795. doi: 10.2165/00003495-199142050-00005. [DOI] [PubMed] [Google Scholar]
- 73.Boor PJ. Allylamine cardiotoxicity: metabolism and mechanism. Adv Exp Med Biol. 1983;161:533–541. doi: 10.1007/978-1-4684-4472-8_32. [DOI] [PubMed] [Google Scholar]
- 74.Sun XX, Dai MS, Lu H. Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. J Biol Chem. 2008;283(18):12387–12392. doi: 10.1074/jbc.M801387200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer specific activation of p53. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Drygin D, Rice WG, Grummt I. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu Rev Pharmacol Toxicol. 2010;50:131–156. doi: 10.1146/annurev.pharmtox.010909.105844. [DOI] [PubMed] [Google Scholar]
- 77.Derenzini M, Donati G, Mazzini G, Montanaro L, Vici M, Ceccarelli C, et al. Loss of retinoblastoma tumor suppressor protein makes human breast cancer cells more sensitive to antimetabolite exposure. Clin Cancer Res. 2008;14(7):2199–2209. doi: 10.1158/1078-0432.CCR-07-2065. [DOI] [PubMed] [Google Scholar]
- 78.Derenzini M, Brighenti E, Donati G, Vici M, Ceccarelli C, Santini D, et al. The p53-mediated sensitivity of cancer cells to chemotherapeutic agents is conditioned by the status of the retinoblastoma protein. J Pathol. 2009;219(3):373–382. doi: 10.1002/path.2612. [DOI] [PubMed] [Google Scholar]
- 79.Donati G, Brighenti E, Vici M, Mazzini G, Treré D, Montanaro L, et al. Selective inhibition of rRNA transcription downregulates E2F-1: a new p53-independent mechanism linking cell growth to cell proliferation. J Cell Sci. 2011;124(Pt 17):3017–3028. doi: 10.1242/jcs.086074. [DOI] [PubMed] [Google Scholar]
- 80.Negi SS, Brown P. rRNA synthesis inhibitor, CX-5461, activates ATM/ATR pathway in acute lymphoblastic leukemia, arrests cells in G2 phase and induces apoptosis. Oncotarget. 2015;6(20):18094–18104. doi: 10.18632/oncotarget.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Quin J, Chan KT, Devlin JR, Cameron DP, Diesch J, Cullinane C, et al. Inhibition of RNA polymerase I transcription initiation by CX-5461 activates non-canonical ATM/ATR signaling. Oncotarget. 2016;7(31):49800–49818. doi: 10.18632/oncotarget.10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N Engl J Med. 2015;373(16):1541–1552. doi: 10.1056/NEJMra1400972. [DOI] [PubMed] [Google Scholar]
- 83.Canellos GP, Anderson JR, Propert KJ, Nissen N, Cooper MR, Henderson ES, et al. Chemotherapy of advanced Hodgkin's disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med. 1992;327(21):1478–1484. doi: 10.1056/NEJM199211193272102. [DOI] [PubMed] [Google Scholar]
- 84.Fisher RI, Gaynor ER, Dahlberg S, Oken MM, Grogan TM, Mize EM, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin's lymphoma. N Engl JMed. 1993;328(14):1002–1006. doi: 10.1056/NEJM199304083281404. [DOI] [PubMed] [Google Scholar]
- 85.Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235–242. doi: 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- 86.Pfreundschuh M, Trümper L, Osterborg A, Pettengell R, Trneny M, Imrie K, et al. MabThera International Trial Group. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol. 2006;7(5):379–391. doi: 10.1016/S1470-2045(06)70664-7. [DOI] [PubMed] [Google Scholar]
- 87.Chihara D, Fanale MA. Management of Anaplastic Large Cell Lymphoma. Hematol Oncol Clin North Am. 2017;31(2):209–222. doi: 10.1016/j.hoc.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 88.Vose J, Armitage J, Weisenburger D. International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–4130. doi: 10.1200/JCO.2008.16.4558. [DOI] [PubMed] [Google Scholar]
