

Abstract
The matrix metalloproteinases (MMPs) are a family of proteolytic enzymes that degrade multiple components of the extracellular matrix. A large body of experimental and clinical evidence has implicated MMPs in tumor invasion, neoangiogenesis and metastasis, and therefore they represent ideal pharmacological targets for cancer therapy. From the 1990's to early 2000's synthetic inhibitors of MMPs (MMPIs) were studied in various cancer types. Unexpectedly, despite strongly promising preclinical data, all trials were unsuccessful in reducing tumor burden or improving overall survival; in addition, MMPIs had unforeseen, severe side effects. Two main reasons can explain the failure of MMPIs in clinical trials. It has now become apparent that some MMPs have anti-tumor effects; therefore, the broad-spectrum MMPIs used in the initial trials might block these MMPs and result in tumor progression. In addition, although MMPs are involved in the early stages of tumor progression, MMPIs were tested in patients with advanced disease, beyond the stage when these compounds could be effective. As more specific MMPIs are now available, MMP-targeting could be reconsidered for cancer therapy; however, new trials should be designed to test their anti-metastatic properties in early-stage tumors, and endpoints should focus on parameters other than decreasing metastatic tumor burden.
Keywords: Cancer, Metastasis, Matrix Metalloproteinases, MMPs, MMP Inhibitors
Introduction
Cancer remains a leading cause of mortality worldwide, in some estimates accounting for more deaths than coronary artery disease or stroke. In the US, over 1.5 million new cases were diagnosed in 2016, leading to 595,000 deaths (1). Patients die of metastatic disease; therefore prevention of metastasis and treatment of micrometastatic disease is most important to improve cure rates. The mechanisms by which tumors metastasize are complex and involve numerous interactions between tumor cells and their microenvironment. A malignant cell invades into the surrounding tissue, enters the vasculature, and extravasates at distant sites. Proteolytic enzymes are essential for this process, degrading the extracellular matrix (ECM) and allowing for tumor dissemination (2–4). While hundreds of proteinase genes have been identified, the Matrix Metalloproteinases (MMPs) have been heavily implicated in metastatic spread (5).
The MMPs are a family of 24 endopeptidases that control the physiological turnover of the ECM. High levels of MMP correlate with unfavorable prognosis in multiple cancers (5). Therefore, clinical trials of synthetic MMP inhibitors (MMPIs) were performed during the late 1990s and early 2000s (6–8). However, these studies failed due to lack of efficacy and severe side effects. This review will discuss the preclinical data that indicated the potential efficacy of MMPIs in cancer, the clinical trials and what led to their failure, and offer a perspective on potential trial designs.
History and Biology of Matrix Metalloproteinases
The MMPs mediate the constant remodeling the extracellular matrix. While their substrates include collagens, gelatins, proteoglycans and elastin, they have wide-reaching effects on many other proteins(2). The first vertebrate matrix metalloproteinase described was the collagenase associated with the resorption of the tadpole tail, in 1962 (9). Human collagenase (now known as MMP-1) was identified in the skin 5 years later, and similar enzymes were further characterized across species (10). Initially MMPs were categorized based on their substrate specificity (e.g. collagenase). However, as further MMPs were discovered it became evident that many substrates are degraded by multiple MMPs, and each MMP can degrade multiple substrates; therefore, a sequential numbering system was adopted reflecting the order in which MMPs were discovered. The MMPs have also been classified per their structure and function into eight groups that comprise secreted and membrane-bound MMPs (membrane-type, or MT-MMPs) (11).
As MMPs cleave numerous substrates, their activity heavily impacts the extracellular environment and, left unchecked, their action can be disastrous (12). Their activity is therefore strictly regulated to prevent excessive ECM degradation. MMP synthesis is first controlled at the level of transcription and translation, and post-translational modifications also regulate MMP activity (11). Like all extracellular proteinases, MMPs are secreted as pro-enzymes, or zymogens, rendered inactive by the interaction between the zinc ion in the catalytic domain and a cysteine-sulphydryl group in the N-terminal (pro) domain. Activation requires removal of this interaction, a mechanism termed “cysteine switch” which can occur either after secretion or intracellularly by pro-hormone convertases (furin). Following the cysteine switch, MMPs are only partially activated; complete activation is achieved by a process of autocatalysis, in which the proteinase cleaves its pro domain. The enzyme can further degrade and inactivate itself, a mechanism of regulation in multiple MMPs.
MMP proteolytic activity is further controlled by specific protein inhibitors, the tissue inhibitors of metalloproteinases (TIMPs), comprising a family of four proteins (TIMP-1 to -4) that reversibly bind to the MMP catalytic site in a stoichiometric manner (2,13). MMPs can also be inhibited by nonspecific inhibitors including α2-macroglobulin, thrombospondin-1 and -2(13).
Matrix Metalloproteinases in Malignancy
Role in Metastatic Spread
MMPs exert profound effects on the extracellular microenvironment and are therefore highly regulated in normal physiology. Invasive malignancies can “deregulate” these proteinases to spread beyond their microenvironment in the complex, multistep metastatic process (5). Highly motile, invasive tumor cells egress from the primary tumor in either a collective pattern in which cell-cell interactions are closely maintained and cells move in broad sheets, or in a streaming pattern in which cells maintain a loose connection moving along the same pathway (11). Single-cell migration also occurs, where by cells move by adopting an amoeboid-like phenotype or a mesenchymal phenotype, a process that mimics the epithelial-mesenchymal transition (EMT) that occurs during embryo development. This transition involves a decrease in E-cadherin expression with a concomitant increase in expression of N-cadherin. Indeed, multiple tumors show decreased E-cadherin levels, which reflect a decrease in synthesis and/or degradation by several MMPs, including MMP-9, -10 and-15 (14,15).
Whatever the mode of local invasion, tumor cells must breach histological barriers, basement membrane, stroma, and vascular basal lamina, to move into the blood stream and spread to distant sites (Fig. 1). This process requires the degradation of their molecular components, and multiple studies have shown that MMPs play an important role (5,13,16–19). After entering the bloodstream, tumor cells invade again through the vascular basal lamina to extravasate into distant tissues. Multiple MMPs including MMP-2, -9, and -14 can degrade the basal lamina of capillary vessels and have been implicated in tumor cell extravasation (20). MMPs also have complex effects on growth factors and cytokines (13). Upregulation of the COX-2 pathway is associated with increased blood-brain barrier permeability and breast cancer cell entry into the CNS (21). Experimental studies of human melanoma have shown that MMP-2 upregulates tumor cell secretion of vascular endothelial growth factor-A (VEGF-A), which in turn activates the vascular endothelium favoring melanoma cell interaction with the blood vessel lining and their extravasation (21,22).
Fig. 1. Roles of MMPs in tumor progression, invasion and metastases.
