

Abstract
Purpose
The clinical utility of circulating tumor DNA (ctDNA) monitoring has been shown in tumors that harbor highly recurrent mutations. Leiomyosarcoma (LMS) represents a type of tumor with a wide spectrum of heterogeneous genomic abnormalities; thus, targeting hotspot mutations or a narrow genomic region for ctDNA detection may not be practical. Here we demonstrate a combinatorial approach that integrates different sequencing protocols for the orthogonal detection of single nucleotide variants (SNVs), small indels and copy number alterations (CNAs) in ctDNA.
Experimental design
We employed Cancer Personalized Profiling by deep Sequencing (CAPP-Seq) for the analysis of SNVs and indels, together with a genome-wide interrogation of CNAs by Genome Representation Profiling (GRP). We profiled 28 longitudinal plasma samples and 25 tumor specimens from 7 patients with LMS.
Results
We detected ctDNA in 6 of 7 of these patients with >98% specificity for mutant allele fractions down to a level of 0.01%. We show that results from CAPP-Seq and GRP are highly concordant, and the combination of these methods allows for more comprehensive monitoring of ctDNA by profiling a wide spectrum of tumor-specific markers. By analyzing multiple tumor specimens in individual patients obtained from different sites and at different times during treatment, we observed clonal evolution of these tumors that was reflected by ctDNA profiles.
Conclusions
Our strategy allows for a comprehensive monitoring of a broad spectrum of tumor-specific markers in plasma. Our approach may be clinically useful not only in LMS but also in other tumor types that lack recurrent genomic alterations.
Keywords: circulating tumor DNA, liquid biopsy, leiomyosarcoma, tumor heterogeneity
INTRODUCTION
Recent improvements in next generation sequencing platforms have paved the way for the highly sensitive detection of ctDNA in plasma specimens. Current strategies for ctDNA analysis may be divided into three categories: 1) patient-specific approaches that utilize personalized assays (1–4); 2) tumor type-specific targeted sequencing that do not require patient-specific optimization (5–7); 3) tumor type-independent genome-wide analyses (8–11). The first approach is highly sensitive but is technically challenging and expensive. The second approach involves targeted sequencing methods such as CAPP-Seq (5, 6). It is highly sensitive, but is most practical in patients with tumor types that harbor highly recurrent aberrations that can be sequenced with a capture panel of a relatively limited size. The third approach, while broadly applicable, does not reach the sensitivity of the first two targeted approaches. In cancer types that are characterized by intermediate levels of recurrent single nucleotide variants (SNVs), small insertions and deletions (indels), or copy number aberrations (CNAs), a combination of multiple assays may improve the sensitivity of ctDNA detection while still retaining the benefits of broad applicability and cost-effectiveness. Leiomyosarcoma (LMS) is a suitable disease to explore the potential of such a combination approach for the orthogonal detection of multiple classes of alterations because these tumors are characterized by a wide range of DNA abnormalities spread across the whole genome. These include complex CNAs, as well as point mutations affecting multiple tumor suppressor genes such as TP53, RB1, ATM, ATR, ATRX and PTEN (12, 13).
LMS patients, like many other cancer patients, could greatly benefit from a non-invasive monitoring of tumor burden by liquid biopsies. Currently, the decision to initiate adjuvant treatment in LMS patients is based on the assessment of multiple prognostic factors related to patient performance, stage of the disease and type of surgery, as well as the potential benefits and side effects of the treatment. LMS ctDNA testing may improve the patients’ clinical outcome through earlier identification of candidates for adjuvant therapy. Longitudinal monitoring of ctDNA may also complement imaging-based regimens for long-term surveillance of LMS patients for disease recurrence.
