

Abstract
Vascular smooth muscle cells (VSMCs) derived from cardiovascular progenitor cell (CVPC) lineage populate the tunica media of the aortic root. Understanding differentiation of VSMCs from CVPC will further our understanding of the molecular mechanisms contributing to aortic root aneurysms, and thus, facilitate the development of novel therapeutic agents to prevent this devastating complication. It is established that the yes-associated protein (YAP) and Hippo pathway is important for VSMC proliferation and phenotype switch. To determine the role of YAP in differentiation of VSMCs from CVPCs, we utilized the in vitro monolayer lineage specific differentiation method by differentiating human embryonic stem cells into CVPCs, and then, into VSMCs. We found that expression of YAP decreased during differentiation of VSMC from CVPCs. Overexpression of YAP attenuated expression of VSMC contractile markers and impaired VSMC function. Knockdown of YAP increased expression of contractile proteins during CVPC-VSMCs differentiation. Importantly, expression of YAP decreased transcription of myocardin during this process. Overexpression of YAP in PAC1 SMC cell line inhibited luciferase activity of myocardin proximal promoter in a dose dependent and NKX2.5 dependent manner. YAP protein interacted with NKX2.5 protein and inhibited binding of NKX2.5 to the 5’-proximal promoter region of myocardin in CVPC-derived VSMCs. In conclusion, YAP negatively regulates differentiation of VSMCs from CVPCs by decreasing transcription of myocardin in a NKX2.5-dependent manner.
Keywords: Yes-associated protein, stem cells, vascular smooth muscle cell, cardiovascular progenitor cell, differentiation
Graphical Abstract
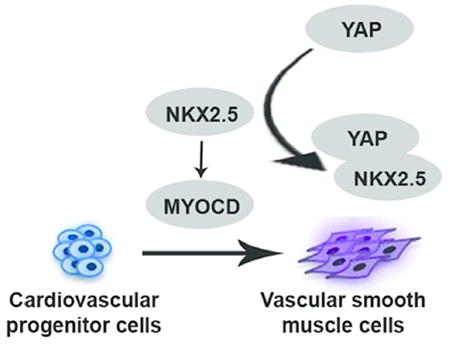
Our study demonstrated a negative regulation of YAP in the cardiovascular progenitor cells (CVPC)-VSMC differentiation process. We found YAP blocked NKX2.5’s binding to myocardin promoter and inhibited transcription of myocardin during this process. We identified novel modulators in VSMC differentiation and provided a potential target for understanding the development of aortic root aneurysm.
Introduction
Aortic aneurysms are associated with aortic dissection and rupture, which are lethal. There is no medical treatment to prevent or reverse aortic aneurysm. Aortic aneurysms could develop from the loss of vascular smooth muscle cells (VSMCs) in the media layer of the aortic wall leading to progressive aortic dilation. Sixty percent of aortic aneurysms involve the aortic root [1]. Specifically, the subgroup of VSMCs located in the media layer of aortic root is differentiated from second heart field-derived cardiovascular progenitor cells (CVPCs), thereby playing a critical role in the pathological progression of aortic root aneurysms [2]. There is a need to further elucidate the mechanisms in CVPC-derived VSMC differentiation so that novel therapeutic strategies can be developed to prevent aortic root aneurysm.
Yes-associated protein (YAP) is a key co-transcription factor involved in the Hippo signaling that includes upstream kinases (NF2, MST1/2, LATS1/2), major co-transcription factors (YAP/TAZ), and transcription factors TEAD1-4 [3]. Our group, as well as evidence from other groups, found that Yap was a central regulator of the VSMC phenotype switch and neointima formation after artery injury in adult rodent models [4, 5]. Neural crest-specific deletion of Yap and Taz [6, 7] caused deficiency of neural crest development and subsequent VSMC differentiation. Conditional deletion of Yap with a Sm22α-Cre in mice showed that knockout of Yap was sufficient to impede VSMC proliferation resulting in vessel wall thinning and vascular morphological defects [8]. These studies suggest that YAP plays an important role in VSMC repair and vascular development. However, it is unknown how YAP regulates the process of VSMCs differentiation from cardiovascular progenitor cell lineage.
The development of human embryonic and induced pluripotent stem cells [9, 10] has provided new sources to sizably derive VSMCs and offers valuable advantages for identifying novel factors in controlling VSMCs-lineage differentiation. As a newly developed humanized in vitro tool, in vitro monolayer VSMC differentiation demonstrated advantages in differentiation lineage, origin, and stage controlling [11, 12]. Importantly, differentiated VSMCs exhibited transcription and metabolomics profile similar to primary VSMCs [13]. Therefore, in vitro lineage and stage controlled human VSMC differentiation models can facilitate ways to dissect the modulators and pathways involved in different lineages and stages. It also can be utilized to improve quality of tissue engineered vascular grafts for future patient treatment. [14, 15]
In this study, we adopted the in vitro monolayer VSMC differentiation system by differentiating human embryonic stem cells (hESCs) H1 cell line into VSMCs through cardiovascular progenitor cells (CVPCs) lineage [12, 15]. Since myocardin is the most potent driver for VSMC differentiation [16-19] and Yap has been showed to abolish myocardin’s function majorly by protein-protein interaction [4], we hypothesized that Yap would inhibit the differentiation of CVPCs to VSMCs by inhibiting the function of myocardin.
Materials and Methods
Cell culture and VSMC differentiation
Human embryonic stem cell line H1 was cultured monolayer by using TeSR™-E8™ feeder free culture medium. VSMC differentiation protocol from H1 ESCs was modified from published papers and our previous report [12, 15]. As VSMC differentiation from CVPC stage was our research interest, it is illustrated in Figure 1A. Briefly, The hESCs were digested with Versene (Life Technologies) into single cells and plated onto Matrigel-coated culture dishes at a density of 5×104 cells/cm2 in CVPC induce medium (DMEM/F12, 1×B27 supplement without vitamin A (Life Technologies), 50 μg/mL ascorbic acid (Sigma), 25 ng/mL BMP4 (R&D Systems), 3 μM GSK3 inhibitor CHIR99021 (Stemgent),1% pen/strep, and 400 μM 1-thioglycerol (Sigma)) for three days. In order to enhance cell survival, 10 μM ROCK inhibitor Y27632 (Stemgent) was added to the medium and incubated overnight after cell digestion. Medium was replenished daily. For VSMCs terminal differentiation, CVPCs were digested into single cells by Accutase (Stemcell technologies) and plated on Matrigel-coated dishes at density of 2×104cells/cm2 with VSMCs differentiation medium (DMEM/F12, 1×B27 supplement without vitamin A, 2 ng/mL TGF-β1 (PeproTech)) for seven days, and Y27632 (10μM) was added to the medium overnight after seeding individualized CVPCs. Primary human aortic VSMCs and fibroblasts were isolated by following previous report [4, 20] and cultured in 5% fetal bovine serum (Sigma). Primary VSMCs were treated with identical VSMC differentiation medium for 2 days before being compared to CVPC-VSMCs.
