

Abstract
To understand the genetic drivers of immune recognition and evasion in colorectal cancer (CRC), we analyzed 1,211 CRC primary tumor samples, including 179 classified as microsatellite instability-high (MSI-high). This set includes The Cancer Genome Atlas CRC cohort of 592 samples, completed and analyzed here. MSI-high, a hypermutated, immunogenic subtype of CRC, had a high rate of significantly mutated genes in important immune modulating pathways and in the antigen presentation machinery, including biallelic losses of B2M and HLA genes due to copy number alterations and copy-neutral loss of heterozygosity (CN-LOH). WNT/β-catenin signaling genes were significantly mutated in all CRC subtypes, and activated WNT/β-catenin signaling was correlated with the absence of T-cell infiltration. This large-scale genomic analysis of CRC demonstrates that MSI-high cases frequently undergo an immunoediting process that provides them with genetic events allowing immune escape despite high mutational load and frequent lymphocytic infiltration, and furthermore, that CRC tumors have genetic and methylation events associated with activated WNT signaling and T-cell exclusion.
INTRODUCTION
Despite major advances in technologies to monitor complex molecular profiles of cancer, translating molecular observations into treatment decisions remains challenging in routine patient care due to cancer heterogeneity and the rapidly expanding repertoire of treatment options, including immunotherapy (1,2). Precision medicine advances have been fueled by technological advances, such as targeted sequencing, and also by multi-omic sequencing efforts, such as The Cancer Genome Atlas (TCGA)(3), which are powered to provide guidance for targeting molecular observations in the form of estimates of the frequencies of drivers to consider.
Colorectal cancer (CRC) in particular represents a heterogenous group of dynamic diseases with differing sets of genetic events, accompanying immune response, and influences of exogenous factors, providing a challenge for personalized therapeutic approaches. The initial TCGA effort to molecularly profile CRC reported on 276 primary samples, providing a deep look at the common non-hypermutated chromosome unstable subset, but only a preliminary consideration of 35 hypermutated cases due to the low overall number of cases with microsatellite instability high (MSI-high) (4). The National Cancer Institute Genetics and Epidemiology of Colorectal Cancer Consortium (NCI-GECCO) effort, which relies on the TCGA CRC data as a basis for integration of epidemiology data and GWAS data from 40,000 participants to make recommendations regarding CRC prevention (5), pursued the systematic completion of the TCGA CRC cohort, which now includes 592 cases with tumor exome, transcriptome, methylation and copy number alteration, as well as key subtyping and intermediate data as a resource for the community, including molecular pathologists. We present this resource here.
To further power the study of CRC, we integrated exome data on 619 previously published primary tumor cases from the Nurses’ Health Study (NHS) and the Health Professionals Follow-up Study (HPFS)(6), yielding an unprecedented sample set of 1,211 primary CRC molecularly characterized cases for identifying significantly mutated genes, pathways and potential oncogenic hot spots. The large number of specimens, including a high number of hypermutated MSI-high cases (N = 179, or 15%) and POLE (16 cases), make it possible to carefully analyze genetic alterations in these hypermutation subtypes.
We were particularly interested in applying this large multi-omic CRC cohort to understand drivers of immune infiltration, since recent advances in immunotherapy have necessitated a deeper consideration of the less frequent MSI-high CRC, which have been shown to respond to immune checkpoint blockade, while the much more common microsatellite stable (MSS) CRC do not usually respond to these immunotherapies (7,8). The U.S. Food and Drug Administration recently approved pembrolizumab and nivolumab, two programmed death receptor-1 (PD-1) immune checkpoint blocking antibodies for the treatment of patients with MSI-high (or mismatch repair deficient (MMR-D)) CRC and in the case of pembrolizumab, all MSI-high or MMR-D treatment refractory solid malignancies. It is hypothesized that a higher somatic mutational load leading to increased presentation of neoepitopes mediates immunotherapy responses in MSI-high tumors (6–8). However, it has been observed previously that MSI-high or immune-infiltrated tumors have evolved mutations that may confer resistance to recognition by the immune system in untreated samples (i.e. immunoediting) (9). On the other hand, it has been postulated that a lower mutational load and poorly characterized effector T cell exclusion processes underlie immunotherapy resistance in MSS CRC (10). Here we demonstrate that there are other factors besides mutation load affecting T-cell infiltration in both MSI-high and MSS. Specifically, we show that MSI-high tumors frequently undergo immunoediting through complete disruption of both alleles of key genes in the MHC-antigen presentation pathway and that both MSS and MSI-high cancers have lower T cell infiltration when molecular events that up-regulate the WNT pathway are present.
RESULTS
CRC subtypes of MSI-high, POLE and MSS analyzed in the TCGA cohort
MSI-high status was available for the previously published 276 TCGA samples, using the clinical grade classification which uses PCR on a set of microsatellite markers (4). To assess MSI-high status on the remaining samples for which we did not have coverage on these microsatellite markers we relied on a benchmarked bioinformatics approach called mSINGS (11,12), which considers whether there is an increased insertion and deletion (indel) rate at microsatellite tracts throughout the exome (Supplementary Table 1 and Methods). MSI-high samples are driven by disruptive mutations of mismatch repair genes or epigenetic silencing of MLH1, and we confirmed these driving events in all but 11 samples, increasing our confidence in our assessment of MSI-high status (Supplementary Figure 1a). We also confirmed increased mutation load in MSI-high tumors with increased substitutions (P < 0.001) and indels (P < 0.001), often at microsatellite tracts due to MMR-D in MSI-high cases (Supplementary Figure 1b). Three Lynch syndrome cases were identified by manual inspection of the germline sequenced reads aligned to the human genome to identify disruptive mutations in MMR genes (Supplementary Table 1). These cases were typical of other MSI-high cases with respect to mutational load for number of substitutions and indels and were treated that way in downstream analysis. The 75 TCGA MSI-high cases presented here included transcriptome, CNA and methylation data, in addition to exome data, making multi-omic analyses possible for these cases.
An additional rare hypermutation subtype exists in CRC that could be potentially targetable by immune checkpoint inhibitors based on having an unusually high mutation load (13): POLE mutated cases that have mutations in the exonuclease domain of the catalytic subunit of the polymerase epsilon, and which have an outlier number of substitutions, but not indels (Supplementary Figure 2). We identified 16 of these cases on the basis of mutations in the gene POLE and an outlier number of substitutions (Figure 1 and Supplementary Tables 1 and 2). Three cases were also identified that were POLE and MSI-high, based on mutations in POLE, mSINGS results, and epigenetic silencing of MLH1. All of these cases had an extremely high mutation load, including both indels and substitutions: the largest mutation load in the series was case TCGA-3821 with both a POLE mutation and MSI-high resulting in 10,088 substitutions and 143 indels.
Figure 1. Frequently mutated genes in CRC.
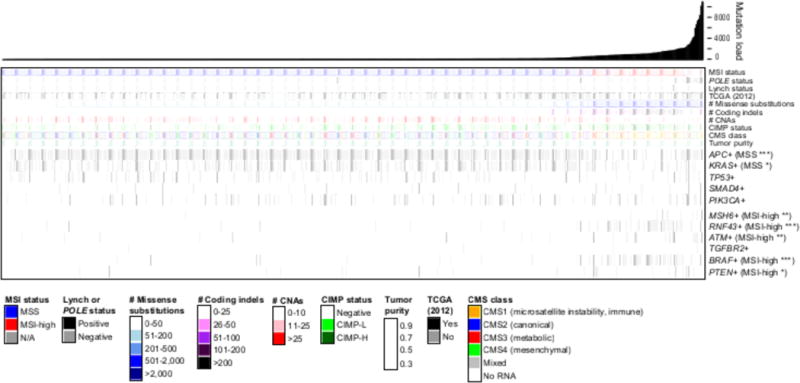
Mutation landscape showing the subtypes of the 592 TCGA tumor samples, sorted by increasing mutation load, including MSI status, POLE status and Lynch status. Samples reported on previously by TCGA (2012) are indicated. Molecular subtypes (see Methods) enriched in MSI-high or MSS are indicated: * P<0.01, ** P<1.0×10−4, *** P<1.0×10−6. Consensus Molecular Subtypes (CMSs) are shown; MSI-high is enriched for CMS1, the microsatellite instability immune subset (P<1×10−15). Mutations defining genomic subtypes are in Methods.
Next, we subtyped the MSI-high and MSS samples further to validate that the resulting TCGA CRC 592 sample cohort recapitulated previously reported features of MSS and MSI-high CRC. CpG Island Methylator Phenotype (CIMP) status was called using an array based methylation method (14). Genomic subtypes, including BRAF+, KRAS+, APC+, TP53+, CTNNB1+, were called using somatic mutation data generated as described in Methods. In addition, the CRC Consensus Molecular Subtype (CMS) for each sample was generated using the messenger RNA (15) (Supplementary Table 1). Figure 1 shows that the CRC data in this cohort were consistent with previously described genetic and molecular subtypes of MSI-high and MSS status (4,6,16). As previously reviewed (16), MSI-high tumors were enriched for hypermethylation (CIMP-high, P < 1×10−15), mutations in BRAF (P < 1×10−15), and RNF43 (P < 1×10−15). On the other hand, MSS tumors were enriched for mutations in APC (P < 1×10−15) and KRAS (P = 0.004) and increased chromosomal instability, including CNAs (P < 0.001) and recurrent high gains and losses (Supplementary Figure 1c and Supplementary Table 3). These differences between MSS and MSI-high tumors reflect differences in precursor lesion and etiology (17) that may contribute to responses to immune checkpoint blockade therapy. A complete description of the data analysis methods, including somatic mutation calling, CNA calling, CN-LOH calling, HLA Class I typing, along with available data may be found in Methods.
WNT signaling and immune-related genes and pathways significantly mutated in in the combined 1,211 CRC cases
To get a deeper look at the mutational landscape of CRC, we combined the somatic mutation data from the 592 fresh frozen exomes from TCGA with the somatic mutations data from the formalin-fixed paraffin embedded exomes from the 619 cases from the Nurses’ Health Study (NHS) and the Health Professionals Follow-up Study (HPFS) (6), yielding a sample set of 1,211 CRC molecularly characterized cases. The combined large number of specimens, including 179 MSI-high cases (104 from the NHS/HPFS cohort typed for MSI-high status using a clinical grade test), provided sufficient power to identify significantly mutated genes and pathways in MSS and MSI-high tumors. This large data set also had the power to identify rare mutation hot spots. For example, we identified the known activating mutation IDH1-R132H (18) in five cases and AKT1-E17K (19) in six cases. Figure 2a and Supplementary Table 4 detail which data set was used for which analyses: the somatic mutations from the NHS/HPFS cohort were additionally used to identify potential drivers, while the multi-omic TCGA data was used to explore further the functional roles of these drivers.
Figure 2. Data sets and summary of significantly mutated genes.
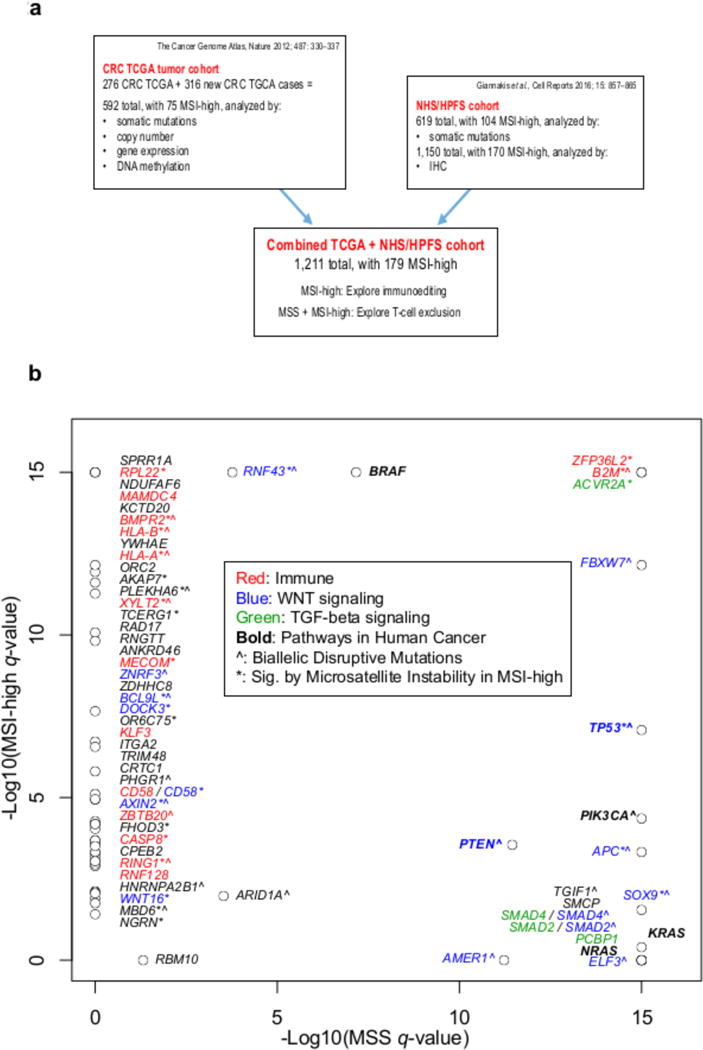
a, Sources of data, breakdown of subtypes, and types of analyses performed on each cohort. b, Significantly mutated genes (by MutsigCV) in MSS relative to MSI-high; all genes with Q<0.01 in the combined TCGA+NHS/HPFS set and at least one of the individual cohorts are shown. Genes are labeled by functional class(es), prevalence of microsatellite mutations (*: significant with Q<0.01 in indel-specific calculation), and incidence of biallelic disruptive mutations (ˆ: present in at least one sample).