Here we describe a proof-of-principle study to determine the feasibility of ctDNA analysis in patients diagnosed with tumors of moderate genomic complexity, through a simultaneous application of two separate methods, Cancer Personalized Profiling by deep Sequencing (CAPP-Seq) and Genome Representation Profiling (GRP) in LMS. The former is a deep, targeted exome sequencing approach optimized for ctDNA detection, which is ideal for the ultrasensitive quantitative analysis of SNVs, indels and fusion breakpoints. The clinical utility of monitoring ctDNA by CAPP-Seq has been previously demonstrated in patients with lung cancer and diffuse large B cell lymphoma (5, 14–16). The second approach, GRP, is based on shallow whole genome sequencing for the assessment of genome-wide copy number alterations and has been shown to detect ctDNA in patients with ovarian carcinoma, Hodgkin lymphoma and follicular lymphoma (9). Successful monitoring of CNAs in plasma has been also described previously in prostate cancer patients (11). In the present study, we demonstrate that the combination of these two techniques enables the reliable monitoring of a wide spectrum of molecular markers in ctDNA and this approach has a significant translational potential in LMS and other cancer types characterized with a comparable genomic complexity.
MATERIALS AND METHODS
LMS patient cohort
Nine LMS patients treated at the Stanford Cancer Institute provided informed consent to participate in the study and donated serial blood samples throughout the course of their treatment. The study was approved by the Stanford University Institutional Review Board (approvals IRB-31067 and IRB-31596). Clinical features of the patients included in this study are summarized in Supporting File: Table S1. Data from two patients have been excluded from the analysis due to failed QC or the absence of SNV/indels in tumor within the genomic region covered by CAPP-Seq panel. The data from the remaining 7 LMS patients have been used for the final analysis comparing CAPP-Seq and GRP. All LMS patients in the ctDNA monitoring analysis had either a primary tumor or metastatic disease confirmed by imaging at all blood collection time points.
Healthy donors
Blood specimens from 24 healthy donors used for CAPP-Seq analysis were collected into EDTA tubes (Beckton Dickinson). Plasma specimens from 428 volunteers (214 females and 214 males) used for GRP analysis were collected into Cell-free DNA BCT tubes (Streck). Collection of plasma from these asymptomatic donors was approved by the local Institutional Review Boards.
LMS-specific CAPP-Seq selector design
Whole exome sequencing data from 77 matched tumor-normal specimens from LMS patients from The Cancer Genome Atlas (TCGA) were used to design an LMS-specific CAPP-Seq capture panel. The analyses presented in the current publication are based on the use of study data downloaded from the dbGaP web site, under phs000178.v8.p7 (17). Paired-end sequencing reads were aligned to the human reference genome (GRCh37/hg19) using BWA-MEM (version 0.7.13) with the default settings (37). SAMtools (version 1.3) was used for converting SAM to BAM format, sorting and indexing the alignments (38). Picard (version 1.96) was used for the removal of duplicate reads (39). The GATK framework (version 3.3-0) was used for the local realignment and base call recalibration (39). SNVs and indels were identified using VarScan2 (version 2.3.7) (40). Variants identified by Varscan2 were annotated with ANNOVAR (41) and filtered for exonic or splice site non-synonymous SNVs, frameshift indels, stopgain and stoploss variants, requiring 0% VAF in the matched germline DNA. All variants were required to be present on at least one forward and one reverse sequencing read. Variants reported at >0.01% frequency in 1000Genomes and ExAC databases were excluded from the analysis. In addition, genes that are likely to produce false positive variant calls in next generation sequencing data were filtered out (including MUC, GOLG, NBP, ZNF, OR, WDR family genes). Somatic variants obtained after this pre-filtering were used to identify all exons mutated in at least two LMS patients. This output was further filtered based on the effective size of each exon, i.e. based on the recurrence index (5) defined as the number of patients with the mutation divided by the exon length in kb. Only exons with recurrence index > 0.5 were retained. Exons with poor mappability according to the Uniqueness of 35bp Windows from ENCODE/OpenChrom (Duke) track in UCSC Genome Browser GRCh37/hg19 were excluded (42). This analysis yielded a panel of 281 exons from 82 genes that were recurrently mutated in TCGA LMS cases. This panel covered 98.7% (76/77 cases) of the TCGA cohort with a median of 3 SNVs/indels per patient. This panel was extended with 25 exons from 7 genes (MED12, KRAS, CDKN2A, CDH1, KIT, HRAS, KDM6A) that carried mutations reported in 114 LMS patients in the COSMIC database v74 (accessed on 10/23/15). The final LMS-specific selector for CAPP-Seq covered the region of 184kb and included 306 exons from 89 genes (Supporting File: Table S2). A custom SeqCap EZ Choice Library (Roche) capture panel was designed using NimbleDesign software version 3.0 (Roche), with the maximum mismatches set to 1. The estimated coverage of the input genomic region of 184,871bp was 98.1%.
