

Abstract
Background
Differential DNA methylation as measured in blood is a promising marker of bladder cancer susceptibility. However, previous studies have exclusively used post-diagnostic blood samples, meaning that observed associations may be markers of disease rather than susceptibility.
Methods
Genome-wide methylation was measured in pre-diagnostic blood samples, using the Illumina Infinium HumanMethylation450 Bead Array, among 440 bladder cancer cases with the transitional cell carcinoma (TCC) subtype and 440 matched cancer-free controls from the Women’s Health Initiative (WHI) cohort. After normalization and probe filtering, we used conditional logistic regression models to test for associations between methylation measurements at 361,184 CpG sites and bladder cancer risk.
Results
Increased methylation at cg22748573, located in a CpG island within the 5′-UTR/first exon of the CITED4 gene, was associated with an 82% decreased risk of bladder cancer after adjusting for race/ethnicity, smoking status, pack-years of smoking, and leukocyte cell profile and accounting for multiple testing (OR=0.18, q-value = 0.05). The result was robust to sensitivity analyses accounting for time between enrollment and diagnosis, race, tumor subtype, and secondhand smoke exposure.
Conclusions
While results need to be confirmed in additional prospective studies, differential methylation in CITED4, as measured in blood, is a promising marker of bladder cancer susceptibility.
Impact
Identification of biomarkers of bladder cancer susceptibility in easily accessible tissues may allow targeting of screening efforts so as to improve bladder cancer prognosis. This is particularly important among women, who tend to have poorer bladder cancer outcomes than men.
INTRODUCTION
According to 2016 estimates, urinary bladder cancer is the 12th most frequently diagnosed cancer among women in the US and is responsible for approximately 4,570 deaths among women each year (1). The incidence of bladder cancer increases with age (2), and it is most frequently diagnosed between the ages of 55 and 84 (SEER 18 data; 2009-2013). The most common histology of bladder cancer is transitional cell carcinoma (TCC), which originates in the urothelial cells of the inner lining of the bladder (3) and accounts for 94% of bladder cancer cases (4). Based on prognostic characteristics, TCC can be further divided into non-muscle invasive (NMIBC) and muscle-invasive (MIBC) subtypes, which represent 75% and 25% of all newly diagnosed disease, respectively (5). While patients with NMIBC are at high risk of recurrence, patients with MIBC have relatively poor survival rates.
Perhaps due to similarities between urinary tract infections and early bladder cancer symptoms causing delays in seeking clinical evaluation, women are 21% more likely to be diagnosed with advanced bladder cancer than men (6,7). Once diagnosed, the risk of death from bladder cancer is approximately 1.5 times greater for women (8). As such, there is great need for screening indicators that would aid in the early diagnosis of bladder cancer, particularly among women.
Differential DNA methylation is a particularly promising biomarker of cancer susceptibility because of its well-established links to human cancer (9,10). Altered methylation is known to both silence tumor suppressor genes and to activate oncogenes (10), and changes in DNA methylation are common and early events in bladder carcinogenesis (11). For example, Wolff et al. found 526 hypermethylated loci in invasive urothelial tumors as compared to urothelial tissue from cancer-free participants, and non-invasive and invasive urothelial tumors shared 117 differentially hypermethylated loci, indicating these changes are present during the early development of bladder cancer (12).
Only one previous study has evaluated the association of genome-wide methylation, as measured in blood, with bladder cancer risk. While it identified 9 strongly associated loci, the study considered only a limited number of total loci (~27k), included only 112 cases and 118 controls, and used post-diagnostic blood samples collected an average of 1 year after diagnosis (13). As such, DNA methylation levels may have been influenced by the presence of cancer or its treatment (4).
To our knowledge, ours is the first study of genome-wide DNA methylation, as measured in pre-diagnostic blood, and bladder cancer risk. In addition to a much larger sample size, we utilized the Illumina Infinium HumanMethylation450 Bead Array to capture a greater number of CpG sites across the genome. Our study focuses entirely on women, who are understudied population more likely to experience adverse bladder cancer outcomes and would particularly benefit from biomarkers of bladder cancer susceptibility.
MATERIALS AND METHODS
Study Setting
This study was approved by the Institutional Review Board of the Fred Hutchinson Cancer Research Center. The study was nested in the WHI, a prospective cohort of 161,808 postmenopausal women from across the US, recruited from 1993 to 1998, who were between the ages of 50 and 79 at enrollment (14). There are two arms of the WHI, the clinical trials study (CT) and the observational study (OS), with 68,132 and 93,676 participants, respectively. The CT involved concurrent randomized controlled trials of hormone therapy, dietary modification, and calcium/vitamin D, and ended in 2005. Those not able or willing to participate in the clinical trials were asked to participate in the OS. After 2005, WHI participants were invited to enroll in the WHI Extension Studies, which tracked health outcomes for another 10 years.