We utilized the MutSigCV algorithm (20) on the combined data set to identify the significantly mutated genes that were found to be mutated more frequently than expected given the cohort-specific background mutation rate in MSS or MSI-high (6) We identified a total of 62 significantly mutated genes, of which 9 were identified in MSS tumors only, 40 in MSI-high tumors only, and 13 in both (Figure 2b and Supplementary Table 5). Supplementary Table 6 shows which genes in Figure 2b were novel with respect to the four previous large-scale whole exome sequencing studies of CRC (1,4,6,21). The significantly mutated genes in MSS were all previously reported in TCGA (4) or in the NHS/HPFS study (6) except for B2M, which is more commonly mutated and significant in MSI-high, occurring less commonly in MSS (Supplementary Table 6). On the other hand, 27 of the 53 significantly mutated genes in MSI-high were novel, likely resulting from the large increase in the number of MSI-high tumors in this combined series relative to previous studies (1,4,6,21). WNT signaling genes were well represented as significantly mutated genes in both subtypes, while immune-related genes were significantly mutated in MSI-high specifically (Figure 2b).
We used MutsigCV on the TCGA cohort to identify significantly mutated pathways in the MSS cohort using BioCarta, REACTOME, and KEGG gene sets. We identified 135 pathway gene sets with a Q-value less than 0.01 in the MSS cohort: only four involved the immune system, specifically interleukin signaling (Supplementary Table 7). Next, we used MutsigCV to identify six significantly mutated pathways in the MSI-high cohort, including two significantly mutated pathways involved in MHC Class I antigen presenting machinery (APM) (Supplementary Table 8). Antigen presenting genes HLA-A, HLA-B, and B2M contribute a large number of the mutations in these pathways.
Immune-related and WNT signaling genes significantly mutated in MSI-high based on MMR-D
It had been previously observed that MMR-D mutations occur at specific microsatellite sites in CRC genome (22), but the functional impact of these microsatellite hot spots was not previously known (22). A recent study (23) presented a method to use microsatellite indels, which occur at a high rate in MSI-high, to identify cancer drivers. The study identified a JAK1 microsatellite indel as driving immune evasion in endometrial tumors and re-demonstrated RNF43 microsatellite indels as inhibitors of WNT signaling in colon cancer, thereby demonstrating the utility of this approach. We applied a similar approach (see Methods) to the full 179 CRC MSI-high cohort to see whether each of the 53 significantly mutated genes in MSI-high had a higher than expected number of indels at microsatellite sites. We observed 29,429 total indels at 1 billion possible microsatellite tracts in the entire MSI-high cohort. Aggregating the data by gene, we observed 28 out of 53 genes significantly mutated by this MMR-D typical mechanism (Figure 2b). Eight of these genes were related to WNT signaling, and 11 were related to immune pathways (Figure 2b), indicating selection pressure on these genes in MSI-high resulting from the MMR-D mutation rate (Supplementary Table 9).
Biallelic losses of WNT and immune-related genes
To add further evidence to the driver status of the 62 significantly mutated genes, and to overcome concerns about hypermutation rates creating false positives, we integrated data on disruptive mutations, single-copy losses, and CN-LOH in MSS (Figure 3a) and MSI-high tumors and thereby identified cases with biallelic losses in 26 of the 62 significantly mutated genes (Figure 3a and Supplementary Table 10) (short reads limit our ability to check whether both alleles are lost when there are two disruptive mutations, so these cases are less conclusive). Of these, 13 were WNT signaling genes and eight were immune-related genes. These data indicate selection pressure to disrupt WNT genes across all CRC, while immune-related genes are selected for disruption in MSI-high.
Figure 3. Biallelic loss events in significantly mutated genes in CRC broken down by MSI status.
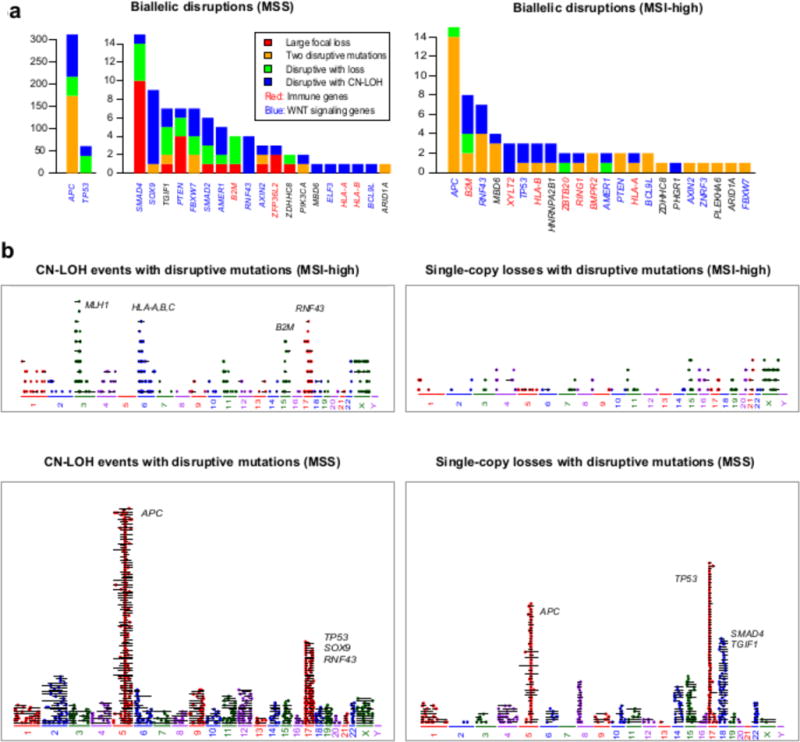
a, Stacked barplot showing genes with recurrent biallelic disruptions in MSS and MSI-high tumors, by number of biallelic disruptions of each type. b, CN-LOH and single copy loss events in MSI-high and MSS. Each panel shows all disruptive somatic mutations (i.e., splice-site mutations, nonsense mutations, start site mutations, and frame-shift indels) that overlap single-copy losses and CN-LOH events. Each horizontal segment represents a single event, with the length of the segment proportional to the length of the alteration; colored dots represent mutations.
While MSI-high cancers have relatively few CNAs (Supplementary Figure 3), we found that CN-LOH events do occur at sites of disruptive mutations, resulting in biallelic loss of a gene and indicating selection pressure to inactivate that gene (Supplementary Figure 4a shows focal CN-LOH events driving biallelic loss of B2M). Figure 3b shows all the CN-LOH and single-copy loss events overlapping disruptive mutations in the TCGA cohort, revealing recurrent CN-LOH events that occur in the HLA-A, HLA-B and B2M regions, providing evidence that in MSI-high cases CN-LOH events cluster in APM genes. Interestingly, a recent study of CN-LOH of the HLA Class I region in lung cancer showed that biallelic loss in this region can drive immune escape (24), while a recent study in melanoma showed that biallelic losses of B2M are enriched in anti-PD1 nonresponders relative to responders (25).
Selection for disruption of immune-related genes in MSI-high tumors
Figure 4a shows the mutation landscape for the 75 MSI-high samples from the TCGA, which has CNA and gene expression data. Fifteen significantly mutated genes (identified using MutsigCV on the combined cohort) have a documented role in the immune system specifically, including 13 significantly mutated in MSI-high tumors, a subtype shown previously to have a high level of T-cell infiltration (6) and two significantly mutated, immune-related genes (ZFP36L2 and B2M) in both MSS and MSI-high (Figs. 2b and 4a). Of these 15 genes, three had known roles in modulating diverse functions in hematopoietic lineage cells and effects beyond antigen presentation but no previously described roles in cancer cells: XYLT2, a gene necessary for dendritic cell traffic and T cell response in the lymph nodes, because of its role in the biosynthesis of heparan sulfate (26); ZFP36L2, a gene involved in thymocyte development (27); and KLF3, a gene involved in B-cell development (28). Four additional significantly mutated immune-related genes had been previously described as being expressed in cancer cells: CD58, a tumor surface protein involved in the activation of NK cells through interaction with CD2 (29) that is concurrently lost with B2M in some cancers (30); CASP8, a tumor-expressed gene involved in innate immunity through its role in apoptosis (31); ZBTB20, a tumor Toll-like receptor mediating innate immune response by repressing IκBα gene transcription (32); and RNF128, a tumor-expressed gene that is essential for IFN-β production and innate antiviral immune response (33). Together, these mutations indicate positive selection for immune escape through other mechanisms beyond mutations in APM.
Figure 4. Mutation landscape of immune-related genes and consequences in MSI-high tumors.
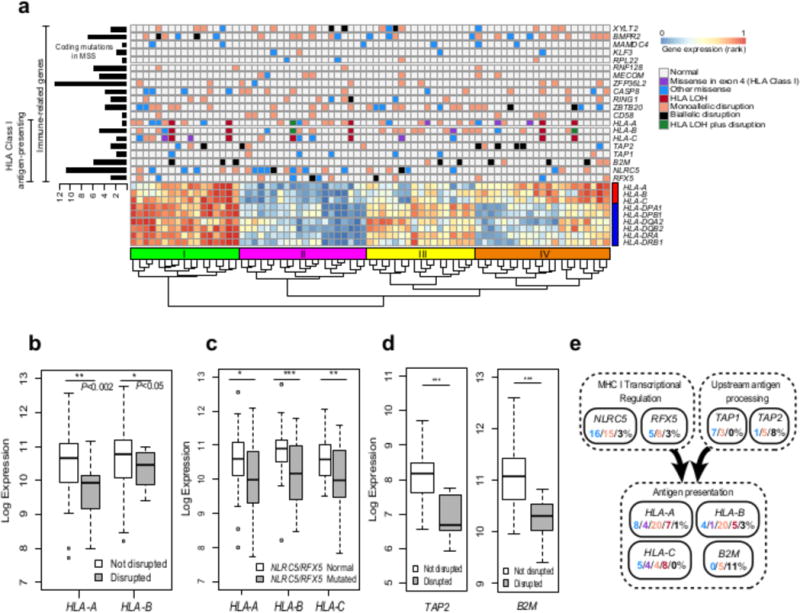
a, Mutation landscape of 9 genes frequently mutated in MSI-high and involved in antigen presentation, as well as 11 significantly mutated immune-related genes that regulate other hematopoietic cell types beyond antigen presentation. Gene expression for HLA Class I (red bar) and Class II (blue bar) genes is shown for comparison. Total number of coding mutations in MSS samples for each gene is shown to the left. Samples were clustered by gene expression; the four main clusters are indicated above the dendrogram: Cluster I (green), Cluster II (purple), Cluster III (yellow), and Cluster IV (orange) bars). b, Decreases in HLA-A and HLA-B expression in samples with disruptive mutations in those genes. c, Decreases in HLA Class I gene expression in samples with mutations in either NLRC5 or RFX5. d, Decreases in TAP2 and B2M expression in samples with disruptive mutations in those genes. (* P<0.01, ** P<0.001, *** P<1.0×10−4 for b-d.) e, Mutation counts in pathways affecting antigen presentation broken down by type.