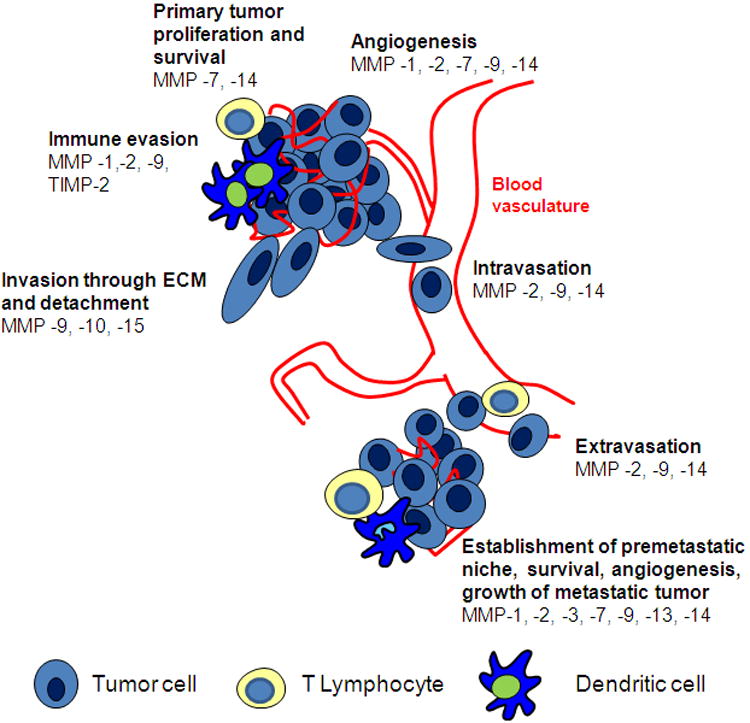
Once tumor cells extravasate, a metastatic niche must be set up to permit tumor cell growth in an unfavorable environment. MMPs promote this process through several mechanisms. Angiogenesis, the formation of capillary blood vessels from pre-existing vasculature, involves multiple interactions between stroma and vascular cells. A number of MMPs, including MMP-1, -2, -7, -9 and -14, contribute to angiogenesis via several mechanisms (23). In addition to mediating the ECM degradation necessary for endothelial cell migration into the tumor to be vascularized, MMPs contribute to the release of proangiogenic factors such as VEGF, fibroblast growth factor-2 (FGF-2), and transforming growth factor (TGF)-β from the ECM (2). These growth factors are sequestered in the stroma, and metastatic foci utilize MMPs to create a favorable metastatic niche by mobilizing these factors to support tumor growth.
ECM-degrading enzymes further influence metastatic cell survival by modulating apoptosis. MMP-7 confers a survival benefit to tumor cells by cleaving Fas ligand, removing it from the cell surface and preventing it from stimulating the Fas death receptor, a potent mediator of innate apoptotic pathways (24). By this mechanism malignant cells evade apoptosis and may also gain resistance to chemotherapeutics (25). Other MMPs such as MMP-14 also promote tumor progression through anti-apoptotic interactions with the surrounding microenvironment (Fig. 1) (26).
The immune system surveilles the body for tumor cells, recognizing and killing malignant cells by recruiting neutrophils, macrophages, and tumor-specific T lymphocytes. Cancer cells have developed multiple mechanisms utilizing MMPs to evade the immune system, thereby ensuring metastatic cell survival. Tumors utilize MMPs to cleave chemokines, preventing inflammatory cell chemotaxis and recruitment to involved tissues (27). In melanoma MMP-2-conditioned dendritic cells prime naïve CD4 T cells to differentiate towards the TH2 helper cell pathway, thereby skewing the immune response (28). MMP -1, -2, and-9 downregulate interleukin receptor on the surface of T cells, further dampening immunity and promoting tolerance of cancer (29). TIMP-2 downregulation has also been implicated in suppressing local immune function, allowing cancer cells to escape (30).
MMP Expression and Modulation in Cancer
MMP overexpression has been well documented in multiple types of solid tumors (31). High levels of MMPs have been correlated with poor overall survival in virtually all solid malignancies (13,31–33). Studies have also shown significant associations between tumor aggressiveness and elevated MMP expression. For example, distant metastases from breast cancer have been correlated with high levels of multiple MMPs including MMP -1, -7, -9, -11, and -13 (34). MMP-13 levels are also increased in lung and prostate malignancies (32,33). MMP-9 overexpression has been strongly associated with poor prognosis in multiple malignancies including breast, lung, colon, gastric, pancreatic, and prostate cancer (33,35–38). However, despite hundreds of observational studies in humans correlating high MMP levels with metastatic spreador recurrence, only MMP-11 (stromelysin) has thus far become part of a prognostic assay, the OncotypeDX™ platform, a clinically validated 21-gene array for prognosticating recurrence and guiding therapy in early hormone receptor-positive, HER2-negative breast cancer (39).
TIMP levels also change as tumors become more aggressive, and ultimately TIMP deregulation contributes to metastatic spread (40). However, while TIMP downregulation is expected to favor tumor progression, evidence shows that more complex mechanisms are in play; some TIMPs are in fact upregulated, while others are silenced. TIMP-1 overexpression is associated with unfavorable prognosis and early recurrence in multiple cancers including breast and prostate carcinoma (41,42). Conversely, the lack of TIMP-1 expression predicts both a favorable prognosis and tumor responsiveness to chemotherapy in some cancers (43). In contrast to TIMP-1 overexpression correlating with poor survival in the metastatic setting, strong data supports that multiple human cancers silence TIMP-3 as they spread (40). This effect seems to imply that TIMP-3 functions as a tumor suppressor gene and that by turning off its expression, tumors are allowed unchecked growth (44). Regardless of which TIMPs are upregulated or silenced, growing evidence shows that their deregulation contributes to metastatic spread of malignancy, and therefore represents a potential therapeutic target.
Clinical Trials of MMPIs
Given the robust experimental and clinical evidence associating MMPs with tumor progression and poor prognosis, several MMPIs were synthesized and trialed from the late 1980s into the early 2000s for various cancer types (Table 1) (45). One of the first drugs developed was batimastat, a small peptidomimetic molecule designed to mimic the most common MMP substrate, collagen. Batimastat showed broad-spectrum inhibition of virtually all MMP family members. Preclinical data indicated a promising anti-tumor effect of the drug; however, early trials showed that its water insolubility resulted in low oral bioavailability (46). Although several phase 1 studies showed efficacy with direct injection of the drug into the pleural or peritoneal space of patients with malignant effusions or ascites, significant toxicity, including pain, pyrexia, transaminitis, dyspnea, cough, and nausea, was observed. Therefore, further testing was not pursued, given the development of a more readily orally bioavailable drug, marimastat (47,48).
