

Abstract
Mutant isocitrate dehydrogenase (IDH) 1/2 converts α-ketoglutarate (α-KG) to D-2 hydroxyglutarate (D-2-HG), a putative oncometabolite that can inhibit α-KG dependent enzymes, including ten-eleven translocation methylcytosine dioxygenase (TET) DNA demethylases. We recently established that miRNAs are components of the IDH1 mutant-associated glioma CpG island methylator phenotype (G-CIMP), and specifically identified MIR148A as a tumor-suppressive miRNA within G-CIMP. However, the precise mechanism by which mutant IDH induces hypermethylation of MIR148A and other G-CIMP promoters remains to be elucidated. In this study, we demonstrate that treatment with exogenous D-2-HG induces MIR148A promoter methylation and transcriptional silencing in human embryonic kidney 293T (293T) cells and primary normal human astrocytes. Conversely, we show that the development of MIR148A promoter methylation in mutant IDH1 over-expressing 293T cells is abrogated via treatment with C227, an inhibitor of mutant IDH1 generation of D-2-HG. Using dot-blot assays for global assessment of 5-hydroxymethylcytosine (5-hmC), we show that D-2-HG treatment reduces 5-hmC levels, whereas C227 treatment increases 5-hmC levels, strongly suggesting TET inhibition by D-2-HG. Moreover, we show that withdrawal of D-2-HG treatment reverses methylation with an associated increase in MIR148A transcript levels and transient generation of 5-hmC. We also demonstrate that RNA Polymerase II binds endogenously to the predicted promoter region of MIR148A, validating the hypothesis that miRNA transcription is driven by an independent promoter.
Keywords: D-2-hydroxyglutarate (D-2-HG), DNA methylation, glioblastoma, isocitrate dehydrogenase (IDH), microRNA (miRNA)
INTRODUCTION
Mutation in the isocitrate dehydrogenase 1 and 2 (IDH 1/2) genes defines a major subset of gliomas (1) and has been identified in a number of human cancers (2–6). The recent discovery of an association between IDH1/2 mutation and the presence of a CpG island methylation phenotype in gliomas (G-CIMP) has led to the widely-accepted notion that mutant IDH induces G-CIMP resulting in the downregulation of key tumor-suppressor genes, ultimately contributing to gliomagenesis (7). This notion is supported by demonstration that stable expression of IDH1 mutant protein in normal astrocytes was sufficient to establish G-CIMP (8). While the wild-type function of IDH1/2 is to convert isocitrate to α-ketoglutarate (α-KG) (9–11), somatic mutations to IDH1/2 result in aberrant enzymatic activity which converts α-KG into the putative oncometabolite D-2-hydroxyglutarate (D-2-HG) (12). IDH1/2 mutant gain of function has been shown to result in a 10 to 100 fold increase in D-2-HG levels in mutant tumors compared with wild type (12–16) and to induce multiple pathophysiological outcomes (17). Specifically, it has been hypothesized that generation of G-CIMP may occur through D-2-HG inhibition of the α-KG-dependent ten-eleven translocation methylcytosine dioxygenases (TETs), which have a key role in the active DNA demethylation process (18,19). Thus, mutant IDH1/2 is thought to induce methylation by diminishing active demethylation mediated by TET. However, inhibition of mutant IDH1 generation of D-2-HG is not sufficient to induce changes in the established global hypermethylation phenotype in glioma (20,21). Overall, the role of D-2-HG in IDH-mutant mediated hypermethylation in G-CIMP has not yet been established.
We recently established that MIRNAs, specifically MIR148A, are components of G-CIMP (22). MIR148A is a member of the MIR148/152 family, which includes three members: MIR148A, MIR148B and MIR152 (23,24). MIR148A plays important roles in the regulation of growth, development, differentiation and tumorigenesis (23). MIR148A down-regulation appears to be related to the maintenance of multipotency in various stem cell populations (25–27), and its upregulation has been observed in early osteogenic differentiation (28), myogenic differentiation (29) and adipogenic differentiation (30). Consistent with our findings, most studies also indicate that MIR148A possesses tumor suppressive activity in a variety of cancer cells (22,24,31–34). In many cases, reduced MIR148A expression in human cancers appears to result as the consequence of promoter CpG hypermethylation (22,35–37). However, to date there has been a paucity of experimental evidence to support the hypothesis that MIR148A transcription is driven by an independent promoter, and the sensitivity of this promoter to DNA CpG methylation has not been confirmed. Thus, this study is aimed at using MIR148A as a reporter to probe the role of D-2-HG in CpG island hypermethylation and at understanding MIR148A silencing as an important factor contributing to gliomagenesis.
MATERIALS AND METHODS
Cell cultures and treatments
The human embryonic kidney 293T cell line (293T cells) was a generous gift from Dr. Paul Mischel (University of California San Diego, La Jolla, CA) and originally obtained from the ATCC. The 293T cell line was maintained in DMEM culture medium with 10%FBS and penicillin/streptomycin at 37°C and 5% CO2 in a tissue culture incubator. The primary human astrocytes (NHA cells) were purchased from Applied Biological Materials (ABM) Inc. (Richmond, BC, CANADA). The culture medium is PriGrow IV purchased from ABM. The culture and sub-culture of NHA cells strictly followed the manufacturer’s protocol. Mycoplasma testing/cell authentication procedures were not applied.
Octyl-D-2-HG, octyl-L-2-HG (the L-enantiomer of 2-hydroxyglutarate), and octyl-α-KG were purchased from Cayman Chemical (Ann Arbor, Michigan) and C227 was from Xcess Biosciences, Inc (San Diego, CA). Each of these chemicals was dissolved in DMSO. 293T and NHA cells were treated with these chemicals at indicated concentrations and time courses, and control cells were treated with equal volume of DMSO. Every two days (one passage) these cells were detached by trysin-EDTA and sub-cultured in medium with fresh chemicals.
Establishment of stable IDH1R132H expressing 293T cell lines
Previously established stable 293T cell lines were used; briefly, to achieve stable expression of IDH1R132H (IDH1MUT) as well as IDH1WT, the 293T cells were infected with the following retroviral constructs respectively: blank-pLPCX, IDH1WT-pLPCX, and IDH1R132H-pLPCX (22). The infected cells were selected by adding puromycin (3 μg/ml) into DMEM and single cell clones were picked up and amplified for later experiments. For each clonal cell line, transfected gene expression was confirmed by immunostaining and western blot. These cells were cultured in normal culture medium plus puromycin, passed every two days upon reaching ~95% confluence and reseeded at ~40% confluence.
Measurement of intracellular D-2-HG content
An enzymatic D-2-HG assay was used to determine the intracellular D-2-HG content (38). This assay is based on the conversion of D-2-HG to α-ketoglutarate (α-KG) in the presence of the enzyme (D)-2-hydroxyglutarate dehydrogenase (HGDH) and nicotinamide adenine dinucleotide (NAD+). The enzyme HGDH was a gift from Dr. von Deimling’s laboratory. In brief, the 293T or NHA cell pellet was harvested in Cell Lysis Buffer (Cell Signaling) and the lysate was divided into two parts, one for protein content determination by Pierce BCA protein assay kit (Thermo Scientific), and another for D-2-HG assay. The lysate for D-2-HG assay was first deproteinized by adding 3 μl of Proteinase K (Qiagen) and incubating for 3 hrs at 37°C. Then, 25 μl of lysate was reacted with 75 μl of assay solution at room temperature for 30 minutes. For each sample a triplicate reaction was done. The assay buffer comprised 100 μM NAD+ (Applichem, Darmstadt, Germany), 0.1 μg HGDH, 5 μM resazurin (Applichem) and 0.01 U/ml diaphorase (MP Biomedical, Irvine, CA, USA) in 100 mM HEPES pH 8.0. Fluorometric detection was carried out in Wallace VICTOR2 1420 MULTILABEL HTS COUNTER (PerkinElmer) with Em540nm/Ex610nm. D-2-HG content was calculated based on a standard curve obtained from a range of concentrations of D-2-HG replacing the cell lysate, and then expressed as D-2-HG pmole/μg protein.
DNA and RNA isolation from 293T and NHA cells
Genomic DNA and total RNA (including microRNA) were isolated from 293T and NHA cells by using the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen). DNA and RNAs were prepared from the same set of cells simultaneously as described by the manufacturer. Thus, the results obtained by using the DNA and RNAs are highly comparable. The concentration of Genomic DNA and total RNA were measured using NanoDrop 1000 (Thermo Fisher Scientific).
Global 5-hydroxymethylcytosine (5-hmC) dot-blot assay
4 μl of the genomic DNA (200 ng) was loaded onto a nitrocellulose membrane (Healthcare Life Sciences). After drying, the membrane was washed with TBS buffer with 0.05% of Triton 100 (TBPT) 3 times and then pre-blocked with 5% nonfat milk and 1% BSA in TBST for 1 hr at room temperature. Rabbit anti-5-Hydroxymethylcytosine polyclonal antibody (ZYMO Res) was used at 1:1000 and goat anti-rabbit IgG-horseradish peroxidase (HRP) (Santa Cruz Biotechnology) was used at 1: 5000. The signal of 5-hmC was generated by using the SuperSignal West Pico Chemiluminescent Substrate (Thermo) according to the manufacturer’s instructions and visualized by exposure to X-ray film.