The observation of mutations consistent with cancer immunoediting motivated us to understand the extent of damage to the APM previously reported in MSI-high or more heavily T cell infiltrated tumors (6–8,34). We found that 57% of MSI-high cases had at least one mutation in HLA-A, HLA-B, or HLA-C, with 39% having a mutation in HLA-A, 33% in HLA-B, and 21% in HLA-C using an HLA mutation calling method that accounts for the polymorphic nature of this region (Figure 4a and Supplementary Table 11). We included in Figure 4a gene expression data for each HLA Class I gene, demonstrating that HLA Class I expression levels vary in CRC primary tumors. We observed both truncating mutations that lower expression of HLA-A and HLA-B (Figure 4b) and missense mutations in exon 4, which disrupt the alpha 3 domain that binds to the CD8 co-receptor of T-cells (35), potentially disrupting antigen presentation. Because the HLA Class I genes are adjacent on chromosome 6, a CN-LOH event in this region, referred to as an HLA LOH event, could simultaneously eliminate up to three maternal or paternal adjacent HLA Class I alleles (Supplementary Figure 5), potentially iteratively reducing the cell’s capacity for antigen presentation while expanding the landscape of possible oncogenic mutations available to the tumor (36). Using a similar method to the one used here, HLA LOH events were observed recently in lung cancer metastasis with evidence that these cases can iteratively drive immune escape (24). Here we found six HLA LOH cases out of 75 primary untreated MSI-high tumors; two of these had additional disruptive mutations, potentially further reducing the number of HLA Class I alleles in those cases (Figure 4a).
Next, we considered significantly mutated genes in MSI-high with a role in disrupting HLA Class I presentation. We identified recurrent mutations in the genes NLRC5 and RFX5, which cooperatively regulate the transcription of HLA Class I genes (37–40). NLRC5 was mutated in 33% of MSI-high samples and RFX5 was mutated in 16% of TCGA MSI-high samples (Figure 4a). Mutations to either of these genes were further found to be associated with decreased expression of HLA-A, HLA-B, and HLA-C (Figure 4c), reducing the expression of each gene by approximately 50%. RFX5 was a significantly mutated gene in the TCGA and NHS/HPFS cohorts independently. While NLRC5 has been previously suggested to be a possible target for immune evasion (41), the size of the gene (1,866 AAs) makes it difficult to identify as significantly mutated by MutSigCV. Instead, NLRC5 was identified here because it was significantly enriched for insertions/deletions at homopolymer and dinucleotide sites in MSI-high based on the MMR deficiency significance calculation performed here (P = 1×10−4).
Antigen presentation machinery genes have been shown to be enriched for mutations in MSI-high relative to MSS tumors (6), in particular TAP2, a gene that helps transport peptides to be presented by the APM, and B2M, a necessary component of HLA Class I complexes on the surface; we found 6/75 MSI-high cases (8%) with biallelic disruption of TAP2 (a gene that is not significantly mutated in MSI-high by itself) and 8/75 MSI-high cases (11%) with biallelic disruption of B2M (Figure 4a & d). Six of the MSI-high cases with biallelic disruption of B2M appeared to be clonal based on the tumor purity, the variant allele fraction, and the CNA or CN-LOH data; and all six showed significantly reduced expression of B2M (Supplementary Figures 4a and 6). Biallelic disruption of B2M has been shown to confer resistance to anti-PD1 (PDCD1) therapy in a melanoma patient undergoing therapy (25,42), as well as lung cancer (43) and colorectal cancer (7).
Figure 6. APC biallelic loss a genomic driver of immunosuppression of TILs by WNT-signaling.
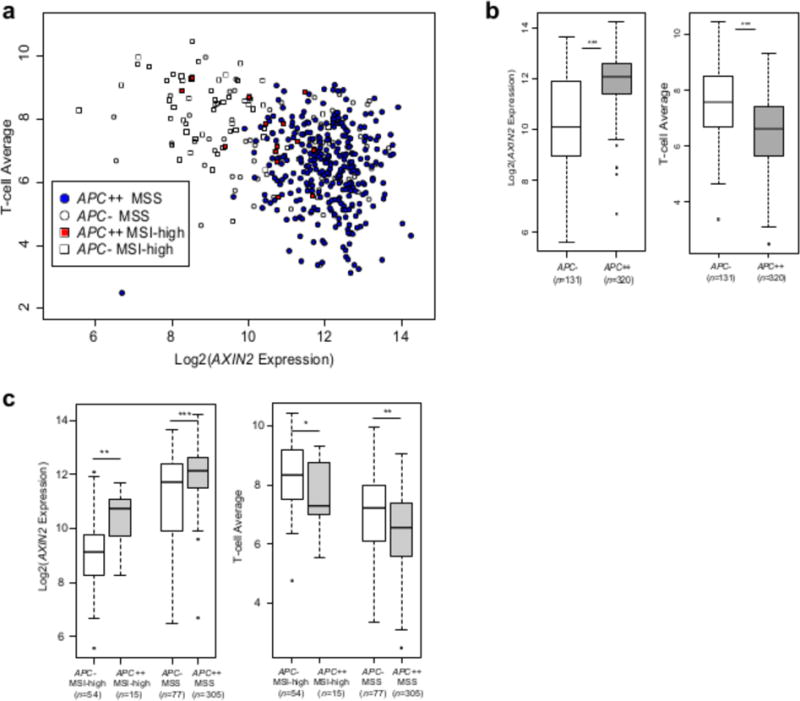
a, Scatterplot showing T-cell average versus AXIN2 gene expression for MSS biallelic APC loss cases (APC++), MSI-high biallelic APC loss cases, and MSS and MSI-high cases without disruptive mutations in APC (APC-). b, Differences in AXIN2 expression and T-cell average for all CRC samples without APC disruptive mutations (APC-) and with biallelic APC disruptive mutations (APC++). c, Differences in AXIN2 expression and T-cell average for samples without APC disruptive mutations and with biallelic APC disruptive mutations separated by MSS and MSI-high status (* P<0.01, ** P<0.001, *** P<1.0×10−4; NS: not significant).
Figure 4a shows that clustering the samples based on the HLA Class I and Class II gene expression yields four clusters: Cluster I has both HLA classes up-regulated, Cluster II has both clusters down-regulated, Cluster III has HLA Class I down-regulated and HLA Class II up-regulated, while Cluster IV has HLA Class I up-regulated and HLA Class II down-regulated, indicating different subtypes within MSI-high based on HLA gene expression. Cluster II is enriched for NLRC5/RFX5 mutated samples (P = 0.01 by a one-sided Fisher’s exact test) consistent with the role of these genes in regulating HLA Class I expression and the role of RFX5 mutations in decreasing HLA Class II expression in MSI-high CRC (44). Clusters I and IV, which have HLA Class I genes up-regulated, are enriched for TAP1, TAP2, and B2M biallelic loss cases (P = 0.02 by a one-sided Fisher’s exact test), indicating that these genes may be selected to be disrupted in cases where HLA Class I gene expression is intact.
Overall, 20/75 (27%) MSI-high primary tumor cases were found to have biallelic disruptions to one of eight genes important for antigen presentation to CD8 T-cells, while another 49/75 (65%) cases where found to have less severe forms of damage to the same gene set (Figure 4e). These mutations are predicted to impact MHC Class I expression, upstream antigen processing, and antigen presentation itself. The biallelic mutations were predominantly mutually exclusive of one another, indicating that these mutations potentially have similar roles in tumor survival. Beyond the biallelic disruption mutations, however, the majority of these mutations likely help tumor survival only marginally, by for example, downregulating MHC Class I epitope presentation by the tumor. In both of these cases, cross-presentation of tumor-derived antigens by dendritic cells, macrophages, and other cell types may remain an avenue for activating an anti-tumor CD8 T-cells response, and characterizing these effects in the context of APM mutations is an important area for future work. Together, these mutations suggest the existence of positive selection for immune escape through the disruption of APM, editing the cancer cell genome likely to avoid the presentation of the most immunogenic neoepitopes resultant from high mutational burden.
Mutation load as a predictor of T-cell infiltration
To assess CD8+ T-cell infiltration levels in the TCGA CRC we applied the expression-based pan T-cell infiltration measurement used on the melanoma TCGA cohort in (45). The T-cell average is the average log-expression of twelve canonical T-cell-associated genes (CCL2, CCL3, CCL4, CXCL9, CXCL10, CD8A, HLA-DOB, HLA-DMB, HLA-DOA, GZMK, ICOS, and IRF1). T-cell average is correlated with tumor infiltrating lymphocyte (TIL) score, a pathology based measure of T-cell infiltration, based on 429 pathology slides available for TCGA samples (see Methods, Supplementary Table 12 and Supplementary Figure 7a). This measurement is highly correlated with CD8A expression by itself (R2 = 0.73) and the “CYT” score (the average of GZMA and PRF1 expression, a measure of immune cytolytic activity based on two genes not present in the T-cell average (46); R2 = 0.67) (Supplementary Figure 7b). We use the measurement with more genes here for robustness and for consistency with recent analyses (45).
This T-cell infiltration measurement was well supported by 1,346 correlated T-cell inflammation signature genes (Supplementary Table 13 and Supplementary Figure 8a). Also, RNAseq-based cell deconvolution of tissue-infiltrating immune and stromal populations using MCP-counter (47) and based on 111 genes, including CD8A and ICOS two genes in the T-cell average measurement, showed that the T-cell average was correlated with all immune cell populations present during immune inflammation, including cytotoxic lymphocytes (R2 = 0.76), monocytic lineage cells such as macrophages (R2 = 0.62), and myeloid dendritic cells (R2 = 0.61) (Supplementary Figure 8b-c and Supplementary Table 14). On the other hand, NK cells were not as correlated with the T-cell average (R2 = 0.21) and a group of samples had outlier high amounts of NK cell expression, one of which was a B2M biallelic loss case (Supplementary Figure 5) possibly attracting NK cells due to the lack of HLA Class I on the surface.
The T-cell average was higher for MSI-high samples, which have on average ~1,000 coding mutations, relative to MSS samples, which have on average ~100 coding mutations. However, the T-cell average was not directly correlated with mutation load (we use mutation load because accurately predicting neoantigens is still an open research problem) within MSS (R2 = 0.001) or MSI-high (R2 = 0.004) (Figure 5b). We sought to identify other explanations for the variability of T-cell average that is not explained by mutation load.
Figure 5. WNT signaling anti-correlated with T-cell infiltration.
