

Abstract
Proteomics experiments commonly aim to estimate and detect differential abundance across all expressed proteins. Within this experimental design, some of the most challenging measurements are small fold changes for lower abundance proteins. While bottom-up proteomics methods are approaching comprehensive coverage of even complex eukaryotic proteomes, failing to reliably quantify lower abundance proteins can limit the precision and reach of experiments to much less than the identified—let alone total—proteome. Here we test the ability of two common methods, a tandem mass tagging (TMT) method and a label-free quantitation method (LFQ), to achieve comprehensive quantitative coverage by benchmarking their capacity to measure 3 different levels of change (3-, 2-, and 1.5-fold) across an entire data set. Both methods achieved comparably accurate estimates for all 3-fold-changes. However, the TMT method detected changes that reached statistical significance three times more often due to higher precision and fewer missing values. These findings highlight the importance of refining proteome quantitation methods to bring the number of usefully quantified proteins into closer agreement with the number of total quantified proteins.
Keywords: proteome quantitation, label-free, TMT, sensitivity, specificity
Graphical Abstract
INTRODUCTION
The success of proteomics experiments is summarily reported as the number of proteins quantified. In a data dependent analysis (DDA) method, the most abundant precursor ions in a survey scan are selected for identification by fragmentation analysis in a second scan. When combined with the stochastic process of precursor selection in a complex sample like a proteome, this leads to higher variability in the detection and quantitation of the lower abundance proteins in particular. While developing computational and experimental1–3 methods to deal with missing values continues to be an important area of research, most common methods of data analysis benefit from—and often require—complete data sets for hypothesis testing.
Advances in detection and quantitation in label free methods have sought to ameliorate the missing values problem for label free methods,4–6 though typically improvements have been benchmarked by evaluating performance with abundant proteins.6 However, as missing values are more common and detrimental for the quantitation of the lower abundance proteins, we sought to evaluate performance across the full spectrum of abundances. Here, we systematically compared two of the most common data-dependent methods for proteome-wide quantitation, isobaric labeling with tandem mass tags (TMT) and label-free (LFQ), using the same standard two-proteome mixture model, to assess their ability to measure 3 different fold changes across the full spectrum of protein abundances in a proteome. We conclude that comparable numbers of quantified proteins across the two methods resulted in markedly different numbers of proteins available for statistical testing of even 3-fold changes.
EXPERIMENTAL PROCEDURES
Creating a Two-Proteome Sample to Assess Method Performance
Cell lysis and protein digestion. Yeast cultures were harvested by centrifugation, and resuspended in lysis buffer −50 mM HEPES pH 8.5, 8 M urea, 75 mM NaCl, protease (complete mini, EDTA-free) inhibitors (Roche, Basel, Switzerland) at 4 °C. Yeast cells were lysed using the MiniBeadbeater (Biospec, Bartlesville, OK) in microcentrifuge tubes with 1 mL zirconium oxide beads at maximum speed for five cycles of 30 s each, with 1 min pauses between cycles to minimize heating the lysates. Human cells (SH-SY5Y) cells were harvested, resuspended in lysis buffer, and lysed by 20 pumps through a 21-gauge needle. After centrifugation, lysates were transferred to new tubes, spun to pellet cell debris, and the supernatants saved. We determined the protein concentration in the lysate using the bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, Waltham, MA).
Proteins disulfide bonds were reduced with 5 mM tris(2-carboxyethyl)-phosphine (TCEP), at room temperature for 25 min, and alkylated with 10 mM iodoacetamide at room temperature for 30 min in the dark). Excess iodoacetamide was quenched with 15 mM dithiotreitol (room temperature, 15 min in the dark). Methanol–chloroform precipitation was performed prior to protease digestion. In brief, four parts neat methanol was added to each sample and vortexed, one part chloroform was added to the sample and vortexed, and three parts water was added to the sample and vortexed. The sample was centrifuged at 4000 rpm for 15 min at room temperature and subsequently washed twice with 100% methanol, prior to air-drying. Samples were resuspended in 50 mM HEPES pH 8.5 and digested at room temperature for 12 h with LysC protease at a 100:1 protein-to-protease ratio. Trypsin was then added at a 100:1 protein-to-protease ratio and the reaction was incubated 6 h at 37 °C. Peptide concentrations in the digests were measured using the Quantitative Colorometric Peptide assay kit (Pierce). Peptides from a LysC/trypsin digest of human or yeast lysates were mixed to create the ratios shown in Figure 1 in at least triplicate (n = 3, 4, 4). Samples were then split for TMT or LFQ workflows.