CpG island bisulfite sequencing (Bis-Seq) and T-A cloning sequencing
Bisulfite converted genomic DNA was prepared by using EZ DNA Methylation-Gold Kit (ZYMO Research) and CpG island sequencing (Bis-Seq) was performed by using a nested PCR protocol as described previously (22,64). The methylation status of the MIR148A and RBP1 promoter CpG islands were assessed as described in (22) and (64), respectively.
The sequence of each sample was reviewed using Chromas Lite 2.33 (Technelysium Pty Ltd), and CpG sites exhibiting a substantial signal for C (as compared with T) were considered methylated. For quantification of MIR148A methylation levels, the T-A cloning method was applied. Briefly, following bisulfite conversion and PCR, the amplified MIR148A CpG island products were ligated into pGEM-Teasy vector (Promega), the ligated products were identified by blue-white screening and amplified by miniprep (Invitrogen), and resulting MIR148A-pGEM-Teasy plasmids were sequenced with SP6 primer.
MIR148A transcript RT-qPCR assay
The level of mature MIR148A was measured as previously described (22) by using the TaqMan MicroRNA Assay (Applied Biosystems). For each sample, expression of RNU6B (as an internal loading control) was measured by using same system. Reverse transcription (RT) of MIR148A and RNU6B was performed at the following conditions: (1) 16°C for 30 min, (2) 42°C for 30 min, and (3) 85°C for 5min. The synthesized RT products of microRNAs were run in qPCR using the TaqMan universal PCR master mix (Applied Biosystems) and LightCycler 480 System (Roche). MIR148A expression was normalized to RNU6b serving as an internal control and presented as the relative Ct method (2ΔΔCt).
Statistical Analysis and Data presentation
All experiments were repeated at least three times independently. For Bis-Seq images and 5-hmC dot-blots, a typical representative figure was shown. For the quantitative data, the results were presented as Mean+SEM, and differences between groups were determined using two-tailed student t-test. Differences in percent CpG island methylation were determined using chi-squared analysis. Statistical significance is indicated in all graphs as follows: * = p<0.05, ** = p<0.01 and *** = p < 0.001.
RESULTS
Octyl-D-2-HG induces MIR148A CpG methylation in 293T cells
To demonstrate the role of D-2-HG in MIR148A CpG island methylation, we treated 293T cells with membrane-permeable octyl-D-2-HG and monitored MIR148A CpG island methylation status over multiple passages of treatment. We first verified that octyl-D-2-HG can be converted to D-2-HG by endogenous cellular esterases using an enzymatic D-2-HG assay (38) that does not detect octyl-D-2-HG (Fig. 1A). When 293T cells were cultured in medium with octyl-D-2-HG (1.0 mM), intracellular D-2-HG achieved a stable level ~20 times that of control cells (Fig. 1B), indicating expected conversion of octyl-D-2-HG to D-2-HG by endogenous cellular esterases. Although the increased D-2-HG level is lower than that found generated by IDH1MUT-293T cells (Fig. 1B), we were restricted in using higher concentrations of octyl-D-2-HG due to observed cytotoxicity of higher octyl-D-2-HG concentrations. The MIR148A CpG region did not show a detectable change in methylation status until roughly passage 30 of octyl-D-2-HG treatment, as assessed by targeted Bis-Seq. At passage 32 of the treatment, the MIR148A bisulfite sequencing (Bis-Seq) chromatogram showed clear T/C double peaks in some CpG sites in octyl-D-2-HG treated 293T cells (Supplementary Fig. S1), indicating partial development of CpG methylation. To quantitatively assess the degree of the methylation, we combined MIR148A Bis-Seq with T-A cloning (Fig. 1C). Untreated 293T cells were unmethylated at all 12 detected CpG sites. When treated with octyl-D-2-HG for 30 passages, some CpG sites became methylated (~10% of total CpGs). After further octyl-D-2-HG treatment to passage 35, additional MIR148A CpG sites became methylated (above 60%). Octyl-D-2-HG treatment beyond passage 35 (up to passage 45) did not result in any further increase in MIR148A methylation levels (Supplementary Fig. S2).
Figure 1. Induction of MIR148A promoter methylation in 293T cells and normal human astrocytes (NHAs) by octyl-D-2-HG and octyl-L-2-HG.
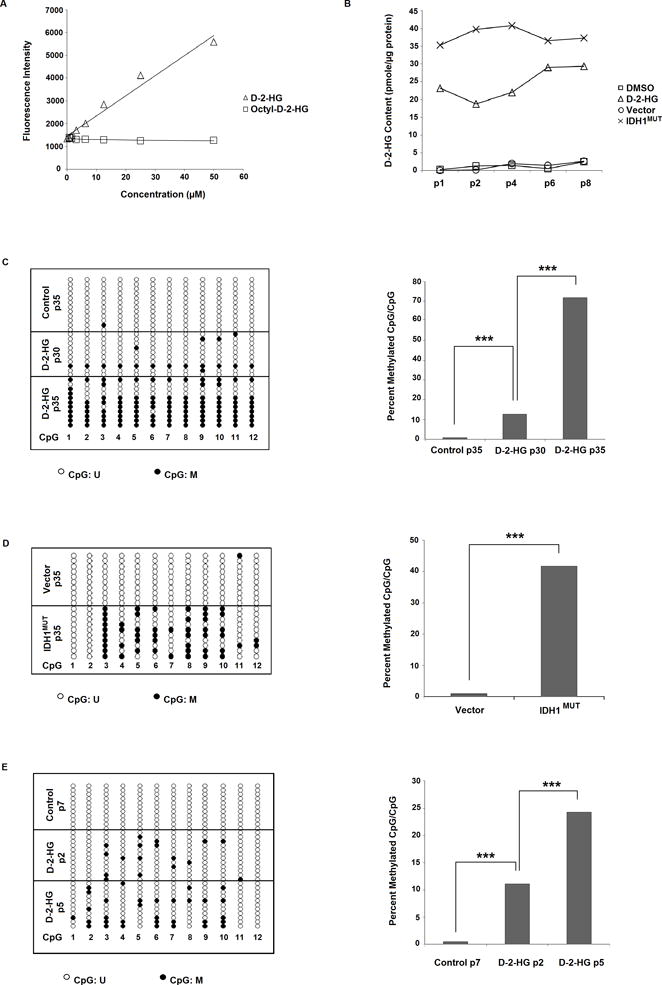
A: Enzymatic D-2-HG assay is not able to detect octyl-D-2-HG. Intensity levels increase with application of D-2-HG, but remain unchanged with increasing concentrations of administered octyl-D-2-HG. B: Intracellular D-2-HG content is elevated with octyl-D-2-HG compared with vehicle-treated 293T cells, and is also elevated in IDH1MUT- versus empty vector-transfected 293T cells. C: Octyl-D-2-HG treatment induces CpG island methylation in 293T cells with >30 passages of treatment. Bis-Seq-TA cloning assay in control 293T cells (35 passages) show that MIR148A is unmethylated, while in octyl-D-2-HG treated cells (30 and 35 passages) MIR148A is partially methylated. Representative lollipop diagram of TA cloning showing methylation status of individual CpG sites (left, n=1). Ratio of methylated CpGs/total CpGs in detected MIR148A promoter region (right). D: IDH1MUT induces miR148a promoter CpG methylation in 293T cells. 293T cells were transfected with empty vector (pLPCX) or pLPCX-IDH1R132H for 35 passages. Representative lollipop diagram of TA cloning showing methylation status of individual CpG sites (left, n=1). Ratio of methylated CpGs/total CpGs in detected MIR148A promoter region (right). E: Octyl-D-2-HG treatment induces rapid CpG island methylation in NHA cells. Lollipop diagram showing MIR148A methylation by Bis-Seq-TA cloning (left, n=1). Figure showing ratio of methylated CpGs/total CpGs (right). Statistical analysis performed using chi-squared analysis. *** indicates p<0.001, ** indicates p<0.01 and * indicates p<0.05 compared with control. U = unmethylated, M = methylated.
To determine if other G-CIMP CpG islands also developed methylation, we performed Bis-Seq on the RBP1 CpG island and observed methylation following 40 passages of treatment with octyl-D-2-HG (Supplementary Fig. S3). These results indicate that D-2-HG is sufficient to induce CpG island methylation of at least two G-CIMP genes.
For reference, we performed quantitative analysis by Bis-Seq/T-A cloning in previously generated 293T cells transfected with empty vector (pLPCX) or pLPCX-IDH1R132H (IDH1MUT) after 35 passages (22). T-A cloning revealed significantly greater CpG methylation in mutant versus vector cells, comparable to cells subjected to long-term octyl-D-2-HG treatment (Fig. 1D).