Blood sample collection and processing
Peripheral blood was collected into EDTA tubes (Beckton Dickinson) and plasma was separated by centrifugation at 2,500g for 20 minutes at room temperature, and stored at -80°C. The cell pellet containing white blood cells was frozen and banked for the germline DNA extraction. cfDNA was extracted from a median of 5mL plasma (range 3 – 5 mL) using QIAamp Circulating Nucleic Acid kit (Qiagen). Germline DNA was extracted from 200μl of PBLs using DNeasy Blood & Tissue kit (Qiagen). cfDNA and germline DNA was quantified using Quant-iT dsDNA High Sensitivity Assay (ThermoFisher Scientific).
Tumor specimens
Twenty-eight formalin fixed paraffin embedded (FFPE) tumor specimens (a median of 3 specimens per patient) from 9 LMS patients, collected between 2013 and 2015, were used for extraction of genomic DNA (Supporting File: Table S3). The analyzed specimens included 8 primary tumors and 20 metastatic/recurrent tumors. Diagnosis was confirmed by a surgical pathologist (MvdR). Tumor specimens selected for CAPP-Seq (n = 24) and SNP array (n = 26) analysis contained a median of 95% tumor cells (range 60 - 100%). Genomic DNA was extracted using AllPrep DNA/RNA FFPE kit (Qiagen). Twenty and twenty-two samples from 7 LMS patients passed the QC by CAPP-Seq and SNP array analysis, respectively (Supporting File: Table S3).
RNA sequencing of LMS tumors used for validation of CAPP-Seq selector
RNA-seq was performed on tumor specimens collected from twenty LMS patients treated at Stanford University Medical Center (19 FFPE and 1 frozen tumor specimen). These specimens included 8 uterine, 5 extremity and 7 thoracic/abdominal/retroperitoneal LMS from 11 female and 9 male patients. Libraries were prepared using TruSeq RNA Sample Prep Kit V2 (Illumina) and sequenced on a HiSeq2000 instrument (Illumina) in 2×101bp mode.
Paired-end reads were mapped to the human reference genome (GRCh37/hg19) using STAR (version 2.3.0.1) (43). Duplicate reads were removed using Picard (version 1.96) (39). The GATK framework (version 3.3-0) was used for Split’N’Trim, indel realignment and base recalibration (39). SNPs and indels were identified using HaplotypeCaller in the RNA-seq mode. Variants were annotated using ANNOVAR (41) and filtered to identify exonic, non-synonymous SNPs and indels.
Within the 184kb region of the LMS-specific CAPP-Seq panel, we identified a median of 7 SNVs/indels per patient in 18/20 patients from our validation cohort. The higher number of variants identified in the RNA-seq data compared to exome sequencing data from TCGA is most likely due to calling both germline and somatic mutations, as no germline control was analyzed by RNA-seq.