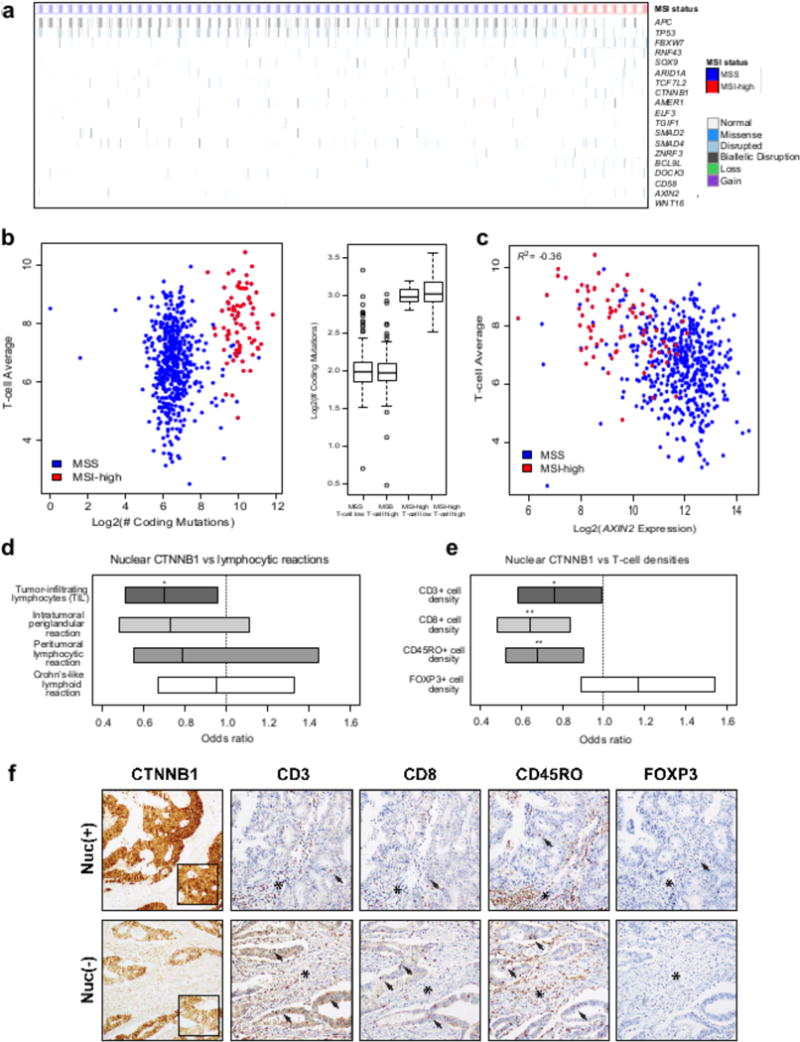
a, Mutation landscape of significantly mutated genes involved in WNT signaling in MSS versus MSI-high tumors. b, Scatterplot showing T-cell average versus the number of coding mutations in MSS and MSI-high samples. Box and whisker plot of the number of coding mutations versus T-cell-inflamed status (“T-cell high” samples are those with T-cell average greater than the median for MSS and MSI-high.) c, Scatterplot showing the T-cell average versus AXIN2 gene expression in MSS and MSI-high samples. d, Immunohistochemical analysis of T lymphocyte infiltrates according to nuclear CTNNB1 (beta-catenin) status. Box plot showing that nuclear CTNNB1 is anti-correlated with tumor-infiltrating lymphocytes (P=0.027). Boxes are 95% confidence intervals; lines are estimated odds ratios. Crohn’s-like reaction was defined as transmural lymphoid reaction. Peritumoral lymphocytic reaction was defined as discrete lymphoid reactions surrounding tumor. Intratumoral periglandular reaction was defined as lymphocytic reaction in tumor stroma within tumor mass. TIL was defined as lymphocytes on top of cancer cells. e, Box plot showing that nuclear CTNNB1 is anti-correlated with CD3+ density (P=0.054), CD8+ density (P=0.0019), CD45RO+ density (P=0.0080), but not FOXP3+ density (P>0.1). (* P<0.1, ** P<0.01 for d-e.) f, The top row depicts a tumor with positive nuclear staining for CTNNB1 (left panel, highlighted by inset) that harbors a low level of CD3, CD8, and CD45RO-positive tumor infiltrating lymphocytes (arrows) compared to the tumor in the bottom row that shows a membranous-only expression pattern for CTNNB1 and harbors higher levels of CD3, CD8, and CD45RO-positive tumor infiltrating lymphocytes. In contrast, levels of FOXP3-positive T lymphocytes are lower than the other T cell subsets and do not show a significantly different density based on nuclear CTNNB1 status. Note that the degree of periglandular lymphocytic response (marked by asterisks) within the stroma is not significantly impacted by nuclear CTNNB1 status.
WNT signaling is inversely correlated with T-cell infiltration in the TCGA CRC cohort
Recently, it has been demonstrated that high WNT signaling excludes T cell infiltration in melanoma primary lesions (45), and that T cells failed to control a melanoma tumor with over expression of CTNNB1 (48). Further evidence has demonstrated a role of WNT signaling in shaping immunoresponse in ovarian (49) and prostate cancer (50). We identified 19 significantly mutated genes using MutsigCV (20) in overall CRC with a role in WNT signaling (Figures 2b and 5a). APC, TP53, RNF43, FBXW7, SOX9, ARID1A, and TCF7L2 are significantly mutated genes in all CRC tumors. On the other hand, AMER1, TGIF1 (51), ELF3 (52), SMAD4, and SMAD2 are significantly mutated in MSS tumors specifically; and ZNRF3 (53), BCL9L, DOCK3 (54), CD58 (55), AXIN2, and WNT16 are significantly mutated in MSI-high tumors. CTNNB1 is recurrently mutated with activating driver mutations (see Methods) in 16 tumors. Using MutsigCV, we also identified KEGG_WNT_SIGNALING_PATHWAY as a significantly mutated pathway in overall CRC (Q < 1.0×10−15) and MSS samples (Q < 3.4×10−14). In MSI-high the WNT signaling pathway was significantly mutated based on the microsatellite indel significance calculation using the BioCarta (Q < 1.0×10−15) and REACTOME (Q < 9.8×10−4) gene sets, but not by MutsigCV (Supplementary Table 15).
To investigate whether, like in primary melanomas (56), WNT signaling is inversely correlated with TILs in CRC, we tested the correlation between WNT target gene expression and the T-cell average. While the role of AXIN2 in cancer is still being understood, here we are using it as a biomarker of WNT signaling expression, since it is a WNT target gene whose gene expression level has been shown to be up-regulated in tumors with high nuclear CTNNB1 protein levels (57). In the TCGA cohort, AXIN2 gene expression was significantly inversely correlated with the T-cell average (R = −0.36, Q = 4.8×10−18; rank = 1,754, Figure 5c). Supplementary Figure 9 shows a heatmap of the T-cell average and the expression level of eleven other WNT target genes (58–63) that were significantly inversely correlated with the T-cell average: RNF43 (Q = 8.1×10−18) and ZNRF3 (Q = 1.5×10−16) (53,64), NKD1 (Q = 1.6×10−11), TCF7 (Q = 4.0×10−15), APCDD1 (Q = 9.3×10−3), ASCL2 (Q = 1.7×10−9), NOTUM (Q = 2.9×10−8), EPHB2 (Q = 2.7×10−14), ETS2 (Q = 2.5×10−7), MYB (Q = 7.4×10−6), and MYC (Q = 0.57). These findings support our conclusion that WNT signaling, as shown using the expression levels of 12 WNT target genes in CRC, is anti-correlated with T-cell infiltration.
Nuclear CTNNB1 (beta-catenin) expression is inversely associated with pathologically-determined immune infiltration in CRC
Given our findings of the inverse association of WNT-signaling with activated T-cell transcriptional signatures, we sought to confirm this result using well-validated immunohistochemical markers of protein expression and immune infiltration. Specifically, we tested 1,150 CRC formalin-fixed paraffin-embedded cases, procured from the NHS/HPFS cohorts, for nuclear CTNNB1 as a marker of activation of the WNT-signaling pathway (65). To reduce bias resulting from spatial heterogeneity, we assessed HE whole tissue sections and four TMAs per tumor. Next we assessed spatially and functionally distinct immune infiltrates in CRC through histopathologic examination of four lymphocytic reaction patterns from: tumor infiltrating lymphocytes, intratumoral periglandular reaction, peritumoral lymphocytic reaction, and Crohn’s-like lymphoid reaction (66) (see Methods), as well as four T-cell functional subsets (CD3+ cells, CD8+ cells, CD45RO+ (PTPRC protein isoform) cells, and FOXP3+ cells), using tissue microarray and immunohistochemistry and image analysis (67).
Consistent with our finding of inverse association between WNT signaling and T-cell average, nuclear CTNNB1 protein expression was inversely associated with TILs (P = 0.027) (Figure 5d and Supplementary Table 16). Intratumoral, peritumoral and Crohn’s-like lymphoid reactions were not significantly associated with nuclear CTNNB1 expression (P > 0.14). As previously shown (66), TILs were more frequently occurring in MSI-high tumors. However, the inverse association between TILs and nuclear CTNNB1 expression was apparent in both MSI-high and MSS cancers (Supplementary Figure 10a and Supplementary Table 17). We further examined the correlation between nuclear CTNNB1 and CD3+ cells (pan T-cells), CD8+ cells (cytotoxic T-cells), CD45RO+ cells (antigen-experienced memory T-cells), and FOXP3+ cells (regulatory T-cells, which suppress cytotoxic T-cells). We observed a significant inverse association between nuclear CTNNB1 expression with CD8+ cells (P = 0.002), CD45RO+ cells (P = 0.008), and CD3+ cells (P = 0.054), but not with FOXP3+ cells (P > 0.1) (Figure 5e and Supplementary Table 18). Figure 5f shows an example of a CRC with nuclear CTNNB1 high and a low level of TILs and another CRC lacking nuclear CTNNB1 expression (cytoplasmic-only) but exhibiting a higher level of TILs. This finding was consistent across both MSI-high and MSS cancers (Supplementary Figure 10b and Supplementary Table 19) and provides independent evidence for attenuation of the anti-tumor immune response in tumors with active WNT signaling.
APC biallelic mutations associate with increased WNT signaling and decreased TILs in MSS and MSI-high
Using the TCGA CRC cohort, we sought to identify specific genomic drivers of increased WNT signaling and decreased T-cell average. We first considered biallelic disruptive mutations in APC, since APC loss results in increased nuclear CTNNB1 thereby driving expression of WNT target genes (68). In samples with biallelic disruptive mutations in APC, AXIN2 expression was significantly increased (P < 2.2×10−16) and T-cell average was significantly decreased (P = 4.0×10−12) relative to samples with no disruptive mutations in APC (Figure 6a-b). This was evident for MSI-high and MSS taken separately (Figure 6c), so that 71% of all CRC cases having APC biallelic loss have lower T-cell infiltration based on the T-cell average measurement.
Super-enhancer hypomethylation of AXIN2 is associated with decreased TILs independent of APC alterations in MSS tumors
Recent work showed that key WNT signaling genes, including AXIN2, are controlled by super-enhancers, which are continuous genomic regions that drive up gene expression when hypomethylated in cancer (69). AXIN2 is over-expressed in tumors relative to normal tissue as a result of hypomethylation of a super-enhancer region located within AXIN2 (chr17:63540803-63558867). A methylation driver of T-cell infiltration is of interest because it has the potential to drive changes in T-cell infiltration much faster than somatic mutations. To assess whether the methylation status of this super-enhancer might be potentially driving T-cell infiltration, we first showed that in the TCGA multi-omic cohort, which has methylation data, AXIN2 expression was inversely correlated with the methylation status of a set of CpG islands near the AXIN2 super-enhancer (Figure 7a). While this region was uniformly hypermethylated in MSI-high and normal samples, in MSS samples, the methylation state of this super-enhancer region is bimodally distributed (Figure 7b; Supplementary Figure 11a-b). AXIN2 was significantly increased (P < 2×10−16) and T-cell average was significantly decreased (P = 1×10−4) in MSS tumors with hypomethylation of the AXIN2 super-enhancer region, even though these samples do not differ significantly in mutation load (P = 0.4) (Figure 7c).
Figure 7. AXIN2 super-enhancer hypomethylation a cis-driver of immunosuppression of TILs by WNT-signaling.