Octyl-D-2-HG and -L-2-HG also induce MIR148A methylation in normal human astrocytes with apparent accelerated time-course
To provide further evidence supporting D-2-HG as an intermediate in IDH1 mutant induced MIR148A methylation, we investigated whether D-2-HG has the same ability to induce methylation in primary normal human brain astrocytes (NHA, Applied Biological Materials (ABM) Inc, Canada). We confirmed that MIR148A was unmethylated in NHA cells. We found that NHA cells could not tolerate long term treatment at concentrations exceeding 0.5 mM octyl-D-2-HG. When compared with 1.0 mM octyl-D-2-HG treated-293T cells (Fig. 1B), the intracellular D-2-HG level achieved in 0.5 mM octyl-D-2-HG treated-NHA cells was ~50% of the former, albeit still ~10 times that found in control NHA cells (Supplementary Fig. S4A). In contrast to untreated control NHA cells in which MIR148A CpG sites remained unmethylated following consecutive passages (Fig.1E and top panel of Supplementary Fig. S4B), NHA cells treated with octyl-D-2-HG or octyl-L-2-HG showed the appearance of small C peaks in Bis-Seq chromatograms after 2 passages of treatment. At passage 4 of continuous octyl-D-2-HG treatment, Bis-Seq chromatograms showed obvious C peaks arising at CpG sites (lower panels of Supplementary Fig. S4B). Through combined T-A cloning and Bis-Seq, we confirmed that ~12% of CpG sites in the evaluable region were methylated after 2 passages of octyl-D-2-HG treatment, which further increased to ~24% at passage 5 (Fig. 1E). In parallel, cells treated with octyl-L-2-HG (0.5 mM) also developed partial CpG methylation (Supplementary Fig. S4C). As a control, cells treated with the same dosage of octyl-α-KG failed to develop methylation. We were unable to prolong the time of octyl-D-2-HG or -L-2-HG treatment at these concentrations, since the NHA cells could not survive beyond passages 6 to 8.
Octyl-D-2-HG and -L-2-HG induce decreased global 5-hmC levels in 293T cells
To provide evidence that TET dioxygenase activity can be inhibited by increased intracellular D-2-HG content, we performed dot-blots on genomic DNA using a specific antibody against 5-hydroxymethylcytosine (5-hmC), the TET-mediated oxidation product of 5-methyl-cytosine (5-mC). At passage 5, 293T cells treated with octyl-D-2-HG showed a marked reduction in global 5-hmC levels (Fig. 2A), indicating inhibition of TET activity by D-2-HG; similar results were also observed in 293T cells with treatment of octyl-L-2-HG. This apparent early inhibition of TET activity as indicated by reduced 5-hmC is surprising given the delay in methylation induction until passage 30. Similarly, after 2 passages of octyl-D-2-HG treatment, global 5-hmC content became significantly lower than controls in NHA cells (Fig. 2B), similarly suggesting TET inhibition. Parallel treatment with octyl-L-2-HG also showed a significant decrease in 5-hmC content, whereas octyl-α-KG had no impact on 5-hmC (Fig. 2B).
Figure 2. Reduction of 5-hmC by octyl-D-2-HG and octyl-L-2-HG and effects of IDH1-mutant inhibitor C227 on D-2-HG and 5-hmC contents in IDH1MUT-transfected 293T cells.
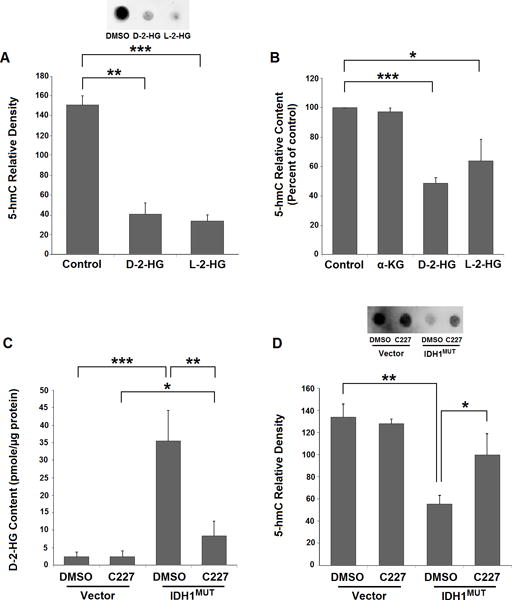
A: Octyl-D-2-HG- or octyl-L-2-HG-treated 293T cells show marked reductions in global 5-hmC at passage 5. Graph shows relative 5-hmC densities with each treatment (n=3, bars show SEM). Representative dot-blot inset at passage 5 shows reductions in 5-hmC levels in DMSO-, octyl-D-2-HG- or octyl-L-2-HG-treated cells. B: Octyl-D-2-HG and octyl-L-2-HG decrease 5-hmC content in NHA cells. Figure shows decrease in global 5-hmC content in NHA cells following two passages of treatment. 5-hmC content of genomic DNA was determined by a 5-hmC ELISA assay (n=3, bars show SEM). Statistical analysis performed using two-tailed student t-test. *** indicates p<0.001, ** indicates p<0.01 and * indicates p<0.05 compared with control. C: C227 reduces D-2-HG in IDH1MUT-293T cells. Figure showing the inhibitory effect of C227 on IDH1MUT-generated D-2-HG with C227 treatment (n=4, bars show SEM). D: C227 treatment restores 5-hmC levels in IDH1MUT-293T cells. Representative dot-blot inset shows 5-hmC levels at passage 20 of C227 treatment, and the results of C227 treatment in IDH1MUT- and vector-293T cells (n=3, bars show SEM). Statistical analysis in all graphs performed using two-tailed student t-test. *** indicates p<0.001, ** indicates p<0.01 and * indicates p<0.05 compared with control. U = unmethylated, M = methylated.
IDH1 mutant inhibitor C227 prevents IDH1MUT-induced MIR148A methylation in 293T cells
To determine whether inhibition of endogenous D-2-HG formation by mutant IDH1 enzymatic activity can prevent IDH1 mutant-induced DNA CpG methylation of MIR148A, we employed an IDH1R132H selective inhibitor, C227 (http://www.xcessbio.com/products/small-molecules/IDH-C227.html) and treated previously established IDH1MUT-293T cells (22) to investigate the effects of development of MIR148A CpG methylation following IDH1MUT transfection. As we previously found (22), the MIR148A promoter region developed CpG methylation after >20 passages (Supplementary Fig. S5). At passage 5 of stable transfection, prior to the induction of MIR148A methylation, 293T cells transfected with IDH1MUT or blank vector were treated with 2.0 μM C227. This dosage effectively inhibits D-2-HG generation by IDH1MUT-293T cells at all tested cell passages (Fig. 2C). We did not observe any detectable cytotoxicity with long term treatment at this concentration. Importantly, in IDH1MUT-293T cells continuously treated with C227, no obvious MIR148A methylation was detected at any time point up to passage 40 (passage 45 of transfection; Supplementary Fig. S6). After C227 treatment, we observed restored 5-hmC levels in IDH1MUT-293T cells to the level of vector-293T cells (Fig. 2D) at passage 20 (passage 25 of transfection), suggesting restoration of TET activity. These results indicate that D-2-HG is necessary for MIR148A CpG island methylation in IDH1MUT-293T cells. Treatment with C227 was also demonstrated to be sufficient to prevent RBP1 methylation, another established G-CIMP member (Supplementary Fig. S7).
MIR148A possesses an independent promoter that interacts with RNA Pol II
While our previous work had shown evidence of promoter activity within the upstream of pre-MIR148A region, the existence of an independent promoter for this had not been further validated in the experimental literature. We thus sought to experimentally confirm the location of the core promoter activity and demonstrate a direct physical interaction between the promoter and DNA-dependent RNA Pol II. Pol II antibody pull-down experiments using 293T cells, which highly express MIR148A, show that an endogenous DNA fragment contained within the previously identified 1.6 kb upstream fragment is associated with Pol II. The specificity of interaction between Pol II and the MIR148A promoter region was confirmed by showing that a ‘control’ DNA fragment upstream of pre-MIR148A gene (−4757 to −4547) was not precipitated by the Pol II antibody (Fig. 3A). Our Pol II ChIP-PCR result is consistent with publicly available data from the UCSC Human Genome Browser (Supplementary Fig. S8), which shows a complete overlap between the CpG island and Pol II pulldown region, suggesting Pol II sensitivity to CpG methylation.
Figure 3. MIR148A transcription is driven by its own independent promoter via Pol II.