- 89.Lunning MA, Vose JM. Management of indolent lymphoma: where are we now and where are we going. Blood Rev. 2012;26(6):279–288. doi: 10.1016/j.blre.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Murphy T, KWL Y. Cytarabine and daunorubicin for the treatment of acute myeloid leukemia. Expert Opin Pharmacother. 2017;18(16):1765–1780. doi: 10.1080/14656566.2017.1391216. [DOI] [PubMed] [Google Scholar]
- 91.Schlenk RF, Döhner K, Krauter J, Fröhling S, Corbacioglu A, Bullinger L, et al. German-Austrian Acute Myeloid Leukemia Study Group. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1909–18. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 92.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374(23):2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goldman JM. Chronic myeloid leukemia: a historical perspective. Semin Hematol. 2010;47(4):302–311. doi: 10.1053/j.seminhematol.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 94.Zenz T, Eichhorst B, Busch R, Denzel T, Häbe S, Winkler D, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010;28(29):4473–4479. doi: 10.1200/JCO.2009.27.8762. [DOI] [PubMed] [Google Scholar]
- 95.Byrd JC, Gribben JG, Peterson BL, Grever MR, Lozanski G, Lucas DM, et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol. 2006;24(3):437–443. doi: 10.1200/JCO.2005.03.1021. [DOI] [PubMed] [Google Scholar]
- 96.Eichhorst B, Hallek M. Prognostication of chronic lymphocytic leukemia in the era of new agents. Hematology Am Soc Hematol Educ Program. 2016;2016(1):149–155. doi: 10.1182/asheducation-2016.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kadia TM, Jain P, Ravandi F, Garcia-Manero G, Andreef M, Takahashi K, et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer. 2016; 10.1002/cncr.30203. [DOI] [PMC free article] [PubMed]
- 98.Forero-Castro M, Robledo C, Benito R, Bodega-Mayor I, Rapado I, Hernández-Sánchez M, et al. Mutations in TP53 and JAK2 are independent prognostic biomarkers in B-cell precursor acute lymphoblastic leukaemia. Br J Cancer. 2017;117(2):256–265. doi: 10.1038/bjc.2017.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 100.Flatt PM, Tang LJ, Scatena CD, Szak ST, Pietenpol JA. p53 regulation of G(2) checkpoint is retinoblastoma protein dependent. Mol Cell Biol. 2000;20(12):4210–4223. doi: 10.1128/MCB.20.12.4210-4223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Treré D, Brighenti E, Donati G, Ceccarelli C, Santini D, Taffurelli M, et al. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Ann Oncol. 2009;20(11):1818–1823. doi: 10.1093/annonc/mdp209. [DOI] [PubMed] [Google Scholar]
- 102.Witkiewicz AK, Ertel A, McFalls J, Valsecchi ME, Schwartz G, Knudsen ES. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res. 2012;18(18):5110–5122. doi: 10.1158/1078-0432.CCR-12-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rassidakis GZ, Lai R, Herling M, Cromwell C, Schmitt-Graeff A, Medeiros LJ. Retinoblastoma protein is frequently absent or phosphorylated in anaplastic large-cell lymphoma. Am J Pathol. 2004;164(6):2259–2267. doi: 10.1016/S0002-9440(10)63782-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fischer K, Bahlo J, Fink AM, Goede V, Herling CD, Cramer P, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127(2):208–215. doi: 10.1182/blood-2015-06-651125. [DOI] [PubMed] [Google Scholar]