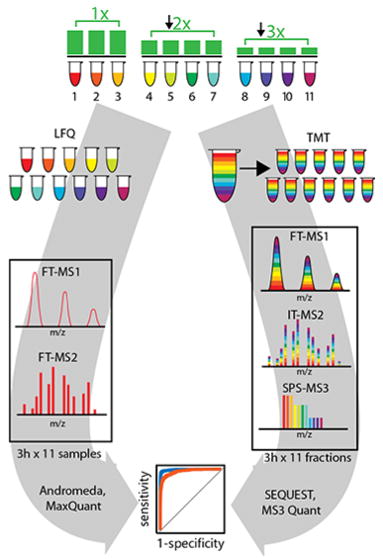
Figure 1.

Experimental design for comparing the ability of a label-free (LFQ) and a tandem mass tag (TMT) dependent method to quantify changes among the less abundant proteins in a proteome. Yeast lysate was spiked into human lysate to 10% of total protein concentration (1× group), 5% (2× group), and 3.3% (3× group) for a total of 11 samples so that the ratio between the first group and second was a 2-fold change, and between the first and third group was a 3-fold change. Samples for TMT were labeled with unique isobaric tags per sample, mixed, and fractionated by basic pH reversed phase HPLC (RP-HPLC). Eleven samples from LFQ and 11 fractions of the TMT set were analyzed on the same Orbitrap Fusion Lumos mass spectrometer with online RP-nHPLC separation with 3 h gradients. Data from label-free samples were processed through MaxQuant using the Andromeda search engine (including match-between-runs and the MaxLFQ algorithm). Data from the TMT fractions were processed using the SEQUEST search engine to identify peptides, and reporter ion abundances were extracted directly from associated SPS-MS3 spectra.
LFQ Pipeline
Samples were desalted via C-18 Stagetips. Eleven, 3-h gradients were collected using an Orbitrap Fusion Lumos instrument coupled to a Proxeon EASY-nLC 1200 liquid chromatography (LC) pump (Thermo Fisher Scientific). Peptides were fractionated on a 100 μm inner diameter microcapillary column packed with ~35 cm of Accucore 150 resin (2.6 μm, 150 Å, ThermoFisher Scientific). For each analysis, we loaded 1 μg per sample onto the column. Peptides were separated using a 3 h gradient of 6 to 26% acetonitrile in 0.125% formic acid at a flow rate of ~400 nL/min. Each analysis used the multinotch MS3-based TMT method18,19 on an Orbitrap Fusion Lumos.
Instrument Settings
FTMS1 resolution (120 000), FTMS2 resolution (15 000), FTMS2 max ion time (100 ms), Isolation window (1.4 m/z), AGC setting (5E5), HCD energy (35%), dynamic exclusion window (90 s). A TOP10 method was used where each FTMS1 scan was used to select up to 10 FTMS2 precursors for interrogation by HCD-MS2 with readout in the orbitrap. An average of 55 000 MS2 scans was collected in each 3 h run.
Samples were searched with MaxQuant (ver. 1.5.8.3) against a combined yeast and human database which was concatenated with their reversed sequences as decoys for FDR determination. Searches restricted the precursor ion tolerance to 20 ppm, and product ion tolerance window was set to 0.5 m/z. Searches allowed up to two missed cleavages, included static carbamidomethylation of cysteine residues (+57.021 Da), and variable oxidation of methionine residues (+15.995 Da). Results were filtered to a 1% FDR at the protein level with MaxQuant.19 Match-between-runs (MBR) was enabled with default settings across the 11 samples (match time window = 0.7 min, alignment time window = 20). Quantification was via MaxQuant’s LFQ algorithm which combines and adjusts peptide intensities into a protein intensity value (Table S2).
TMT Pipeline
The 11 TMT-labeled samples were mixed into a single sample and separated by basic pH RP HPLC. We used an Agilent 1100 pump equipped with a degasser and a photodiode array (PDA) detector (set at 220 and 280 nm wavelength) from Thermo Fisher Scientific (Waltham, MA). Peptides were subjected to a 50 min linear gradient from 5% to 35% acetonitrile in 10 mM ammonium bicarbonate pH 8 at a flow rate of 0.6 mL/min over an Agilent 300Extend C18 column (3.5 μm particles, 4.6 mm ID and 220 mm in length). The peptide mixture was fractionated into a total of 96 fractions which were consolidated into 24 fractions. Samples were subsequently acidified with 1% formic acid and vacuum centrifuged to near dryness. Each eluted fraction was desalted via StageTip, dried via vacuum centrifugation, and reconstituted in 5% acetonitrile, 5% formic acid for LC–MS/MS processing.