Study Subjects
Bladder cancer cases were identified through annual medical questionnaires, and confirmed by blinded physician adjudicators using standard criteria based on pathology, cytology, and operative reports and on hospital discharge information. As of September 2012, 618 WHI participants were diagnosed with bladder cancer. We restricted to the 584 participants diagnosed with the TCC subtype of bladder cancer. We excluded cases lacking eligible controls, cases diagnosed with any cancer before baseline, and cases that did not have a sufficient amount of baseline DNA. We also removed the only American Indian case and her matched control to allow for accurate estimation of the race/ethnicity covariate. Selected cases were followed for a median of 7.22 years until bladder cancer diagnosis. Cancer-free controls were matched to cases on year of enrollment, age at enrollment (+/- 2 years), follow-up time greater than or equal to their matched case, trial component, and DNA extraction method (5-prime, phenol, or PureGene).
Once the methylation data were generated, we checked genetic distance among our study samples and discovered a genetically identical case-control pair. This issue was confined to the single case-control pair, since quality control duplicates located on the same assay plate demonstrated high levels of genetic concordance with their replicates on other plates. The problematic case-control pair was excluded from all analyses. Our final sample size included 440 case-control pairs, where 228 cases were OS participants and 212 were CT participants. Of the 440 cases, 105 were classified as having MIBC.
Data and Biospecimen Collection
During initial screening for the WHI, basic demographic information was collected, and eligible women were invited for a clinic visit. Staff collected physical measurements and blood specimens at baseline. Blood samples were taken after at least 12 hours of fasting, divided into aliquots, centrifuged, and stored at −70°C as buffy coats. Self-administered questionnaires were also collected at baseline to gather information related to risk factors for various health outcomes of interest, including detailed data on smoking. Participants were asked to report whether they had ever smoked at least 100 cigarettes in their lifetime, age at smoking initiation, cigarettes per day, and years of smoking, as well as age at quitting smoking (if applicable), from which smoking status (current, former, never) and pack-years were computed. For OS participants, information on second-hand smoke exposure was captured at baseline by asking the participant if she lived with an inside-smoker as a child, lived with an inside-smoker as an adult, and if she worked in a place where people smoked. Second-hand smoke exposure data were not collected for CT participants.
DNA Methylation Array
We used the Illumina 450K Infinium HumanMethylation beadchip to interrogate methylation status at 485,577 CpG sites. The array covers RefSeq genes (99%) with an average of 17 CpG sites per gene distributed across various regions including the first exon, 3’ and 5’ untranslated regions, the gene body, and close proximity to transcription start sites. Within or near a gene, CpGs can also occur in CpG islands, which are regions with a high density of CpG sites. If occurring within 2 kb of a CpG island, a locus is in the shore of the CpG island. If occurring within 2kb of a CpG shore, a locus is in the shelf of the CpG island (15).
DNA was previously extracted from buffy coats using 5-prime, phenol, or PurGene methods and stored at −70°C. DNA was quantified by Picogreen (Invitrogen, Carlsbad, CA). The EZ DNA Methylation™ kit (Zymo Research, Irvine, CA) was used to treat 500ng of DNA with sodium bisulfate, which converts unmethylated cytosine to uracil. Converted DNA was stored for no more than one month at −80°C prior to being assayed. Following standard Infinium HD Methylation protocols (Illumina, San Diego, CA), 4ul of converted DNA was denatured and neutralized to prepare it for amplification. The denatured DNA was isothermally amplified at 37°C, followed by end-point enzymatic fragmentation and isopropanol precipitation. The DNA was then resuspended in Illumina hybridization buffer. Twelve samples were applied to each Illumina Human Methylation 450k beadchip and were kept separated with an IntelliHyb seal. The prepared beadchip was then incubated in a hybridization oven at 48°C for 16-24 hours with rocking. Unhybridized and non-specifically hybridized DNA was washed away. The beadchip underwent staining and extension in capillary flow-through chambers on a Freedom EVO liquid handling robot (Tecan Trading AG, Switzerland), after which beadchips were scanned using the Illumina iScan+. Twenty-one blind duplicate sample pairs were randomly placed among study samples for quality control assessment.
We used the M-value to measure methylation at each CpG site in order to reduce heteroscedasticity, as compared to the β value (16). The M-value is the base 2 logit of the β-value, where the β-value is the ratio of the methylated signal over the total signal and is interpreted as the percent of methylation at a specific site (17).
Methylation Data Processing
After reading in the raw image files and checking for failed samples, we performed normalization in two steps. First, we corrected the probe intensity levels for background fluorescence, to avoid potential dye-bias, using the normal-exponential convolution with out-of-band probes (noob) method (18). Next, we performed functional normalization to adjust the marginal distribution of methylation levels using summarized information from the control and out-of-band probes (19). This method effectively isolates unwanted variation, including batch effects, and performs well in studies of cancer (19). After normalization, we sequentially excluded probes that had detection p-values greater than or equal to 0.01 in at least 10% of samples (707 probes), probes with a beadcount less than 3 in at least 10% of samples (215 probes), single nucleotide polymorphism (SNP)-related probes (87,706 probes), cross-reactive probes (24,939 probes), probes located on the sex chromosomes (9,578 probes), and non-CpG sites (1,183 probes). Because SNP-related probes may impact probe binding (20), we removed probes with any SNP present at the CpG interrogation or single nucleotide extension site as identified by the Illumina annotation, and probes within 10 base pairs of a common SNP (minor allele frequency > 1%) based on 1000 Genomes data from ‘Illumina450ProbeVariants.db’ (21). Previously identified cross-reactive probes were also excluded (20,22). We performed these steps using the ‘minfi’ and ‘wateRmelon’ packages within R (version 3.4.0) (23,24).