Table 1. Synopsis of the MMP Inhibitors Discussed.
| Name of Inhibitor | Type of Inhibitor | MMPs Targeted | Type of Cancer Studied | Toxicity | Outcome |
|---|---|---|---|---|---|
| Batimastat (BB-94) *5362422 | Hydroxymate (zinc chelator) | Broad, including MMP-1, -2, -3, -7, -9, -14 | Malignant Ascites (Pancreatic, Colorectal, Gastric, Ovarian, Cholangiocarcinoma, Ovarian, Mesothelioma) Malignant Pleural Effusion (Non-Small Cell Lung, Breast, Melanoma, Renal, Mesothelioma) |
Musculoskeletal syndrome, Fever, Liver Function Abnormalities, pleural pain at site of injection, GI upset | Canceled in phase III clinical trials (local toxicity, slow accrual, Marimastat developed) |
| Marimastat (BB-2516) *119031 | Hydroxymate (zinc chelator) | Broad, including MMP-1. -2, -3, -7, -9 | Breast, Non-Small Cell Lung, Colorectal, Pancreatic, Gastric, Prostate, Glioblastoma | Musculoskeletal Syndrome, GI upset | Prolongation of survival in randomized Ph2 in gastric cancer, Canceled in phase III clinical trials |
| Tanomastat (BAY 12-9566)*6918336 | Carboxylate (zinc chelator) | MMP-2, -3, -8, -9, -13 | Pancreatic, Ovarian, Small Cell Lung | Hematologic (anemia, thrombocytopenia), electrolyte abnormalities, hyperbilirubinemia, GI upset | Canceled in phase III clinical trials |
| Prinomastat (AG3340) *466151 | Hydroxymate (zinc chelator) | MMP-2, -3, -9, -13, -14 | Non-Small Cell Lung, Esophageal | Musculoskeletal, Venous Thromboembolism, Hematologic, GI Upset | Canceled in phase III clinical trials |
| Rebimastat (BMS-275291) *9913881 | Sulfhydryl based mercaptoacyl zinc chelator | MMP-1, -2, -3, -8, -9, -13, -14 | Non-Small Cell Lung, Breast, Prostate | Dermatologic, Hypersensitivity | Canceled in phase III clinical trials |
| Andecaliximab (GS-5745) | Monoclonal Antibody | MMP-9 | Gastric, Breast, Pancreatic, Non-Small Cell Lung, Esophageal, Colorectal | Neutropenia, Nausea, Pain, GI Upset | Ongoing phase I, II and III clinical trials |
| AB0041, AB0046, GS-5745 | Monoclonal Antibody | MMP-9 | Colorectal | n/a | Active in preclinical studies |
| DX-2400 | Monoclonal Antibody | MMP-14 | Breast, Melanoma, Fibrosarcoma | n/a | Active in preclinical studies |
| Single Chain Fragment Variables | Monoclonal Antibody | MMP-1 MMP-2 MMP-3 | Breast | n/a | Active in preclinical studies |
PubChem Identification Number
Marimastat was developed as a next-generation oral analogue with a similar mechanism of action as batimastat. It too showed much promise in the preclinical setting, and reached phase 2 and 3 clinical trials in the metastatic setting for multiple solid tumor types including pancreatic, lung, breast, colorectal, brain, and prostate cancer (7,8,49–51). Despite the breadth of these trials, they uniformly failed to demonstrate a survival benefit. Many patients also had a negative impact on their quality of life due to a debilitating “musculoskeletal syndrome” consisting of joint pain, stiffness and inflammation, which forced the discontinuation of the drug in several patients (52). One trial which evaluated the drug for unresectable gastric carcinoma did show a modest survival benefit at 2 years (9% in the treatment arm versus 3% in the placebo group), but again with significant musculoskeletal toxicity (53).
The musculoskeletal syndrome seen in patients treated with batimastat and marimastat has been attributed to the inhibition of two members of the ADAM (a disintegrin and metalloproteinase) family of proteinases, ADAM-10 and -17. These ADAMS are also termed “sheddases” as they cleave the membrane-bound precursor of tumor necrosis factor-α (TNF- α), shedding the active form into the circulation. However, ADAM-10 and-17 are also responsible for the degradation of TNF-α receptors, serving as a regulatory mechanism for TNF-α action (54). Inhibiting the activity of these proteinases therefore disrupts this balance as receptors remain upregulated and activated TNF-α molecules are able to bind to their unregulated receptors, contributing to the musculoskeletal symptoms seen in these patients. Significant fibrosis has also been described in subjects treated with marimastat, due to MMP-1 inhibition. The inhibition of this enzyme prevents interstitial type I collagen remodeling, leading to excessive deposition in the ECM and fibrosis, which may have contributed to some of the severe side effects that led to the failure of marimastat (55).
Other more selective inhibitors that avoided inhibition of ADAM-10 and -17 were then trialed, including tanomastat, a small molecule inhibitor of MMP-2, -3, -8, -9, and -13, prinomastat which inhibits MMP-2, -3, -9, -13, and -14, and rebimastat, an inhibitor of MMP-1, -2, -3, -8, -9, -13, -14 (56). All these inhibitors were studied in the metastatic setting of ovarian, pancreatic, lung, breast, and prostate carcinomas (6,57–63). Unfortunately, despite their narrower inhibitory action, these trials failed to demonstrate a positive effect on survival. While musculoskeletal toxicity was seen less often with these inhibitors, some studies still reported significant joint pain and swelling, as well as bone marrow suppression and venous thromboembolism. Ultimately, further trials of MMPIs were halted after these negative results were published in the mid-2000s (Table 1).
Why did MMPIs block tumor progression in mice but not in man?
Several reasons have been hypothesized to explain why, despite preclinical and clinical evidence implicating MMPs in tumor growth and metastasis, clinical trials of MMPIs were unsuccessful (5,17,19). First, the difference between human and murine biology may at least partially explain the ineffectiveness of these drugs (64). Mice live 2-3 years, a 25-fold shorter lifespan than humans. This leads to many more cell divisions in human cells, allowing them to acquire many more oncogenic mutations than in the mouse (65). In mice, growth and spread of malignancy happens quickly, and aggressive tumors may grow locally before metastasizing late in the course of the disease, killing the animal in a matter of weeks. Conversely, in man cancer takes months or even years to grow to the point of invasion and metastasis, although aggressive human tumors may metastasize more quickly relatively to the mouse.
Most models of malignancy in the mouse used for preclinical studies provide a means to study localized cancer, as cells are injected subcutaneously or (more rarely) into the organ of interest to form a site-specific tumor. This leads to the formation of a primary tumor that can grow to a size of 10% or more of the host's weight in a short time and without metastasizing. In contrast, human cancers grow much more slowly to a much smaller relative size; and clinically undetectable tumors can spread numerous metastases. Most preclinical mouse models of metastatic cancer artificially introduce metastasis by bolus injection of tumor cells into the blood stream, which causes many metastatic sites to develop at once. This contrasts with humans, in which malignant cells are shed slowly and constantly into the blood or lymphatics, and lead to the gradual formation of metastases over time. Many human cancers form through a progressive process of metaplasia leading to dysplasia, malignancy in-situ, and then invasive cancer. While some mouse models of spontaneous malignancy do mimic this process, most MMPIs were trialed preclinically using a tumor bolus to form metastasis, which may explain why preclinical successes failed to translate into successful clinical trials (66).
The genetic setup of mouse models of cancer may also contribute to the lack of success in translating preclinical work. It is well known that human tumors are genetically heterogenous; as tumor cells metastasize, they continue to acquire new mutations, and therapy selects for resistant clones, making metastatic cancer incurable. In contrast, most mouse models of metastatic cancer consist of bolus injection of an immortalized cell line that is genetically homogenous, and the relatively short duration of experiments provides little time for mutations to arise and expand, all of which provides an overly simplistic system in which to trial new therapeutics. The tumor microenvironment is also different in humans and mice, which may lead to a different outcome when MMPs are inhibited preclinically versus clinically.