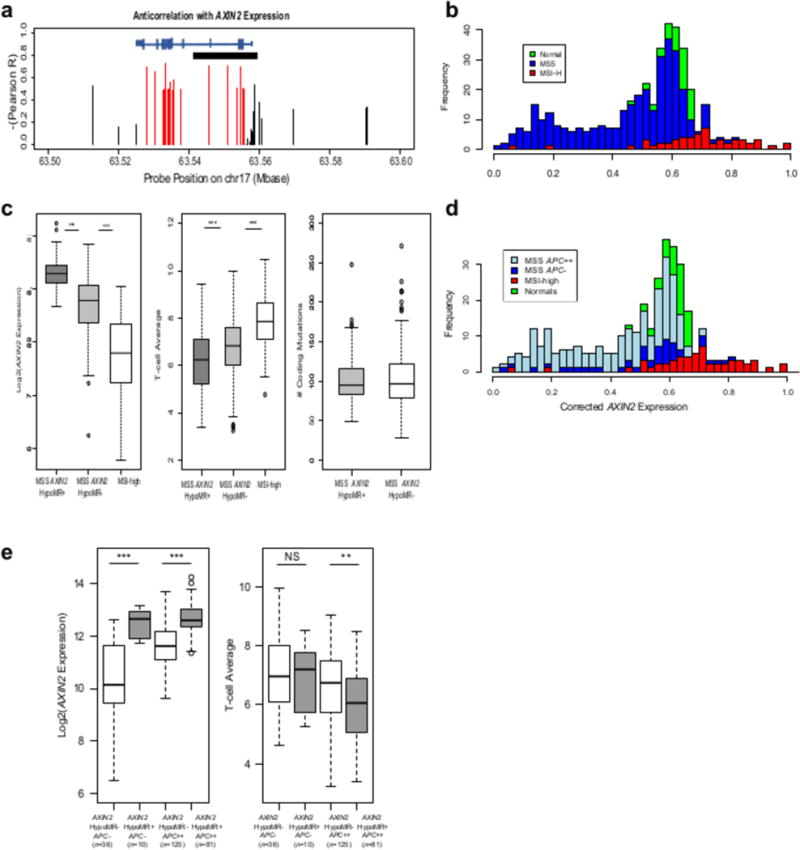
a, Gene structure for AXIN2 showing the location of the super-enhancer (black horizontal bar) and the level of anti-correlation of the CpG sites in this region with the T-cell average. Red CpG sites are averaged to yield a beta-value for the AXIN2 hypomethylated region (HMR); black are not included in the average. b, Histogram showing the AXIN2 HMR beta-value for matched normal (green, N = 35), MSS (blue, N = 322) and MSI-high (red, N = 55) samples. In MSS, 112 samples are hypomethylated and 210 are hypermethylated based on an average beta of 0.4 after adjusting for tumor purity. c, Differences in AXIN2 expression and T-cell average for hypomethylated (HypoMR+) AXIN2 MSS cases, hypermethylated (HypoMR-) AXIN2 MSS cases, and MSI-high cases. Mutation load for hypomethylated and hypermethylated AXIN2 MSS cases. d, Histogram showing the AXIN2 HMR beta-value for matched normal (green, N = 35), MSI-high (red, N = 55), and MSS restricted to APC biallelic loss cases (light blue, N = 206) and cases without APC disruptions (dark blue, N = 46). e, Differences in AXIN2 expression and T-cell average for MSS broken down by AXIN2 hypomethylation status and APC biallelic mutation status.
The methylation state of this super-enhancer varied within APC biallelic loss cases and cases without APC disruptions (Figure 7d), indicating the possibility of it being a driver independent of APC mutation status. Figure 7e shows that AXIN2 super-enhancer hypomethylation significantly increased AXIN2 expression within the set of cases without APC disruptions and also within the set of cases with APC biallelic losses. However, T-cell average was only significantly decreased within the APC cases with biallelic losses. Together, these observations indicate that there may be multiple pathways to immune resistance in CRC by way of altering WNT signaling.
DISCUSSION
At a time when cancer immunotherapy is becoming a mainstream treatment for patients with several cancer types, it is important to understand how cancer cells interact with the immune system and avoid being eliminated by it. As a cancer evolves from its normal cell of origin with progressively accumulated mutations, it needs to evade immune recognition and prevent tumor antigen-specific T-cells from infiltrating and attacking it. A main mechanism by which cancers avoid immune attack is by triggering immune checkpoints that are inhibitory pathways crucial for limiting self-attack and minimizing collateral damage in normal tissues (70). Antibody therapy can be used to block the CTLA4 or PD-1 (PDCD1) checkpoints, releasing the T-cells to attack the tumor (71,72). These T-cells likely recognize neoepitopes created by non-synonymous somatic mutations (73), and result in tumors with higher mutational loads being more likely to respond to immunotherapy (74,75), regardless of tumor type or site of origin (7,8). Among CRCs, MMR-D tumors are the major molecular subtype with a high mutational load, and these are the patients that preferentially respond to anti-PD-1 therapy (7,8). However, not all patients with CRC with high mutational load respond to immune checkpoint inhibitor therapy, suggesting that the cancer cells may have evolved alternate mechanisms to evade immune recognition prior to treatment. Others respond initially and then develop resistance.
CRC provides a clear opportunity to identify immune resistance mechanisms present in untreated samples, since MSI-high is a subtype that does respond to immune blockade therapy and MSS does not. As a result of the large number of MSI-high samples in this cohort and the use of analysis techniques that account for the high MMR-D mutation rate, we were able to identify 11 significantly mutated immune-related genes with roles in modulating diverse hematopoietic cell types and effects beyond antigen presentation, suggesting potential novel therapeutic targets. Additional genes with a role in the immune system were found to be frequently mutated by these techniques. Of particular relevance to this study, NLRC5 is a large gene that was only identified in the MMR-D significance calculation. It regulates HLA Class I gene expression (40) and has been shown to activate WNT signaling in hepatocellular carcinoma (76), suggesting that master regulators connecting tumor biology and immune response can still be discovered.
In our recent study (6), we observed an increased number of mutations in HLA Class I and APM genes in tumors with higher numbers of TILs. Here, we show enrichment of APM and other immune-related genes in MSI-high versus MSS tumors. These findings potentially explain why not all CRCs respond to immune checkpoint blockade therapy or go on to progress later. Similarly, HLA LOH has been recently reported in 40% of mostly smoking-induced non-small cell lung cancers (NSCLCs) with a high mutational load, being associated with a high subclonal neoantigen burden and increased T cell infiltration. The frequency of HLA LOH was enriched in metastatic sites (24). Taken together with the data here on MSI-high CRC, this further supports that highly mutated cancers develop escape mechanism upon strong immune selection pressures.
Another opportunity presented by large-scale cancer genomics studies like this one is the ability to assess the frequency of established drivers of resistance to immunotherapy in the untreated CRC population. Our study showed that 11% of untreated MSI-high patients with CRC harbor biallelic loss of B2M, which is the subunit of MHC required for the stable surface expression of all HLA Class I molecules. Loss of B2M, long known to be an escape mechanism to cancer immunotherapy strategies (77–79), has been recently shown to confer resistance to anti-PD1 antibody therapy (25,42)(43).
In addition, our study showed that the majority of MSI-high cases had at least one mutation likely to play a role in decreasing antigen presentation. While in most cases these initial mutations are not predicted to be sufficient to confer resistance to immune checkpoint blockade therapy, they indicate that immune editing is happening prior to treatment and that patient tumors are on a resistance continuum that needs to be monitored in advance of treatment. For example, while a B2M mutation may be sufficient to cause resistance due to a loss of all HLA Class I antigen presenting alleles, a patient whose cancer has four HLA Class I parental alleles disrupted may be farther along the path to resistance than a patient with all HLA Class I parental alleles intact. In fact, we observed here that 55% of cases (41 of 75) had multiple alterations in the HLA Class I antigen presenting pathway. This does not suggest that these cases will not respond to immune blockade, when a large percentage do. It means that these cancers are hypermutating and evolving a means of immune escape even before treatment and that we need to consider other approaches. For example, HLA Class II antigen presenting genes were expressed and not significantly mutated in MSI-high tumors, providing evidence that there is a population for which HLA Class II-based therapies, currently being pursued (80), might prove effective.
Many CRCs, particularly MSS tumors, are non-T-cell-infiltrated (274 of 490 MSS cases). A study in melanomas (45) has shown that WNT signaling up-regulation may suppress immune infiltration. Because mutations in WNT signaling pathway genes, including APC and CTNNB1, are commonly observed in CRC, we sought to assess whether there was an anti-correlation between the activation of WNT signaling and T-cell infiltration. Both by transcriptome analyses in the TCGA set and by immunohistochemical evaluation of T-cell infiltration and CTNNB1 nuclear localization in independent tumor samples, it was evident that CRCs with high-level WNT signaling exhibit low levels of T-cell infiltration independent of mutation load. In addition, our data shows that biallelic loss of APC (a gene which up-regulates WNT signaling), occurring in 62% of MSS cases and 20% of MSI-high cases, is associated with down-regulation of T-cell infiltration.
In summary, our findings suggest that MSI-high tumors display strong evidence of cancer hypermutation of immune genes to evolve a means of escape even before immune blockade treatment allowing these highly mutated tumors to grow unimpeded by immune recognition. On the other hand, MSS tumors which harbor a lower mutational load exhibit less cancer immunoediting while exhibiting higher WNT pathway signaling and decreased T-cell infiltration.
METHODS
TCGA data access
The results published here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. All the raw sequence data for TCGA exomes and transcriptomes, as well as the SNP6 array data, are available through dbGAP access. The raw exome data for this study are available through The Cancer Genome Atlas using the barcodes provided in Supplementary Table 1. Access to methylation array data is available through the TCGA GDC at https://portal.gdc.cancer.gov.
Data harmonization yields a 592-sample TCGA CRC cohort
The original TCGA CRC publication provided an analysis of 276 colorectal samples (4). In the current study, we added the 242 prior samples that had complete multi-omic data to 350 unpublished TCGA CRC samples to produce a cohort of 592 TCGA CRC cases with exome sequencing, DNA copy number, methylation, and RNA sequencing for multi-omic analyses (4). As a first step, we assessed tumor purity with InfiniumPurify (81) using methylation array data. We chose this method because the other popular method, ABSOLUTE (82), relies on copy number alterations, and MSI-high cases (N = 75) have few to no copy number alterations. To minimize tumor heterogeneity, TCGA assessed the tumor mass area. Next, we called somatic mutations from all 592 whole exome sequencing data ensuring consistency with prior calls using a previously published and validated method (83) that has been used in several settings (84–86) (Supplementary Table 2). We benchmarked our variant calling against Mutect2 on the 528 TCGA samples from our cohort to which the GDC has applied their Mutect2 pipeline (GDC results available at https://portal.gdc.cancer.gov). Our overall coverage of Mutect2 results was high, with 95% of that method’s reported coding mutations either called by our method or specifically flagged by one of our filters (see Supplementary Table 20 for sample-by-sample details). Finally, we provided additional evidence for the somatic mutations by providing variant allele fractions for the matched tumor transcriptome samples, in addition to the exomes. This data shows that missense mutations are typically expressed, while disruptive mutations result in reduced expression of the variant due to nonsense mediated decay, consistent with (87) (Supplementary Figure 12 and Supplementary Table 2).
Calling indels continues to present challenges due to insufficient coverage, alignment quality, the presence of repeats, short interspersed elements and homopolymer/dimers, microsatellite instability, that result in false negatives (88). Prior calling methods filtered out indels at homopolymer runs likely in order to decrease false positives due to polymerase slip errors during massively parallel sequencing (64). We did not exclude such indels and instead aggregated the indels with respect to the microsatellite runs for downstream analyses, including but not limited to annotating biallelic losses and identifying significantly mutated genes based on MMR-D at the level of microsatellite sites (see below).
We also generated copy number alteration (CNA) calls using the TCGA SNP array data using a method previously published and validated (83–86). CN-LOH events were called as described in Methods. We call CNA and copy neutral loss of heterozygosity (CN-LOH) events conservatively by not adjusting for tumor purity, instead setting uniform tighter cut-offs that decrease the likelihood of false positives. Supplementary Tables 21 and 22 detail all the CNAs and CN-LOH events we generated for all 592 TCGA samples with information on the amplitude of the gain or loss and the genes affected where possible. To call HLA Class I typing, we used POLYSOLVER (35) when applicable to the available exome data type and seq2HLA (89). Once the HLA Class I type was identified, we remapped the data and called point mutations, CNA events, and CN-LOH for the HLA region using the approaches cited above (Supplementary Table 11). Figure 2a shows the analyses for which we use this data and Supplementary Table 4 indicates how we integrated cohorts. We assessed our overall accuracy by checking whether the data recapitulates known subtype relationships and frequencies (Figure 1).
The Cancer Genome Atlas sample collection
The Cancer Genome Atlas cohort was collected and molecularly characterized by the U.S. National Cancer Institute as part of a multi-institute effort. The CRC cohort includes 592 primary colorectal adenocarcinomas, 48 normal adjacent tissues used for gene expression and methylation controls, and 592 germline DNA used for somatic mutation, CNA and CN-LOH calling. All patients were untreated by chemo- or radiation therapy and provided informed consent; tissue collection was approved by the Institutional Review Board.
MSI-high status calling
We compared the mSINGS calls and the clinical grade calls on the previously annotated and published 276 samples using the default settings and 0.27 cutoff for the score. This gave us agreement with the MSI-PCR results, where they existed, for all but three samples. For the samples with disagreements, we checked the other indicators, which consistently pointed toward the clinical classifications being correct: case TCGA-AA-3715 (NEG by mSINGS; but called MSI-H by clinical grade testing, with MLH1 epigenetic silencing, high mutation rate, and MMR mutations), case TCGA-AA-A02R (NEG by mSINGS; but called MSI-H with clinical grade testing, with MLH1 epigenetic silencing, high mutation rate, and MMR mutations), and case TCGA-AA-A00L (POS by mSINGS; but MSS by clinical grade testing, high MLH1 expression, low mutation rate, no MMR mutations). We ran mSINGS on the remaining 316 TCGA samples using 0.27 as a cutoff for the score, verifying the resulting classification with MLH1 epigenetic status, MMR-D mutation status, and mutation load for missense mutations and indels. As an independent verification, we were able to identify confirming MMR gene disruptions in all but 11 cases in the samples classified as MSI-high and none of the MSS cases had MMR gene disruptions.