Imputation of Missing Data
We used multivariate imputation by chained equations (MICE) to impute values for missing covariate data (25). Using default imputation model specifications, we generated 5 datasets (10 iterations per dataset) with complete covariate information. As the first step in generating each dataset, we multiply imputed missing values for the pack-years and race/ethnicity covariates. Our predictor set for the initial phase of imputation included these variables as well as: age at baseline, WHI arm (CT, OS), ever smoking status, cell type composition, case/control status, and methylation levels at 123 CpG sites. These sites were identified by including the top 100 sites associated with current smoking and the top 100 sites correlated with pack-years among current smokers based on a large study by Ambatipudi et al. (26). While there were 155 unique sites from these lists, we only used the 123 loci that remained in our dataset after applying our processing pipeline. As the second step, within each dataset with imputed values for pack-years and race/ethnicity, we added smoking status at baseline to the predictor set, and then singly imputed missing values for current or former smoking status among smokers (pack-years > 0) and assumed never smoking status among non-smokers (pack-years = 0).
Statistical Analysis
We used conditional logistic regression models to assess the association between bladder cancer case-control status and each continuous M-value after adjusting for potential confounders, including race/ethnicity (Asian/Pacific Islander, black/African American, Hispanic/Latino, non-Hispanic white, other), smoking status (never, former, current), pack-years of smoking (continuous), as well as the estimated proportion of CD4+ T-cells (continuous), CD8+ T-cells (continuous), natural killer cells (continuous), granulocytes (continuous), B-cells (continuous), and monocytes (continuous). The relative contributions of the six major white blood cell types were reconstructed using the Houseman et al. method (27). To account for multiple testing, we calculated q-values by adjusting p-values using the false-discovery rate (FDR). Associations with bladder cancer at a q-value less than or equal to 0.05 were deemed statistically significant.
Since we performed multiple imputation, the conditional logistic regression analysis for each site was repeated in all of the imputed datasets. To determine the combined parameter estimates and their variance, the results from each of the five complete data analyses were pooled using the approach of Rubin (28) and implemented in the ‘mice’ R package (25).
Sensitivity Analyses
To address the potential influence of early, undiagnosed cancer at the time of blood draw on DNA methylation, we performed a sensitivity analysis restricted to cases diagnosed at least 3 years after enrollment and their matched controls. To address any possible residual confounding by race/ethnicity, we also completed an analysis restricting to case-control pairs that had matching races/ethnicities. We also performed an analysis restricted to the MIBC bladder cancer subtype to explore the impact of clinically heterogeneous bladder cancer subtypes on our primary results. To address the potential impact of secondhand smoke exposure, we restricted to OS participants and additionally adjusted for secondhand smoke exposure at home as a child (yes, no), secondhand smoke exposure at home as an adult (yes, no), and secondhand smoke exposure at work (yes, no). Given the importance of smoking as a potential confounder, we also completed our analyses restricting to never smokers. In addition, we completed an unmatched, genome-wide analyses with the M-value of each CpG site as the outcome to check for associations with smoking status (current, never), pack-years of smoking and second-hand smoke exposure. The absence of associations among our top hits with these smoking variables would help address concerns that our primary findings were driven by residual confounding from incomplete adjustment for tobacco smoke exposure.
RESULTS
Genome-wide Analysis of DNA Methylation
Based on the 65 SNP probes included on the 450k array, the genetic distance between the 21 blind duplicate pairs in our study ranged from 0.005 to 0.026, verifying that these samples were genetically identical. The Pearson correlation coefficients between the methylation measurements (β-values) across all 361,184 loci were at least 0.995 for each duplicate pair, indicating suitable precision of the assay.
The distribution of selected variables is presented by case-control status in Table 1. Compared to controls, a greater proportion of cases were white, were more likely to be past and current smokers and to have greater pack years of smoking. While OS cases were equally likely as controls to be exposed to secondhand smoke at home as children and at work, they were slightly more likely than controls to be exposed to secondhand smoke at home as adults. Inferred cell-type composition was similar across cases and controls.
TABLE 1.