MMPI specificity has also been challenged as a possible reason for failure of the clinical trials. Early MMPIs such as batimastat were nonspecific and inhibited virtually all MMPs. Even later, more specific MMPIs still targeteda number of MMPs. While most MMPs have been associated with poor prognosis and metastatic spread, over the past decade it has become apparent that some MMPs have anti-tumorigenic activity; that is, they are drug anti-targets whose beneficial actions should not be contrasted. MMP-3, -8, -9, -11, -12, -19 and -26 have been validated as anti-targets in vivo; they inhibit angiogenesis and metastasis in experimental models; and low levels of these MMPS are associated with shorter survival in cancer patients (67). Deregulation of TIMP family members also has an effect on these protective MMPs, and broad MMP inhibition by MMPIs and natural inhibitors may have contributed to the failure of clinical trials.
MMPs have wide-reaching effects. Inhibiting physiological ECM remodeling led to unforeseen side effects such as the musculoskeletal syndrome, observed to some degree with nearly all the MMPIs tested (55). As the effect was found to be reversible, some later trials used lower doses than the early trials, which may have led to suboptimal dosing strategies.
Perhaps most importantly, the clinical trials were performed without regard for disease stage. MMPs act in the earliest stages of tumor progression when primary tumor cells begin spreading. Preclinical testing reflected this concept, successfully inhibiting early-stage cancers and hematogenous metastases, while having less effect on large tumors. However, clinical trials were performed almost exclusively in the metastatic, refractory setting, beyond a time when MMP inhibition is expected to be effective (68). To mimic treatment in the premetastatic setting as early as possible after diagnosis and before surgical excision, in the “window-of-opportunity,” we designed a preclinical murine model of aggressive triple negative breast carcinoma. The animals were treated with SD-7300, a specific inhibitor of MMP-2, -9, and -13, or control vehicle for 7 days after the primary tumor became detectable. We then excised the tumor and sacrificed the mice for analysis of lung metastases one month later. This “window-of-opportunity” treatment significantly decreased metastatic burden and increased survival relative to vehicle-treated controls (17). Therefore, to obtain a therapeutic benefit MMPIs should be trialed in the earliest, premetastatic setting, where MMPs act (13,19).
What can we do next?
New, Selective MMPIs; Patient Selection Based on Individual MMP Expression, Novel Clinical Trial Design
Our knowledge of the biochemistry and biology of MMPs has grown considerably in the 15 years since the clinical trials of MMPIs were halted. New MMPs have been discovered and new roles of already known MMPs – including inflammation and protection against cancer – have been revealed (67). New molecular genetics techniques such as CRISPR-Cas9 and the availability of genetic tools for the tissue-and time-specific silencing of genes, make the generation of mouse models easier, faster and cheaper than 15 years ago. The combination of conditional MMP knockout models with spontaneous tumor models can afford clear, unambiguous target validation before MMPIs are generated and brought into preclinical and clinical studies. Detailed analyses of MMP molecular structures have provided accurate information of the determinants of their substrate specificity, paving the path to the design of novel, highly selective and potent MMPIs based on differing mechanisms of action (69). These advances can allow us to overcome the limitations that potentially caused the failure of the clinical trials, and reconsider MMPI treatment of metastatic cancer in a new light.
The first-generation MMPIs were designed to target the MMP catalytic site, which is highly conserved (i.e. very similar) in all members of the MMP family. This approach resulted in a generation of drugs that could effectively block MMP-mediated proteolysis but lacked the ability to selectively inhibit the specific MMP(s) associated with a given tumor. In the light of today's knowledge that some MMPs have anti-tumor effects in some types of cancers, considerable effort has been, and is being put into the design of MMPIs that are highly selective and possibly inhibit only deleterious functions of specific, tumor-associated MMPs. A turning point in this effort was determined over a decade ago by findings that MMP activity can be inhibited specifically by targeting molecular structures outside of the catalytic domain (so-called “exosites”) (69). Unlike the catalytic domains, different exosites are present in different MMPs, and their targeting with synthetic, low-molecular weight compounds or antibodies can result in the selective inhibition of even a specific function of a single MMP. This approach has led to the generation of highly selective monoclonal antibodies to MMP-9, AB0041 and AB0046, and the humanized version of AB0041, GS-5745, which have shown efficacy in a mouse xenograft model of colorectal carcinoma (69). A set of monoclonal antibodies to exosites of MMP-14 (LEM-2/15, -2/63, and-1/58) have shown high selectivity for MMP-14. Importantly, LEM-2/15 specifically inhibits MMP-14 degradation of gelatin and collagen type I without affecting its capacity to activate proMMP-2, an important function of MMP-14. Conversely, another antibody to MMP-14 (9E8), which is also highly selective for this MMP, has no effect on MMP-14 proteolytic activity but inhibits proMMP-2 activation, showing that specific MMP functions can be selectively inhibited (69).
Selective MMP inhibition can also be achieved by the use of “endogenous” or “intrinsic” MMP inhibitors. Like all extracellular proteinases, MMPs are secreted as inactive proenzymes whose proteolytic activity is inhibited by the intramolecular interaction of the catalytic domain with the N-terminal “pro” domain. Removal of the prodomain by limited proteolysis or other mechanisms results in activation of the proMMP. Unlike the highly conserved catalytic domains, the pro domains differ from one MMP to another, a difference that can be exploited to generate specific protein inhibitors. Exogenous addition of the pro domains of the sheddases ADAM10 and ADAM17 results in selective inhibition of the respective enzyme, without cross-reactivity in spite of the high similarity of the two ADAMs (69).
Other approaches to the development of selective and specific MMPIs have used sophisticated biochemical techniques such as protein engineering and directed evolution to improve the inhibitory activity of antibodies and TIMPs. Anti-MMP-14 antibodies that effectively reduce tumor growth and metastasis in preclinical models have been generated by selection of a phage display library of single-chain variable fragments (scFv), followed by protein engineering to increase their affinity and inhibitory activity. A monoclonal antibody to MMP-14, DX-2400, was selected by screening a human Fab-phage library for candidates binding selectively the MMP-14 catalytic domain. DX-2400 is a high-affinity, highly selective inhibitor of MMP-14 that retards tumor growth and metastasis in several in vivo mouse models of breast cancer and melanoma, both as single agent and in combination with paclitaxel or bevacizumab. Monoclonal antibody fragments (scFv) have also been developed to MMP-1, MMP-2 and MMP-3 by a combination of phage-display library screening and combinatorial mutagenesis (69).
Protein engineering has been used to generate TIMP variants that specifically inhibit MMP-14. Point mutations in the sequence of TIMP-2 increase binding to and inhibition of MMP14 by 9 - 14 folds. TIMP mutants with inhibitory activity towards MMPs that were not their native targets were developed by protein engineering, directed evolution and computational design. By these methods a mutant of TIMP-2 was generated which selectively blocks MMP-14 activity with an inhibition constant of 0.9 pM, the strongest inhibitor of this MMP thus far generated (69).