- 105.Van Dyke DL, Shanafelt TD, Call TG, Zent CS, Smoley SA, Rabe KG, et al. A comprehensive evaluation of the prognostic significance of 13q deletions in patients with B-chronic lymphocytic leukaemia. Br J Haematol. 2010;148(4):544–550. doi: 10.1111/j.1365-2141.2009.07982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 107.Crocker J, Nar P. Nucleolar organizer regions in lymphomas. J Pathol. 1987;151(2):111–118. doi: 10.1002/path.1711510203. [DOI] [PubMed] [Google Scholar]
- 108.De Keersmaecker K, Atak ZK, Li N, Vicente C, Patchett S, Girardi T, et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat Genet. 2013;45(2):186–190. doi: 10.1038/ng.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hofman IJ, van Duin M, De Bruyne E, Fancello L, Mulligan G, Geerdens E, et al. RPL5 on 1p22.1 is recurrently deleted in multiple myeloma and its expression is linked to bortezomib response. Leukemia. 2017;31(8):1706–1714. doi: 10.1038/leu.2016.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hofman IJF, Patchett S, van Duin M, Geerdens E, Verbeeck J, Michaux L, et al. Low frequency mutations in ribosomal proteins RPL10 and RPL5 in multiple myeloma. Haematologica. 2017;102(8):e317–e320. doi: 10.3324/haematol.2016.162198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fancello L, Kampen KR, Hofman IJ, Verbeeck J, De Keersmaecker K. The ribosomal protein gene RPL5 is a haploinsufficient tumor suppressor in multiple cancer types. Oncotarget. 2017;8(9):14462–14478. doi: 10.18632/oncotarget.14895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19:368–371. doi: 10.1038/nm.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rao S, Lee SY, Gutierrez A, Perrigoue J, Thapa RJ, Tu Z, et al. Inactivation of ribosomal protein L22 promotes transformation by induction of the stemness factor, Lin28B. Blood. 2012;120:3764–3773. doi: 10.1182/blood-2012-03-415349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Novetsky AP, Zighelboim I, Thompson DM, Jr, Powell MA, Mutch DG, Goodfellow PJ. Frequent mutations in the RPL22 gene and its clinical and functional implications. Gynecol Oncol. 2013;128(3):470–474. doi: 10.1016/j.ygyno.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ferreira AM, Tuominen I, van Dijk-Bos K, Sanjabi B, van der Sluis T, van der Zee AG, et al. High frequency of RPL22 mutations in microsatellite-unstable colorectal and endometrial tumours. Hum Mutat. 2014;35:1442–1445. doi: 10.1002/humu.22686. [DOI] [PubMed] [Google Scholar]
- 117.Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ljungström V, Cortese D, Young E, Pandzic T, Mansouri L, Plevova K, et al. Whole-exome sequencing in relapsing chronic lymphocytic leukemia: clinical impact of recurrent RPS15 mutations. Blood. 2016;127(8):1007–1016. doi: 10.1182/blood-2015-10-674572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nieminen TT, O’Donohue MF, Wu Y, Lohi H, Scherer SW, Paterson AD, et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology. 2014;147:595–598. doi: 10.1053/j.gastro.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Onofrillo C, Galbiati A, Montanaro L, Derenzini M. The pre-existing population of 5S rRNA effects p53 stabilization during ribosome biogenesis inhibition. Oncotarget. 2017;8(3):4257–4267. doi: 10.18632/oncotarget.13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Donati G, Bertoni S, Brighenti E, Vici M, Treré D, Volarevic S, et al. The balance between rRNA and ribosomal protein synthesis up- and downregulates the tumour suppressor p53 in mammalian cells. Oncogene. 2011;30(29):3274–3288. doi: 10.1038/onc.2011.48. [DOI] [PubMed] [Google Scholar]