Somatic mutation calling
We called somatic mutations for the whole TCGA cohort from the alignments available from The Cancer Genome Atlas (4). We used the published and validated methodology in (83) checking to ensure consistency with the TCGA somatic mutation calls available in (4). Mapped tumor and matched normal whole-exome sequencing data was obtained from TCGA, with reads mapped to the human genome (either hg18 or hg19). Functional annotation was performed relative to the full set of Refseq transcripts, as obtained from the UCSC Genome Browser database on 14 Mar 2016. During the process of calling somatic mutations we identified 38 samples that were excluded to yield the final set of 592 samples, because they did not pass QC measures due to missing files, low coverage, or contamination; details are provided in Supplementary Table 23. The final set of barcodes for accessing the raw sequence data through TCGA are in Supplementary Table 1.
HLA typing and somatic mutation calling of HLA Class I genes
HLA allele types were first determined for the HLA Class I genes for each sample, using either POLYSOLVER (35) or seq2HLA (89). For those samples already analyzed in (35), we used the HLA types provided by (S.S.) and (C.W.); for the remaining samples, we applied seq2HLA to RNAseq data when correctly matepair-labeled paired-end RNAseq data was available, and applied POLYSOLVER to whole-exome data in all other cases. The TCGA CRC cohort had to be analyzed using both approaches because some of the data was generated using the SOLiD sequencing platform, which cannot be run using POLYSOLVER. We then remapped all reads that were originally mapped to the HLA region of chromosome 6 to the consensus sequence for the HLA alleles determined by HLA typing (consensus sequences were obtained from the IMGT/HLA Database; see http://www.hla-alleles.org), using bowtie2 (90) to perform the realignment. Somatic variants, CNA events, and CN-LOH events for HLA-A, HLA-B, and HLA-C were then called based on the realigned reads using the method described above.
Copy number alteration (CNA) assessment
We evaluated the presence or absence of CNAs on the basis of SNP6 arrays, when available, or whole-exome sequencing data using the published and validated method in (83). To assess if the segment is gained or lost, we calculate the copy-number ratio for the segment by taking the average of the log2 copy-number ratio of the individual SNP6 probes (resp., exon depths) for that segment. Segments for which the copy-number ratio increased (decreased) by at least 30% are called single-copy gains (losses); similarly, increases (decreases) of 60% defined two-copy gains (losses). We did not account for tumor purity directly, instead we set cut-offs sufficiently high to ensure that gains and losses were present. This approach increases the number of false negatives relative to methods that account for tumor purity. However, copy number alterations that are identified are more likely to be real. Two-copy gains and losses were considered to be focal when they included ten or fewer genes. This method was applied to HLA Class I genes after typing and remapping data to the correct allele(s).
Copy-neutral LOH assessment
To identify CN-LOH events, we first restricted attention to SNPs in the SNP6 panel that appeared heterozygous in the matched normal sample (B-allele frequency at least 25%). When SNP6 data was not available, we instead used probable coding SNPs identified in exome sequencing data using the method described above for calling variants and identifying SNPs variants that had a B-allele frequency at least 25% in the tumor and the normal. For each such SNP, we calculated its expected B-allele frequency in the tumor sample based its observed copy number and assuming a mono-allelic gain or loss. We call the difference between this quantity and the actual B-allele frequency the “B-allele deficit.” The B-allele deficit, defined in this way, should be positive for actual instances of CN-LOH, zero for mono-allelic gains or losses, and negative for gains involving both alleles. We segmented the B-allele deficits for each of the samples using the Circular Binary Segmentation Algorithm (91). This resulted in a set of segments for each sample, each corresponding to a piece of DNA with consistent B-allele deficit; we averaged the log2 copy number ratio and the B-allele deficit over each such segment. Segments that were copy neutral (i.e., less than 30% gain or loss) and had a mean B-allele deficit of at least 0.15 were called as CN-LOH. Once a segment is assessed as having CN-LOH, then all the genes on that segment are considered to be CN-LOH. This method was applied to HLA Class I genes after typing and remapping data to the correct allele(s).
Subtyping using somatic mutation data
To identify a sample as positive for a particular genomic subtype we used somatic mutation data. For TP53, APC, PTEN, RNF43, ZNRF3, AXIN2, AXIN1, ATM, MSH6, TGFBR2, SMAD4, MLH1, MSH2, MSH6, PMS2, MLH3, MSH3, any disruptive mutation was used as a positive identification. For KRAS, any mutations at codons 12, 13, 59, 61, 117, 146 were used. For BRAF, any mutations at codons 600, 601, 466, 469 were used. For PIK3CA, any mutations at codons 88, 110, 111, 345, 542, 545, 546, 1043, 1046, 1047 were used. For CTNNB1, any mutations at codons 31-35, 37, 40, 41, 45, 383, 387 were used.
Identification of MLH1 epigenetically silenced cases
We assessed the MLH1 DNA methylation status of each sample following the method used by (4), which relied on the specific probe (cg00893636) in the promoter of MLH1.
Tumor purity assessment using methylation data
To assess tumor purity, we used DNA methylation levels from a specific set of CpG probes that are differentially methylated between cancer and normal cells, as described in (81) and published as InfiniumPurify. We used this approach because copy number based approaches, like ABSOLUTE (82), are not effective on samples with few copy number alterations, like MSI-high cases. The published methodology contains a list of ~1000 differentially methylated probes (DMPs) on the Illumina 450k platform, each specific to a different cancer type, including colorectal cancer specific DMPs that we used for this analysis. In order to apply InfiniumPurify to the maximum number of TCGA samples, including the cases with 27k methylation arrays, we restricted the colorectal cancer DMP list to the 46 probes shared between the 27k and 450k methylation microarrays and confirmed, using samples where both 27k and 450k data was available, that this restriction had a minimal effect on the estimated tumor purity relative to running the full set of colorectal cancer DMPs. We then used this restricted set of DMPs for the cases with only 27k methylation arrays and the full set of DMPs on the cases with 450k methylation arrays. Other than this modification, we calculated tumor purities using the published method, making a final linear adjustment suggested by (81) to improve agreement between InfiniumPurify and ABSOLUTE.
Significantly mutated genes and pathways using MutsigCV
We ran MutsigCV on the set of coding variants using the MutsigCV package as described in (20) with minor adjustments. We used the co-variates file provided by the Broad that accounts for the effect that DNA replication time, chromatin state (open/closed), and general level of transcription activity on the background mutation rate for each gene. To generate an accurate coverage file we used the exome kits, Roche v2 and VCRome 2.1, used on the samples run on Solid and Illumina respectively, taking the union of the exons included in each kit to define the gene, and the reading frame for each exon provided by the UCSC Genome Browser for the corresponding Refseq transcript. A gene was included in the list of significantly mutated genes if it was significantly mutated in the TCGA cohort and the combined cohort, the NHS/HPFS cohort and the combined cohort, or the TCGA cohort, the NHS/HPFS and the combined cohort. This was done to ensure that we did not include genes for which increasing the number of samples did not decrease the evidence that the gene is significantly mutated. Also, to avoid artifacts the cohorts were considered independently because the TCGA samples were fresh frozen and the NHS/HPFS cohort were FFPE and each was done on a different exome kit. To identify significantly mutated pathways we did that same analysis after treating each pathway as a union of genes. A few aspects of the MutsigCV that are gene specific will not be run in this mode, including DNA replication time, chromatin state, and transcription activity.
Significantly mutated genes and pathways by microsatellite instability resulting from MMR-D
To assess significantly mutated genes on the basis of MMR-D-induced mutations alone, we developed a modified version of the original Mutsig algorithm (20). We located all homopolymer runs at least 3 bases long (about 4.2 million) and all dinucleotide repeats at least 4 bases long (about 2.5 million) within the coding exons of any Refseq genes. These constitute our potential MMR-D mutation sites of interest. Because the base MMR-D mutation rate differs significantly depending on the run length and content, we partitioned the sites into 48 different contexts (length 3, 4, 5, 6, 7, 8, 9, or >= 10, for homopolymer base A/T or C/G or dinucleotide pair AT, AC/GT, AG/CT, or CG), and determined the background mutation rate (BMR) for each context as the number of run-lengthening or run-shortening indels observed in that context divided by the number of candidate sites within a particular set of samples, in this case all MSI-high cases in TCGA and NHS/HPFS.
If each site were mutated independently at random with the appropriate BMR, then the number of MMR-D indels within gene G would be Poisson-distributed with expected value EG = sum_c BMR_c * sites_c(G), where the sum is over contexts. We assign each gene a Z-score on the basis of its actual observed number of run-lengthening or run-shortening somatic indels, NG, as compared to the expected number: ZG = (NG – EG) / sqrt(EG). To determine the unlikeliness of a given Z-score, we repeatedly shuffle the observed MMR-D indels at random through the available sites, preserving their contexts, and recompute Z-scores for all genes; doing this 1000 times gives an empirical Z-score distribution with > 107 samples, and we assign p-values with respect to this empirical distribution. In other words, the p-value for gene G is the probability that an arbitrary gene would achieve a Z-score as high as ZG if the indels in each context were randomly distributed among the available sites. To identify significantly mutated pathways we did that same analysis after treating each pathway as a union of genes. A similar method for using microsatellites to identify drivers was published recently (23).
T-cell inflammation score and identification of T-cell inflammation signature genes
We denoted the average log-expression of twelve canonical T-cell-associated genes (CCL2, CCL3, CCL4, CXCL9, CXCL10, CD8A, HLA-DOB, HLA-DMB, HLA-DOA, GZMK, ICOS, and IRF1) as the “T-cell average” for each sample with expression data (45,92,93). To identify T-cell inflammation signature genes that are correlated with T-cell average we followed the method in (56). We partitioned the samples into eight classes of T-cell inflammation, with divisions at the 12.5th, 25th, 37.5th, etc., percentiles of the T-cell average within the set of MSS and MSI-high samples. Samples with T-cell average below (above) the median are designated as T-cell non-inflamed (inflamed). We restricted attention to protein-coding genes that were expressed with at least 1.0 FPKM in at least 80% of the MSS and MSI-H samples; this constituted around 15000 genes. We fit the log-expression of each gene to its T-cell class with a linear model; we reported those genes that changed expression by at least two-fold between the inflamed and non-inflamed sets and for which the relation had a p-value of < 1×10−4 by ANOVA as T-cell inflamed signature genes (Supplementary Table 13).
Prospective cohort study dataset
We utilized incident CRC cases identified within two prospective cohort studies in the U.S., the Nurses’ Health Study (NHS, which has followed 121,701 women aged 30-55 years at 1976) and the Health Professionals Follow-up Study (HPFS, which has followed 51,529 men aged 40-75 years at 1986). We collected formalin-fixed paraffin-embedded (FFPE) tumor tissue blocks from hospitals throughout the U.S. where patients with CRC had undergone surgical resection (94). Cases with available tissue showed characteristics similar to cases without available tissue (94). Among participants diagnosed as CRC until 2012 in the NHS and HPFS, we analyzed 1,150 cases with available data on nuclear CTNNB1 status and immune response in tumor tissue samples. After whole exome sequencing on DNA from tumor and normal FFPE tissue pairs underwent an assay validation using 185 CRC cases in the NHS and HPFS cohorts (64), we conducted whole exome sequencing in additional cases with available ample tissue materials. We did not have gene expression data for these cases, because they were FFPE.