Eleven fractions were used for 3-h gradient separations as described above, but with the SPS-MS3 method on an Orbitrap Fusion Lumos.20,25 Parameter settings: FTMS1 resolution (120 000), ITMS2 isolation window (0.4 m/z), ITMS2 max ion time (120 ms), ITMS2 AGC (2E4), ITMS2 CID energy (35%), SPS ion count (up to 10), FTMS3 isolation window (0.4 m/z), FTMS3 max ion time (150 ms), FTMS3 AGC (1.5E5), FTMS3 resolution (50 000). A TOP10 method was used where each FTMS1 scan was used to select up to 10 MS2 precursors for interrogation by CID-MS2 with readout in the ion trap. Each MS2 was used to select precursors (SPS ions) for the MS3 scan which measured reporter ion abundance for the 11 samples simultaneously,20,25 which has been shown to reduce ion interference compared to MS2 quantification.43 An average of 27 500 MS2/MS3 paired scans was collected in each run.
Data Analysis
Samples were searched with the Sequest algorithm (Ver. 28) against a combined yeast and human database which was concatenated with their reversed sequences as decoys for FDR determination. Searches restricted the precursor ion tolerance to 50 ppm, and product ion tolerance window was set to 0.9 m/z. As for the LFQ analysis, searches allowed up to two missed cleavages, included static modification of lysine residues and peptide N-termini with TMT tags (+229.163 Da), static carbamidomethylation of cysteine residues (+57.021 Da), variable oxidation of methionine residues (+15.995 Da). Results were filtered to a 1% FDR at the peptide and protein levels using linear discriminant analysis and the target-decoy strategy. MS3 spectra were processed as signal-to-noise ratios for each reporter ion based on noise levels within a 25 Th window. Proteins were quantified by summing reporter ion intensities across all matching PSMs. PSMs with low isolation specificity (<0.7) or MS3 spectra with TMT reporter summed signal-to-noise ratio that is less than 200 were removed (Table S2). Equal human protein starting amounts was enforced by normalizing to the sum of all human peptides for each of the 11 channels (Table S1).
Reciprocal analysis of the two data sets using the alternate data analysis pipeline produced nominally equivalent data sets, as demonstrated by their matching number of total proteins, total peptides, yeast proteins and yeast peptides (Table S3).
Statistical Analysis
The data sets from both pipelines (Table S1) were imported into the Perseus (version 1.5.3.2) program.1 Reverse database hits and contaminants were removed before performing two-sided t tests separately for 3-fold, 2-fold, and 1.5-fold changes across the data set with multiple hypothesis correction of p-values (Benjamini–Hochberg FDR). A minimum of 2 measurements per group was required. The human proteins were expected to have no changes while every yeast protein should contain the expected changes in the expected channels. In Figure S2d, missing values were also imputed in Perseus using random values derived from a distribution of all other log2-transformed protein values from that sample. The distribution for drawing values was created from the default settings (mean down shift = 0.3, standard deviation down shift = 1.8) so that the final matrix was devoid of missing values.
Data Availability
Raw files and metadata are publicly available on ProteomeXchange at project accession PXD007683.
RESULTS
Experimental Overview
We first sought to create samples where known differences for thousands of proteins could be present in an even larger unchanging background. To accomplish this, we spiked yeast lysate into human lysate to 10% of total protein concentration (1× group), 5% (2× group), and 3.3% (3× group) for a total of 11 samples so that the ratio between the yeast proteins in the first group and second was a 2-fold change, between the first and third group was a 3-fold change, and between the second group and the third group was a 1.5-fold change. Human proteins formed an unchanging background across all samples (Figure 1). The resultant mixture distributes yeast proteins across the entire range of protein abundances but with a distinct enrichment among the lower abundance proteins (Figure S1).
In this work, we examined two very common workflows for protein expression profiling (Figure 1). Both methods rely on data-dependent acquisition via shotgun sequencing to collect proteome-wide measurements. For clarity, we highlight the workflow for each separately below.