Figure 1. Characterization of VSMCs differentiated from ESCs derived CVPCs: highly efficient and comparable with primary human VSMCs.
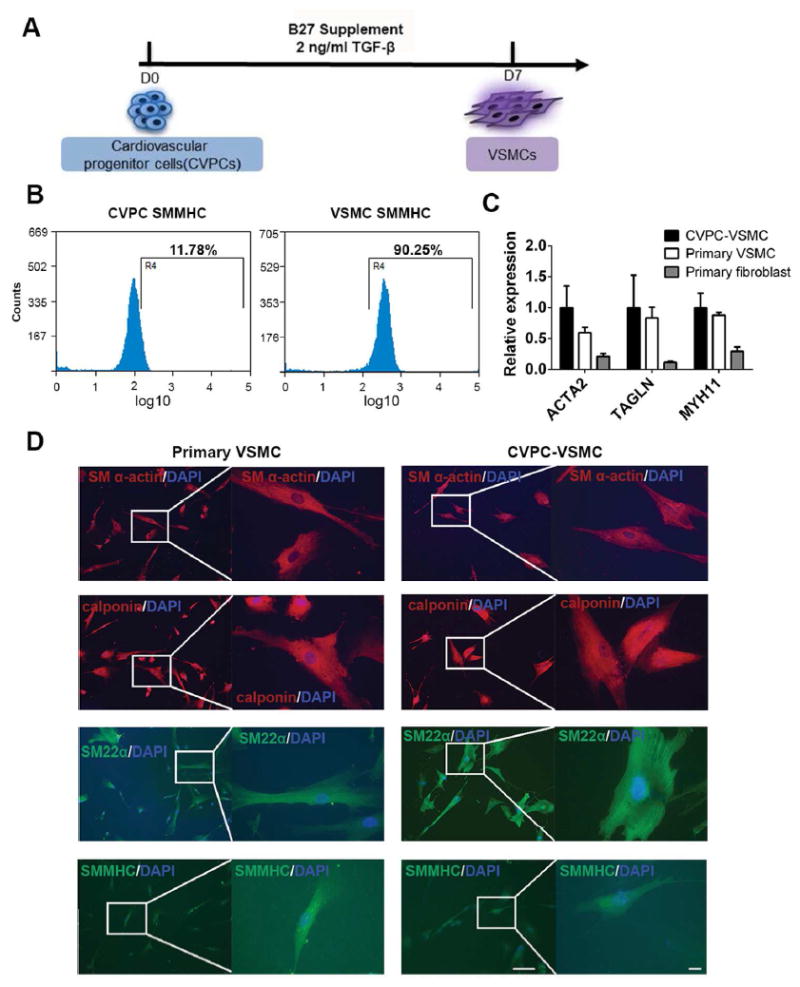
(A): Strategy of VSMC differentiation from CVPCs. (B): Flow cytometry analysis of SMMHC positive cells on CVPC and day7 VSMC. (C): Compare expression of contractile markers between CVPC-VSMC, primary human VSMC and primary human fibroblast by RT-qPCR. (D): Compare expression of contractile markers and cell morphology between CVPC-VSMC and primary human VSMC by immunofluorescence with low magnification (Left, scale bar=100 μm) and high magnification (Right, scale bar=20 μm). RT-qPCR was shown as average ± SEM (n=3). TGF-β: transforming growth factor β; CVPC: cardiovascular progenitor cell; VSMC: vascular smooth muscle cell; SMMHC: smooth muscle myosin heavy chain.
RNA extraction, reverse transcription and real-time quantitative polymerase – chain reaction
Total RNA from cultured cells was extracted with the RNeasy Plus Micro and Mini kit (Qiagen), and cDNA was synthesized with the Superscript IV first-strand synthesis system (Invitrogen). Quantitative realtime PCR amplification was carried out using custom primers with iTaq SYBR supermix (Bio-Rad) as described in our previous reports [15].The housekeeping gene GAPDH was served as internal control.
Western blot analysis, immunofluorescence staining and flow cytometry
Western blot analysis was carried out as we described previously [4]. Whole cell lysate samples were prepared using the RIPA buffer (Thermo Fisher Scientific) supplemented with a protease inhibitor mixture (Roche Applied Science). Antibodies against GAPDH (1:1000, Santa Cruz), smooth muscle α-actin (SM α-actin) (1:1000, sigma), Smooth muscle Myosin heavy chain 11(SMMHC) (1:500, Abcam), SM22α (1:1000; Abcam), calponin1 (1:1000, Sigma), YAP (1:1000, Cell signal technology) and NKX2.5 (1:200, Santa Cruz) were used to examine individual protein expression. Immuno-activity and band density were visualized by the Odyssey system (LI-COR Biosciences) according to the manufacturer’s instructions.
Immunofluorescence staining was performed according to a method described previously [15]. The following primary antibodies against SM α-actin (1:250, sigma), Smooth muscle Myosin heavy chain 11(SMMHC) (1:200, Abcam), SM22α (1:250; Abcam), calponin1 (1:250, Sigma), YAP (1:250 Cell signal technology) were used. DAPI was stained with ProLong® Gold Antifade Mountant with DAPI (Life technologies). Stained cells were observed on an OLYMPUS microscope.
Flow cytometry was carried out with previous protocol [15]. Primary antibody against Smooth muscle Myosin heavy chain 11(SMMHC) (1:200, Abcam) was used and Isotype IgG was used as the negative control.
Infection with pLX304-lentivirus and shRNA lentivirus
Overexpression of YAP was achieved by infection with lentivirus-based YAP expressing vector, pLX304-YAP (generous gift from Dr. William Hahn, Dana Farber Cancer Institute, Boston, MA [21], the control vector pLX304 is generous gift from Dr. David Root, Broad Institute of Harvard and Massachusetts Institute of Technology, Cambridge, MA [22]). Knockdown of YAP, TAZ or NKX2.5 experiments were carried out by infection with scrambled control or YAP shRNA lentivirus as described previously [4] (YAP shRNA is generous gift from Dr. Kunliang Guan, University of California, San Diego, CA), TAZ shRNA or NKX2.5 shRNA lentivirus (shRNA plasmids are ordered from Sigma). Lentivirus was titrated with Lenti-X™ qRT-PCR titration kit (Clontech). For overexpression of YAP or knockdown of YAP, TAZ, and NKX2.5 in VSMC differentiation from CVPCs, differentiated CVPCs were infected twice with above viruses at day1 and day2 of VSMC differentiation (Figure 1A), and then infected cells were differentiated with VSMC differentiation medium for another three days before analysis. Overexpression of YAP in PAC1 cell line was also achieved with lentivirus pLX304-YAP and pLX304 as controls.