- 122.Daftuar L, Zhu Y, Jacq X, Prives C. Ribosomal proteins RPL37, RPS15 and RPS20 regulate the Mdm2-p53-MdmX network. PLoS One. 2013;8(7):e68667. doi: 10.1371/journal.pone.0068667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Spector DL, Ochs RL, Busch H. Silver staining, immunofluorescence, and immunoelectron microscopic localization of nucleolar phosphoproteins B23 and C23. Chromosoma. 1984;90(2):139–148. doi: 10.1007/BF00292451. [DOI] [PubMed] [Google Scholar]
- 124.Biggiogera M, Fakan S, Kaufmann SH, Black A, Shaper JH, Busch H. Simultaneous immunoelectron microscopic visualization of protein B23 and C23 distribution in the HeLa cell nucleolus. J Histochem Cytochem. 1989;37(9):1371–1374. doi: 10.1177/37.9.2768807. [DOI] [PubMed] [Google Scholar]
- 125.Borer RA, Lehner CF, Eppenberger HM, Nigg EA. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. 1989;56(3):379–390. doi: 10.1016/0092-8674(89)90241-9. [DOI] [PubMed] [Google Scholar]
- 126.Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6(7):493–505. doi: 10.1038/nrc1885. [DOI] [PubMed] [Google Scholar]
- 127.Lindström MS. NPM1/B23: A Multifunctional Chaperone in Ribosome Biogenesis and Chromatin Remodeling. Biochem Res Int. 2011;2011:195209. doi: 10.1155/2011/195209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lindström MS, Zhang Y. B23 and ARF: friends or foes? Cell Biochem Biophys. 2006;46(1):79–90. doi: 10.1385/CBB:46:1:79. [DOI] [PubMed] [Google Scholar]
- 129.Colombo E, Alcalay M, Pelicci PG. Nucleophosmin and its complex network: a possible therapeutic target in hematological diseases. Oncogene. 2011;30(23):2595–2609. doi: 10.1038/onc.2010.646. [DOI] [PubMed] [Google Scholar]
- 130.Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, et al. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003;12(5):1151–1164. doi: 10.1016/S1097-2765(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 131.Szebeni A, Olson MO. Nucleolar protein B23 has molecular chaperone activities. Protein Sci. 1999;8:905–912. doi: 10.1110/ps.8.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chan LW, Lin X, Yung G, Lui T, Chiu YM, Wang F, et al. Novel structural co-expression analysis linking the NPM1-associated ribosomal biogenesis network to chronic myelogenous leukemia. Sci Rep. 2015;5:10973. doi: 10.1038/srep10973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Derenzini M, Sirri V, Pession A, Trerè D, Roussel P, Ochs RL, et al. Quantitative changes of the two major AgNOR proteins, nucleolin and protein B23, related to stimulation of rDNA transcription. Exp Cell Res. 1995;219(1):276–282. doi: 10.1006/excr.1995.1228. [DOI] [PubMed] [Google Scholar]
- 134.Derenzini M, Sirri V, Trerè D, Ochs RL. The quantity of nucleolar proteins nucleolin and protein B23 is related to cell doubling time in human cancer cells. Lab Invest. 1995;73(4):497–502. [PubMed] [Google Scholar]
- 135.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. GIMEMA Acute Leukemia Working Party. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352(3):254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 136.Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc_ AML): biologic and clinical features. Blood. 2007;109(3):874–885. doi: 10.1182/blood-2006-07-012252. [DOI] [PubMed] [Google Scholar]
- 137.The Cancer Genome Atlas Research Network Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Colombo E, Martinelli P, Zamponi R, Shing DC, Bonetti P, Luzi L, et al. Delocalization and destabilization of the Arf tumor suppressor by the leukemia-associated NPM mutant. Cancer Res. 2006;66(6):3044–3050. doi: 10.1158/0008-5472.CAN-05-2378. [DOI] [PubMed] [Google Scholar]