The MaxLFQ method is an end-to-end workflow that has been used successfully in dozens of studies.7–18 It includes single-shot analyses of trypsin digested lysates using long gradients to identify as many peptides as possible. In our case, the 11 single-shot LC–MS/MS analyses (33 h total) resulted in 454 545 matched peptides using Andromeda and a 1% protein-level false discovery rate (FDR). In addition, the match-between-runs (MBR) feature within MaxQuant allows for the potential transfer of missed identifications from one sample to any of the other samples.5,19 Using MBR on our 11 runs increased the total number of peptides matched to 627 579 (38% increase). Combining the measured intensities from all matched peptides into proteins is then performed by the MaxLFQ algorithm.5 This is a very challenging step in the workflow. The mean accuracy of the fold-change measurements based on MaxLFQ was excellent (within 15% of expected, Table S1). Finally, the MaxLFQ values for each protein were analyzed for statistical changes in the Perseus environment. This workflow of single-shots → Andromeda → MBR → MaxLFQ → Perseus is commonly found today in studies using data-dependent quantitative proteomics.1 Because of its rapid and inexpensive sample preparation, accessible instrumentation, and ability to scale to an arbitrarily large number of samples this workflow is the method of choice for many experiments.
The TMT11-plex workflow makes use of chemical labeling and sample multiplexing to combine the 11 trypsin-digested lysates into a single complex peptide mixture. This single sample is then fractionated via basic pH reverse-phase separation, and 11 samples are again analyzed with 3 h gradients (33 h total). A strategy that overcomes the issue of ratio distortion in TMT-based experiments was used, termed the SPS-MS3 method.20 The MS2 scan is not used for quantification but rather several precursor ions are selected simultaneously using a notched waveform in the ion trap called synchronous precursor selection (SPS). The 11 TMT reporter ions are then measured in the ensuing MS3 scan. This method is slower than LFQ ones since both MS2 (identification) and MS3 (quantification) are collected for each precursor. Peptide sequence identification was via Sequest and 112 663 peptides were matched at a 1% protein-level FDR. Reporter ion signal for peptides derived from the same protein are summed (Table S1). The protein-level reporter ion signals were then passed for statistical analysis. For simplicity, we also used the Perseus suite to perform the t tests with multiple hypothesis correction. This workflow of TMT-labeling → Fractionation → SPS-MS3 analysis → Sequest → Statistical analysis has been applied in dozens of studies for data-dependent proteome-wide quantification.20–32 Isobaric labeling methods such as this one share the major advantage of (1) allowing more complex sample preparation like fractionation without a proportional increase in technical variation, and (2) increased quantitation across samples.
Data Set Summary
Figure 2a presents the major quantitative statistics. Both methods utilized 33 h of instrument time and analyzed the same 11 lysates and dilution factors. The LFQ-method identified 4 fold more total peptides and 3 fold more yeast peptides. Yet, the number of sequence unique, nonredundant peptides observation still favored the TMT method due to the ability to fractionate the combined sample. The TMT-method quantified 20% more total proteins (9312 vs 7731) including 38% more yeast proteins (1306 vs 945). Typically the number of quantified proteins is reported as a single number, often the union of proteins quantified across samples. This simple nomenclature, however, glosses over a complex difference in the structure of the data sets, namely the distribution of missing values among those quantified proteins. Missing values were present to some extent across the 11 samples in nearly 50% of all LFQ protein measurements, with a surprising 24% retaining quantitation in only a single sample (Figure 2b). The case is even starker among yeast proteins specifically, with more than 50% of yeast proteins missing quantitation in at least one sample, and over a quarter retaining quantitation in a single sample (Figure 2c). These values are comparable to datasets published using this method.11
Figure 2.