Carbachol and angiotensin II contractility assays
Differentiated VSMCs were stimulated with 100 μM Carbachol (Sigma) or 10 μM angiotensin II (MP Biomedicals). To test the specificity of angiotensin II induced contraction was through angiotensin II type 1 receptor, VSMCs were pretreated with inhibitor losartan (10μM, Sigma) for 1 h and then stimulated with 10 μM angiotensin II (MP Biomedicals). The contractions of cells were recorded with Nikon microscope and changes of cell surface area after 30 min were analyzed using ImageJ software.
Luciferase Activity Assay
Rat smooth muscle cell line PAC1 cells (generous gift from Dr. Li Li, Wayne State University) were seeded in 24-well plates for 24 h. The cells then were transiently transfected by Lipofectamine 2000 (Invitrogen) with pGL3-MYOCD promoter-luciferase reporter (MYOCD WT) or the mutated promoter reporter without NKX2.5-binding element (MYOCD NKE mut) [23] (generous gift from Dr. Shiyou Chen, The University of Georgia). Thymidine kinase Renilla luciferase reporter vector was used for dual-luciferase assay. pcDNA-YAP plasmid was transfected for YAP overexpression in PAC-1. Forty-eight hours post-transfection, cells were lysed, and luciferase activity was detected with the Dual-Luciferase reporter assay system (Promega).The activity of thymidine kinase Renilla served as an internal standard.
Chromatin Immunoprecipitation Assay (ChIP)
ChIP assays were performed in both PAC1 and CVPC-derived VSMCs by using EZ-ChIP™ kit (EMD Millipore) according to manufacturer’s manual. Chromatin complexes from YAP lentivirus infected cells were immunoprecipitated with 1 μg of Nkx2.5 antibody or IgG (negative control). Quantitative realtime PCR were performed to amplify the Myocd promoter region containing Nkx2.5-binding element (NKE) using the following primer set: for PAC1 SMC cell line: 5-TTCCGCCTCCTCTCTGATTC-3 (forward) and 5-CCTTTTCCTCCTCCTCTGGG-3 (reverse); for CVPC-VSMCs: 5-TCAGGTGATTCTCCCACCTC-3 (forward) and 5-TCACACCTGTAATCCCAGCA-3 (reverse).
Co-immunoprecipitation (Co-IP)
For HEK293 cells, pcDNA-YAP-Myc (Addgene) and pcDNA-Nkx2.5 (generous gift from Dr. Shiyou Chen, The University of Georgia) were co-transfected into HEK293 cells, and cells were harvested 48 h post-transfection. For the process of CVPC-VSMCs differentiation, seeded CVPCs were infected with pLX304-YAP or pLX304-Vector virus at day1 and day2, and cells were harvested 3 days later. The Co-IP assay was performed as described in a previous report [24]. Briefly, cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.8, 137 mM NaCl, 1 mM EDTA, 0.1% Triton X-100 with a protease inhibitor mixture). After centrifugation at 12,000 rpm for 10 min at 4°C, the supernatants were collected and pre-cleared with protein A/G plus-agarose for 1 h at 4°C. Anti-Myc-Tag (Cell signal technology) or anti-YAP (Cell signaling technology) antibodies were added into pre-cleared supernatants and incubated overnight at 4°C. Normal IgG was used for a negative control. The protein complex was pulled down by incubation of A/G plus-agarose for 1 h at 4°C and washed three times with wash buffer (0.2mM EDTA, 100mM KCl, 2mM Mgcl2, 0.1% Tween-20 and 10% glycerol). The immunoprecipitants were analyzed by Western Blotting. Immuno-activity and band density were visualized by the Odyssey system (LI-COR Biosciences) according to the manufacturer’s instructions.
Results
Characterization of VSMCs differentiated from ESCs derived CVPCs: highly efficient and comparable with primary human VSMCs
We previously established a method involving highly specific induction of CVPCs from human embryonic stem cells [12, 15]. In this study, we were able to further differentiate VSMCs through induced CVPCs by treatment with TGF-β only. The differentiation of VSMCs using TGF-β only was as good as that using TGF-β and PDGF-BB (Figure 1A, Supplement figureS1A). Flow cytometry analysis showed approximately 90% of differentiated VSMCs were positive by staining with the anti-human smooth muscle heavy chain 11 (SMMHC) antibody, which is the most specific differentiation marker for smooth muscle maturation (Figure 1B). The efficiency was consistent to the differentiation efficiency by using other SMC markers as we previously reported [15]. Furthermore, CVPC-VSMCs and the primary VSMCs shared a comparable mRNA expression level of VSMC contractile markers including ACTA2, TAGLN and MYH11, and all the contractile markers were higher than primary aortic fibroblast (Figure 1C). Primary VSMCs and CVPC-VSMCs showed similar and typical VSMC cell morphology and contractile fiber structure by immunofluorescent staining. Similar expression levels of contractile proteins including smooth muscle α-actin (SM α-actin), calponin, SM22α, and smooth muscle heavy chain 11 (SMMHC) were detected between human primary VSMCs and CVPC-VSMCs by immunofluorescent staining (Figure 1D). To exclude the possibility of differentiation of CVPCs to cardiomyocytes and endothelial cells during the VSMC differentiation, we examined expression of MYH6, MYH7, CD31 and CD34 (Supplemental figureS1B and S1C). We found MYH7 was undetectable (data not shown) and other markers of cardiomyocytes and endothelial cells decreased during CVPC-VSMC differentiation.
Expression of YAP decreased during VSMCs differentiation from CVPCs
To determine role of YAP in VSMC differentiation, mRNAs transcription patterns of VSMCs markers and Hippo signaling were profiled during the VSMC differentiation from CVPCs. As shown in Figure 2A, myocardin, VSMC contractile markers, and SMTNB (the specific marker distinguish vascular SMCs and visceral SMCs) increased gradually from day 0 (CVPCs) to day 7 (mature VSMCs), while expression of YAP, TAZ, and TEAD4 decreased (Figure 2B, Supplemental figureS2). Moreover, expression of upstream kinase LATS2 and NF2 increased with no change in TEAD1/2/3 (Supplemental figureS2). These findings suggest that YAP’s function was decreased during CVPC-VSMC differentiation. These findings were validated by Western blot (Figure 2C) and immunofluorescent analysis (Figure 2D). Taken together, we found a negative correlation between expression of YAP and myocardin/VSMC markers in CVPC-VSMC differentiation, indicating YAP was a negative regulator for VSMC differentiation from CVPCs.
Figure 2. Expression of YAP decreased during VSMCs differentiated from CVPCs.