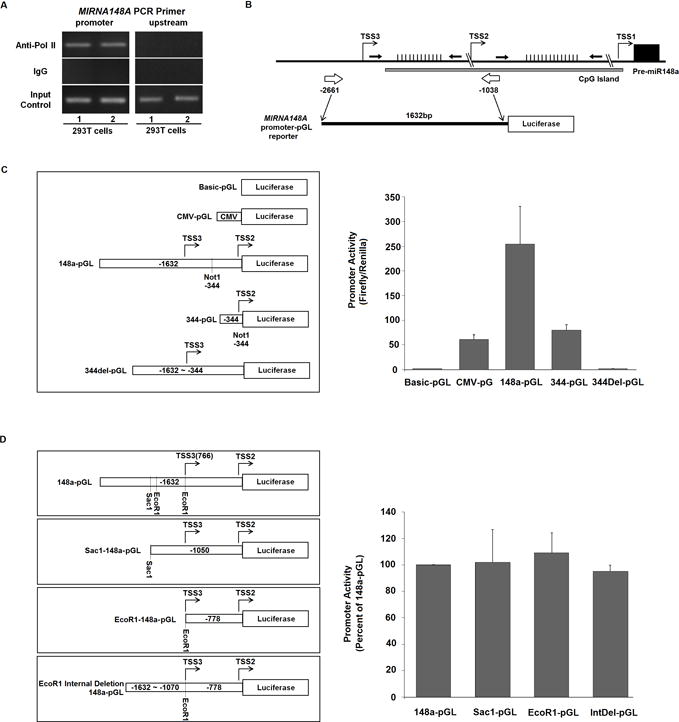
A: Specific recruitment of Pol II to the MIR148A promoter visualized by anti-Pol II ChIP followed by 148a promoter PCR in 293T cells. IgG antibody control shows no activity (n=3). B: Schematic diagram of MIR148A genomic location, CpG island, and promoter-reporter construct. Black arrows indicate Bis-Seq regions, and white arrows indicate the region cloned into the pGL vector. C: The 344 bp region surrounding the predicted TSS2 promoter is required for basic promoter activity. Schematic diagram illustrating MIR148A promoter reporter constructs (left). Figure showing promoter assay results in 293T cells with different pGL reporter constructs (right; n=3, bars show SEM). D: Deletion of the TSS3 upstream region does not impact MIR148A promoter activity. Various reporter constructs with deletion of predicted TSS3 upstream regions achieved by restrictive enzyme digestion (left). Figure showing results of luciferase assay of 293T cells expressing different pGL reporter constructs (right; n=3, bars show SEM).
However, due to limitations of the ChIP-PCR technique in mapping precise promoter structure, we were not able to characterize the core promoter region in more detail through endogenous experiments, and therefore proceeded to perform luciferase reporter assays. The MIR148A gene is localized to p15.2 of chromosome 7 (7p15.2), and is 200 kb upstream and 250 kb downstream from the nearest protein coding genes, strongly suggesting that MIR148A is an intergenic miRNA (UCSC Human Genome Browser; Fig. 3B, Supplementary Fig. S8). Unlike intragenic miRNAs, which generally lack an independent promoter and are instead driven by the promoters of neighboring protein-encoding genes, intergenic miRNAs possess independent promoters that drive their transcription. Indeed, our initial studies indicated that the 1.6 kb upstream fragment (−1038 to −2661 bp) of the pre-MIR148A gene can drive luciferase expression, suggesting functional promoter activity (22). The 1.6 kb fragment between −1038 to −2661bp has two potential transcriptional start sites (TSS2 and TSS3) predicted by Promoter 2.0 Prediction Server (http://www.cbs.dtu.dk/services/Promoter/), implying two potential promoters. Although another potential transcription start site, TSS1, was also predicted at −203 bp of pre-MIR148A by the program (Fig. 3B, Supplementary Fig. S8), based on the low score as well as the location within the pri-MIR148A coding region, the predicted TSS1 has a low probability to represent a transcriptional start site; thus we did not construct a promoter reporter for the TSS1 region, and focused our attention to TSS2 and TSS3.
To determine whether TSS2 or 3 were possible transcriptional start sites, we mapped the core promoter of MIR148A to a 344 bp region (−1038 to −1382 bps) upstream of pre-MIR148A gene by testing a series of deletion constructs in a luciferase reporter assay. Although deletion of the portion upstream from −1382 to −2661 bp resulted in significantly reduced promoter activity, the remaining 344 bp fragment (−1038 to −1382 bps) still possessed transcriptional activity comparable to CMV promoter activity (Fig. 3C). Moreover, once the 344 bp fragment was deleted, the remaining upstream region completely lost transcriptional activity. These results indicated that the 344 bp region around TSS2 (−1113 bp) possesses basic promoter activity. Another potential promoter may be located near the predicted region TSS3 (−1713; Fig. 3B, Supplementary Fig. S8). However, when the region immediately upstream of TSS3 was removed, luciferase expression was unaffected (Fig. 3D), indicating that the TSS3 region lacks transcriptional activity. Overall, these results strongly suggest that MIR148A transcription is driven by its own independent promoter, and that significant (core) promoter activity is contained within a region −1038 to −1382 bp upstream of the pre-MIR148A gene location.
MIR148A promoter methylation reduces transcription by abrogating Pol II binding
Using 293T cells, we investigated the role of CpG methylation in the regulation of MIR148A promoter function. We found that Pol II recruitment to the MIR148A promoter is decreased in IDH1MUT-293T cells compared to control 293T cells (Fig. 4A). In addition, as we similarly reported earlier (22), we observed a corresponding reduction of MIR148A transcripts in IDH1MUT-293T cells, compared to 293T cells transfected with blank vector as determined by RT-qPCR (Fig. 4B). Moreover, after 4 days of transfection of the MIR148A-pGL reporter, MIR148A-promoter driven luciferase expression is partially reduced in later passage IDH1MUT-293T cells as compared to vector-293T cells and IDH1WT-293T cells (Fig. 4C). To further verify the ability of MIR148a promoter methylation to silence transcription, we generated a fully methylated MIR148A-pGL reporter by in vitro treatment with the methyltransferase, SssI. When the methylated construct was transfected into 293T cells, its luciferase activity almost completely abrogated (Fig. 4D). In contrast, control constructs did not show substantial inhibition with SssI enzymatic methylation. Taken together, these results strongly suggest that MIR148A promoter activity is negatively regulated by CpG island methylation.
Figure 4. Epigenetic regulation of MIR148A promoter activity by DNA CpG methylation.
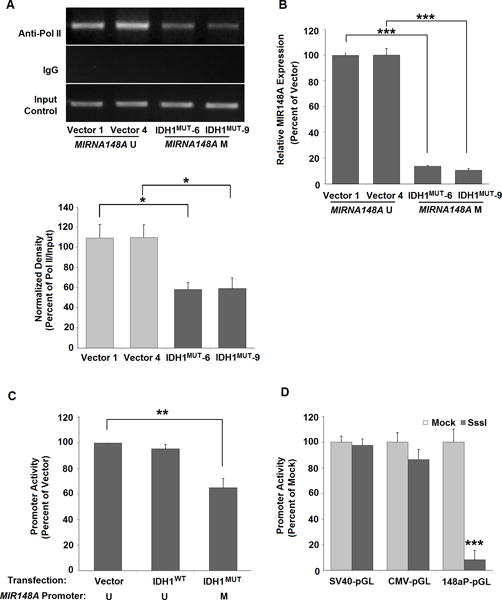
A: Decreased recruitment of Pol II to the MIR148A promoter after development of MIR148A methylation in IDH1MUT-293T cells compared to vector-293T cells at passage 30. Bar graph shows MIR148A ChIP-PCR relative density measured via ChIP-PCR. For each transfection, two clonal cell lines were tested (n=2, bars show SEM). B: Figure showing decreased MIR148A expression in IDH1MUT-293T cells compared to vector-293T cells at passage 30 as detected by RT qPCR (n=2, bars show SEM). C: Methylation reduces MIR148A promoter-pGL activity in IDH1MUT-293T cells. Figure shows blank vector-, IDH1WT- and IDH1MUT - 293T cells at passage >40 as determined by luciferase assay (n=3, bars show SEM). D: MIR148A promoter activity is decreased by in vitro methylation with SssI treated miR148a-pGL (n=3), CMV-pGL (n=2) and SV40-pGL transfection (n=2; all bars show SEM). Each was either pre-methylated by in vitro SssI reaction or treated with mock reaction without SssI, and activity was determined by luciferase assay. Statistical analysis for all data performed using two-tailed student t-test. *** indicates p<0.001, ** indicates p<0.01 and * indicates p<0.05 compared with control. U = unmethylated, M = methylated.
D-2-HG downregulates MIR148A transcription in 293T and NHA cells
To confirm that octyl-D-2-HG treatment downregulates MIR148A transcription, we measured cellular MIR148A transcript levels in 293T and NHA cells by both RT-PCR and RT-qPCR. At passage 34-36, octyl-D-2-HG treatment reduced MIR148A expression compared to DMSO treated cells (Fig. 5A). At earlier passages, octyl-D-2-HG produced no change in MIR148A expression (Supplementary Fig. S9A).
Figure 5. Treatment with octyl-D-2-HG and octyl-L-2-HG reduces MIR148A expression.
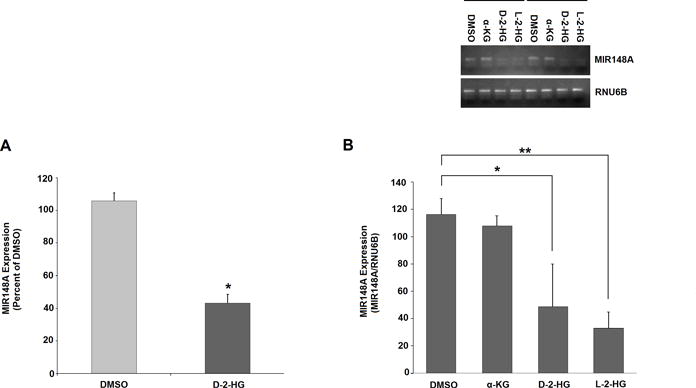
A: Treatment with octyl-D-2-HG (1.0 mM) significantly reduces MIR148A expression in 293T cells. Transcript levels in cells treated by vehicle and octyl-α-KG determined at passage 34-36 by RT-qPCR (n=2, bars show SEM). B: Treatment with octyl-D-2-HG and octyl-L-2-HG significantly reduces MIR148A expression in NHA cells. Transcript levels in cells treated by vehicle, octyl-α-KG, -D-2-HG and -L-2-HG determined by RT-PCR (inset above, n=2, combined on single gel) and RT-qPCR (below; n=3, bars show SEM). RUN6B transcription was used as an internal control for RT-PCR. Statistical analysis in all graphs performed using two-tailed student t-test. *** indicates p<0.001, ** indicates p<0.01 and * indicates p<0.05 compared with control.