Comparing the two methods found an advantage for the TMT approach in breadth of coverage and depth of quantified proteins without missing values. (a) Summary of the two data sets. The TMT-method quantified 20% more total proteins and 38% more yeast proteins, while the label-free method identified over 4 times as many total peptides and over 3 times as many yeast peptides. (b) The percent of total proteins missing quantitation in a sample plotted as a histogram shows that almost half of all proteins from the LFQ method were missing at least 1 value, with a surprising 24% retaining quantitation in only a single sample. No peptides with missing values were present in the TMT set following filtering for greater than 70% isolation specificity and a combined signal-to-noise value greater than 200 across all 11 channels. (c) Similar profiles characterized the missing values for yeast proteins, with over half of proteins missing one or more values. (d) Stacked barchart comparing the number of proteins quantified in two or more samples within a group for total proteins (lighter color) and yeast proteins (darker color) found the TMT-method reached ~2 times as many total proteins and ~2.5 times as many yeast proteins. Box heights mark the number of proteins in each set, with the dashed boxes denoting the size of the original data set (7700 proteins). (e) Less abundant proteins were more likely to be missing values in the LFQ data set. The percent missing values per protein for each group was calculated and plotted as a heatmap from 100% missing (black) to 0% missing (yellow). Proteins were rank ordered by the sum LFQ values and sampled around the deciles producing 10 heatmaps, demonstrating a difference in missing values for the least abundant proteins compared to the most abundant. Yeast protein names are shown in red. *Match-Between-Runs increases the Total Peptides for MaxQuant searches to 627 579 and the Yeast Peptides to 43 605 for the LFQ data. Protein numbers unchanged.
The effect of the missing values within LFQ samples was large. For example, out of 945 yeast protein detected, only 58%, 47%, and 40% of proteins in the 1×, 2×, and 3× groups, respectively, still maintained at least 2 values per group (sufficient information for the most basic statistical hypothesis testing) (Figure 2d). To understand the relationship between missing values and protein abundance, we created 10 heat maps each representing 25 proteins surrounding each decile of abundance from lowest (10%) to highest (100%). Each of the three groups was plotted as yellow if no missing values were present across all of that group’s sample members (Figure 2e). Missing values were very common up through the bottom ~40% of the data set sorted by abundance. For the top half of the data set, most proteins reported all 11 measurements. Yeast proteins are shown in red text, and they were more frequent in the lower half of the data set.
Accuracy and Precision
We next compared precision and accuracy among proteins measured in at least 2 samples/group (Figure 3). Both methods achieved comparably high levels of accuracy (within 15% of expected for 3-, 2-, and 1.5-fold changes—Figure 3a). Precision was assessed by comparing the coefficient of variation (CV) for each method (Figure 3b). While the average measurements were accurate, the variance associated with these measurements was ~3× greater for the LFQ method (4% vs 12% CV).
Figure 3.

The two methods showed comparable average accuracy, but the TMT approach had 3-fold higher average precision. (a) Accuracy assessments. Scatterplots of the average fold change are shown for each protein for 3-fold (blue) 2-fold (orange), and 1.5-fold (green) against the total PSM per protein for the two groups in a given fold change comparison for proteins with at least 2 measurements. Good accuracy was achieved for both methods, even at the 1.5 fold change. (b) Precision for the three groups as measured by the coefficient of variation (CV) and plotted as histograms and boxplots showed a 3-fold higher average CV for the label-free method in all three groups.
Sensitivity and Selectivity
Ultimately a proteome-wide measurement technique is only as valuable as the number of proteins for which useful measurements can be made. Here, this is the number of yeast proteins whose relative abundance could be determined to be statistically different between sample groups (among the human proteins of unchanging abundance). Proteins are typically chosen for follow-up experiments and validation based on the significance of their p-values. Thus, we compared the two method’s receiver operating characteristic (ROC) curves based on p-values generated in the Perseus software environment1 from a 2-sided t test (after correcting for multiple hypothesis testing) for each of the fold changes. The goal was to assess each method’s capacity to correctly detect changes in relative protein abundance. Focusing on the largest (3-fold) change for yeast proteins (group 1 compared to group 3) we see that both the TMT and the LFQ methods have very high diagnostic ability, with area under the curve (AUC) values of 0.987 and 0.975, respectively. The probabilistic interpretation of these AUC values is that the methods would provide a lower p-value for a true 3-fold change than for a false signal an impressive 98.7% or 97.5% of the time (Figure 4a). On the same ROC curves, zooming in to the false positive rate (FPR) region between 0 and 0.05 (Figure 4b) highlights the proteins with the lowest p-values for a fold change. These would be the proteins often chosen as targets for follow-up experiments. Within this region the TMT method outperformed the LFQ method by a larger margin. In constructed examples such as this one, the true positives are known, and the rate or number of true positives recovered at a 1% false positive rate (FPR) can be used to compare the methods diagnostic ability among their respective best predictions. We found the TMT method had a 20% higher true positive rate, but more importantly, 4 times as many true positives at the 1% FPR (1245 vs 288). Even more pronounced detection capabilities were observed for the TMT-method at the 2-fold and 1.5-fold changes (Figure S2b,d).