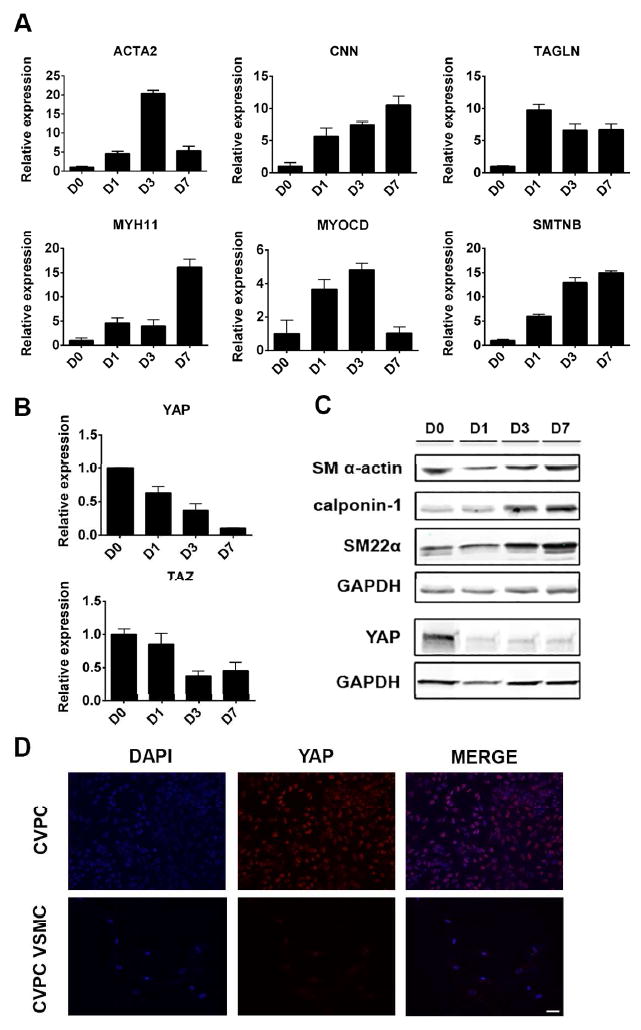
(A): Expression of VSMC markers from CVPC (D0) to VSMC (D7) by RT-qPCR. (B): Expression of YAP and TAZ from CVPC (D0) to VSMC (D7) RT-qPCR. (C): Validation of (A, B) by western blot. (D): Expression of YAP in CVPC and VSMC by immunofluorescence, scale bar=50 um. RT-qPCR was shown as average ± SEM of three independent replicates. D: day; CVPC: cardiovascular progenitor cell; VSMC: vascular smooth muscle cell.
Overexpression of YAP decreased expression of VSMCs markers and its contractile function, and knockdown of YAP/TAZ increased expression of VSMC markers
To further confirm YAP as a negative regulator for VSMC differentiation from CVPCs, the effect of YAP overexpression during VSMC differentiation from CVPCs was examined. As expected, overexpression of YAP significantly decreased mRNA and protein levels of multiple VSMC differentiation markers, including ACTA2, CNN, TAGLN, MYH11 and SMTNB (Figure 3A, 3B). With treatment of carbachol and angiotensin II, a significant decrease in contractile function was found in the YAP overexpressed VSMCs by quantifying the reduction of cell surface area (Figure 3C, 3D) (Supplemental VideoS1 and S2; S3 and S4). Pretreating VSMCs with losartan abolished angiotensin II induced contraction in both YAP-overexpression and control groups, confirming a specific response of VSMCs to angiotensin II (Figure 3D).
Figure 3. Overexpression of YAP attenuated differentiation of VSMCs.
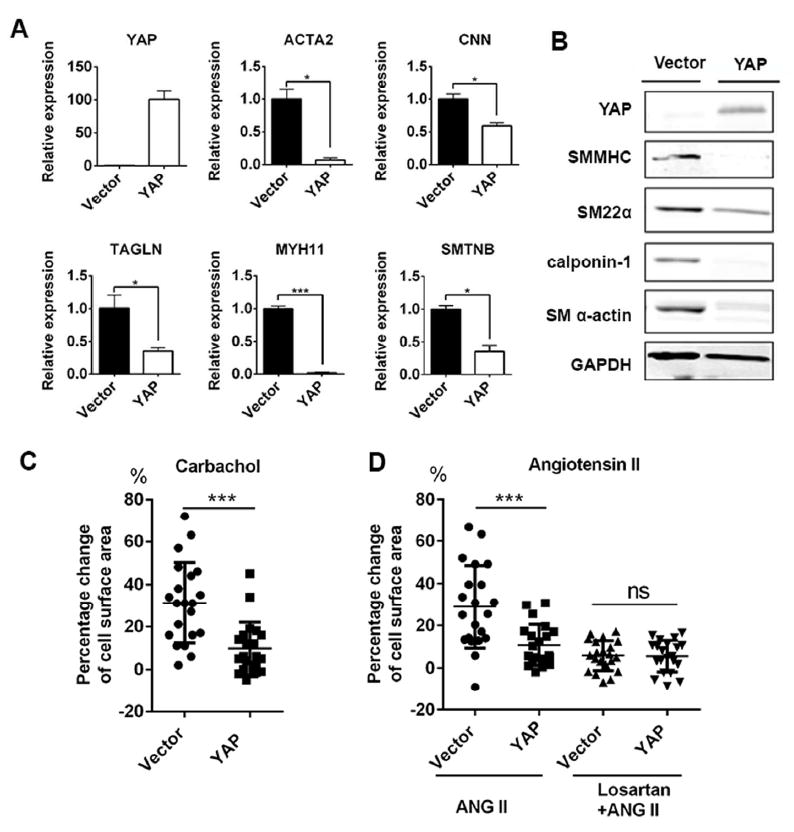
(A)(B): Overexpression of YAP decreased expression of VSMC contractile markers by RT-qPCT (A) and western blot (B). (C): Quantification of decreased cell surface of VSMCs in Vector and YAP overexpressed group after Carbachol 100 uM for 30 min. (D): Quantification of decreased cell surface of VSMCs in Vector and YAP overexpressed group after 30 min angiotensin II 10 uM treatment in normal VSMCs or losartan 10 uM pretreated VSMCs (Losartan+ANG II). Results were shown as average ± SEM with three independent replicates and statistic analyzed with unpaired t-test, *p<0.05, ***p<0.001. ANG II: angiotensin II.
Based on the results that gain-of-function of YAP attenuated CVPCs-derived VSCMs differentiation, we further determined if loss-of-function of YAP would abolish its inhibitory effects on VSMCs differentiation from CVPCs. Given that YAP and TAZ behaved complementarily and overlapping [3], knockdown of YAP and TAZ together was carried out with shRNA lentivirus. Compared to the scramble control group, expression of YAP and TAZ were significantly inhibited in shYAP and shTAZ VSMCs (Figure 4A, 4B). For VSMC markers, knockdown of YAP and TAZ resulted in a significant increase in expression of VSMC contractile markers by both RT-qPCR and Western blot (Figure 4A, 4B). Western blot was repeated three times and relative expression level compared to GAPDH of each protein was quantified (Figure 4C).