Distribution of relevant demographic and clinical characteristics among bladder cancer cases and controls nested within the Women’s Health Initiative.
| BC Cases | Controls | |
|---|---|---|
| Age (mean, SD) | 65.12 (7.04) | 65.12 (7.03) |
| Race (N, %) | ||
| Asian/Pacific Islander | 4 (1%) | 12 (3%) |
| Black/African American | 25 (6%) | 45 (10%) |
| Hispanic/Latino | 8 (2%) | 16 (4%) |
| Non-Hispanic White | 400 (91%) | 360 (82%) |
| Other | 2 (<1%) | 7 (2%) |
| Missing | 1 (<1%) | 0 (0%) |
| Smoking (N, %) | ||
| Never Smoked | 156 (35%) | 236 (54%) |
| Past Smoker | 218 (50%) | 178 (40%) |
| Current Smoker | 56 (13%) | 19 (4%) |
| Missing | 10 (2%) | 7 (2%) |
| Pack-Years (N, %) | ||
| Never Smoker | 156 (35%) | 236 (54%) |
| < 5 | 36 (8%) | 68 (15%) |
| 5 - <20 | 63 (14%) | 46 (10%) |
| >= 20 | 162 (37%) | 78 (18%) |
| Missing | 23 (5%) | 12 (3%) |
| Child Secondhand Smoke Exposure at Home (N, %)a | ||
| No | 80 (18%) | 78 (18%) |
| Yes | 141 (32%) | 145 (33%) |
| Don’t Know | 4 (1%) | 4 (1%) |
| Missing | 215 (49%) | 213 (48%) |
| Adult Secondhand Smoke Exposure at Home (N, %)a | ||
| No | 48 (11%) | 70 (16%) |
| Yes | 178 (40%) | 155 (35%) |
| Missing | 214 (49%) | 215 (49%) |
| Secondhand Smoke Exposure at Work (N, %)a | ||
| No | 46 (10%) | 54 (12%) |
| Yes | 180 (41%) | 173 (39%) |
| Missing | 214 (49%) | 213 (48%) |
| Cell-type Composition (mean, SD)b | ||
| B-Cells | 0.06 (0.03) | 0.06 (0.03) |
| CD8 T-Cells | 0.09 (0.05) | 0.09 (0.04) |
| CD4 T-Cells | 0.19 (0.08) | 0.17 (0.07) |
| Natural Killer Cells | 0.09 (0.05) | 0.09 (0.04) |
| Granulocytes | 0.50 (0.13) | 0.51 (0.13) |
| Monocytes | 0.11 (0.03) | 0.11 (0.03) |
| Tumor Behavior (N, %) | ||
| Non-muscle Invasive Bladder Cancer | 335 (76%) | N/A |
| Muscle Invasive Bladder Cancer | 105 (24%) | N/A |
Information about exposure to secondhand smoke was only available for participants in the Observational Study arm, so the 212 case-control pairs from the clinical trials arm were missing this data.
Cell-type composition was calculated using the Houseman method (27).
As shown in Table 1, 35 participants were missing pack-years of smoking data and 17 of those missing pack-year data were also missing smoking status data. A single participant was missing race/ethnicity data. The imputed datasets are summarized in Supplementary Table 1.
Results for the 10 CpG sites with the lowest q-values in association with bladder cancer are summarized in Table 2, while results for all 361,184 CpG loci are provided in Supplementary Table 2. These results are also summarized as a volcano plot in Figure 1, with the top association highlighted. After adjusting for multiple comparisons, increased methylation at cg22748573 was statistically significantly associated with a reduced risk of bladder cancer (OR=0.18 per unit increase in M-value, q-value = 0.05). cg22748573 is located within the chr1:41326949-41328285 CpG island in the 5-’UTR of the CBP/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 4 (CITED4) gene, and it had an average methylation level of 6% in controls. Although not statistically significant, increased methylation at cg20010635 was strongly associated with risk of bladder cancer (OR = 8.42 per unit increase in M-value, q-value = 0.40). The cg20010635 CpG site is located in the body of the calmodulin binding transcription activator 1 (CAMTA1) gene and had an average methylation of 95% among controls.
TABLE 2.
Top 10 CpG sites in tests of association between methylation level and bladder cancer risk in pre-diagnostic blood from the Women’s Health Initative.
| Illumina ID | Chra | Position | Gene | Gene Groupa | Relation to CpG Islandb | Average β-value in Controls | Odds Ratio | 95% CIc | P-value | Q-valued |
|---|---|---|---|---|---|---|---|---|---|---|
| cg22748573 | chr1 | 41327924 | CITED4 | 5′UTR;1stExon | Island | 0.058 | 0.18 | (0.09, 0.34) | 1.5×10−7 | 0.05 |
| cg20010635 | chr1 | 7811409 | CAMTA1 | Body | OpenSea | 0.952 | 8.42 | (3.48, 20.33) | 2.2×10−6 | 0.40 |
| cg25955565 | chr14 | 71863756 | SNORD56B | TSS1500 | OpenSea | 0.945 | 9.29 | (3.59, 24.08) | 4.5×10−6 | 0.41 |
| cg26492847 | chr11 | 96063104 | MAML2 | Body | OpenSea | 0.929 | 7.24 | (3.01, 17.38) | 9.5×10−6 | 0.41 |
| cg06414161 | chr16 | 53534040 | AKTIP | Body | Shelf | 0.931 | 8.93 | (3.36, 23.72) | 1.1×10−5 | 0.41 |
| cg27627854 | chr11 | 108158382 | ATM | Body;1stExon | OpenSea | 0.905 | 4.19 | (2.21, 7.98) | 1.2×10−5 | 0.41 |
| cg02393449 | chr10 | 70743010 | DDX21 | 3′UTR | OpenSea | 0.929 | 6.91 | (2.89, 16.53) | 1.4×10−5 | 0.41 |
| cg26695157 | chr12 | 109985283 | OpenSea | 0.917 | 3.71 | (2.05, 6.72) | 1.5×10−5 | 0.41 | ||
| cg17803089 | chr6 | 35419793 | FANCE | TSS1500 | Shore | 0.036 | 0.18 | (0.08, 0.39) | 1.8×10−5 | 0.41 |
| cg26729242 | chr10 | 96980135 | C10orf129 | Body | OpenSea | 0.906 | 3.00 | (1.81, 4.96) | 1.9×10−5 | 0.41 |
Chr = Chromosome
Functional region of gene as indicated in v1.2 Illumina annotation: TSS1500 = 200–1500 bases upstream of the transcription start site; TSS200 = 0–200 bases upstream of the transcription start site; 5′UTR = Within the 5 prime untranslated region; 1stExon = First segment of gene coding for peptide sequence; Body = Between the ATG and stop codon; 3′UTR = Between the stop codon and poly A signal; Multiple listings indicate a locus in a region with multiple splice variants.