Figure 4.

Evaluation of hypothesis-testing using each method. (a) ROC plots of the adjusted p-values from a 2-sided t test for the 3-fold change comparison for proteins with >2 quant values highlighting the sensitivity and specificity for the TMT (blue) and LFQ (red) methods, which at the whole proteome level were both quite good at distinguishing proteins that changed in abundance between groups (yeast) from proteins that did not (human). (b) In the false positive rate (FPR) range from 0 to 0.05, which contains the proteins typically chosen for follow-up experiments, the TMT approach outperformed the LFQ method. Green filled circles represent the actual point where the adjusted p-value was 0.05. (c) Summary plot showing quantified yeast proteins and the outcome for statistical testing using Perseus with a 2-sided t test (multiple hypothesis correction included with FDR < 0.05) for each method at each fold change. The LFQ method detected less than a third of identified yeast proteins as significantly changing. (d) Summary statistics for each method’s performance at a common significance threshold (p < 0.05 after multiple hypothesis-testing correction) demonstrate the substantial difference in sensitivities between these two methods. TP = true positive, TPR = true positive rate, AUC = area under curve.
In a real-world experiment the true positives are, of course, unknown, with their prediction being the goal. This is often achieved by thresholding the rank-ordered p-value lists to an arbitrary level of stringency, typically p < 0.05. Green filled circles in the inset of the ROC curves (Figures 4b, S2b,d) denote the actual points where the p-values were exactly 0.05. The size of the set of proteins beneath this threshold would typically be used for further inference and validation, making it one of the more useful summary statistics for the value of a proteome-wide measurement method. Comparing the number of yeast proteins reported as quantified compared to the number that ultimately reach statistical significance in each of the three different fold-change comparisons shows a remarkable difference in the predictive power of quantified proteins between the methods (Figure 4c). Out of 945 quantified yeast proteins by the LFQ-method, fewer than 300 were found significantly changed even among 3-fold changes (t test, FDR < 0.05). While this represents 80% of the proteins with sufficient values to perform a t test (372 had 2 or more values), it is less than 33% of the 945 quantified proteins. Similar trends were observed for the 2-fold and 1.5-fold changes, with fewer than 30% of proteins quantified in the LFQ method showing a detectable 1.5-fold change.
DISCUSSION
In this study, we examined the accuracy and sensitivity of an LFQ and a TMT-MS3 method for an 11-sample data set (n = 3, n = 4, n = 4) at 3 expected fold change ratios. This is a very common workflow for three conditions with replicates. The use of known mixtures of human and yeast at different levels enabled data set-wide method assessment. For these experiments, yeast protein content in each sample was present at a maximum of 10% to represent changes in proteins of lower abundance. Both methods provided accurate median ratios even for 1.5-fold changes. However, the sensitivity of the LFQ method was severely reduced due to missing values and the larger measurement error compared to the TMT method.
Missing values occur frequently in data dependent analyses because shotgun sequencing does not select the identical precursor peptides reliably from run to run with a bias toward lower abundance precursors being less likely to be observed. MaxQuant’s match-between-runs (MBR) algorithm attempts to transfer stochastic identifications to the remaining samples where MS/MS identification was not attempted or successful. This by no means solves the missing values problem. In our case, 454 545 peptides were identified across the 11 label-free samples and MBR increased this number to 627 579 (38%). However, even after MBR, the LFQ values returned by the MaxLFQ algorithm still reported 25% of all possible peptide values as missing. These missing values had a profound effect, such that performing t-tests was not even possible on a large fraction of the data set. For lower abundance proteins at least two-thirds of identified proteins in LFQ experiments using replicates would not be expected to result in the detection of a significant difference even if a 3-fold difference existed.
One possible solution to missing values is imputation. Perseus allows for imputation of all missing values in a column based on randomly drawing intensities from a collapsed distribution of the remaining values in the column. We performed imputation on the LFQ data set in Figure S2 using default settings so that there were no missing values. While this doubled the number of proteins on which ttests could be performed, the final number of detected changes among yeast proteins was almost identical (Figure S2d), but the number of false negatives markedly increased (Figure S3).
Data-independent acquisition methods (e.g., SWATH) are also commonly based on label free quantification,3,33–42 but with a dramatic reduction in the number of missing values between samples. This is because fewer, but identical, peptides are generally used for quantification across all samples, overcoming a major hurdle for single-shot data dependent proteomics. In general, however, data independent acquisition methods are still not capable of the same depth of proteome analysis as the TMT method used here, in part because they do not routinely employ fractionation as this would affect quantification in a label-free environment. Future advances in SWATH methodologies may allow this promising technique to bridge that gap.