Thus, a number of novel MMPIs have been and continue to be engineered with the high affinity, specificity and selectivity that earlier-generation MMPIs lacked. These features can circumvent not only the potentially deleterious inhibition of protective MMPs but also avoid the onset of the musculoskeletal syndrome.
Novel molecular biology techniques afford relatively fast and inexpensive analysis of MMP expression in very small biological samples or even single-cells. Diagnostic bioptic material can provide sufficient tumor tissue to analyze the expression of the MMPs expressed by the individual patient's tumor. Relatively low numbers of tumor cells can be identified in peripheral blood and analyzed for MMP expression; tumor cell DNA can also be detected in the circulation, providing a potential surrogate of metastasis. These techniques can allow identification of the MMP(s) produced by a tumor, rapid assessment of the treatment efficacy, and therefore a precision medicine approach to anti-MMP treatment.
However, to effectively test MMPIs, a fundamental shift in clinical trial design is necessary. Currently, investigational cancer drugs are first tested in advanced cancer patients with overt metastatic disease. As MMPIs act most effectively (and almost exclusively) in the pre- and peri-metastatic setting, these clinical trials can only be ineffective. To effectively study MMPIs, new trials should be designed, incorporating early-stage patients in the pre-metastatic setting.
Traditionally, in neoadjuvant and adjuvant trials recurrence-free survival or freedom from metastatic disease is the required primary endpoint for approval of therapies in the early disease setting. These trials are costly as they require enrollment of many patients and outcome readouts take years or even decades. Therefore, to meaningfully study the effectiveness of systemic cytotoxic drugs anti-tumor efficacy is first is determined in the metastatic setting, then the drug is moved to neoadjuvant stages and surrogates of early response, such as pathologic complete response (pCR), are assessed. This strategy has proven useful, for instance, in aggressive breast cancers, where pCR rates correlate with freedom from metastases and survival, supporting testing of cytotoxic compounds in this setting.
As MMPIs are not expected to decrease tumor size alternative surrogates must be tested, such as decrease in circulating tumor cells (as indicator of decrease in micrometastases) and/or decrease in tumor-associated MMP activity. MMPIs should therefore be studied in two stages of a phase 2 trial. In stage one, surrogate markers should be used as an endpoint instead of recurrence-free survival, to demonstrate target inhibition (which could also direct dose finding) and possibly early effectiveness. For example, a preclinical model in mice used markers such as change in target mRNA expression to show drug effectiveness, and correlated this outcome with a decrease in bone metastases (70). In the case of MMPI therapy, target MMP activity could be used as surrogate marker. A decrease in circulating tumor cells is also a possible endpoint and several studies in lung, breast and castration-resistant prostate cancer have correlated a decrease in this marker and an improvement in metastatic burden (71). If stage one using a surrogate marker shows a positive outcome, the trial could then move on to stage two, expanding in size and evaluating a more traditional and clinical endpoint such as freedom from metastatic disease (Figure 2a).
Fig. 2.
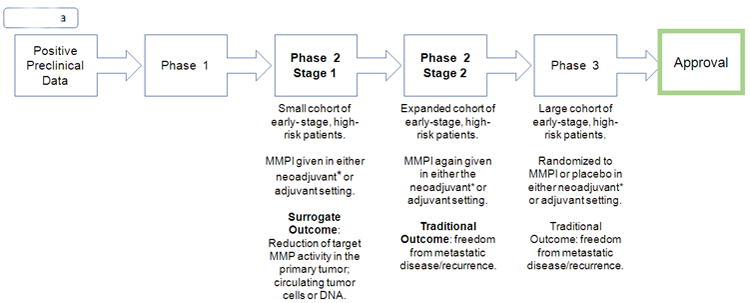
a. Modified Trial Design
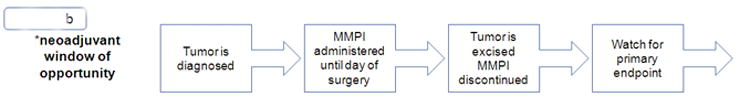
b. Neoadjuvant Window of Opportunity
Unlike previous trials of MMPIs, which primarily studied patients with stage IV disease, new trials should enroll patients with high-risk disease that is not yet clinically or pathologically metastatic, or patients with high-risk precursor lesions. The drug should be given prior to surgery, in the so called “window-of-opportunity” between the time of diagnosis and surgical excision, or post-operatively in the adjuvant setting as microscopic residual disease may not have developed the mutations necessary to fully metastasize (Figure 2b). While clinical trials with MMPIs haven't been conducted in the pre-metastatic setting, there are ongoing trials of other drugs with a similar design. For example, the ongoing D-Care study is investigating denosumab, a drug already approved for the prevention of pathologic fracture in breast cancer patients with osseous metastasis, in the neoadjuvant or adjuvant setting for patients with stage II or III breast cancer at high risk of recurrence. Primary outcome includes bone-metastasis free survival, which, if positive, would be a successful confirmation of this novel trial design and outcome (72).
As discovered in the past decade, inhibition of tumor growth is largely dependent on the individual MMP targeted, and the mechanism of action of novel MMPIs must be more specific than earlier generations'. Specific inhibitors have recently been and are currently being developed. The monoclonal antibodies discussed above are perhaps the most promising of the new MMPIs. Ideally, personalized therapies can be envisaged in which only MMPs expressed by the individual tumor are targeted. Such an approach is now feasible thanks to multiple techniques that allow the analysis of gene expression in few, or even single cells that can be identified by laser capture microscopy.
Since the failure of the last trials in the mid-2000s, compounds have been shelved and trials have been on hold. Given the enormous costs of drug development, most manufacturers have been hesitant to reopen the door on trialing MMPIs and a search of active clinical trials in the US yields few results (73). While a few pharmaceutical companies are beginning to trial highly selective MMPIs, these trials are still being conducted in the metastatic setting, and it remains unclear what benefit may be gained by this approach (74,75). Clearly, a culture shift is needed if the true effects of MMPIs are to be revealed. A first step may be to perform “window of opportunity” trials in early cancers, identifying and validating biomarkers of enzymatic inhibition and metastasis as proxy for clinical success.
Acknowledgments
Financial support: This work was supported by NIH grants R01 CA136715 and 3R01CA136715-05S1 to P. Mignatti.