Position relative to CpG island as indicated in v1.2 Illumina annotation: Island = Within CpG island (CG content > 50%, Obs/Exp CpG ratio > 0.60, and length > 200 bps); OpeanSea = Non-island region; Shore = 0–2 kb flanking CpG Island; Shelf = 2–4 kb flanking CpG Island.
95% confidence interval for odds ratio estimate.
Q-values represent p-values adjusted for multiple testing using the FDR method.
Figure 1.
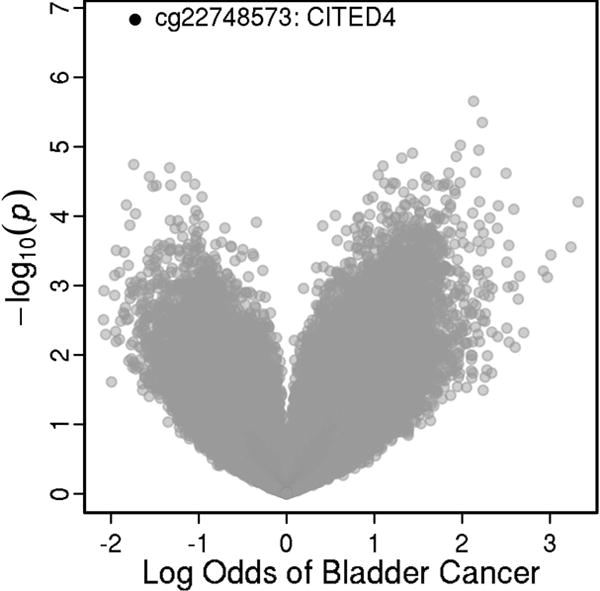
Figure 1 is a volcano plot of results from the analysis of 361,184 CpG sites in association with risk of bladder cancer in the Women’s Health Initative. The figure plots statistical significance versus effect size for the association of CpG sites with bladder cancer. The locus with the most statistically significant association (cg22748573) is highlighted.
Sensitivity Analyses
Most of the sensitivity analyses yielded odds ratios for cg22748573 that were similar to or slightly stronger than the result from the main analysis, suggesting little bias in the odds ratio estimate. However, as expected, statistical significance was reduced due to the decreased sample size in each of these analyses. When examining the potential impact of undiagnosed disease, we excluded 74 case-control pairs where the case was diagnosed with bladder cancer within 3 years of study entry, and cg22748573 remained the most significant finding (OR = 0.15, q-value = 0.06). In the race/ethnicity sensitivity analysis, we included only the 337 case-control pairs with matching race/ethnicity, and cg22748573 was no longer statistically significantly associated with bladder cancer; however, cg22748573 still had the 3rd lowest p-value (OR = 0.19, q-value = 0.47). With very small numbers in various non-white race/ethnic groups, the MIBC analysis had to be restricted to 96 case-control pairs with white or black race/ethnicity for our statistical models to converge. While the analysis produced null results, the cg22748573 locus in CITED4 was the 2nd most significant locus and the magnitude of the association was much stronger than in the overall TCC analysis (OR = 0.03, q-value = 0.71).
After restricting to OS participants and additionally imputing the secondhand smoke exposure variables for 8 subjects, the sensitivity analysis for secondhand smoke exposure was also null, but had cg22748573 as the 11th most significant locus (OR = 0.13, q-value = 0.87). With only 85 case-control pairs in the analysis restricted to never smokers, cg22748573 was only the 530th most significant locus (OR = 0.08, q-value = 0.93), but the magnitude of the association with bladder cancer was stronger than the result from our primary analysis. In our analyses of associations between genome-wide DNA methylation and the various tobacco smoke exposure variables, no statistically significant associations were observed with any of the differentially methylated CpG sites highlighted in Table 2 (unpublished observations).