Figure 4. Knockdown of YAP/TAZ further increased expression of VSMC markers.
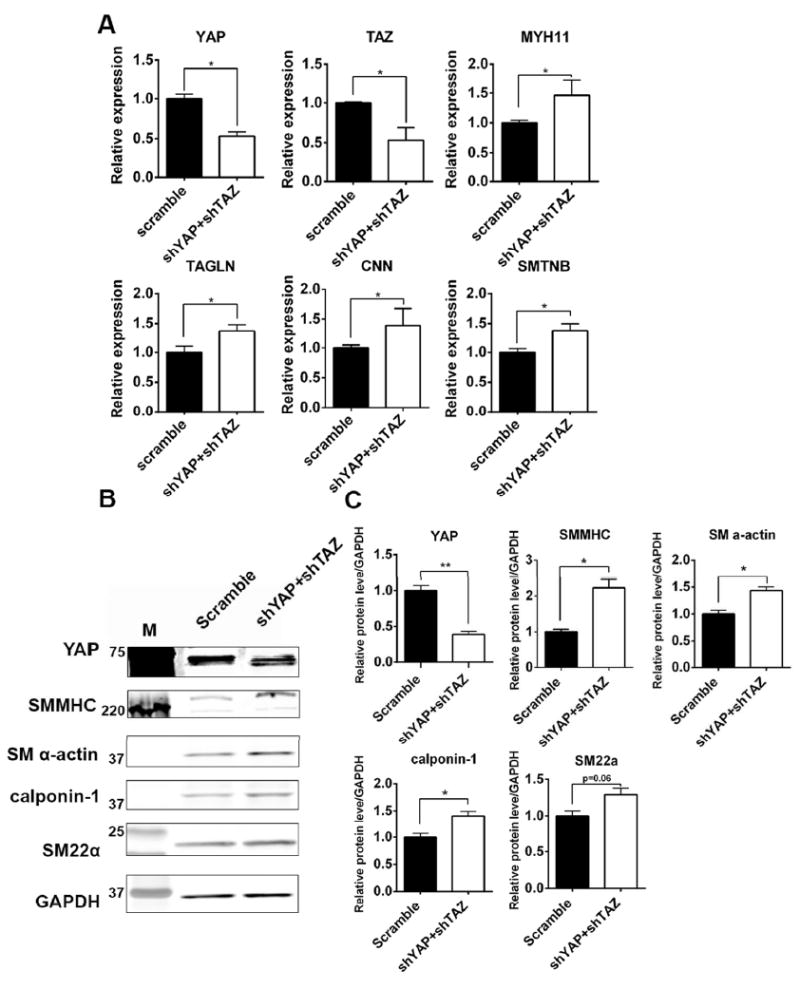
(A-B): Expression of VSMC contractile markers in scramble group and shYAP+shTAZ group by RT-qPCR (A) and western blot (B). Bands for YAP in (B) were overexposed to show successful knockdown. M: molecular mass markers. (C): Quantification of each band’s density to GAPDH in with three independent western blots replicate, results were shown as average ± SEM and statistic analyzed with unpaired t-test. *p<0.05, **p<0.005
YAP inhibited myocardin transcription by preventing NKX2.5 binding to myocardin promoter
To further examine how YAP regulates the differentiation of VSMCs, we analyzed the expression of myocardin, the master regulator of VSMC differentiation. We found that expression of myocardin in differentiated VSMCs was down-regulated in YAP overexpressed group (Figure 5A) and upregulated in YAP and TAZ knockdown group (Figure 5B) compared to the control groups. To identify the potential cis-elements located in myocardin promoter that was involved in YAP-mediated inhibition, we compared YAP effects on both luciferase activities of wild type myocardin promoter and its mutant (illustrated in Figure 5C) in PAC1 rat SMC cell line. First, we examined the activities of luciferase reporters driven by the wild type 1.9kb 5’-proximal upstream DNA fragments in the myocardin gene.
Figure 5. YAP inhibited myocardin transcription by preventing NKX2.5 binding to myocardin promoter.
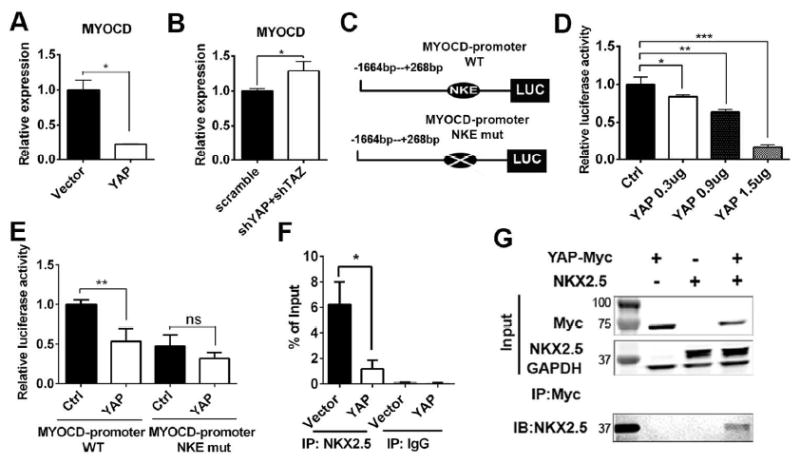
(A): Expression of myocardin in Vector control and YAP overexpressed group by RT-qPCR. (B): Expression of myocardin in scramble control and shYAP+shTAZ knockdown group by RT-qPCR. (C): Schematic of designs of luciferase reporters. (D): Luciferase activity of MYOCD-promoter WT by different dose of pCDNA-YAP in PAC1 cells. (E): Luciferase activity of MYOCD-promoter WT and NKE mut between pCDNA control and pCDNA-YAP in PAC1 cells. (F): Chip-PCR assay between Vector control and YAP overexpressed groups in PAC1 cells. (G): Co-IP assay in HEK 293 cells by overexpressing YAP-Myc and Nkx2.5 as indicated. Results were shown as average ± SEM and statistic analyzed with unpaired t-test.*p<0.05, **p<0.005, ***p<0.001, ns: no significance. WT: wild-type; NKE: NKX2.5 binding element; IP: immunoprecipitation; IB: immunoblot. M: molecular mass markers.