CAPP-Seq library construction
CAPP-Seq libraries were prepared from 32 ng of cfDNA (if less than 32 ng was available all the cfDNA obtained was input into library preparation, input range: 18.3 – 32ng) and 50ng of genomic tumor DNA (with the exception of 14.6 – 100ng input DNA used from 4 tumor specimens). For CAPP-Seq libraries from germline DNA, the whole yield of DNA obtained from 200μl of PBLs was used (input range: 28.8 – 64.6ng). Tumor and germline DNA was sheared before library construction using Covaris S2 instrument to obtain ~170bp fragments. Libraries were prepared using unique molecular identifiers (UMIs) as described before (6). Post-capture enrichment was evaluated by qPCR using Power SYBR Green PCR Master Mix (ThermoFisher Scientific) with primers specific for ARID1A, ATRX (genes included in the selector), B2M (as a negative control) and internal quality controls for the NimbleGen SeqCap capture panel. Seven to twelve libraries were pooled and sequenced using 2×101bp mode on HiSeq2000 or HiSeq4000 instruments (Illumina).
CAPP-Seq data analysis
Sequencing data was processed using a custom bioinformatics pipeline and SNV/indel calling was performed as previously described with minor modifications (6). Briefly, sequencing reads were demultiplexed using a 4bp sample index and de-duplicated using molecular barcodes. For cfDNA samples, background polishing was performed to reduce stereotypical base substitution errors. SNV/indel calling was performed as previously described (6) with the following modifications. We defined a “blacklist” as the genes in our panel that were found to be recurrently mutated in the plasma sequencing data from 24 healthy controls. Alterations were recurrently observed in four genes in our panel: MLL2, APOBR, PPR21, and DSPP. One or more alterations was observed in each of these genes in >90% of healthy plasma samples and thus these genes were removed from consideration for variant calling in tumor or plasma samples in LMS patients. For tumor genotyping, we applied additional requirements to identify high-confidence somatic variant calls, to account for the possible artifacts in DNA extracted from FFPE tissue (44, 45). We required ≥3 supporting duplex reads, a positional depth in tumor and germline ≥ 25% of the selector wide median depth, ≥5% mutant AF in the tumor, ≤1 read in matched germline, and no overlap with the UCSC RepeatMasker track (42, 46).
For CAPP-Seq-based ctDNA analysis, cfDNA samples were sequenced to a median de-duplicated depth of 2,031× (Supporting File: Table S4) and only somatic mutations that were present in one or more tumor samples were considered.
Genome representation profiling (GRP) of plasma specimens
Sequencing libraries were prepared with the TruSeq Chip preparation kit (Illumina), indexed, and 23 samples were pooled for multiplex sequencing across both lanes of an Illumina HiSeq2500 flow cell. Sequencing was performed in a 1×50bp mode and at least 10 million reads per sample were required from the LMS plasma specimens.
Sequencing reads were mapped to the GRCh37/hg19 reference genome using BWA-MEM with the default settings (version 0.7.10) (37). The pseudo-autosomal region on chromosome Y was masked in the reference genome. Duplicate reads were removed using SAMtools (version 0.1.18) (38, 47). The sequencing summary statistics are included in Supporting File: Table S5. Copy number variants in cfDNA were identified using the depth-of-coverage Plasma-Seq algorithm version 0.6 (11), with the following modifications: 1) we used sequencing reads of 50 bp; 2) genome was divided into 100,000, where each window contains the same amount of mappable reads; 3) the average length of the bins was 28 kb; 4) data from 189 female and 189 male healthy donors were used as the non-tumor controls. Next we applied Plasma-Seq algorithm with these settings to an independent group of 50 healthy donors, to define the genome-wide segmented Z-scores that set the specificity for LMS analysis at 98% (allowing 1/50 healthy donors to carry a CNA in cfDNA). The genome-wide Z-score was set at < −5.44 and > 5.44 for the significantly under- and overrepresented regions in LMS specimens, respectively.