In NHA cells, at passage 4, both octyl-D-2-HG and -L-2-HG significantly inhibited MIR148A transcription (Fig. 5B). However, octyl-α-KG did not impact MIR148A expression. At passages 1 and 2, MIR148A content in NHA cells treated with octyl-D-2-HG, -L-2-HG or -α-KG showed no change compared to untreated cells (Supplementary Fig. S9B). The occurrence of CpG methylation in the MIR148A promoter region (passage 2) immediately prior to MIR148A transcriptional inhibition (passage 4) suggests a causal role for methylation in transcriptional regulation. These results indicate that, in 293T and NHA cells, D- or L-2-HG can induce MIR148A promoter hypermethylation and reduce MIR148A expression.
C227 prevents methylation-induced silencing of miRNA148a in IDH1 mutant overexpressing 293T cells
We then investigated the role of long-term C227 treatment on MIR148A transcription in 293T cells. At passage 10 of C227 treatment (passage 15 of transfection), as was expected given that this is prior to the development of promoter methylation, MIR148A transcript levels were similar between IDH1MUT- and vector-293T cells with or without application of C227 (Fig. 6A, left). However, beyond ~ passage 20 of IDH1MUT transfection, chronic C227 application prevented MIR148A transcript reduction occurring with the establishment of MIR148A promoter methylation in IDH1MUT-293T cells (Fig. 6A, right). At passage 40 of C227 treatment (passage 45 of transfection), parallel vector-293T cells, with or without C227 treatment, retained an unmethylated status at the MIR148A promoter (Supplementary Fig. S6) and maintained a similar level of MIR148A transcription as shown in Fig. 6A (right). At passage 35 of C227 treatment (passage 40 of IDH1MUT transfection), IDH1MUT-293T cells still retained an unmethylated status at the MIR148A promoter compared to the methylated status in vehicle-treated IDH1MUT-293T cells (Supplementary Fig. S6). In these 293T cells, we assessed Pol II interaction with MIR148A promoter by ChIP-PCR. We did not observe an effect of C227 on Pol II binding with the unmethylated MIR148A promoter in vector-293T cells (Fig. 6B). Similar to passage 30 (see Fig. 4A), untreated IDH1MUT-293T cells at passage 40 of transfection still display a decrease in Pol II binding with MIR148A promoter compared to vector-293T cells, but treatment with C227 significantly prevents the reduction of Pol II recruitment in IDH1MUT-293T cells (Fig. 6B). These results further validate the effect of IDH1MUT generated D-2-HG on MIR148A promoter methylation status and transcriptional regulation.
Figure 6. Treatment with C227 restores MIR148A expression and Pol II promoter binding.
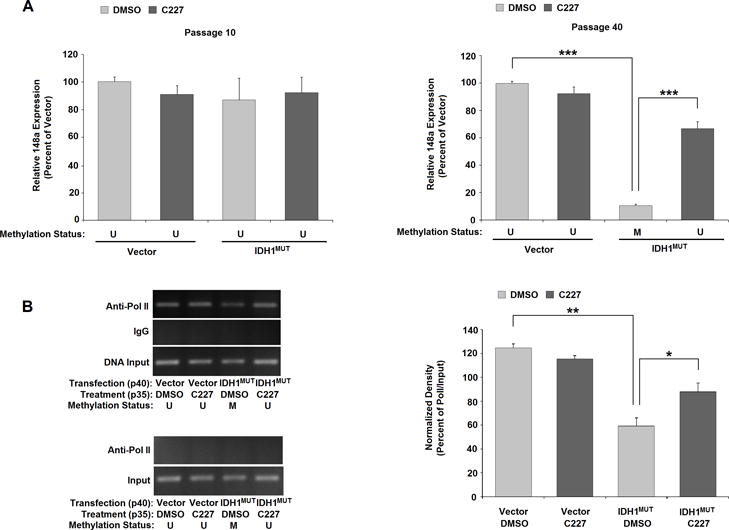
A: C227 restores miRNA148a expression in IDH1MUT-293T cells. MIR148A transcription determined by RT-qPCR in vector- or IDH1MUT-293 cells with or without C227 treatment at passage 10 (left) and at passage 40 of treatment (right; n=3 in both graphs, bars show SEM). B: C227 restores promoter recruitment in C227-treated IDH1MUT-293T cells, but has no effect in empty vector-transfected cells (n=2). Images of ChIP-PCR for determining Pol II recruitment to the MIR148A region in Vector- or IDH1MUT-293T cells with or without C227 treatment at passage 35 (top gel). No effect is observed in empty vector-transfected cells (n=2). Pol II does not bind to the upstream -4943 to −4737 region of the pre-MIR148A gene (bottom gel). C: Relative binding density normalized to input. Statistical analysis in all graphs performed using two-tailed student t-test. *** indicates p<0.001, ** indicates p<0.01 and * indicates p<0.05 compared with control. U = unmethylated, M = methylated.
D-2-HG treatment withdrawal reverses D-2-HG-induced MIR148A promoter methylation in 293T cells
Lastly, we sought to investigate whether D-2-HG-induced CpG methylation in the MIR148A promoter is reversible after withdrawal of octyl-D-2-HG treatment from 293T cells. To confirm the hypothesis that octyl-D-2-HG withdrawal will restore TET demethylation activity, we divided octyl-D-2-HG-treated 293T cells in which MIR148A promoter methylation had been established (passage 35) into two treatment groups: one with continued octyl-D-2-HG treatment and the other with discontinued octyl-D-2-HG treatment (de-D-2-HG). Following another 4 passages in culture, DNA and RNA were extracted from control, octyl-D-2-HG and de-D-2-HG –treated 293T cells to analyze MIR148A DNA methylation and RNA expression. As previously shown (Fig. 1C), 293T cells with 39 passages of octyl-D-2-HG treatment demonstrated MIR148A promoter hypermethylation compared to parallel control cells, but cells in which octyl-D-2-HG treatment had been discontinued for 4 passages showed a dramatic decrease in 5-mC sites (Figure 7A).
Figure 7. D-2-HG-induced MIR148A promoter methylation is reversible after removal of octyl-D-2-HG treatment.
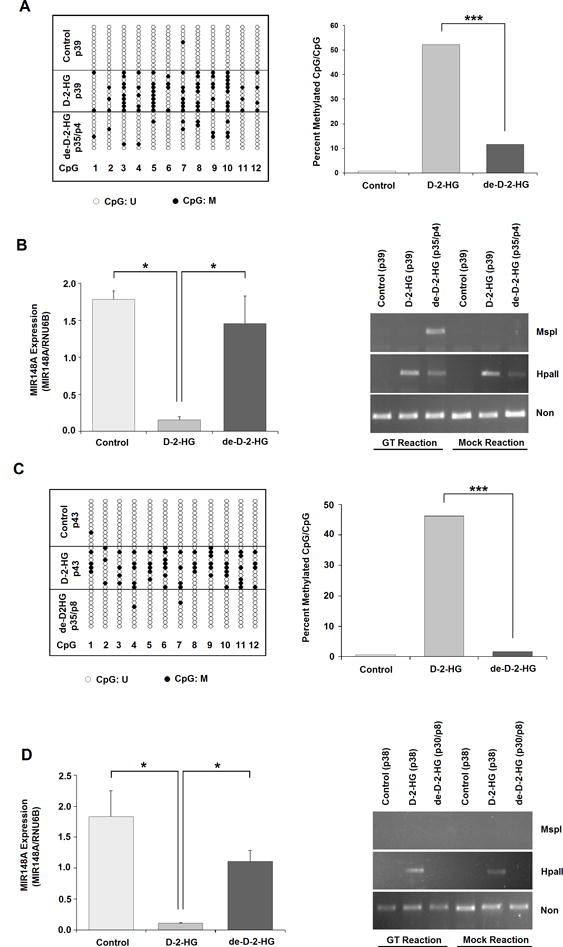
A: CpG sites reverted to an unmethylated state 4 passages after discontinuation of octyl-D-2-HG treatment (de-D-2-HG) in 293T cells following 35 passages of octyl-D-2-HG treatment, compared with cells in which treatment was continued (D-2-HG). g-DNAs were used to detect MIR148A methylation by Bis-Seq-TA cloning (n=1). B: MIR148A expression measured by RT-qPCR and 5-hmC detected using 5-hmC glucosylation-MSP1 digestion assay after 4 passages of discontinued D-2-HG treatment (de-D-2-HG). Total RNA and g-DNAs were used for these measurements. C: CpG site demethylation 8 passages after discontinuation of octyl-D-2-HG treatment (de-D-2-HG) in 293T cells following 35 passages of treatment, compared with cells in which treatment was continued (D-2-HG). g-DNAs were used to detect MIR148A methylation by Bis-Seq-TA cloning (n=1). D: MIR148A expression measured by RT-qPCR and 5-hmC detected using 5-hmC glucosylation-MSP1 digestion assay after 8 passages of discontinued octyl-D-2-HG treatment (de-D-2-HG). Total RNA and g-DNAs were used for these measurements. Bars show SEM. Statistical analysis in all graphs performed using two-tailed student t-test. *** indicates p<0.001, ** indicates p<0.01 and * indicates p<0.05 compared with control.