Footnotes
Competing financial interests: The authors declare no competing financial interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Kapoor C, Vaidya S, Wadhwan V, Hitesh, Kaur G, Pathak A. Seesaw of matrix metalloproteinases (MMPs) J Cancer Res Ther. 2016;12(1):28. doi: 10.4103/0973-1482.157337. [DOI] [PubMed] [Google Scholar]
- 3.Nabeshima K, Inoue T, Shimao Y, Sameshima T. Matrix metalloproteinases in tumor invasion: role for cell migration. Pathol Int. 2002;52(4):255–64. doi: 10.1046/j.1440-1827.2002.01343.x. [DOI] [PubMed] [Google Scholar]
- 4.López-Otín C, Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol. 2002;3(7):509–19. doi: 10.1038/nrm858. [DOI] [PubMed] [Google Scholar]
- 5.Coussens LM. Matrix Metalloproteinase Inhibitors and Cancer : Trials and Tribulations. Siecnce. 2009;295(5564):2387–93. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 6.Hirte H, Vergote IB, Jeffrey JR, Grimshaw RN, Coppieters S, Schwartz B, et al. A phase III randomized trial of BAY 12-9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: a National Cancer Institute of Canada Clinical Trials G. Gynecol Oncol. 2006;102(2):300–8. doi: 10.1016/j.ygyno.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 7.Bramhall SR, Schulz J, Nemunaitis J, Brown PD, Baillet M, Buckels JaC. A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br J Cancer. 2002;87(2):161–7. doi: 10.1038/sj.bjc.6600446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sparano JA, Bernardo P, Stephenson P, Gradishar WJ, Ingle JN, Zucker S, et al. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group Trial E2196. J Clin Oncol. 2004;22(23):4631–8. doi: 10.1200/JCO.2004.08.054. [DOI] [PubMed] [Google Scholar]
- 9.Gross J, Lapiere CM. Collagenolytic Activity in Amphibian Tissues: a Tissue Culture Assay. Proc Natl Acad Sci U S A. 1962;48(6):1014–22. doi: 10.1073/pnas.48.6.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisen A, Jeffery J, Gross J. Human Skin Collagenase, Isolation And Mechanism of Attack on the Collagen Molecule. Biochim Biophys Acta. 1967;151(1968):637–45. doi: 10.1016/0005-2744(68)90010-7. [DOI] [PubMed] [Google Scholar]
- 11.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69(3):562–73. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Tallant C, Marrero A, Gomis-Rüth FX. Biochim Biophys Acta - Mol Cell Res. 1. Vol. 1803. Elsevier B.V; 2010. Matrix metalloproteinases: Fold and function of their catalytic domains; pp. 20–8. [DOI] [PubMed] [Google Scholar]
- 13.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 14.Lamouille Samy, Xu Jian, Derynck R. Molecular Mechanisms of Epithelial-Mesenchymal Transition. Nat Rev Mol Cell Biol. 2014;15(3):178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dreymueller D, Theodorou K, Donners M, Ludwing A. Fine Tuning Cell Migration by A Disintegrin And Metalloproteinases. Mediators Inflamm. 2016;13 doi: 10.1155/2017/9621724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hadler-Olsen E, Winberg JO, Uhlin-Hansen L. Matrix metalloproteinases in cancer: Their value as diagnostic and prognostic markers and therapeutic targets. Tumor Biol. 2013;34(4):2041–51. doi: 10.1007/s13277-013-0842-8. [DOI] [PubMed] [Google Scholar]
- 17.Winer A, Janosky M, Harrison B, Zhong J, Moussai D, Siyah P, et al. Inhibition of breast cancer metastasis by presurgical treatment with an oral matrix metalloproteinase inhibitor: a preclinical proof-of-principle study. Mol Cancer Ther. 2016:1–8. doi: 10.1158/1535-7163.MCT-16-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheng WuJ, Liang S, Tang XY. The role of tumor microenvironment in collective tumor cell invasion. Futur Oncol. 2017 doi: 10.2217/fon-2016-0501. fon-2016-0501. [DOI] [PubMed] [Google Scholar]
- 19.Overall CM, López-Otín C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2(9):657–72. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 20.Voura E, English J, Yu H, Ho A, Subarsky P, Hill R, Hojilla C, Khokha R. Proteolysis during Tumor Cell Extravasation In Vitro: Metalloproteinase Involvement across Tumor Cell Types. PLoS One. 2013;8(10):e78413. doi: 10.1371/journal.pone.0078413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee KY, Kim YJ, Yoo H, Lee SH, Park JB, Kim HJ. Human brain endothelial cell-derived COX-2 facilitates extravasation of breast cancer cells across the blood-brain barrier. Anticancer Res. 2011;31(12):4307–13. [PubMed] [Google Scholar]
- 22.Desch A, Strozyk EA, Bauer AT, Huck V, Niemeyer V, Wieland T, et al. Am J Pathol. 2. Vol. 181. Elsevier Inc; 2012. Highly Invasive Melanoma Cells Activate the Vascular Endothelium via an MMP-2/Integrin–Induced alpha v beta5-Induced Secretion of VEGF-A; pp. 693–705. [DOI] [PubMed] [Google Scholar]
- 23.Deryugina EI, Quigley JP. Matrix Biol [Internet] Elsevier B.V; 2015. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature; pp. 44–46.pp. 94–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strand S, Vollmer P, van den Abeelen L, Gottfried D, Alla V, Heid H, et al. Cleavage of CD95 by matrix metalloproteinase-7 induces apoptosis resistance in tumour cells. Oncogene. 2004;23(20):3732–6. doi: 10.1038/sj.onc.1207387. [DOI] [PubMed] [Google Scholar]
- 25.Mitsiades N, Yu WH, Poulaki V, Tsokos M, Stamenkovic I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001;61(2):577–81. [PubMed] [Google Scholar]
- 26.Maquoi E, Assent D, Detilleux J, Pequeux C, Foidart JM, Noël a. MT1-MMP protects breast carcinoma cells against type I collagen-induced apoptosis. Oncogene. 2012;31(4):480–93. doi: 10.1038/onc.2011.249. [DOI] [PubMed] [Google Scholar]
- 27.McQuibban G, Gong J, Tam E, McCulloch C, Clark-Lewis I, Overall C. Infalmmation Dampened by Gelatinase A Cleavage Of Monocyte Chemoattractant Protein-3. Science (80-) 2000;289:1202–5. doi: 10.1126/science.289.5482.1202. [DOI] [PubMed] [Google Scholar]
- 28.Godefroy E, Manches O, Dréno B, Hochman T, Rolnitzky L, Labarrière N, et al. Matrix Metalloproteinase-2 Conditions Human Dendritic Cells to Prime Inflammatory TH2 Cells via an IL-12- and OX40L-Dependent Pathway. Cancer Cell. 2011;19(3):333–46. doi: 10.1016/j.ccr.2011.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheu BC, Hsu SM, Ho HN, Lien HC, Huang SC, Lin RH. A novel role of metalloproteinase in cancer-mediated immunosuppression. Cancer Res. 2001;61(1):237–42. [PubMed] [Google Scholar]