DISCUSSION
We identified a differentially methylated locus in pre-diagnostic blood that was statistically significantly associated with an 82% reduction in bladder cancer risk. The cg22748573 locus resides in a CpG island within the putative promoter region of CITED4. Though methylation is often tissue-specific, methylation levels are more likely to be conserved across various tissues in CpG islands. Price et al. observed that probes differentially methylated between tissues were depleted in Illumina-annotated islands (29), and Eckhardt et al. reported that only 13% of tissue-specific differentially methylated regions in the 5’-UTR were located within CpG islands (30). Though speculative, this suggests that the methylation status of cg22748573 in blood may reflect that of normal bladder tissue.
There is evidence that CITED4 hypermethylation is associated with better cancer outcomes (31), which is unexpected, since CITED genes have been identified as tumor suppressors (32–34), and methylation of CpG loci in promoter regions is typically associated with transcriptional silencing (35–37). However, based on data from subjects in The Cancer Genome Atlas (TCGA) with both Illumina 450k array methylation and Illumina HiSeq RNA-Seq gene expression data (38), we found that methylation at cg22748573 was not significantly associated with CITED4 expression in normal bladder tissue (Spearman correlation = −0.22, p-value = 0.31). Therefore, the importance of the methylation change at cg22748573 may not be its impact on CITED4 expression. Instead, this methylation change may be a marker of decreased susceptibility to the development of bladder cancer through other mechanisms. As an example, hypermethylation of cg22748573 has been closely associated with deletion of 1p and 19q in oligodendrogliomas (31), where tumors with 1p/19q tumor deletions often lack TP53 mutations (39) and have better patient prognoses (31,40).
Tews et al. observed CITED4 hypermethylation in glioma patients to be associated with longer recurrence-free and overall survival, but hypermethylation was not observed in matched leukocyte DNA (31). However, no comparisons between normal brain tissue and blood were made. Given the genomic instability of tumors, one would expect the correlation with DNA methylation levels to be lower for tumor than for normal tissue. Even if blood does not reflect the methylation state of CITED4 in bladder tissue, differential methylation of CITED4, a gene which has been shown to play a central role in blood cell differentiation in mice (41), may reflect systemic changes due to factors like inflammation or environmental exposures that are associated with bladder cancer. In this context, differential methylation of CITED4 could still be a valuable marker of susceptibility to bladder cancer.
While not statistically significant at the genome-wide level, the second-most significant association was one of increased bladder cancer risk with increased methylation at the cg20010635 locus, occurring in the gene body of CAMTA1. CAMTA1 has been shown to act as an oncogene when fused with WWTR1 in epithelioid hemangioendothelioma (42). Since CAMTA1 is usually only expressed in the brain (42), its role in bladder carcinogenesis is uncertain.
Though not among the top 10 associations highlighted in Table 2, we also observed a particularly strong association between increased methylation at cg13413384 and risk of bladder cancer (OR = 20.30 per unit increase in M-value, q-value = 0.44). This locus is found in the body of the retinoid X receptor-α (RXRA) gene, an oncogene found to be significantly altered in some bladder cancers (43). In our study, the observed methylation change occurred in the RXRA gene body, where increased methylation levels have a well-supported association with higher expression levels (44–47).
Marsit et al. conducted the first study of loci-specific genome-wide DNA methylation of bladder cancer in blood (48). This study identified the top 9 loci associated with bladder cancer among 112 cases and 118 controls. While CITED4 was not among their top hits, the Marsit et al. study was comparatively underpowered and utilized post-diagnostic blood samples.
To our knowledge, ours is the first study to investigate the potential link between genome-wide DNA methylation in blood and future risk of bladder. Being nested in the WHI, we benefited from a relatively large sample size with detailed baseline information on potential confounders.
Even with pre-diagnostic blood samples, it is possible that methylation measurements in blood reflected early tumor development (49). However, our sensitivity analysis restricted to cases diagnosed at least 3 years after enrollment suggests that the association observed with cg22748573 is not driven by the presence of early cancer. Sensitivity analyses also suggested that residual confounding due to inadequate control of exposure to tobacco smoke did not drive our primary result.
Our results suggest that subtypes of TCC may share some methylation susceptibility markers; however, we were underpowered to specifically examine genome-wide associations between DNA methylation and MIBC. We will explore efforts to pool data with future studies of DNA methylation and bladder cancer in an effort to increase power to evaluate subtype-specific effects.
Ideally, pre-diagnostic measurements of DNA methylation would be measured in the tissue of interest. Accessing bladder tissue, however, involves invasive procedures. While urine samples can provide epithelial cells from the bladder, they also include epithelial cells from the rest of the urinary tract. The contribution of cells from the various tissues in the urinary tract can be highly variable (50), and urine samples were only collected from three of the clinical sites in the WHI.
Overall, we observed differential methylation of cg22748573 in pre-diagnostic blood to be strongly and significantly associated with bladder cancer risk among post-menopausal women. If confirmed in additional prospective studies, molecular studies should be conducted to determine the exact role of this locus in bladder carcinogenesis. Future prospective studies should involve both men and women and should evaluate the contribution of methylation measurements at this locus to risk prediction models that account for smoking history and germline genetic polymorphisms previously associated with bladder cancer risk.