Pathological evaluation of tumor immunity status
In the NHS and HPFS, a single pathologist (S.O.), blinded to other data, reviewed hematoxylin and eosin-stained tissue sections, confirmed diagnosis of colorectal carcinoma, and recorded pathological features including four components of lymphocytic reaction [tumor-infiltrating lymphocytes (TIL), intratumoral periglandular reaction, peritumoral lymphocytic reaction, and Crohn’s-like lymphoid reaction] as previously described (66). In the TCGA dataset, a single pathologist (K.I.), blinded to other data, reviewed publicly-available images of hematoxylin and eosin-stained tissue sections, and recorded pathological features including TIL. TIL was precisely defined as lymphocytes on top of neoplastic epithelial cells, as has been defined in the literature. Intratumoral periglandular reaction was defined as lymphocytic reaction in tumor stroma within a tumor mass. Peritumoral lymphocytic reaction was defined as discrete lymphoid reaction surrounding a tumor mass. Crohn’s-like lymphoid reaction was defined as transmural lymphoid reaction. Each of these features was graded as negative/low, intermediate, or high (66).
Immunohistochemistry
In the NHS and HPFS, we constructed tissue microarrays to include up to four cores from CRC and up to two cores from normal tissue blocks. We conducted immunohistochemical analysis for CTNNB1 using an anti-CTNNB1 antibody (clone 14, dilution 1:400; BD Transduction Laboratories, NJ, USA), as previously described (95). A single pathologist (T.M.) graded cytoplasmic, nuclear, and membrane CTNNB1 expression status separately as no expression, weak expression, or moderate/strong expression. Cytoplasmic and nuclear CTNNB1 positivity was defined as the presence of moderate/strong expression, and loss of membrane CTNNB1 as no or weak membrane expression. A selected set of tumors (N = 292) was independently examined by a second pathologist (S.O.), and the concordance between the two pathologists was 0.90 for nuclear CTNNB1 (κ=0.80), 0.78 for cytoplasmic CTNNB1 (κ=0.54), and 0.86 for membrane CTNNB1 (κ=0.72), indicating good to substantial agreement (all P < 0.0001) (95). We performed immunohistochemistry for CD3, CD8, CD45RO, and FOXP3 as previously described (67). We used an automated scanning microscope and the Ariol image analysis system (Genetix, CA, USA) to measure densities (cells/mm2) of CD3+ cells, CD8+ cells, CD45RO+ cells, and FOXP3+ cells in CRC tissue.
Analyses of MSI, DNA methylation, and KRAS, BRAF, and PIK3CA mutations in the NHS and HPFS
DNA was extracted from FFPE tissue blocks using QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). MSI status was determined using 10 microsatellite markers (D2S123, D5S346, D17S250, BAT25, BAT26, BAT40, D18S55, D18S56, D18S67, and D18S487), and MSI-high was defined as presence of instability in ≥ 30% of the markers, as previously described (96). U sing bisulfite-treated DNA, methylation status of eight CpG island methylator phenotype (CIMP)-specific promoters (CACNA1G, CDKN2A, CRABP1, IGF2, MLH1, NEUROG1, RUNX3, and SOCS1) and long interspersed nucleotide element-1 (LINE-1) was analyzed (97,98). CIMP-high was defined as methylation in ≥ 6 of eight promoters (97). Polymerase chain reaction and pyrosequencing were performed for KRAS (codons 12, 13, 61, and 146) (99), BRAF (codon 600) (96), and PIK3CA (exons 9 and 20) (94).
Statistical analysis NHS & HPFS IHC cohort
We used logistic regression analyses to assess the association of nuclear CTNNB1 status (positive vs. negative) with immune response to tumor. We assessed four lymphocytic reaction patterns (tumor infiltrating lymphocytes, intratumoral periglandular reaction, peritumoral lymphocytic reaction, and Crohn’s-like lymphoid reaction) and four T cell subsets (CD3+ cells, CD8+ cells, CD45RO+ cells, and FOXP3+ cells) in tumor tissue. Multivariable logistic regression models initially included age (continuous), sex, year of diagnosis (continuous), family history of CRC (present vs. absent), tumor location (proximal colon vs. distal colon vs. rectum), MSI status (high vs. low/MSS), CpG island methylator phenotype-specific promoter (CIMP) status (high vs. low/negative), long interspersed nucleotide element-1 (LINE-1) methylation level (continuous), KRAS (mutant vs. wild-type), BRAF (mutant vs. wild-type), and PIK3CA (mutant vs. wild-type). A backward elimination with a threshold of P = 0.05 was used to select variables for the final model. The cases with missing data (family history of CRC [0.9%], tumor location [0.4%], MSI status [4.0%], CIMP status [7.7%], KRAS [3.7%], BRAF [3.0%], and PIK3CA [10.5%]) were included in the majority category of a given categorical covariate to limit the degrees of freedom of the models. For cases with missing data on LINE-1 methylation level (5.0%), we assigned a separate indicator variable. We confirmed that excluding cases with missing data in any of the variables did not substantially alter our results. We assessed statistical interactions between nuclear CTNNB1 status (positive vs. negative) and microsatellite instability (MSI) status (high vs. low/microsatellite stable [MSS]) in relation to tumor immunity status using the Wald test for the cross-product in logistic regression models. The proportional odds assumption was assessed using the ordinal logistic regression model. We observed evidence for violation of this assumption in two of four lymphocytic reaction patterns (intratumoral periglandular reaction and Crohn’s-like lymphoid reaction) and therefore, used binary histological lymphocytic reaction variables as outcome variables for logistic regression analyses. To compare characteristics between subgroups according to nuclear CTNNB1 status, we used the chi-square test for categorical variables, and the unpaired t-test for continuous variables.
All statistical analyses were performed using SAS software (version 9.4, SAS Institute, NC, USA), and all P values were two-sided.
Supplementary Material
SIGNIFICANCE.
This multi-omic analysis of 1,211 colorectal cancer primary tumors reveals that it should be possible to better monitor resistance in the 15% of cases that respond to immune blockade therapy and also to use WNT signaling inhibitors to reverse immune exclusion in the 85% of cases that currently do not.
Acknowledgments
We would like to thank the participants and staff of the Nurses’ Health Study and the Health Professionals Follow-up Study for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data.
Funding: This work was supported by U.S. National Institutes of Health (NIH) grants U01 CA137088 to U.P.; U54 HG003067 to E.S.L. and S.B.G.; P01 CA87969 to M. J. Stampfer; UM1 CA186107 to M. J. Stampfer; P01 CA55075 to W. C. Willett; UM1 CA167552 to W. C. Willett; P50 CA127003 to C.S.F.; R35 CA197735 to S.O.; R01 CA151993 to S.O.; K07 CA190673 to R.N.; 1R01CA194663-01 W.M.G.; R35 CA197633 and P01 CA168585 to A.R. and C.S.G.; Nodal Award from the Dana-Farber Harvard Cancer Center to S.O.; The Parker Institute for Cancer Immunotherapy, the Ressler Family Fund, the Samuels Family Fund and the Garcia-Corsini Family Fund to A.R. and C.S.G.; and by grants from the Project P Fund, The Friends of the Dana-Farber Cancer Institute, Bennett Family Fund, and the Entertainment Industry Foundation through National Colorectal Cancer Research Alliance. M.G. was supported by a KL2/Catalyst Medical Research Investigator Training award NIH Award KL2 TR001100. M.G. and C.S.F. were supported by the Stand Up to Cancer Colorectal Cancer Dream Team Translational Research Grant (Grant Number SU2C-AACR-DT22-17). Stand Up to Cancer is a program of the Entertainment Industry Foundation and the research grant is administered by the American Association for Cancer Research, a scientific partner of SU2C. S.H.-L. was supported by a Career Development Award from the American Society of Clinical Oncology (ASCO), a Tower Cancer Research Foundation Grant, a Dr. Charles Coltman Fellowship Award from the Hope Foundation, and a UCLA KL2 Award. D.S.S. was supported by the Tumor Immunology 4T32CA009120-40 training grant and 2016 Conquer Cancer Foundation ASCO Young Investigator Award. T.J.H. and S.H.Z. were supported by the Ontario Institute for Cancer Research, through generous support from the Ontario Ministry of Research and Innovation. B.N. was supported by the Biomedical Big Data training grant from the NIH-NLM National Cancer Institute; PHS grant number 5T32LM012424-03. D.J.M. was supported by NIAMS 3P50AR063020-03S1. W.M.G. was supported by R.A.C.E. Charities and the Gensch family, STTR Translational Research grant. This research was supported by funds from the Parker Institute for Cancer Immunotherapy (PICI), grant number 20163828
Footnotes
M. Leiserson is a consultant with Microsoft. C. Wu is a co-founder of Neon Therapeutics, and a member of their scientific advisory board. C. Fuchs is a consultant for Eli Lilly, Entrinsic Health, Genentech, Merck, Sanofi, Five Prime Therapeutics, Merrimack, Bayer, Agios, Taiho, Kew, Bain Capital, and a board member of CytomX. L. Garraway is a Senior Vice President of Global Development and Affairs at Eli Lilly. The other authors declare no potential conflicts of interest.
Supplementary Information is available in the online version of the paper.