- 30.Guedez L, Jensen-Taubman S, Bourboulia D, Kwityn CJ, Wei B, Caterina J, et al. TIMP-2 targets tumor-associated myeloid suppressor cells with effects in cancer immune dysfunction and angiogenesis. J Immunother. 2012;35(6):502–12. doi: 10.1097/CJI.0b013e3182619c8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rydlova M, Holubec L, Ludvikova M, Kalfert D, Franekova J, Povysil C, et al. Biological activity and clinical implications of the matrix metalloproteinases. Anticancer Res. 2008;28(2B):1389–97. [PubMed] [Google Scholar]
- 32.Hsu CP, Shen GH, Ko JL. Matrix metalloproteinase-13 expression is associated with bone marrow microinvolvement and prognosis in non-small cell lung cancer. Lung Cancer [Internet] 2006;52(3):349–57. doi: 10.1016/j.lungcan.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 33.Morgia G, Falsaperla M, Malaponte G, Madonia M, Indelicato M, Travali S, et al. Matrix metalloproteinases as diagnostic (MMP-13) and prognostic (MMP-2, MMP-9) markers of prostate cancer. Urol Res. 2005;33(1):44–50. doi: 10.1007/s00240-004-0440-8. [DOI] [PubMed] [Google Scholar]
- 34.Vizoso F, González L, Rodríguez J, Vázquez J, Lamelas M, Junquera S, et al. Study of matrix metalloproteinases and their inhibitors in breast cancer. Br J Cancer. 2007;96:903–11. doi: 10.1038/sj.bjc.6603666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng WJ, Zhang JQ, Wang BX, Pan HF, Lu MM, Wang J. Clin Chim Acta [Internet] Vol. 413. Elsevier B.V; 2012. Prognostic value of matrix metalloproteinase 9 expression in patients with non-small cell lung cancer; pp. 13–14.pp. 1121–6. [DOI] [PubMed] [Google Scholar]
- 36.Yang B, Tang F, Zhang B, Zhao Y, Feng J, Rao Z. Matrix metalloproteinase-9 overexpression is closely related to poor prognosis in patients with colon cancer. World J Surg Oncol. 2014;12:1–6. doi: 10.1186/1477-7819-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen S, Yao H, Zhu S, Li Q, Guo G, Yu J. Expression levels of matrix metalloproteinase-9 in human gastric carcinoma [Internet] Oncology Letters. 2014:915–9. doi: 10.3892/ol.2014.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Y, Li Z, Jiang P, Wu G, Chen K, Zhang X, et al. The co-expression of MMP-9 and Tenascin-C is significantly associated with the progression and prognosis of pancreatic cancer. Diagn Pathol Diagnostic Pathology. 2015;10(1):211. doi: 10.1186/s13000-015-0445-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sparano JA, Gray RJ, Makower DF, Pritchard KI, Albain KS, Hayes DF, et al. Prospective Validation of a 21-Gene Expression Assay in Breast Cancer. N Engl J Med [Internet] 2015;373(21) doi: 10.1056/NEJMoa1510764. 150927220039001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson HW, Defamie V, Waterhouse P, Khokha R. Nat Publ Gr [Internet] 1. Vol. 17. Nature Publishing Group; 2016. TIMPs: versatile extracellular regulators in cancer; pp. 38–53. [DOI] [PubMed] [Google Scholar]
- 41.Dos Reis ST, Viana NI, Iscaife A, Pontes-Junior J, Dip N, Antunes AA, et al. Loss of TIMP-1 immune expression and tumor recurrence in localized prostate cancer. Int Braz J Urol. 2015;41(6):1088–95. doi: 10.1590/S1677-5538.IBJU.2014.0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dechaphunkul A, Phukaoloun M, Kanjanapradit K, Graham K, Ghosh S, Santos C, et al. Prognostic significance of tissue inhibitor of metalloproteinase-1 in breast cancer. Int J Breast Cancer. 2012;2012:290854. doi: 10.1155/2012/290854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuvaja P, Talvensaari-Mattila A, Pääkkö P, Turpeenniemi-Hujanen T. The absence of immunoreactivity for tissue inhibitor of metalloproteinase-1 (TIMP-1), but not for TIMP-2, protein is associated with a favorable prognosis in aggressive breast carcinoma. Oncology. 2005;68(2–3):196–203. doi: 10.1159/000086774. [DOI] [PubMed] [Google Scholar]
- 44.Rettori MM, De carvalho AC, Bomfim longo AL, De oliveira CZ, Kowalski LP, Carvalho AL, et al. Prognostic significance of TIMP3 hypermethylation in post-treatment salivary rinse from head and neck squamous cell carcinoma patients. Carcinogenesis. 2013;34(1):20–7. doi: 10.1093/carcin/bgs311. [DOI] [PubMed] [Google Scholar]
- 45.Cathcart JM, Cao J, Brook S. MMP Inhibitors : Past, present and future. Front Biosci (Landmark Ed) 2015;20:1164–78. doi: 10.2741/4365. [DOI] [PubMed] [Google Scholar]
- 46.Rasmussen HS, McCann PP. Matrix metalloproteinase inhibition as a novel anticancer strategy: A review with special focus on Batimastat and Marimastat. Pharmacol Ther. 1997;75(1):69–75. doi: 10.1016/s0163-7258(97)00023-5. [DOI] [PubMed] [Google Scholar]
- 47.Parsons SL, Watson SA, Steele RJ. Phase I/II trial of batimastat, a matrix metalloproteinase inhibitor, in patients with malignant ascites. Eur J Surg Oncol [Internet] 1997;23(6):526–31. doi: 10.1016/s0748-7983(97)93077-8. [DOI] [PubMed] [Google Scholar]
- 48.Macaulay VM, O'Byrne KJ, Saunders MP, Braybrooke JP, Long L, Gleeson F, et al. Phase I study of intrapleural batimastat (BB-94), a matrix metalloproteinase inhibitor, in the treatment of malignant pleural effusions. Clin Cancer Res. 1999;5(3):513–20. [PubMed] [Google Scholar]
- 49.King J, Zhao J, Clingan P, Morris D. Randomised Double Blind Placebo Control Study of Adjuvant Treatmetn with the Metalloproteinase Inhibitor, Marimastat in Patients with Inoperable Colorectal Hepatic Metastases: Significant Survival Advantage in Patients with Musculoskeletal Side-Effects. Anticancer Res. 2003;23:639–46. [PubMed] [Google Scholar]
- 50.Rosenbaum E, Zahurak M, Sinibaldi V, Carducci MA, Pili R, Laufer M, et al. Marimastat in the Treatment of Patients with Biochemically Relapsed Prostate Cancer: A Prospective Randomized, Double-Blind, Phase I/II Trial. Clin Cancer Res. 2005;11(12):4437–43. doi: 10.1158/1078-0432.CCR-04-2252. [DOI] [PubMed] [Google Scholar]
- 51.Levin VA, Phuphanich S, Yung WKA, Forsyth PA, Del Maestro R, Perry JR, et al. Randomized, double-blind, placebo-controlled trial of marimastat in glioblastoma multiforme patients following surgery and irradiation. J Neurooncol. 2006;78(3):295–302. doi: 10.1007/s11060-005-9098-5. [DOI] [PubMed] [Google Scholar]