Copy number profiling in LMS tumor specimens
Seventy-five nanograms of genomic DNA was used for genome-wide copy number and allelic ratio profiling with the OncoScan FFPE Assay by the Affymetrix Research Services Laboratory (Affymetrix, Santa Clara, CA). Results were visualized and analyzed with NexusExpress for OncoScan3 software using SNP-FASST2 algorithm (BioDiscovery, El Segundo, CA). Specimens with the Median Absolute Pairwise Difference (MAPD) value above 0.3 were excluded from the analysis. Diploid recentering was performed manually in all samples and copy number gain and loss was defined as log2 ratio > 0.25 and <−0.25, respectively. A minimum of 200 probes in the segment was used to call CNAs.
Availability of data
GRP sequencing data have been deposited in the European Nucleotide Archive (ENA) and have study accession number PRJEB22076 (www.ebi.ac.uk/ena/data/view/PRJEB22076). Raw data from CAPP-Seq experiments is available upon request.
RESULTS
Overview of patient cohort
In this study, we report the CAPP-Seq, GRP and SNP array results from 60 samples from 7 female patients treated for LMS at Stanford University Medical Center (Figure 1A, B) (Supplementary Table S1). These included 28 longitudinal plasma samples (median of 4 samples per patient), 25 tumor specimens (median of 3 samples per patient) and 7 specimens of peripheral blood cells (PBLs) for germline DNA analysis. In this proof-of-principle study, we focused primarily on those genomic aberrations that could be detected in matching tumor and plasma specimens.
Figure 1.
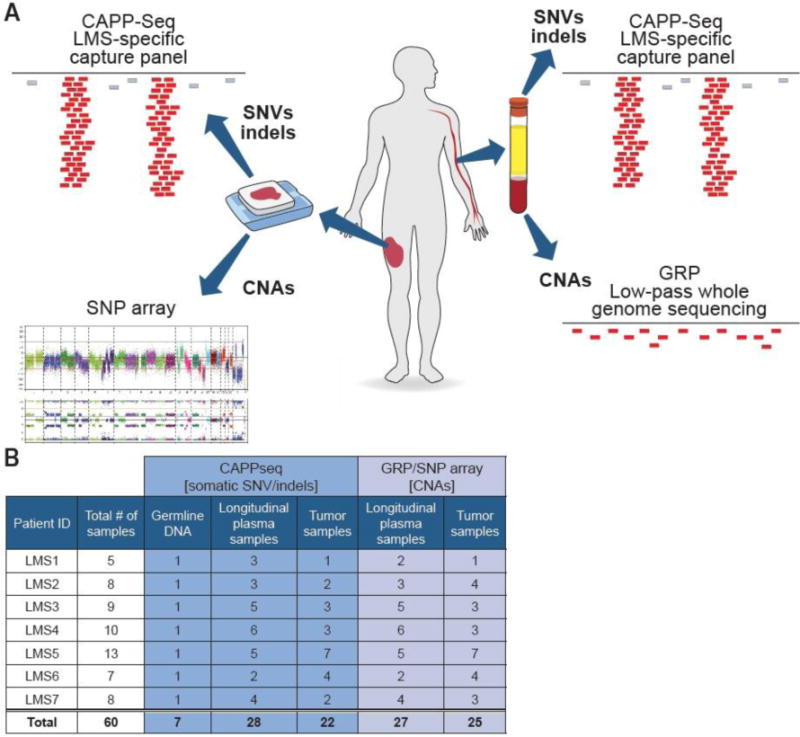
Study design and a summary of the analyzed specimens. A) Types of assays applied to study tumor and plasma specimens. B) Summary of all tumor, plasma and peripheral blood cell specimens analyzed by CAPP-Seq, GRP and SNP arrays.