By co-transfecting the wild type myocardin promoter luciferase plasmid and pcDNA-YAP construct, we found that YAP inhibited luciferase activity of the myocardin promoter in a dose-dependent manner compared to the pcDNA vector control group (Figure 5D). Transcription factor NKX2.5 was demonstrated to be an activator for myocardin expression both in cardiomyocytes [25] and SMCs [23, 26] and a NKX2.5 binding element (NKE) was conserved in the myocardin promoter. Myocardin promoter luciferase plasmid with mutated NKX2.5 binding element was co-transfected with pcDNA-YAP construct. Results showed that YAP-mediated inhibitory effects were abolished in the NKE-mutated myocardin promoter luciferase reporter group (Figure 5E), indicating YAP inhibited the transcription of myocardin through NKX2.5. We also performed Chip-PCR on PAC1 SMC cells between YAP overexpression group and vector control group to explore the effects of YAP on NKX2.5’s binding to endogenous myocardin promoter. We found that YAP overexpression significantly attenuated NKX2.5’s binding to the endogenous myocardin promoter (Figure 5F). To explore the possibility that protein-protein interaction between YAP and NKX2.5 may contribute to YAP-mediated inhibitory effects on myocardin expression, we overexpressed YAP-Myc and NKX2.5 in HEK 293 cells and performed immunoprecipatation with an anti-Myc antibody. Immunoblotting on NKX2.5 demonstrated that YAP-NKX2.5 protein physically interacted with each other (Figure 5G), which indicated that YAP was able to bind with NKX2.5 and interfere NKX2.5’s transcription activation on myocardin.
YAP interfered with NKX2.5 protein during CVPC-VSMC differentiation and attenuated VSMC differentiation in a NKX2.5 dependent manner
Consistent with results from PAC1 cells, we found that overexpression of YAP inhibited NKX2.5’s binding to the myocardin promoter during CVPC-VSMC differentiation (Figure 6A, 6B). Moreover, knockdown of NKX2.5 decreased expression of myocardin and VSMC markers, which was validated by two shNKX2.5 constructs (Figure 6C). Based on our finding, we hypothesized that during CVPC-VSMC differentiation, knockdown of NKX2.5 could block the positive effects of YAP and TAZ knockdown on VSMC marker expression. As expected, further knockdown of NKX2.5 in YAP/TAZ knockdown cells showed (Figure 6D, Supplemental figureS3) decreased expression of myocardin and VSMC markers compared to the YAP and TAZ knockdown only group. These results indicated that YAP attenuates VSMC differentiation, at least in part, in a NKX2.5 dependent manner.
Figure 6. YAP interfered with NKX2.5 protein during CVPC-VSMC differentiation and attenuates VSMC differentiation in a NKX2.5 dependent manner.
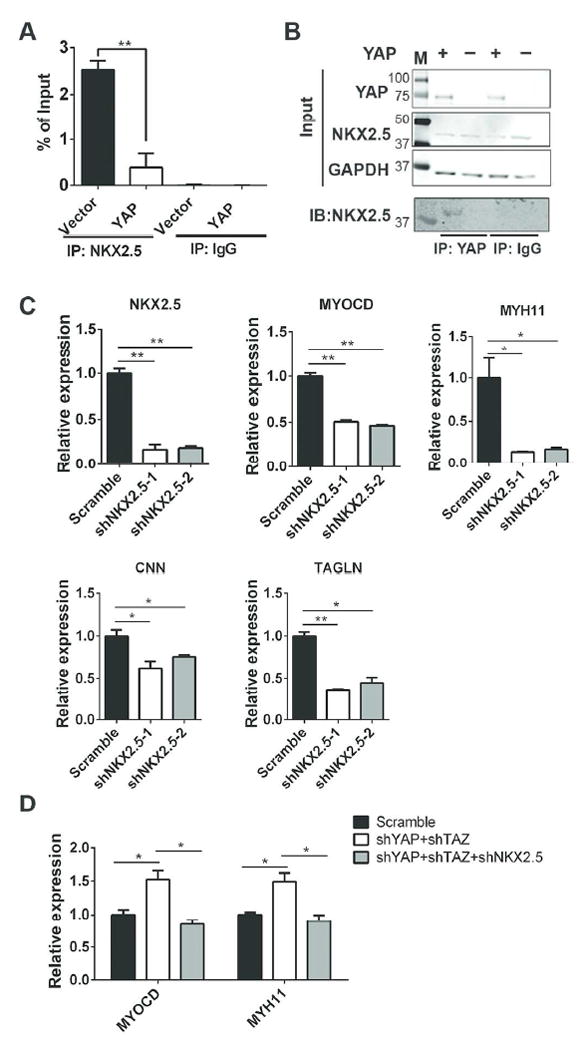
(A): Chip-PCR assays between Vector control and YAP overexpressed groups were confirmed in CVPC-derived VSMCs. (B): CO-IP assay on CVPC-derived VSMCs to detect protein-protein interaction between overexpressed YAP and endogenous NKX2.5. (C): Knockdown of NKX2.5 during CVPC-VSMC differentiation by different shRNAs: shNKX2.5-1 and shNKX2.5-2 while sh-scramble was used as control and MYOCD and VSMC markers were detected by RT-qPCR. (D): Knockdown of NKX2.5 abolished effect of YAP+TAZ knockdown during CVPC-VSMC differentiation. Results were shown as average ± SEM and statistic analyzed with unpaired t-test. *p<0.05, **p<0.01. IP: immunoprecipitation; IB: immunoblot. M: molecular mass markers.
Discussion
In this study, we examined the function of YAP in the CVPC-VSMCs differentiation process and underlying mechanism. Our results showed that expression of YAP decreased when CVPCs were differentiated into VSMCs (Figure 2). Overexpression and knockdown of YAP demonstrated that YAP inhibited the expression of contractile proteins and contractile function of CVPC-derived VSMCs (Figure 3, 4). Finally, we found that YAP decreased transcription of myocardin by interfering with NKX2.5’s binding to the 5’-proximal promoter region of myocardin gene.
To our knowledge, this was the first study to identify that YAP inhibited the expression of VSMCs contractile markers through NKX2.5 during the differentiation process from CVPCs. Our finding is consistent with preclinical data showing forced expression of Yap in rodent matured SMCs down-regulated the expression of SMCs markers and promoted synthetic phenotypic switch [4, 5, 27]. However, different from our finding that knockdown of YAP and TAZ increased expression of myocardin and VSMC markers, significant increases of VSMC markers were not found in the SM22α-Cre Yap deletion mice [8]. The possible reason for this difference includes increased Taz expression in Yap deletion mice may have compensated for the lack of Yap. As a matter of fact, this compensatory increase of Taz has been previously reported by Finch-Edmondson et al, when Yap was knocked out [28].