- 139.Colombo E, Bonetti P, Lazzerini Denchi E, Martinelli P, Zamponi R, et al. Nucleophosmin is required for DNA integrity and p19Arf protein stability. Mol Cell Biol. 2005;25(20):8874–8886. doi: 10.1128/MCB.25.20.8874-8886.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.den Besten W, Kuo ML, Williams RT, Sherr CJ. Myeloid leukemia-associated nucleophosmin mutants perturb p53-dependent and independent activities of the Arf tumor suppressor protein. Cell Cycle. 2005;4(11):1593–1598. doi: 10.4161/cc.4.11.2174. [DOI] [PubMed] [Google Scholar]
- 141.Döhner K, Schlenk RF, Habdank M, Scholl C, Rücker FG, Corbacioglu A, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106(12):3740–3746. doi: 10.1182/blood-2005-05-2164. [DOI] [PubMed] [Google Scholar]
- 142.Falini B, Brunetti L, Martelli MP. Dactinomycin in NPM1-Mutated Acute Myeloid Leukemia. N Engl J Med. 2015;373(12):1180–1182. doi: 10.1056/NEJMc1509584. [DOI] [PubMed] [Google Scholar]
- 143.Werner MT, Zhao C, Zhang Q, Wasik MA. Nucleophosmin-anaplastic lymphoma kinase: the ultimate oncogene and therapeutic target. Blood. 2017;129(7):823–831. doi: 10.1182/blood-2016-05-717793. [DOI] [PubMed] [Google Scholar]
- 144.Cui YX, Kerby A, McDuff FK, Ye H, Turner SD. NPM-ALK inhibits the p53 tumor suppressor pathway in an MDM2 and JNK-dependent manner. Blood. 2009;113(21):5217–5227. doi: 10.1182/blood-2008-06-160168. [DOI] [PubMed] [Google Scholar]
- 145.Rassidakis GZ, Thomaides A, Wang S, Jiang Y, Fourtouna A, Lai R, et al. p53 gene mutations are uncommon but p53 is commonly expressed in anaplastic large-cell lymphoma. Leukemia. 2005;19(9):1663–9. doi: 10.1038/sj.leu.2403840. [DOI] [PubMed] [Google Scholar]
- 146.Xu-Monette ZY, Møller MB, Tzankov A, Montes-Moreno S, Hu W, Manyam GC, et al. MDM2 phenotypic and genotypic profiling, respective to TP53 genetic status, in diffuse large B-cell lymphoma patients treated with rituximab-CHOP immunochemotherapy: a report from the International DLBCL Rituximab-CHOP Consortium Program. Blood. 2013;122(15):2630–2640. doi: 10.1182/blood-2012-12-473702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zenz T, Vollmer D, Trbusek M, Smardova J, Benner A, Soussi T, et al. TP53 mutation profile in chronic lymphocytic leukemia: evidence for a disease specific profile from a comprehensive analysis of 268 mutations. Leukemia. 2010;24(12):2072–2079. doi: 10.1038/leu.2010.208. [DOI] [PubMed] [Google Scholar]
- 148.Zenz T, Kröber A, Scherer K, Häbe S, Bühler A, Benner A, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood. 2008;112(8):3322–3329. doi: 10.1182/blood-2008-04-154070. [DOI] [PubMed] [Google Scholar]
- 149.Guièze R, Robbe P, Clifford R, de Guibert S, Pereira B, Timbs A, et al. Presence of multiple recurrent mutations confers poor trial outcome of relapsed/refractory CLL. Blood. 2015;126(18):2110–2117. doi: 10.1182/blood-2015-05-647578. [DOI] [PubMed] [Google Scholar]
- 150.Stengel A, Schnittger S, Weissmann S, Kuznia S, Kern W, Kohlmann A, et al. TP53 mutations occur in 15.7% of ALL and are associated with MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood. 2014;124(2):251–258. doi: 10.1182/blood-2014-02-558833. [DOI] [PubMed] [Google Scholar]
- 151.Seifert H, Mohr B, Thiede C, Oelschlägel U, Schäkel U, Illmer T, et al. The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia. 2009;23(4):656–663. doi: 10.1038/leu.2008.375. [DOI] [PubMed] [Google Scholar]