Development of LMS-specific CAPP-Seq approach for ctDNA monitoring
To assess the feasibility of detecting ctDNA in LMS patients using CAPP-Seq, we designed a custom hybrid capture panel (i.e. “selector”) to target the most frequently mutated genomic regions in LMS based on the analysis of matched tumor and germline whole exome sequencing data from 77 LMS patients from The Cancer Genome Atlas (TCGA) (data downloaded from the dbGaP web site, under phs000178.v8.p7) (Figure 2A) (17). In addition, our selector included exons with mutations reported in 114 LMS patients in the COSMIC database v74 (18). The final LMS-specific selector targets 184kb and includes 306 exons from 89 genes (Supplementary Table S2). This panel covered 98.7% (76/77) of the patients in the TCGA cohort with a median of 3 SNVs/indels per patient. Consistent with prior LMS studies (12, 19–21), the most frequently mutated genes in the TCGA cohort included TP53, RB1, and ATRX (Figure 2B).
Figure 2.
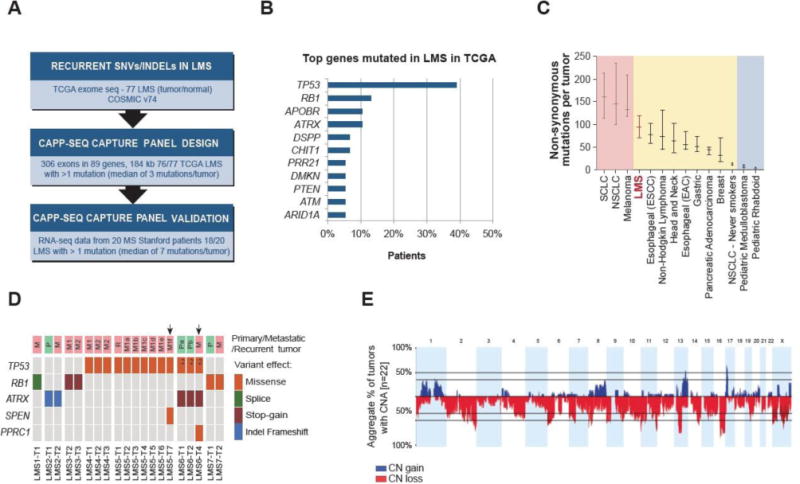
Mutational landscape of LMS and design of LMS-specific CAPP-Seq selector.
A) Design and validation of LMS-specific CAPP-Seq capture panel based on TCGA, COSMIC and Stanford sequencing data. B) The most frequently mutated genes in 77 LMS TCGA cases, according to the analysis described in the present study. C) The median number of exonic somatic mutations in LMS based on the analysis of TCGA cohort (n = 77) compared to selected types of cancer based on the studies reviewed by Vogelstein et al. (22). Horizontal bars indicate 25 and 75% quartiles. D) SNVs and indels detected by CAPP-Seq in 20 LMS tumor specimens analyzed in the present study. Arrows indicate tumor specimens with subclonal SNVs. Index 2 indicates two different somatic mutations in the same gene. M1, M2 indicate two different metastatic tumors. Pa, Pb and M1a, M1b etc. indicate different regions of the same tumor. E) Cumulative representation of copy number alterations identified by SNP array in 22 LMS tumor specimens analyzed in the present study. SCLC – small cell lung cancer; NSCLC – non-small cell lung cancer; ESCC - esophageal squamous cell carcinoma; EAC – esophageal adenocarcinoma; CN – copy number, CNA – copy number alteration.
Previous studies have shown a wide spectrum of the prevalence of somatic mutations across human cancer types (22, 23). Based on TCGA data, we calculated that LMS patients carried a median of 96 non-synonymous mutations per tumor, which classifies LMS as a tumor type with an intermediate level of SNVs and indels, as compared to the other cancer types (Figure 2C) (23). As such, we anticipated that the number of SNVs/indels detectable in ctDNA of LMS patients may be substantially lower than it would be for a cancer with higher mutational burden, such as lung cancer, for which a similar approach to selector design and comparable selector size yielded a panel covering an average of 8 mutations per patient (6).