The TMT method identified 4 fold fewer peptides across the 11 samples, yet the number of nonredundant (unique) peptide sequences was greater for the TMT method (75k vs 59k). The greater number of unique peptides can be attributed to the use of fractionation in the TMT workflow. This allowed for an increase in the total amount of each peptide that could be loaded onto the instrument since 1 μg of each fraction was loaded instead of 1 μg of each lysate. Optimally peptides only appear in one fraction which allows lower abundance peptides to be sequenced even though the spectrum acquisition rate is lower.
For the largest fold change measured here (3 fold) the LFQ method detected only 396 out of 945 yeast proteins (31%) as significantly different. In contrast, for the lowest fold change measured (1.5 fold), the TMT method detected 1065 out of 1306 yeast proteins (80%) as significantly different. Thus, the TMT method detected 3 times more significant changes for 2 times smaller changes. The majority of these differences could be attributed directly to a lack of sufficient values to perform statistical tests in the LFQ data set. On the basis of these findings, real hazards exist in simply reporting the number of proteins identified/quantified in data sets for single-shot LFQ experiments. These results clearly demonstrate that a large fraction of the data set does not contain sufficient information to allow for routine statistical testing due to missing values. Finally, we demonstrate that the TMT workflow brings the number of usefully quantified proteins into closer agreement with the number of total quantified proteins largely by higher precision and more complete quantitation across samples, particularly among less abundant proteins.
While this study compared at proteome scale two commonly used workflows, the raw mass spectrometry files (11 LFQ mass spectrometry runs, and 11 TMT mass spectrometry runs) are available to be reprocessed by any alternative workflow. Importantly, the ground truth within the sample is always known (i.e., every quantified yeast protein has altered levels and every human protein does not). Thus, this work can serve as a resource for the community to improve the sensitivity or the accuracy or both within LFQ- and TMT-based approaches.
Supplementary Material
Acknowledgments
We would like to thank the many members of the Gygi lab for their constructive feedback and encouragement. This work was supported by NIH grant (GM67945) to S.P.G. and K01 DK098285 to J.A.P.
ABBREVIATIONS
- LFQ
label-free quantitation
- TMT
tandem mass tag
- FDR
false discovery rate
Footnotes
Author Contributions
J.D.O., J.A.P. performed experiments; J.D.O., J.A.P, J.J.O., S.P.G. analyzed data and conceptualized the work; and J.D.O., S.P.G. wrote the manuscript. J.J.O worked on the use and interpretations of metrics for comparing different methodologies.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteo-me.8b00016.
Figures S1–S3; Table S3 (PDF)
Table S1 (XLSX)
Table S2 (XLSX)
References
- 1.Tyanova S, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 2.Navarro P, et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat Biotechnol. 2016;34:1130–1136. doi: 10.1038/nbt.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Röst HL, et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014;32:219–23. doi: 10.1038/nbt.2841. [DOI] [PubMed] [Google Scholar]
- 4.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 5.Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry – based shotgun proteomics. Nat Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 6.Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kulak Na, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11:319–24. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- 8.Gilbert LA, et al. Resource Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoshino A, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–35. doi: 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rieckmann JC, et al. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat Immunol. 2017;18:583–593. doi: 10.1038/ni.3693. [DOI] [PubMed] [Google Scholar]
- 11.Sacco F, et al. Glucose-regulated and drug-perturbed phosphoproteome reveals molecular mechanisms controlling insulin secretion. Nat Commun. 2016;7:29. doi: 10.1038/ncomms13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geyer PE, et al. Proteomics reveals the effects of sustained weight loss on the human plasma proteome. Mol Syst Biol. 2016;12:901. doi: 10.15252/msb.20167357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeiler M, Moser M, Mann M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol Cell Proteomics. 2014;13:3435–3445. doi: 10.1074/mcp.M114.038513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sacco F, et al. Deep Proteomics of Breast Cancer Cells Reveals that Metformin Rewires Signaling Networks Away from a Pro-growth State. Cell Syst. 2016;2:159–171. doi: 10.1016/j.cels.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Bianchi G, et al. Fasting induces anti-Warburg effect that increases respiration but reduces ATP-synthesis to promote apoptosis in colon cancer models. Oncotarget. 