Supplementary Material
Acknowledgments
Financial Support
Research reported in this publication was supported by the American Cancer Society under award number 125299-RSG-13-100-01-CCE. K.M. Jordahl was partially supported by the National Cancer Institute of the National Institutes of Health under award number R25 CA094880.
Footnotes
Disclosure of Potential Conflicts of Interest
No conflicts of interest to report.
References
- 1.American Cancer Society. Cancer Facts and Figures 2016. Atlanta: American Cancer Society; 2016. [Google Scholar]
- 2.Cancer of the Urinary Bladder - SEER Stat Fact Sheets [Internet] [cited 2016 Aug 26]. Available from: http://seer.cancer.gov/statfacts/html/urinb.html.
- 3.What is bladder cancer? [Internet] [cited 2016 Aug 29]. Available from: http://www.cancer.org/cancer/bladdercancer/detailedguide/bladder-cancer-what-is-bladder-cancer.
- 4.Zhang Y, Zhu C, Curado MP, Zheng T, Boyle P. Changing patterns of bladder cancer in the USA: evidence of heterogeneous disease. BJU Int. 2012;109:52–6. doi: 10.1111/j.1464-410X.2011.10283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanli O, Dobruch J, Knowles MA, Burger M, Alemozaffar M, Nielsen ME, et al. Bladder cancer. Nat Rev Dis Primer. 2017;3:17022. doi: 10.1038/nrdp.2017.22. [DOI] [PubMed] [Google Scholar]
- 6.Cárdenas-Turanzas M, Cooksley C, Pettaway CA, Sabichi A, Grossman HB, Elting L. Comparative outcomes of bladder cancer. Obstet Gynecol. 2006;108:169–75. doi: 10.1097/01.AOG.0000223885.25192.91. [DOI] [PubMed] [Google Scholar]
- 7.Dobruch J, Daneshmand S, Fisch M, Lotan Y, Noon AP, Resnick MJ, et al. Gender and Bladder Cancer: A Collaborative Review of Etiology, Biology, and Outcomes. Eur Urol. 2016;69:300–10. doi: 10.1016/j.eururo.2015.08.037. [DOI] [PubMed] [Google Scholar]
- 8.Scosyrev E, Noyes K, Feng C, Messing E. Sex and racial differences in bladder cancer presentation and mortality in the US. Cancer. 2009;115:68–74. doi: 10.1002/cncr.23986. [DOI] [PubMed] [Google Scholar]
- 9.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 10.Kulis M, Esteller M. 2 - DNA Methylation and Cancer. In: Ushijima ZH, T, editors. Adv Genet [Internet] Academic Press; 2010. pp. 27–56. [cited 2015 Oct 19] Available from: http://www.sciencedirect.com/science/article/pii/B9780123808660600022. [DOI] [PubMed] [Google Scholar]
- 11.Besaratinia A, Cockburn M, Tommasi S. Alterations of DNA methylome in human bladder cancer. Epigenetics Off J DNA Methylation Soc. 2013;8:1013–22. doi: 10.4161/epi.25927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolff EM, Chihara Y, Pan F, Weisenberger DJ, Siegmund KD, Sugano K, et al. Unique DNA methylation patterns distinguish noninvasive and invasive urothelial cancers and establish an epigenetic field defect in premalignant tissue. Cancer Res. 2010;70:8169–78. doi: 10.1158/0008-5472.CAN-10-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marsit CJ, Koestler DC, Christensen BC, Karagas MR, Houseman EA, Kelsey KT. DNA Methylation Array Analysis Identifies Profiles of Blood-Derived DNA Methylation Associated With Bladder Cancer. J Clin Oncol. 2011;29:1133–9. doi: 10.1200/JCO.2010.31.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hays J, Hunt JR, Hubbell FA, Anderson GL, Limacher M, Allen C, et al. The Women’s Health Initiative recruitment methods and results. Ann Epidemiol. 2003;13:S18–77. doi: 10.1016/s1047-2797(03)00042-5. [DOI] [PubMed] [Google Scholar]
- 15.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–95. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 16.Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dedeurwaerder S, Defrance M, Bizet M, Calonne E, Bontempi G, Fuks F. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief Bioinform. 2014;15:929–41. doi: 10.1093/bib/bbt054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright ML, Dozmorov MG, Wolen AR, Jackson-Cook C, Starkweather AR, Lyon DE, et al. Establishing an analytic pipeline for genome-wide DNA methylation. Clin Epigenetics. 2016;8:45. doi: 10.1186/s13148-016-0212-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fortin J-P, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:503. doi: 10.1186/s13059-014-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–9. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butcher L. Illumina 450ProbeVariants.db: Annotation Package combining variant data from 1000 Genomes Project for Illumina HumanMethylation450 Bead Chip probes. R Package. 2013 [Google Scholar]
- 22.Nordlund J, Bäcklin CL, Wahlberg P, Busche S, Berglund EC, Eloranta M-L, et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013;14:r105. doi: 10.1186/gb-2013-14-9-r105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pidsley R, Wong YCC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Buuren S. Groothuis-Oudshoorn K. mice: Multivariate Imputation by Chained Equations in R. J Stat Softw [Internet] 2011 [cited 2017 Jul 6];45. Available from: http://www.jstatsoft.org/v45/i03.