To determine alterations in MIR148A transcription, we isolated mRNA in parallel from the various experimentally treated cells and performed RT-qPCR assays. We found that following octyl-D-2-HG withdrawal, MIR148A transcripts were significantly increased at passage compared to the cells with continuing octyl-D-2-HG treatment (passage 39), and were not significantly different compared with control 293T cells at the same passages (Fig. 7B). These results provide further evidence for D-2-HG epigenetic regulation of MIR148A promoter activity. Based on our T-A cloning data, following withdrawal of octyl-D-2-HG for 4 passages, the MIR148A promoter retained a low level of apparent methylated (5-mC) sites compared with control cells, but MIR148A transcription increased to levels approaching those of control cells (Fig. 7B).
After 8 passages of octyl-D-2-HG withdrawal, the MIR148A promoter became unmethylated to a degree comparable with that of control cells at the same passage, whereas octyl-D-2-HG-treated 293T cells retained their hypermethylation status (Fig. 7C). A similar independent result was obtained via bulk sample Bis-Seq (without T-A cloning) by starting with 293T cells previously treated with octyl-D-2-HG for 40 passages (Supplementary Fig. S10). Following 8 passages of octyl-D-2-HG withdrawal, MIR148A transcripts also remained significantly increased compared to the cells with continuing octyl-D-2-HG treatment (passage 43), and were not significantly different compared with control 293T cells at the same passages (Fig. 7D). However, although the MIR148A promoter was completely unmethylated, MIR148A transcript levels did not increase markedly compared with passage 4, and in fact appears slightly reduced although this difference was not significant. In light of the fact that both 5-mC and 5-hmC are resistant to bisulfite conversion, and therefore indistinguishable by Bis-Seq, this result suggests that the residual 5-mC signals following withdrawal of octyl-D-2-HG for 4 passages are not entirely comprised exclusively of 5-mC, but may contain 5-hmC.
To test this idea, we selected the −546 to −746 bp region of pre-MIR148A (~250 bp downstream of TSS2) to dissect 5-hmC signals from 5-mC signals by using the EpiMark 5-hmC and 5-mC Analysis Kit (New England Biolab). This DNA region contains two CCGG sites which can be cut by MspI (5-mc/5-hmC non-resistant) and Hpa II (5-mc/5-hmC resistance). If the CpGs of both CCGG sites are 5-hydroxymethylated, the hydroxyls of 5-hmCs can be glucosylated to 5-gluco-hmC (5-ghmC) by the T4 Phage β-glucosyltransferase (T4-BGT), which leads to resistance to MspI digestion and therefore can be detected by PCR amplification. As expected, in control cells (passage 39 and 45), MspI or HpaII digestion completely prevented PCR amplification of the 200 bp fragment, indicating that neither 5-mC nor 5-hmC was present. In octyl-D-2-HG treated cells at the same passage, although HpaII resistance was present, glucosylation did not prevent MSP1 digestion, indicating presence of 5mC of both CCGG sites. However, following withdrawal of octyl-D-2-HG for 4 passages, we observed the protection of glucosylation on MSP digestion, indicating that both CCGG sites contained 5-hmC signal (Figure 7B). The 5-hmC signals disappeared following 8 passages of discontinued octyl-D-2-HG treatment (Figure 7D). Because 5-hmC is a positive epigenetic marker for gene transcription, the temporal 5-hmC increase (albeit non-significant) may explain why passage 4 cells may have higher miR148a transcripts than passage 8 cells.
DISCUSSION
By inhibiting α-KG dependent TET activity, D-2-HG has been widely hypothesized to alter the dynamic balance between DNA methylation and demethylation resulting in the DNA CpG island hypermethylation phenotype associated with IDH1/2 mutant gliomas (G-CIMP) (7,8). Turcan and colleagues reported that expression of the IDH1R132H mutation in immortalized human astrocytes can induce development of this phenotype (8). However, to date there has been limited direct experimental evidence to prove a causal role for D-2-HG in the establishment of G-CIMP. IDH1 mutation causes multiple metabolic abnormalities independent of D-2-HG, such as inactivation of NADPH-dependent reductive carboxylation (39), stimulation of glutamine metabolism under hypoxia (40), inhibition of ATP synthase (41), and alteration of glutathione and TCA cycle metabolite levels (42). Further adding complexity, D-2-HG does not mimic all IDH mutation effects (43), and accumulation of IDH1MUT-generated D-2-HG is able to alter and/or dysregulate a variety of other pathways through D-2-HG-mediated inhibition of other dioxygenases, including histone lysine demethylases (44), regulators of the HIF pathway (45), and mediators of collagen protein maturation (46).
In this study, we used the MIR148A gene as a ‘reporter’ to probe the role of D-2-HG in CpG island hypermethylation, and additionally, we further characterized the miRNA148a promoter region. Our results clearly indicate that: (1) octyl-D-2-HG treated 293T and NHA cells develop MIR148A promoter CpG methylation; (2) 5-hmC levels in cells treated with octyl-D-2-HG or -L-2-HG exhibit significant decreases, reflecting D- or L-2-HG inhibition of TET demethylation activity; (3) 2-HG-treated cells display a reduction in MIR148A expression following development of MIR148A promoter methylation; and (4) 2-HG withdrawal reverses methylation, increases 5-hMC, and allows transcription. These results establish that the putative oncometabolite is sufficient to induce methylation of a G-CIMP gene promoter likely via dysregulation of TET demethylating activity. That methylation could be induced in 293T cells and NHA cells is consistent with the notion that IDH1 mutation induction of CIMP is not astrocyte-specific, given that IDH1-associated CIMPs are found in other malignancies such as AML (47). Upon initiation of octyl-D-2-HG treatment, NHAs, even with only 50% of D-2-HG concentration, exhibited MIR148A promoter methylation in an apparent accelerated time course compared with 293T cells (2-5 versus 30 passages). In IDH1MUT-293T cells, we observed a similar delay in methylation. We hypothesize these differences in sensitivity derive from inherent differences in basal methylation and demethylation activity, which could offer a potential explanation for observed variations in IDH1/2 mutation frequency among distinct tissue types.
To investigate whether D-2-HG was necessary for CpG island methylation of MIR148A in IDH1MUT 293T cells, we used C227 to block IDH1R132H mutant activity. C227 is a potent and selective IDH1R132H-mutant inhibitor first reported in patent WO2012009678 (48). Rohle and colleagues demonstrated that AGI-5198, a compound comparable to C227, inhibits D-2-HG production and proliferation of IDH1MUT glioma cells but not IDH1WT glioma cells in vitro and in vivo (xenograft model) (20). Our previous work has also demonstrated that stable IDH1MUT-293T cells exhibit CpG methylation of the MIR148A promoter (22). Using this cell model, we show in this study that initiation of long-term C227 treatment at an early stage of IDH1MUT transfection normalizes global 5-hmC content, and prevented both development of MIR148A promoter methylation and inhibition of MIR148A transcription, presumably through reduced D-2-HG levels. A separate series of experiments demonstrated D-2-HG-mediated induction of promoter methylation in another gene associated with G-CIMP, RBP1, underscoring the role of D-2-HG in establishment of a global methylation phenotype. Promoter methylation of this gene was also prevented by treatment with C227. Thus, our results strongly suggest that D-2-HG is necessary for mutant IDH1 induced methylation of miRNA148a, and this effect likely extends to the other G-CIMP promoters. These results corroborate previous data indicating that octyl-D-2-HG treatment leads to transcriptional repression of the miRNA 200 family in HCT116 and MCF-10A cells, although methylation was not assessed in this study (49). Despite evidence indicating inability of IDHMUT inhibitors to reverse established methylation (20,21), it remains to be determined whether C227 can reverse MIR148A methylation once established via inhibition of D-2-HG. Interestingly, reversal of IDH1 mutation-induced methylation by similar inhibitors was observed in an erythroleukemia model system (50).