Previous studies indicate that Yap1 [4] and TEAD1 proteins [29] were able to interfere with the function of myocardin and abolish its activation of smooth muscle cell contractile markers through protein-protein interaction. Similarly, our results showed YAP negatively regulated function of myocardin and YAP attenuated function of myocardin by inhibiting transcription of myocardin in a dose and NKX2.5 dependent manner. As indicated herein, Nkx2.5 was an activator of myocardin transcription and important in the maturation of VSMCs, which is consistent with previous findings [23, 26]. Importantly, we found that protein-protein interaction between YAP and NKX2.5 mediated YAP’s inhibition on myocardin transcription and myocardin-driven VSMC differentiation. YAP is characterized with one or two WW domains (two splicing variants, YAP1 and YAP2, respectively) [30]. The WW domain is a protein-protein interaction module, which prefers to bind to ligands containing proline-rich sequences. Interestingly, a proline-rich sequence is conserved in human, rat, and mouse NKX2.5 proteins. It is quite likely that the motifs in YAP discussed above contribute to the binding to NKX2.5. Future studies are warranted to identify the specific protein domain responsible for binding between YAP and NKX2.5.
Besides regulating VSMCs differentiation, YAP also played an important role in VSMC-progenitor cells development. Recent studies showed that neural crest-specific deletion of Yap and Taz with Wnt1-Cre or WntCre2SOR [6, 7] abrogated Notch signaling and caused severe neural tube vessel regression. Differentiation of VSMC from neural crest was interfered and resulted in aortic arch aneurysm and hemorrhaging. Moreover, a Tead2 binding element in the myocardin distal promoter has also been identified in early cardiovascular development [31], suggesting that the Yap-Tead complex may be important during the progenitor stage of VSMC differentiation. We also found that expression of YAP and TAZ increased when H1 ESCs differentiated into CVPCs (data not shown), indicating that YAP functions variably during distinct differentiation stages. Taken together, YAP is very important in the development of the vascular system and YAP’s function may vary during different developmental stages. For aortic aneurysm development, besides the findings in rodent models [6-8], it was suggested that YAP may be a potential target for development of human ascending aortic aneurysms [32]. However, future studies with a larger human sample cohort are needed to identify the role of YAP in both aortic root and ascending aortic aneurysm development.
Conclusion
In conclusion, by using the in vitro ESC-CVPC-VSMCs monolayer differentiation model, our study demonstrated a negative regulation of YAP in the CVPC-VSMC differentiation process. We found YAP blocked NKX2.5’s binding to myocardin promoter and inhibited transcription of myocardin during this process (Figure 7). We identified novel modulators in VSMC differentiation and provided a potential target for understanding the development of aortic root aneurysm.
Figure 7.
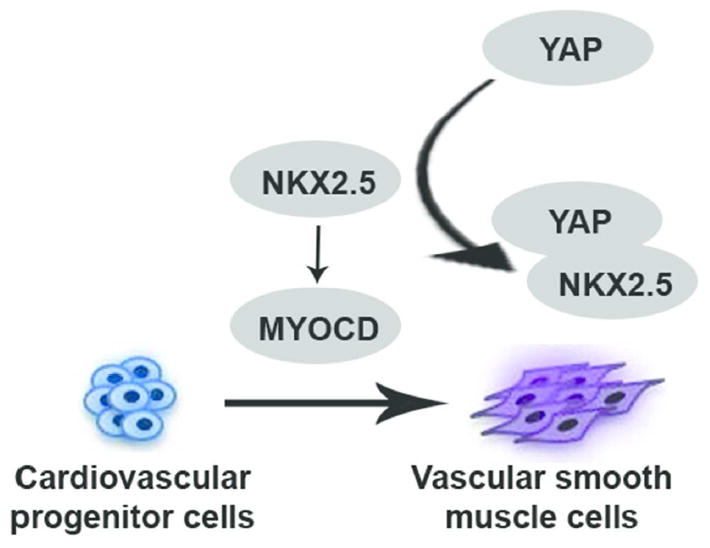
Working hypothesis which elaborates role of YAP in differentiation of VSMC from CVPCs
Supplementary Material
(A): Expression of VSMC contractile markers between TGFβ and TGF β pluses PDGF-BB groups. M: molecular mass marker. (B)(C): Expression of cardiomyocyte marker (B) and endothelial cell markers (C) in CVPC-VSMC differentiation. RT-qPCR was shown as average ± SEM of three independent replicates. TGF-β: transforming growth factor β; PDGF-BB: platelet-derived growth factor BB; CVPC: cardiovascular progenitor cell; SMC: smooth muscle cell.
Expression of other key factors in Hippo pathway from CVPC (D0) to VSMC (D7) differentiation by RT-qPCR, results are shown as average ± SEM of three independent replicates.
(A): Successful knockdown of NKX2.5, YAP and TAZ by shRNAs by RT-qPCR. (B): Expression of VSMC markers TAGLN and CNN by RT-qPCR. Results are shown as average ± SEM and statistic analyzed with unpaired t-test. *p<0.05, ns: no significant.
Acknowledgments
This study was supported by AATS Graham foundation, Thoracic Surgery Foundation of Research and Education, and McKay Award, CVC Aiken’s Award, Phil Jenkins Breakthrough Fund, University of Michigan and the National Institute of Health Grant (HL114038: PXM and YEC) and Grant R01HL068878 (YEC), and continuous financial support from Mr. Steven J. Szatmari and Mrs. Darlene Szatmari. We would like to thank supports from University of Michigan Medical School (UMMS) and Central South University Xiangya School of Medicine (CSU-XYSM) Training Program. We appreciate the generosity of Dr. Shiyou Chen (The University of Georgia, Athens, GA) and Dr. Kunliang Guan (University of California, San Diego, CA) and Dr. William Hahn, (Dana Farber Cancer Institute, Boston, MA) Dr. Li Li (Wayne State University, Detroit, MI) in sharing the plasmids and cells. We appreciate Dr. Whitney Hornsby and Aroosa Malik for their contributions related to the drafting of this manuscript.
Footnotes
1,2Lunchang Wang: Conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing
1Ping Qiu: Conception and design, data interpretation, manuscript writing
1Jiao Jiao: Conception and design, manuscript writing
1Hiroyuki Hirai: Conception and design, administrative support
1Wei Xiong: Conception and design
1Jifeng Zhang: Conception and design, administrative support
1Tianqing Zhu: Administrative support
3Peter X. Ma: Conception and design, final approval of manuscript
1Y. Eugene Chen: Conception and design, final approval of manuscript
1Bo Yang: Conception and design, financial support, data interpretation, manuscript writing, final approval of manuscript
Conflicts of Interest: The authors declare that they have no conflict of interests.