- 152.Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J Clin Oncol. 2015;33(33):3911–3920. doi: 10.1200/JCO.2014.59.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jardin F, Jais JP, Molina TJ, Parmentier F, Picquenot JM, Ruminy P, et al. Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: a GELA study. Blood. 2010;116(7):1092–1104. doi: 10.1182/blood-2009-10-247122. [DOI] [PubMed] [Google Scholar]
- 154.Alhejaily A, Day AG, Feilotter HE, Baetz T, Lebrun DP. Inactivation of the CDKN2A tumor-suppressor gene by deletion or methylation is common at diagnosis in follicular lymphoma and associated with poor clinical outcome. Clin Cancer Res. 2014;20(6):1676–1686. doi: 10.1158/1078-0432.CCR-13-2175. [DOI] [PubMed] [Google Scholar]
- 155.Iacobucci I, Ferrari A, Lonetti A, Papayannidis C, Paoloni F, Trino S, et al. CDKN2A/B alterations impair prognosis in adult BCR-ABL1-positive acute lymphoblastic leukemia patients. Clin Cancer Res. 2011;17(23):7413–23. doi: 10.1158/1078-0432.CCR-11-1227. [DOI] [PubMed] [Google Scholar]
- 156.Mian M, Scandurra M, Chigrinova E, Shen Y, Inghirami G, Greiner TC, et al. Clinical and molecular characterization of diffuse large B-cell lymphomas with 13q14. 3 deletion. Ann Oncol. 2012;23(3):729–735. doi: 10.1093/annonc/mdr289. [DOI] [PubMed] [Google Scholar]
- 157.Ouillette P, Collins R, Shakhan S, Li J, Li C, Shedden K, et al. The prognostic significance of various 13q14 deletions in chronic lymphocytic leukemia. Clin Cancer Res. 2011;17(21):6778–6790. doi: 10.1158/1078-0432.CCR-11-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Okamoto R, Ogawa S, Nowak D, Kawamata N, Akagi T, Kato M, et al. Genomic profiling of adult acute lymphoblastic leukemia by single nucleotide polymorphism oligonucleotide microarray and comparison to pediatric acute lymphoblastic leukemia. Haematologica. 2010;95(9):1481–1488. doi: 10.3324/haematol.2009.011114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Scheijen B, Boer JM, Marke R, Tijchon E, van Ingen Schenau D, Waanders E, et al. Tumor suppressors BTG1 and IKZF1 cooperate during mouse leukemia development and increase relapse risk in B-cell precursor acute lymphoblastic leukemia patients. Haematologica. 2017;102(3):541–551. doi: 10.3324/haematol.2016.153023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cao B, Fang Z, Liao P, Zhou X, Xiong J, Zeng S, et al. Cancer-mutated ribosome protein L22 (RPL22/eL22) suppresses cancer cell survival by blocking p53-MDM2 circuit. Oncotarget. 2017;8(53):90651–90661. doi: 10.18632/oncotarget.21544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Savage KJ, Harris NL, Vose JM, Ullrich F, Jaffe ES, Connors JM, et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008;111(12):5496–5504. doi: 10.1182/blood-2008-01-134270. [DOI] [PubMed] [Google Scholar]
- 162.Drygin D, O'Brien SE, Hannan RD, McArthur GA, Von Hoff DD. Targeting the nucleolus for cancer-specific activation of p53. Drug Discov Today. 2014;19(3):259–265. doi: 10.1016/j.drudis.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 163.Quin JE, Devlin JR, Cameron D, Hannan KM, Pearson RB, Hannan RD. Targeting the nucleolus for cancer intervention. Biochim Biophys Acta. 2014;1842(6):802–816. doi: 10.1016/j.bbadis.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 164.Brighenti E, Treré D, Derenzini M. Targeted cancer therapy with ribosome biogenesis inhibitors: a real possibility? Oncotarget. 2015;6(36):38617–38627. doi: 10.18632/oncotarget.5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jin R, Zhou W. TIF-IA: An oncogenic target of pre-ribosomal RNA synthesis. Biochim Biophys Acta. 2016;1866(2):189–196. doi: 10.1016/j.bbcan.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.