All three members of the TET family possess a common core catalytic domain at the carboxyl terminus, comprised of a double-stranded β-helix (DSBH) domain and a cysteine-rich domain. α-KG binds with the DSBH and is necessary for TET demethylase activity (51,52). Our 5-hmC data in response to octyl-D-2-HG exposure and withdrawal, coupled with prior evidence that that 2-HG can bind TETs competitively with a-KG and block TET activity in vivo and in vitro (19,53), strongly indicate inhibition of TET by D-2-HG. Direct confirmation of 2-HG and TET2 physical interaction will await further studies. TETs oxidize 5-mC to 5-hmC, 5-hmC to 5-fC, and then 5-fC to 5-caC in a three-step process. Since the first step is more rapid than the latter two steps, 5-hmC signals can transiently accumulate in the TET-associated demethylation process, and can function as a positive epigenetic marker of gene transcription (52). Expression levels of TET enzymes are largely decreased in various cancers, which is also consistent with a low level of 5-hmC (54). However, TET enzymes (55–57) and 5-hmC (58–60) are known to be present at high levels in neuronal cells, potentially contributing to differences in basal demethylating activity which could be responsible for increased sensitivity in NHA cells.
Our results clearly indicate that discontinuation of octyl-D-2-HG treatment induced an increase in MIR148A transcript and, using a 5-hmC glycosylation protection assay, a transient generation of 5-hmC in at least two CCGG sites, implicating 5-hmC as a positive transcriptional regulator. However, technical limitations prevent elucidation of details on 5-hmC distribution within the MIR148A promoter region/CpG island. Despite our inability to determine 5-hmC changes throughout the CpG island, emerging evidence indicates processivity of TET enzymes, (52) which suggest that the entire CpG island may undergo demethylation and thus display 5-hmC markings throughout.
In previous work, we established the existence of a CpG island in close proximity with the MIR148A coding region as a component of the glioma-associated methylation phenotype, G-CIMP. This data provided primary evidence that MIR148A possesses tumor suppressive activity and is epigenetically silenced in IDH1/2 mutant gliomas (22). Analysis of the pre-MIR148A 5′ upstream region using a promoter prediction program (Promoter 2.0 Prediction Server, http://www.cbs.dtu.dk/services/Promoter/), identified three potential transcriptional start sites (TSSs) at −203 bp (TSS1), −1113 bp (TSS2) and −1713 bp (TSS3) respectively (Fig. 3B, Supplementary Fig. S8). Data provided by Li et al (37) indicate that there is methylation-sensitive transcriptional regulatory activity in the region located at −155 to +218. Based on its location downstream of TSS-1, their data does not indicate involvement of TSS-1 in transcriptional regulation of miRNA148a. Therefore, we did not attempt to identify promoter activity near the predicted TSS-1 because its borderline score and location with the pri-miRNA transcript coding sequence strongly argues against TSS-1 being the true start site. Instead, we focused our attention to TSS2 and TSS3.
Previously, we cloned a 1623 bp DNA fragment upstream of the MIR148A coding region which contains TSS-2 and TSS-3 and has been verified to possess strong promoter activity (22). Thus, in the present study, we used ChIP-PCR assay to establish the presence of physical interaction between Pol II and a putative MIR148A promoter region in 293T cells (Fig. 3 and 4), providing greater evidence that MIR148A transcription is driven by an independent promoter. RNA polymerase II (Pol II) catalyzes DNA transcription to form the precursors of a majority of miRNA transcripts as well as mRNAs (61). These Pol II ChIP-PCR experiments demonstrate that the endogenous promoter region of MIR148A, in 293T cells, is physically occupied by Pol II (Fig. 3A); that occupancy is sensitive to CpG methylation (Fig. 4A); and that decreasing occupancy was followed by a decrease in endogenous MIR148A expression (Fig. 4B). This assay provides the most direct evidence to mark an active promoter site by probing the interaction between the DNA promoter and DNA-dependent RNA transcriptase (62). Moreover, using a serial deletion assay of the promoter-luciferase reporter expressed in 293T cells, a 344bp fragment around TSS2 was mapped as a core region of MIR148A promoter activity, while the DNA fragment around TSS3 did not have detectable transcriptional activity (Fig. 3C–D). Our identified active region for the MIR148A promoter overlaps with a CpG island identified upstream of MIR148A by the UCSC Human Genome Browser (see Fig. 3B, Supplementary Figure S8). Previously, we established a 293T cell model with stable IDH1MUT expression in which we observed MIR148A CpG island methylation compared to blank vector transfected 293T cells (22). Using this model, we observed that CpG island methylation abolished Pol II-promoter binding in the 293T expressing IDH1MUT protein (Fig. 4A), suggesting that MIR148A CpG island methylation impairs Pol II-mediated MIR148A transcription. Moreover, when a pre-methylated MIR148A promoter-reporter created in vitro using CpG methyltransferase SssI was transfected into 293T cells, the promoter activity was reduced by >80% compared to the mock reporter, indicating a direct causal consequence of MIR148A promoter methylation on its transcriptional activity. Taken together, these results strongly support the conclusion that MIR148A expression is driven by an independent, methylation-sensitive promoter.
In conclusion, the present study demonstrates that D-2-HG, the IDHMUT-generated oncometabolite, is necessary and sufficient to induce methylation of a G-CIMP gene promoter: this study provides insight into the mechanisms by which mutant IDH dysregulates global methylation patterns. These findings support the widely-held notion by which the IDH1/2 mutation induces oncogenesis via epigenetic silencing of important tumor suppressive genes such as MIR148A.
Supplementary Material
Implications.
Establishment of D-2-HG as a necessary and sufficient intermediate by which mutant IDH1 induces CpG island methylation of MIR148A will help with understanding the efficacy of selective mutant IDH1 inhibitors in the clinic.
Acknowledgments
This study is supported by NIH (R01CA179071 to A. Lai, UCLA SPORE in Brain Cancer (P50 CA211015) *, and the Art of the Brain Foundation.
*The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest disclosure: The authors declare no potential conflicts of interest.
References
- 1.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM, Carmichael L, Eldred JM, Harris CC, Walker J, Peck JB, Du F, Dukes AF, Sanderson GE, Brummett AM, Clark E, McMichael JF, Meyer RJ, Schindler JK, Pohl CS, Wallis JW, Shi X, Lin L, Schmidt H, Tang Y, Haipek C, Wiechert ME, Ivy JV, Kalicki J, Elliott G, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson MA, Baty J, Heath S, Shannon WD, Nagarajan R, Link DC, Walter MJ, Graubert TA, DiPersio JF, Wilson RK, Ley TJ. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hemerly JP, Bastos AU, Cerutti JM. European journal of endocrinology/European Federation of Endocrine Societies. 2010;163:747–755. doi: 10.1530/EJE-10-0473. [DOI] [PubMed] [Google Scholar]
- 4.Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O’Donnell P, Grigoriadis A, Diss T, Eskandarpour M, Presneau N, Hogendoorn PC, Futreal A, Tirabosco R, Flanagan AM. The Journal of pathology. 2011;224:334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 5.Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, Kwak EL, Clark JW, Ryan DP, Deshpande V, Dias-Santagata D, Ellisen LW, Zhu AX, Iafrate AJ. The oncologist. 2012;17:72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E, Saada V, Mylonas E, Quivoron C, Popovici-Muller J, Saunders JO, Salituro FG, Yan S, Murray S, Wei W, Gao Y, Dang L, Dorsch M, Agresta S, Schenkein DP, Biller SA, Su SM, de Botton S, Yen KE. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 7.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, Tigchelaar W, Troost D, Vandertop WP, Bardelli A, Van Noorden CJ. Acta neuropathologica. 2010;119:487–494. doi: 10.1007/s00401-010-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J, Kim JI, Jang HS, Park JW, Park KM. Free radical research. 2011;45:759–766. doi: 10.3109/10715762.2011.577426. [DOI] [PubMed] [Google Scholar]
- 11.Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, Lee YS, Jeong KS, Kim WB, Park JW, Song BJ, Huh TL. The Journal of biological chemistry. 2001;276:16168–16176. doi: 10.1074/jbc.M010120200. [DOI] [PubMed] [Google Scholar]
- 12.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F, McCarthy S, Fantin VR, Straley KS, Lobo S, Aston W, Green CL, Gale RE, Tirabosco R, Futreal A, Campbell P, Presneau N, Flanagan AM. Nature genetics. 2011;43:1262–1265. doi: 10.1038/ng.994. [DOI] [PubMed] [Google Scholar]