- 52.Vandenbroucke, Roosmarijn E, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev drug Discov. 2014;13(12):904–27. doi: 10.1038/nrd4390. [DOI] [PubMed] [Google Scholar]
- 53.Bramhall S, Hallissey M, Whiting J, Tierney G, Stuart R, Hawkins R, McCulloch P, Maughan T, Baillet M, Fielding J. Marimastat as maintenance therapy for patients with advanced gastric cancer - a randomised trial. Br J Cancer. 2002;86:1864–70. doi: 10.1038/sj.bjc.6600310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ge L, Baskic D, Bassse P, Vujanovic L, Unlu S, Toshie Y, Vujanovic A, Han J, Bankovic D, Szczepanski M, Hunt J, Herberman R, Gollin S, Ferris R, Whiteside T, Myers E, Vujanovic N. Sheddase Activity of Tumor Necrosis Factor-Alpha Converting Enzyme is Increased and Prognostically Valuable in Head and Neck Cancer. Cancer Epidemiol Biomarkers Prev. 2009;18(11):2913–22. doi: 10.1158/1055-9965.EPI-08-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fingleton B. MMPs as therapeutic targets - still a viable option? Semin cell Dev Biol cell Dev Biol. 2009;19(1):61–8. doi: 10.1016/j.semcdb.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lombard M, Wallace T, Kubicek M, Petzold G, Mitchell M, Hendges S, et al. Synthetic Matrix Metalloproteinase Inhibitors and Tissue Inhibitor of Metalloproteinase (TIMP)-2, but not TIMP-1, Inhibit Shedding of Tumor Necrosis Factor-α Receptors in a Human Colon Adenocarcinoma (Colo 205) Cell Line. Cancer Res. 1998;58(17):4001–7. [PubMed] [Google Scholar]
- 57.Moore BMJ, Hamm J, Dancey J, Eisenberg PD, Dagenais M, Fields A, et al. Comparison of Gemcitabine Versus the Matrix Metalloproteinase Inhibitor BAY 12-9566 in Patients With Advanced or Metastatic Adenocarcinoma of the Pancreas : A Phase III Trial of the National Cancer Institute of Canada. J Clin Oncol. 2003;21(17):3296–302. doi: 10.1200/JCO.2003.02.098. [DOI] [PubMed] [Google Scholar]
- 58.Lara PN. A Randomized Phase II Trial of the Matrix Metalloproteinase Inhibitor BMS-275291 in Hormone-Refractory Prostate Cancer Patients with Bone Metastases. Clin Cancer Res. 2006;12(5):1556–63. doi: 10.1158/1078-0432.CCR-05-2074. [DOI] [PubMed] [Google Scholar]
- 59.Leighl NB. Randomized Phase III Study of Matrix Metalloproteinase Inhibitor BMS-275291 in Combination With Paclitaxel and Carboplatin in Advanced Non-Small-Cell Lung Cancer: National Cancer Institute of Canada-Clinical Trials Group Study BR.18. J Clin Oncol. 2004;23(12):2831–9. doi: 10.1200/JCO.2005.04.044. [DOI] [PubMed] [Google Scholar]
- 60.Miller KD, Saphner TJ, Waterhouse DM, Chen TT, Rush TA, Sparano JA, et al. A randomized phase II feasibility trial of BMS-275291 in patients with early stage breast cancer. Clin Res. 2004;10(6):1971–5. doi: 10.1158/1078-0432.ccr-03-0968. [DOI] [PubMed] [Google Scholar]
- 61.Behrendt CE, Ruiz RB. Venous thromboembolism among patients with advanced lung cancer randomized to prinomastat or placebo, plus chemotherapy. Thromb Haemost. 2003;90(4):734–7. doi: 10.1160/TH03-01-0041. [DOI] [PubMed] [Google Scholar]
- 62.Bissett D, O'byrne KJ, Von PJ, Gatzemeier U, Price A, Nicolson M, et al. Phase III Study of Matrix Metalloproteinase Inhibitor Prinomastat in Non-Small-Cell Lung Cancer. J Clin Oncol. 2005;23(4):842–9. doi: 10.1200/JCO.2005.03.170. [DOI] [PubMed] [Google Scholar]
- 63.Heath EI, Burtness BA, Kleinberg L, Salem RR, Yang SC, Heitmiller RF, et al. Phase II, parallel-design study of preoperative combined modality therapy and the matrix metalloprotease (mmp) inhibitor prinomastat in patients with esophageal adenocarcinoma. Invest New Drugs. 2006;24(2):135–40. doi: 10.1007/s10637-006-5934-5. [DOI] [PubMed] [Google Scholar]
- 64.Slawomir WP, Dickson R, Hawkins M. Matrix metalloproteinase inhibitors. Invest New Drugs. 1997;15:61–75. doi: 10.1023/a:1005722729132. [DOI] [PubMed] [Google Scholar]
- 65.Rangarajan A, Weinberg RA. Opinion: Comparative biology of mouse versus human cells: modelling human cancer in mice. Nat Rev Cancer [Internet] 2003;3(12):952–9. doi: 10.1038/nrc1235. [DOI] [PubMed] [Google Scholar]
- 66.Karim BO, Huso DL. Mouse models for colorectal cancer. Am J Cancer Res. 2013;3(3):240–50. [PMC free article] [PubMed] [Google Scholar]
- 67.Dufour A, Overall CM. Trends Pharmacol Sci. 4. Vol. 34. Elsevier Ltd; 2013. Missing the target: Matrix metalloproteinase antitargets in inflammation and cancer; pp. 233–42. [DOI] [PubMed] [Google Scholar]
- 68.Steeg P. The right trials. Nature. 2012;485:S59. doi: 10.1038/485S58a. [DOI] [PubMed] [Google Scholar]
- 69.Levin M, Udi Y, Solomonov ISI. Next generation matrix metalloproteinase inhibitors — Novel strategies bring new prospects. Biophys Acta - Mol Cell Res. 2017;1864(11):1927–39. doi: 10.1016/j.bbamcr.2017.06.009. [DOI] [PubMed] [Google Scholar]
- 70.Gustavsson H, Jennbacken K, Welén K, Damber JE. Altered expression of genes regulating angiogenesis in experimental androgen-independent prostate cancer. Prostate. 2008;68(2):161–70. doi: 10.1002/pros.20672. [DOI] [PubMed] [Google Scholar]
- 71.Wu LiY, Bai SF. Semin Cell Dev Biol. Elsevier Ltd; 2017. Molecular characterization of circulating tumor cells—from bench to bedside. [DOI] [PubMed] [Google Scholar]
- 72.Study of Denosumab as Adjuvant Treatment for Women With High Risk Early Breast Cancer Receiving Neoadjuvant or Adjuvant Therapy (D-CARE). Clinicaltrials.gov. 2917 [cited 2017 Oct 9].
- 73.Chakravarthy R, Cotter K, DiMasi J, Milne CP, Wendel N. public and private sector contributions to the research and development of the most transformational drugs of the last 25 years. Bost Tufts Cent Study Drug Dev. 2015:1–43. doi: 10.1177/2168479016648730. [DOI] [PubMed] [Google Scholar]
- 74.Shah M, Starodub A, Wainberg Z. Results of a phase I study of GS-5745 in combination with mFOLFOX in patients with advanced unresectable gastric/GE junction tumors. ASCO Annu. 2016:2016–7. [Google Scholar]
- 75.Bendell JC, Starodub A, Walnberg ZA, Wu M, Werner D, Maltzman JD, et al. A phase 3 randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of GS-5745 combined with mFOLFOX6 as first-line treatment in patients with advanced gastric or gastroesophageal junction adenocarcinoma. J Clin Oncol. 2016;34:2–3. [Google Scholar]