2015;6:11806–19. doi: 10.18632/oncotarget.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azimifar SB, Nagaraj N, Cox J, Mann M. Cell-type-resolved quantitative proteomics of murine liver. Cell Metab. 2014;20:1076–1087. doi: 10.1016/j.cmet.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Geyer PE, et al. Plasma Proteome Profiling to Assess Human Health and Disease. Cell Syst. 2016;2:185–195. doi: 10.1016/j.cels.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 18.Sharma K, et al. Cell type - and brain region - resolved mouse brain proteome. Nat Neurosci. 2015;18:1–16. doi: 10.1038/nn.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 20.Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods. 2011;8:937–40. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isasa M, et al. Multiplexed, Proteome-Wide Protein Expression Profiling: Yeast Deubiquitylating Enzyme Knockout Strains. J Proteome Res. 2015;14:5306–5317. doi: 10.1021/acs.jproteome.5b00802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perera RM, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–5. doi: 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chick JM, et al. Defining the consequences of genetic variation on a proteome-wide scale. Nature. 2016;534:500–505. doi: 10.1038/nature18270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paulo JA, et al. Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J Proteomics. 2016;148:85–93. doi: 10.1016/j.jprot.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McAlister GC, et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weekes MP, et al. Quantitative temporal viromics: An approach to investigate host-pathogen interaction. Cell. 2014;157:1460–1472. doi: 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fielding CA, et al. Control of immune ligands by members of a cytomegalovirus gene expansion suppresses natural killer cell activation. eLife. 2017;6:e22206. doi: 10.7554/eLife.22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulo JA, Gaun A, Gygi SP. Global Analysis of Protein Expression and Phosphorylation Levels in Nicotine-Treated Pancreatic Stellate Cells. J Proteome Res. 2015;14:4246–4256. doi: 10.1021/acs.jproteome.5b00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy JP, Stepanova E, Everley RA, Paulo JA, Gygi SP. Comprehensive Temporal Protein Dynamics during the Diauxic Shift in Saccharomyces cerevisiae. Mol Cell Proteomics. 2015;14:2454–2465. doi: 10.1074/mcp.M114.045849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wühr M, et al. The Nuclear Proteome of a Vertebrate. Curr Biol. 2015;25:2663–2671. doi: 10.1016/j.cub.2015.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erickson BK, et al. Evaluating multiplexed quantitative phosphopeptide analysis on a hybrid quadrupole mass filter/linear ion trap/orbitrap mass spectrometer. Anal Chem. 2015;87:1241–1249. doi: 10.1021/ac503934f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dephoure N, et al. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. eLife. 2014;3:e03023. doi: 10.7554/eLife.03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Methods. 2004;1:39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 34.Navarro P, et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat Biotechnol. 2016;34:1130–1136. doi: 10.1038/nbt.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins BC, et al. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14–3-3 system. Nat Methods. 2013;10:1246–53. doi: 10.1038/nmeth.2703. [DOI] [PubMed] [Google Scholar]
- 36.Selevsek N, et al. Reproducible and Consistent Quantification of the Saccharomyces cerevisiae Proteome by SWATH-mass spectrometry. Mol Cell Proteomics. 2015;14:739–749. doi: 10.1074/mcp.M113.035550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shao S, et al. Minimal sample requirement for highly multiplexed protein quantification in cell lines and tissues by PCT-SWATH mass spectrometry. Proteomics. 2015;15:3711–3721. doi: 10.1002/pmic.201500161. [DOI] [PubMed] [Google Scholar]
- 38.Tsugawa H, et al. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat Methods. 2015;12:523–526. doi: 10.1038/nmeth.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lambert JP, et al. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat Methods. 2013;10:1239–45. doi: 10.1038/nmeth.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsou CC, et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods. 2015;12:258–264. doi: 10.1038/nmeth.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Egertson JD, et al. Multiplexed MS/MS for improved data-independent acquisition. Nat Methods. 2013;10:744–6. doi: 10.1038/nmeth.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Distler U, et al. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat Methods. 2014;11:167–70. doi: 10.1038/nmeth.2767. [DOI] [PubMed] [Google Scholar]
- 43.Paulo JA, O’Connell JD, Gygi SP. A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J Am Soc Mass Spectrom. 2016;27:1620–1625. doi: 10.1007/s13361-016-1434-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw files and metadata are publicly available on ProteomeXchange at project accession PXD007683.