- 14.Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, Dang L, Fantin VR, Mak TW. The Journal of experimental medicine. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward PS, Cross JR, Lu C, Weigert O, Abel-Wahab O, Levine RL, Weinstock DM, Sharp KA, Thompson CB. Oncogene. 2012;31:2491–2498. doi: 10.1038/onc.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB. Cancer cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horbinski C. Acta neuropathologica. 2013;125:621–636. doi: 10.1007/s00401-013-1106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Jiang W, Liu J, Zhao S, Xiong J, Mao Y, Wang Y. Journal of neuro-oncology. 2012;109:253–260. doi: 10.1007/s11060-012-0914-4. [DOI] [PubMed] [Google Scholar]
- 19.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Cancer cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, Kunii K, Pedraza A, Schalm S, Silverman L, Miller A, Wang F, Yang H, Chen Y, Kernytsky A, Rosenblum MK, Liu W, Biller SA, Su SM, Brennan CW, Chan TA, Graeber TG, Yen KE, Mellinghoff IK. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, Wiederschain D, Bedel O, Deng G, Zhang B, He T, Shi X, Gerszten RE, Zhang Y, Yeh JR, Curry WT, Zhao D, Sundaram S, Nigim F, Koerner MV, Ho Q, Fisher DE, Roider EM, Kemeny LV, Samuels Y, Flaherty KT, Batchelor TT, Chi AS, Cahill DP. Cancer cell. 2015;28:773–784. doi: 10.1016/j.ccell.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S, Chowdhury R, Liu F, Chou AP, Li T, Mody RR, Lou JJ, Chen W, Reiss J, Soto H, Prins R, Liau LM, Mischel PS, Nghiemphu PL, Yong WH, Cloughesy TF, Lai A. Clin Cancer Res. 2014;20:5808–5822. doi: 10.1158/1078-0432.CCR-14-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Song YX, Wang ZN. Molecular cancer. 2013;12:43. doi: 10.1186/1476-4598-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Song Y, Wang Z, Yue Z, Xu H, Xing C, Liu Z. Journal of gastrointestinal surgery: official journal of the Society for Surgery of the Alimentary Tract. 2010;14:1170–1179. doi: 10.1007/s11605-010-1202-2. [DOI] [PubMed] [Google Scholar]
- 25.Merkerova M, Vasikova A, Belickova M, Bruchova H. Stem cells and development. 2010;19:17–26. doi: 10.1089/scd.2009.0071. [DOI] [PubMed] [Google Scholar]
- 26.Giraud-Triboult K, Rochon-Beaucourt C, Nissan X, Champon B, Aubert S, Pietu G. Physiological genomics. 2011;43:77–86. doi: 10.1152/physiolgenomics.00077.2010. [DOI] [PubMed] [Google Scholar]
- 27.Gao J, Yang T, Han J, Yan K, Qiu X, Zhou Y, Fan Q, Ma B. Journal of cellular biochemistry. 2011;112:1844–1856. doi: 10.1002/jcb.23106. [DOI] [PubMed] [Google Scholar]
- 28.Schoolmeesters A, Eklund T, Leake D, Vermeulen A, Smith Q, Force Aldred S, Fedorov Y. PloS one. 2009;4:e5605. doi: 10.1371/journal.pone.0005605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, Ying ZZ, Tang ZL, Long LQ, Li K. The Journal of biological chemistry. 2012;287:21093–21101. doi: 10.1074/jbc.M111.330381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.John E, Wienecke-Baldacchino A, Liivrand M, Heinaniemi M, Carlberg C, Sinkkonen L. Nucleic acids research. 2012;40:4446–4460. doi: 10.1093/nar/gks025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Joshi P, Jeon YJ, Lagana A, Middleton J, Secchiero P, Garofalo M, Croce CM. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:8650–8655. doi: 10.1073/pnas.1500886112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lehmann U, Hasemeier B, Christgen M, Muller M, Romermann D, Langer F, Kreipe H. The Journal of pathology. 2008;214:17–24. doi: 10.1002/path.2251. [DOI] [PubMed] [Google Scholar]
- 33.Li L, Chen YY, Li SQ, Huang C, Qin YZ. Medical science monitor: international medical journal of experimental and clinical research. 2015;21:1155–1161. doi: 10.12659/MSM.892940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xia J, Guo X, Yan J, Deng K. Journal of cancer research and clinical oncology. 2014;140:1451–1456. doi: 10.1007/s00432-014-1649-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu A, Xia J, Zuo J, Jin S, Zhou H, Yao L, Huang H, Han Z. Med Oncol. 2012;29:2701–2709. doi: 10.1007/s12032-011-0134-3. [DOI] [PubMed] [Google Scholar]
- 36.Hanoun N, Delpu Y, Suriawinata AA, Bournet B, Bureau C, Selves J, Tsongalis GJ, Dufresne M, Buscail L, Cordelier P, Torrisani J. Clinical chemistry. 2010;56:1107–1118. doi: 10.1373/clinchem.2010.144709. [DOI] [PubMed] [Google Scholar]
- 37.Li HP, Huang HY, Lai YR, Huang JX, Chang KP, Hsueh C, Chang YS. Oncotarget. 2014;5:7610–7624. doi: 10.18632/oncotarget.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balss J, Pusch S, Beck AC, Herold-Mende C, Kramer A, Thiede C, Buckel W, Langhans CD, Okun JG, von Deimling A. Acta neuropathologica. 2012;124:883–891. doi: 10.1007/s00401-012-1060-y. [DOI] [PubMed] [Google Scholar]
- 39.Leonardi R, Subramanian C, Jackowski S, Rock CO. The Journal of biological chemistry. 2012;287:14615–14620. doi: 10.1074/jbc.C112.353946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reitman ZJ, Duncan CG, Poteet E, Winters A, Yan LJ, Gooden DM, Spasojevic I, Boros LG, Yang SH, Yan H. The Journal of biological chemistry. 2014;289:23318–23328. doi: 10.1074/jbc.M114.575183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nalbandian A, Llewellyn KJ, Gomez A, Walker N, Su H, Dunnigan A, Chwa M, Vesa J, Kenney MC, Kimonis VE. Mitochondrion. 2015;22:1–8. doi: 10.1016/j.mito.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, He Y, Bigner DD, Vogelstein B, Yan H. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3270–3275. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oizel K, Gratas C, Nadaradjane A, Oliver L, Vallette FM, Pecqueur C. Cell death & disease. 2015;6:e1704. doi: 10.1038/cddis.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A. EMBO reports. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, Ligon KL, Verhaak RG, Yan H, Kaelin WG., Jr Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II, Cairns RA, McCracken S, Wakeham A, Haight J, Ten AY, Snow B, Ueda T, Inoue S, Yamamoto K, Ko M, Rao A, Yen KE, Su SM, Mak TW. Genes & development. 2012;26:2038–2049. doi: 10.1101/gad.198200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes LA, Melotte V, de Schrijver J, de Maat M, Smit VT, Bovee JV, French PJ, van den Brandt PA, Schouten LJ, de Meyer T, van Criekinge W, Ahuja N, Herman JG, Weijenberg MP, van Engeland M. Cancer research. 2013;73:5858–5868. doi: 10.1158/0008-5472.CAN-12-4306. [DOI] [PubMed] [Google Scholar]
- 48.Chen J, Yang J, Cao P. Mini reviews in medicinal chemistry. 2016;16:1344–1358. doi: 10.2174/1389557516666160609085520. [DOI] [PubMed] [Google Scholar]
- 49.Grassian AR, Lin F, Barrett R, Liu Y, Jiang W, Korpal M, Astley H, Gitterman D, Henley T, Howes R, Levell J, Korn JM, Pagliarini R. Journal of Biological Chemistry. 2012;287:42180–42194. doi: 10.1074/jbc.M112.417832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kernytsky A, Wang F, Hansen E, Schalm S, Straley K, Gliser C, Yang H, Travins J, Murray S, Dorsch M, Agresta S, Schenkein DP, Biller SA, Su SM, Liu W, Yen KE. Blood. 2015;125:296–303. doi: 10.1182/blood-2013-10-533604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu X, Zhang Y. Nature reviews Genetics. 2017;18:517–534. doi: 10.1038/nrg.2017.33. [DOI] [PubMed] [Google Scholar]
- 53.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Cancer cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yin X, Xu Y. Advances in experimental medicine and biology. 2016;945:275–302. doi: 10.1007/978-3-319-43624-1_12. [DOI] [PubMed] [Google Scholar]
- 55.Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, Bruckl T, Biel M, Carell T. PloS one. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, Wang J, Zhang L, Looney TJ, Zhang B, Godley LA, Hicks LM, Lahn BT, Jin P, He C. Nature biotechnology. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munzel M, Globisch D, Bruckl T, Wagner M, Welzmiller V, Michalakis S, Muller M, Biel M, Carell T. Angew Chem Int Ed Engl. 2010;49:5375–5377. doi: 10.1002/anie.201002033. [DOI] [PubMed] [Google Scholar]
- 59.Ruzov A, Tsenkina Y, Serio A, Dudnakova T, Fletcher J, Bai Y, Chebotareva T, Pells S, Hannoun Z, Sullivan G, Chandran S, Hay DC, Bradley M, Wilmut I, De Sousa P. Cell research. 2011;21:1332–1342. doi: 10.1038/cr.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szwagierczak A, Bultmann S, Schmidt CS, Spada F, Leonhardt H. Nucleic acids research. 2010;38:e181. doi: 10.1093/nar/gkq684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. The EMBO journal. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bartel DP. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 63.Li S, Chou AP, Chen W, Chen R, Deng Y, Phillips HS, Selfridge J, Zurayk M, Lou JJ, Everson RG, Wu KC, Faull KF, Cloughesy T, Liau LM, Lai A. Neuro Oncol. 2013;15:57–68. doi: 10.1093/neuonc/nos261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chou AP, Chowdhury R, Li S, Chen W, Kim AJ, Piccioni DE, Selfridge JM, Mody RR, Chang S, Lalezari S, Lin J, Sanchez DE, Wilson RW, Garrett MC, Harry B, Mottahedeh J, Nghiemphu PL, Kornblum HI, Mischel PS, Prins RM, Yong WH, Cloughesy T, Nelson SF, Liau LM, Lai A. Journal of the National Cancer Institute. 2012;104:1458–1469. doi: 10.1093/jnci/djs357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.