References
- 1.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–87. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research N. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters U, Jiao S, Schumacher FR, Hutter CM, Aragaki AK, Baron JA, et al. Identification of Genetic Susceptibility Loci for Colorectal Tumors in a Genome-Wide Meta-analysis. Gastroenterology. 2013;144:799–807. doi: 10.1053/j.gastro.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016;15:857–65. doi: 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity. 2016;44:698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 10.Basile D, Garattini SK, Bonotto M, Ongaro E, Casagrande M, Cattaneo M, et al. Immunotherapy for colorectal cancer: where are we heading? Expert Opin Biol Ther. 2017;17:709–21. doi: 10.1080/14712598.2017.1315405. [DOI] [PubMed] [Google Scholar]
- 11.Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC. Microsatellite instability detection by next generation sequencing. Clin Chem. 2014;60:1192–9. doi: 10.1373/clinchem.2014.223677. [DOI] [PubMed] [Google Scholar]
- 12.Pritchard CC, Morrissey C, Kumar A, Zhang X, Smith C, Coleman I, et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun. 2014;5:4988. doi: 10.1038/ncomms5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nebot-Bral L, Brandao D, Verlingue L, Rouleau E, Caron O, Despras E, et al. Hypermutated tumours in the era of immunotherapy: The paradigm of personalised medicine. Eur J Cancer. 2017;84:290–303. doi: 10.1016/j.ejca.2017.07.026. [DOI] [PubMed] [Google Scholar]
- 14.Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22:271–82. doi: 10.1101/gr.117523.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–6. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer. 2017;17:79–92. doi: 10.1038/nrc.2016.126. [DOI] [PubMed] [Google Scholar]
- 17.Langner C. Serrated and non-serrated precursor lesions of colorectal cancer. Dig Dis. 2015;33:28–37. doi: 10.1159/000366032. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Huang J, Huang F, Jin Q, Zhu H, Wang X, et al. Decreased expression of IDH1-R132H correlates with poor survival in gastrointestinal cancer. Oncotarget. 2016;7:73638–50. doi: 10.18632/oncotarget.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hechtman JF, Sadowska J, Huse JT, Borsu L, Yaeger R, Shia J, et al. AKT1 E17K in Colorectal Carcinoma Is Associated with BRAF V600E but Not MSI-H Status: A Clinicopathologic Comparison to PIK3CA Helical and Kinase Domain Mutants. Mol Cancer Res. 2015;13:1003–8. doi: 10.1158/1541-7786.MCR-15-0062-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–4. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. 2016;22:1342–50. doi: 10.1038/nm.4191. [DOI] [PubMed] [Google Scholar]
- 23.Maruvka YE, Mouw KW, Karlic R, Parasuraman P, Kamburov A, Polak P, et al. Analysis of somatic microsatellite indels identifies driver events in human tumors. Nat Biotechnol. 2017;35:951–9. doi: 10.1038/nbt.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell. 2017;171:1259–71. doi: 10.1016/j.cell.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8:1136. doi: 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin X, Johns SC, Kim D, Mikulski Z, Salanga CL, Handel TM, et al. Lymphatic specific disruption in the fine structure of heparan sulfate inhibits dendritic cell traffic and functional T cell responses in the lymph node. J Immunol. 2014;192:2133–42. doi: 10.4049/jimmunol.1301286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogel KU, Bell LS, Galloway A, Ahlfors H, Turner M. The RNA-Binding Proteins Zfp36l1 and Zfp36l2 Enforce the Thymic beta-Selection Checkpoint by Limiting DNA Damage Response Signaling and Cell Cycle Progression. J Immunol. 2016;197:2673–85. doi: 10.4049/jimmunol.1600854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vu TT, Gatto D, Turner V, Funnell AP, Mak KS, Norton LJ, et al. Impaired B cell development in the absence of Kruppel-like factor 3. J Immunol. 2011;187:5032–42. doi: 10.4049/jimmunol.1101450. [DOI] [PubMed] [Google Scholar]
- 29.Rolle A, Halenius A, Ewen EM, Cerwenka A, Hengel H, Momburg F. CD2-CD58 interactions are pivotal for the activation and function of adaptive natural killer cells in human cytomegalovirus infection. Eur J Immunol. 2016;46:2420–5. doi: 10.1002/eji.201646492. [DOI] [PubMed] [Google Scholar]
- 30.Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, et al. Combined genetic inactivation of beta2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell. 2011;20:728–40. doi: 10.1016/j.ccr.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Philip NH, Dillon CP, Snyder AG, Fitzgerald P, Wynosky-Dolfi MA, Zwack EE, et al. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-kappaB and MAPK signaling. Proc Natl Acad Sci U S A. 2014;111:7385–90. doi: 10.1073/pnas.1403252111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Zhang P, Bao Y, Han Y, Wang Y, Zhang Q, et al. Zinc finger protein ZBTB20 promotes Toll-like receptor-triggered innate immune responses by repressing IkappaBalpha gene transcription. Proc Natl Acad Sci U S A. 2013;110:11097–102. doi: 10.1073/pnas.1301257110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song G, Liu B, Li Z, Wu H, Wang P, Zhao K, et al. E3 ubiquitin ligase RNF128 promotes innate antiviral immunity through K63-linked ubiquitination of TBK1. Nat Immunol. 2016;17:1342–51. doi: 10.1038/ni.3588. [DOI] [PubMed] [Google Scholar]
- 34.Kubo T, Hirohashi Y, Matsuo K, Sonoda T, Sakamoto H, Furumura K, et al. Mismatch Repair Protein Deficiency Is a Risk Factor for Aberrant Expression of HLA Class I Molecules: A Putative “Adaptive Immune Escape” Phenomenon. Anticancer Res. 2017;37:1289–95. doi: 10.21873/anticanres.11446. [DOI] [PubMed] [Google Scholar]
- 35.Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33:1152–8. doi: 10.1038/nbt.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marty R, Kaabinejadian S, Rossell D, Slifker MJ, van de Haar J, Engin HB, et al. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell. 2017;171:1272–83. doi: 10.1016/j.cell.2017.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobayashi KS, van den Elsen PJ. NLRC5: a key regulator of MHC class I-dependent immune responses. Nat Rev Immunol. 2012;12:813–20. doi: 10.1038/nri3339. [DOI] [PubMed] [Google Scholar]
- 38.Meissner TB, Liu YJ, Lee KH, Li A, Biswas A, van Eggermond MC, et al. NLRC5 cooperates with the RFX transcription factor complex to induce MHC class I gene expression. J Immunol. 2012;188:4951–8. doi: 10.4049/jimmunol.1103160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci U S A. 2010;107:13794–9. doi: 10.1073/pnas.1008684107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ludigs K, Seguin-Estevez Q, Lemeille S, Ferrero I, Rota G, Chelbi S, et al. NLRC5 exclusively transactivates MHC class I and related genes through a distinctive SXY module. PLoS Genet. 2015;11:e1005088. doi: 10.1371/journal.pgen.1005088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshihama S, Roszik J, Downs I, Meissner TB, Vijayan S, Chapuy B, et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc Natl Acad Sci U S A. 2016;113:5999–6004. doi: 10.1073/pnas.1602069113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375:819–29. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017;7:1420–35. doi: 10.1158/2159-8290.CD-17-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michel S, Linnebacher M, Alcaniz J, Voss M, Wagner R, Dippold W, et al. Lack of HLA class II antigen expression in microsatellite unstable colorectal carcinomas is caused by mutations in HLA class II regulatory genes. Int J Cancer. 2010;127:889–98. doi: 10.1002/ijc.25106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 46.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31:711–23. doi: 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jimenez-Sanchez A, Memon D, Pourpe S, Veeraraghavan H, Li Y, Vargas HA, et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell. 2017;170:927–38. doi: 10.1016/j.cell.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linch M, Attard G. Prostate cancers that ‘Wnt’ respond to abiraterone. Ann Oncol. 2017;29:290–2. doi: 10.1093/annonc/mdx785. [DOI] [PubMed] [Google Scholar]
- 51.Razzaque MS, Atfi A. TGIF function in oncogenic Wnt signaling. Biochim Biophys Acta. 2016;1865:101–4. doi: 10.1016/j.bbcan.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 52.Wang JL, Chen ZF, Chen HM, Wang MY, Kong X, Wang YC, et al. Elf3 drives beta-catenin transactivation and associates with poor prognosis in colorectal cancer. Cell Death Dis. 2014;5:e1263. doi: 10.1038/cddis.2014.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bond CE, McKeone DM, Kalimutho M, Bettington ML, Pearson SA, Dumenil TD, et al. RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget. 2016;7:70589–600. doi: 10.18632/oncotarget.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caspi E, Rosin-Arbesfeld R. A novel functional screen in human cells identifies MOCA as a negative regulator of Wnt signaling. Mol Biol Cell. 2008;19:4660–74. doi: 10.1091/mbc.E07-10-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu S, Wen Z, Jiang Q, Zhu L, Feng S, Zhao Y, et al. CD58, a novel surface marker, promotes self-renewal of tumor-initiating cells in colorectal cancer. Oncogene. 2015;34:1520–31. doi: 10.1038/onc.2014.95. [DOI] [PubMed] [Google Scholar]
- 56.Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci U S A. 2016;113:E7759–E68. doi: 10.1073/pnas.1609376113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Veloudis G, Pappas A, Gourgiotis S, Falidas E, Dimitriou N, Karavokiros I, et al. Assessing the clinical utility of Wnt pathway markers in colorectal cancer. J BUON. 2017;22:431–6. [PubMed] [Google Scholar]
- 58.Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Goke B, et al. Comprehensive analysis of beta-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/beta-catenin signaling. BMC Genomics. 2014;15:74. doi: 10.1186/1471-2164-15-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hao HX, Jiang X, Cong F. Control of Wnt Receptor Turnover by R-spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer. Cancers (Basel) 2016:8. doi: 10.3390/cancers8060054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Caldwell GM, Jones CE, Soon Y, Warrack R, Morton DG, Matthews GM. Reorganisation of Wnt-response pathways in colorectal tumorigenesis. Br J Cancer. 2008;98:1437–42. doi: 10.1038/sj.bjc.6604327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takahashi M, Fujita M, Furukawa Y, Hamamoto R, Shimokawa T, Miwa N, et al. Isolation of a novel human gene, APCDD1, as a direct target of the beta-Catenin/T-cell factor 4 complex with probable involvement in colorectal carcinogenesis. Cancer Res. 2002;62:5651–6. [PubMed] [Google Scholar]
- 62.Giraldez AJ, Copley RR, Cohen SM. HSPG modification by the secreted enzyme Notum shapes the Wingless morphogen gradient. Dev Cell. 2002;2:667–76. doi: 10.1016/s1534-5807(02)00180-6. [DOI] [PubMed] [Google Scholar]
- 63.Munera J, Cecena G, Jedlicka P, Wankell M, Oshima RG. Ets2 regulates colonic stem cells and sensitivity to tumorigenesis. Stem Cells. 2011;29:430–9. doi: 10.1002/stem.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. 2014;46:1264–6. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCrea PD, Gottardi CJ. Beyond beta-catenin: prospects for a larger catenin network in the nucleus. Nat Rev Mol Cell Biol. 2016;17:55–64. doi: 10.1038/nrm.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ogino S, Nosho K, Irahara N, Meyerhardt JA, Baba Y, Shima K, et al. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin Cancer Res. 2009;15:6412–20. doi: 10.1158/1078-0432.CCR-09-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nosho K, Baba Y, Tanaka N, Shima K, Hayashi M, Meyerhardt JA, et al. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol. 2010;222:350–66. doi: 10.1002/path.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heyn H, Vidal E, Ferreira HJ, Vizoso M, Sayols S, Gomez A, et al. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol. 2016;17:11. doi: 10.1186/s13059-016-0879-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 72.Ribas A. Releasing the Brakes on Cancer Immunotherapy. N Engl J Med. 2015;373:1490–2. doi: 10.1056/NEJMp1510079. [DOI] [PubMed] [Google Scholar]
- 73.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 74.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–9. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peng YY, He YH, Chen C, Xu T, Li L, Ni MM, et al. NLRC5 regulates cell proliferation, migration and invasion in hepatocellular carcinoma by targeting the Wnt/beta-catenin signaling pathway. Cancer Lett. 2016;376:10–21. doi: 10.1016/j.canlet.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 77.Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst. 1996;88:100–8. doi: 10.1093/jnci/88.2.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.D’Urso CM, Wang ZG, Cao Y, Tatake R, Zeff RA, Ferrone S. Lack of HLA class I antigen expression by cultured melanoma cells FO-1 due to a defect in B2m gene expression. J Clin Invest. 1991;87:284–92. doi: 10.1172/JCI114984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sucker A, Zhao F, Real B, Heeke C, Bielefeld N, Mabetaen S, et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin Cancer Res. 2014;20:6593–604. doi: 10.1158/1078-0432.CCR-14-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Desrichard A, Snyder A, Chan TA. Cancer Neoantigens and Applications for Immunotherapy. Clin Cancer Res. 2016;22:807–12. doi: 10.1158/1078-0432.CCR-14-3175. [DOI] [PubMed] [Google Scholar]
- 81.Zhang N, Wu HJ, Zhang W, Wang J, Wu H, Zheng X. Predicting tumor purity from methylation microarray data. Bioinformatics. 2015;31:3401–5. doi: 10.1093/bioinformatics/btv370. [DOI] [PubMed] [Google Scholar]
- 82.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3:111ra21. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. 2015;21:555–9. doi: 10.1038/nm.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fu X, Jeselsohn R, Pereira R, Hollingsworth EF, Creighton CJ, Li F, et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc Natl Acad Sci U S A. 2016;113:E6600–E9. doi: 10.1073/pnas.1612835113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shlien A, Raine K, Fuligni F, Arnold R, Nik-Zainal S, Dronov S, et al. Direct Transcriptional Consequences of Somatic Mutation in Breast Cancer. Cell Rep. 2016;16:2032–46. doi: 10.1016/j.celrep.2016.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jiang Y, Turinsky AL, Brudno M. The missing indels: an estimate of indel variation in a human genome and analysis of factors that impede detection. Nucleic Acids Res. 2015;43:7217–28. doi: 10.1093/nar/gkv677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boegel S, Lower M, Schafer M, Bukur T, de Graaf J, Boisguerin V, et al. HLA typing from RNA-Seq sequence reads. Genome Med. 2012;4(12):102. doi: 10.1186/gm403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–72. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 92.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 93.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;367:1596–606. doi: 10.1056/NEJMoa1207756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kawasaki T, Nosho K, Ohnishi M, Suemoto Y, Kirkner GJ, Dehari R, et al. Correlation of beta-catenin localization with cyclooxygenase-2 expression and CpG island methylator phenotype (CIMP) in colorectal cancer. Neoplasia. 2007;9:569–77. doi: 10.1593/neo.07334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Meyerhardt JA, Loda M, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58:90–6. doi: 10.1136/gut.2008.155473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nosho K, Irahara N, Shima K, Kure S, Kirkner GJ, Schernhammer ES et al. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS One. 2008;3:e3698. doi: 10.1371/journal.pone.0003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Irahara N, Nosho K, Baba Y, Shima K, Lindeman NI, Hazra A, et al. Precision of pyrosequencing assay to measure LINE-1 methylation in colon cancer, normal colonic mucosa, and peripheral blood cells. J Mol Diagn. 2010;12:177–83. doi: 10.2353/jmoldx.2010.090106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Imamura Y, Lochhead P, Yamauchi M, Kuchiba A, Qian ZR, Liao X, et al. Analyses of clinicopathological, molecular, and prognostic associations of KRAS codon 61 and codon 146 mutations in colorectal cancer: cohort study and literature review. Mol Cancer. 2014;13:135. doi: 10.1186/1476-4598-13-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.