References
- 1.Isselbacher EM. Thoracic and abdominal aortic aneurysms. Circulation. 2005;111(6):816–828. doi: 10.1161/01.CIR.0000154569.08857.7A. [DOI] [PubMed] [Google Scholar]
- 2.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arteriosclerosis, thrombosis, and vascular biology. 2007;27(6):1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 3.Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163(4):811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie C, Guo Y, Zhu T, et al. Yap1 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. The Journal of biological chemistry. 2012;287(18):14598–14605. doi: 10.1074/jbc.M111.329268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Hu G, Gao X, et al. The induction of yes-associated protein expression after arterial injury is crucial for smooth muscle phenotypic modulation and neointima formation. Arteriosclerosis, thrombosis, and vascular biology. 2012;32(11):2662–2669. doi: 10.1161/ATVBAHA.112.254730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manderfield LJ, Aghajanian H, Engleka KA, et al. Hippo signaling is required for Notch-dependent smooth muscle differentiation of neural crest. Development. 2015;142(17):2962–+. doi: 10.1242/dev.125807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Xiao Y, Hsu CW, et al. Yap and Taz play a crucial role in neural crest-derived craniofacial development. Development. 2015 doi: 10.1242/dev.126920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Hu GQ, Liu F, et al. Deletion of Yes-Associated Protein (YAP) Specifically in Cardiac and Vascular Smooth Muscle Cells Reveals a Crucial Role for YAP in Mouse Cardiovascular Development. Circulation research. 2014;114(6):957–965. doi: 10.1161/CIRCRESAHA.114.303411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu JY, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 10.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 11.Cheung C, Bernardo AS, Trotter MWB, Pedersen RA, Sinha S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat Biotechnol. 2012;30(2):165–173. doi: 10.1038/nbt.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao N, Liang H, Huang JJ, et al. Highly efficient induction and long-term maintenance of multipotent cardiovascular progenitors from human pluripotent stem cells under defined conditions. Cell Res. 2013;23(9):1119–1132. doi: 10.1038/cr.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patsch C, Challet-Meylan L, Thoma EC, et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol. 2015;17(8):994–U294. doi: 10.1038/ncb3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Hu J, Sorek CE, et al. Fabrication of tissue-engineered vascular grafts with stem cells and stem cell-derived vascular cells. Expert opinion on biological therapy. 2015:1–14. doi: 10.1517/14712598.2016.1118460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu J, Wang Y, Jiao J, et al. Patient-specific cardiovascular progenitor cells derived from integration-free induced pluripotent stem cells for vascular tissue regeneration. Biomaterials. 2015;73:51–59. doi: 10.1016/j.biomaterials.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang ZG, Wang DZ, Pipes GCT, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. P Natl Acad Sci USA. 2003;100(12):7129–7134. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 18.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. Journal of molecular and cellular cardiology. 2002;34(10):1345–1356. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- 19.Miano JM. Myocardin in biology and disease. Journal of biomedical research. 2015;29(1):3–19. doi: 10.7555/JBR.29.20140151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Hu J, Jiao J, et al. Engineering vascular tissue with functional smooth muscle cells derived from human iPS cells and nanofibrous scaffolds. Biomaterials. 2014;35(32):8960–8969. doi: 10.1016/j.biomaterials.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenbluh J, Nijhawan D, Cox AG, et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151(7):1457–1473. doi: 10.1016/j.cell.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang X, Boehm JS, Yang X, et al. A public genome-scale lentiviral expression library of human ORFs. Nature methods. 2011;8(8):659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie WB, Li Z, Miano JM, Long X, Chen SY. Smad3-mediated myocardin silencing: a novel mechanism governing the initiation of smooth muscle differentiation. The Journal of biological chemistry. 2011;286(17):15050–15057. doi: 10.1074/jbc.M110.202747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan Y, Guo Y, Hamblin M, et al. Inhibition of gluconeogenic genes by calcium-regulated heat-stable protein 1 via repression of peroxisome proliferator-activated receptor alpha. The Journal of biological chemistry. 2011;286(47):40584–40594. doi: 10.1074/jbc.M111.232918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ueyama T, Kasahara H, Ishiwata T, Nie Q, Izumo S. Myocardin expression is regulated by Nkx2.5, and its function is required for cardiomyogenesis. Mol Cell Biol. 2003;23(24):9222–9232. doi: 10.1128/MCB.23.24.9222-9232.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi N, Chen SY. Cell division cycle 7 mediates transforming growth factor-beta-induced smooth muscle maturation through activation of myocardin gene transcription. The Journal of biological chemistry. 2013;288(48):34336–34342. doi: 10.1074/jbc.M113.498238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu F, Ahmed AI, Kang XH, et al. MicroRNA-15b/16 Attenuates Vascular Neointima Formation by Promoting the Contractile Phenotype of Vascular Smooth Muscle Through Targeting YAP. Arterioscl Throm Vas. 2015;35(10):2145–2152. doi: 10.1161/ATVBAHA.115.305748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finch-Edmondson ML, Strauss RP, Passman AM, et al. TAZ Protein Accumulation Is Negatively Regulated by YAP Abundance in Mammalian Cells. Journal of Biological Chemistry. 2015;290(46):27928–27938. doi: 10.1074/jbc.M115.692285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu F, Wang X, Hu G, Wang Y, Zhou J. The transcription factor TEAD1 represses smooth muscle-specific gene expression by abolishing myocardin function. The Journal of biological chemistry. 2014;289(6):3308–3316. doi: 10.1074/jbc.M113.515817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sudol M, Bork P, Einbond A, et al. Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. The Journal of biological chemistry. 1995;270(24):14733–14741. doi: 10.1074/jbc.270.24.14733. [DOI] [PubMed] [Google Scholar]
- 31.Creemers EE, Sutherland LB, McAnally J, Richardson JA, Olson EN. Myocardin is a direct transcriptional target of Mef2, Tead and Foxo proteins during cardiovascular development. Development. 2006;133(21):4245–4256. doi: 10.1242/dev.02610. [DOI] [PubMed] [Google Scholar]
- 32.Li HY, Jiang WJ, Ren WH, et al. Downregulation of the Yes-Associated Protein Is Associated with Extracellular Matrix Disorders in Ascending Aortic Aneurysms. Stem Cells Int. 2016 doi: 10.1155/2016/6786184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A): Expression of VSMC contractile markers between TGFβ and TGF β pluses PDGF-BB groups. M: molecular mass marker. (B)(C): Expression of cardiomyocyte marker (B) and endothelial cell markers (C) in CVPC-VSMC differentiation. RT-qPCR was shown as average ± SEM of three independent replicates. TGF-β: transforming growth factor β; PDGF-BB: platelet-derived growth factor BB; CVPC: cardiovascular progenitor cell; SMC: smooth muscle cell.
Expression of other key factors in Hippo pathway from CVPC (D0) to VSMC (D7) differentiation by RT-qPCR, results are shown as average ± SEM of three independent replicates.
(A): Successful knockdown of NKX2.5, YAP and TAZ by shRNAs by RT-qPCR. (B): Expression of VSMC markers TAGLN and CNN by RT-qPCR. Results are shown as average ± SEM and statistic analyzed with unpaired t-test. *p<0.05, ns: no significant.