- 26.Ambatipudi S, Cuenin C, Hernandez-Vargas H, Ghantous A, Le Calvez-Kelm F, Kaaks R, et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8:599–618. doi: 10.2217/epi-2016-0001. [DOI] [PubMed] [Google Scholar]
- 27.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubin DB. Multiple Imputation for Nonresponse in Surveys. Hoboken, NJ, USA: John Wiley & Sons, Inc; 1987. [Google Scholar]
- 29.Price EM, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, et al. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6:4. doi: 10.1186/1756-8935-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–85. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tews B, Roerig P, Hartmann C, Hahn M, Felsberg J, Blaschke B, et al. Hypermethylation and transcriptional downregulation of the CITED4 gene at 1p34.2 in oligodendroglial tumours with allelic losses on 1p and 19q. Oncogene. 2007;26:5010–6. doi: 10.1038/sj.onc.1210297. [DOI] [PubMed] [Google Scholar]
- 32.Fox SB, Bragança J, Turley H, Campo L, Han C, Gatter KC, et al. CITED4 inhibits hypoxia-activated transcription in cancer cells, and its cytoplasmic location in breast cancer is associated with elevated expression of tumor cell hypoxia-inducible factor 1alpha. Cancer Res. 2004;64:6075–81. doi: 10.1158/0008-5472.CAN-04-0708. [DOI] [PubMed] [Google Scholar]
- 33.Huang KT, Takano EA, Mikeska T, Byrne DJ, Dobrovic A, Fox SB. Aberrant DNA methylation but not mutation of CITED4 is associated with alteration of HIF-regulated genes in breast cancer. Breast Cancer Res Treat. 2011;130:319. doi: 10.1007/s10549-011-1657-1. [DOI] [PubMed] [Google Scholar]
- 34.Rogers MA, Kalter V, Marcias G, Zapatka M, Barbus S, Lichter P. CITED4 gene silencing in colorectal cancer cells modulates adherens/tight junction gene expression and reduces cell proliferation. J Cancer Res Clin Oncol. 2016;142:225–37. doi: 10.1007/s00432-015-2011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 36.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12:529–41. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 38.Díez-Villanueva A, Mallona I, Peinado MA. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenetics Chromatin. 2015;8:22. doi: 10.1186/s13072-015-0014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reifenberger G, Louis DN. Oligodendroglioma: toward molecular definitions in diagnostic neuro-oncology. J Neuropathol Exp Neurol. 2003;62:111–26. doi: 10.1093/jnen/62.2.111. [DOI] [PubMed] [Google Scholar]
- 40.Boots-Sprenger SHE, Sijben A, Rijntjes J, Tops BBJ, Idema AJ, Rivera AL, et al. Significance of complete 1p/19q co-deletion, IDH1 mutation and MGMT promoter methylation in gliomas: use with caution. Mod Pathol. 2013;26:922–9. doi: 10.1038/modpathol.2012.166. [DOI] [PubMed] [Google Scholar]
- 41.Yahata T, Takedatsu H, Dunwoodie SL, Bragança J, Swingler T, Withington SL, et al. Cloning of Mouse cited4, a Member of the CITED Family p300/CBP-Binding Transcriptional Coactivators: Induced Expression in Mammary Epithelial Cells. Genomics. 2002;80:601–13. doi: 10.1006/geno.2002.7005. [DOI] [PubMed] [Google Scholar]
- 42.Tanas MR, Sboner A, Oliveira AM, Erickson-Johnson MR, Hespelt J, Hanwright PJ, et al. Identification of a Disease-Defining Gene Fusion in Epithelioid Hemangioendothelioma. Sci Transl Med. 2011;3:98ra82–98ra82. doi: 10.1126/scitranslmed.3002409. [DOI] [PubMed] [Google Scholar]
- 43.Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015;15:25–41. doi: 10.1038/nrc3817. [DOI] [PubMed] [Google Scholar]
- 44.Hellman A, Chess A. Gene Body-Specific Methylation on the Active X Chromosome. Science. 2007;315:1141–3. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 45.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ball MP, Li JB, Gao Y, Lee J-H, LeProust EM, Park I-H, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–8. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–31. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marsit CJ, Koestler DC, Christensen BC, Karagas MR, Houseman EA, Kelsey KT. DNA Methylation Array Analysis Identifies Profiles of Blood-Derived DNA Methylation Associated With Bladder Cancer. J Clin Oncol. 2011;29:1133–9. doi: 10.1200/JCO.2010.31.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu Z, Bolick SCE, DeRoo LA, Weinberg CR, Sandler DP, Taylor JA. Epigenome-wide Association Study of Breast Cancer Using Prospectively Collected Sister Study Samples. J Natl Cancer Inst. 2013;105:694–700. doi: 10.1093/jnci/djt045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng THT, Jiang P, Tam JCW, Sun X, Lee W-S, Yu SCY, et al. Genomewide bisulfite sequencing reveals the origin and time-dependent fragmentation of urinary cfDNA. Clin Biochem. 2017;50:496–501. doi: 10.1016/j.clinbiochem.2017.02.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.