Performance of CAPP-Seq in LMS patients
To assess the performance of our LMS-focused CAPP-Seq approach, we first analyzed 22 DNA samples obtained from spatially and temporally distinct sites of tumors from 7 LMS patients. We identified a median of 1 (range 0 - 4) non-synonymous SNV/indel in 20/22 tumor specimens (Figure 2D). We identified point mutations in numerous genes previously implicated in LMS including TP53, RB1, ATRX, SPEN and PPRC1. TP53, RB1 and ATRX are amongst the most frequently mutated genes in LMS (Figure 2B), and all mutations identified in these genes were present in each tumor specimen analyzed from each patient (Figure 2D), suggesting that these are truncal genetic events, i.e. shared early drivers in the development of LMS. We also found evidence for intra-patient tumor heterogeneity in two patients. In LMS5 and LMS6 we identified mutation in SPEN and PPRC1 that were unique to individual metastatic sites in these patients. Mutations in these genes have been less frequently reported in LMS, and likely represent subclonal events in these patients (Figure 2D).
We next applied LMS-focused CAPP-Seq to a total of 28 serial plasma samples from the 7 patients, as well as plasma samples from 24 healthy controls to assess specificity of ctDNA detection. Data from cfDNA of healthy donors was used to characterize selector-specific error profiles and perform digital error suppression as described before (6). CAPP-Seq demonstrated a baseline sensitivity of ctDNA detection of 86% (defined as detection of ctDNA at the first blood draw in a patient with known disease, Table 1, Supplementary Figure S1) with a specificity of 98.91% (determined using plasma from the 24 healthy donors) (Supplementary Figure S2). The overall sensitivity of ctDNA detection across all analyzed samples was 68% (19/28 positive samples) with a median variant allele fraction (VAF) of 0.27% (range 0 – 31.89%), indicating that ctDNA levels are relatively low in LMS as compared to other solid tumors (7). The CAPP-Seq protocol was initially optimized for the input of 32ng cfDNA but it has been previously shown to perform well with the input as low as 4ng cfDNA (5). We confirm this in LMS patients, as for 7/28 samples we used less than 32ng of cfDNA (range 18.3 – 29.6ng) (Supplementary Table S6) and ctDNA detection by CAPP-Seq was not correlated with the amount of input cfDNA (two-tailed Fisher’s exact test p = 0.65).
Table 1.
Summary of SNV/indels and CNAs detected in ctDNA of LMS patients.
| ctDNA monitoring | ||
|---|---|---|
| Plasma sample ID | CAPP-Seq SNV/indels |
# of CNA regions per GRP |
| LMS1-C1 | RB1 splice site | - |
| LMS1-C2 | - | not analyzed |
| LMS1-C3 | RB1 splice site | - |
| LMS2-C1 | ATRX indel | - |
| LMS2-C2 | ATRX indel | - |
| LMS2-C3 | ATRX indel | 3 |
| LMS3-C1 | RB1 K154* | 194 |
| LMS3-C2 | - | - |
| LMS3-C3 | RB1 K154* | 1 |
| LMS3-C4 | RB1 K154* | 16 |
| LMS3-C5 | RB1 K154* | 4 |
| LMS4-C1 | TP53 V272L | - |
| LMS4-C2 | TP53 V272L | - |
| LMS4-C3 | - | - |
| LMS4-C4 | TP53 V272L | 3 |
| LMS4-C5 | TP53 V272L | - |
| LMS4-C6 | TP53 V272L | 41 |
| LMS5-C1 | TP53 L344P | 40 |
| LMS5-C2 | TP53 L344P | 5 |
| LMS5-C3 | - | 1 |
| LMS5-C4 | TP53 L344P | 130 |
| LMS5-C5 | TP53 L344P | - |
| LMS6-C1 | - | - |
| LMS6-C2 | - | - |
| LMS7-C1 | RB1 D604G | 2 |
| LMS7-C2 | - | - |
| LMS7-C3 | - | - |
| LMS7-C4 | - | - |
Spectrum of copy number alterations and performance of GRP in LMS patients
To obtain a more comprehensive picture of ctDNA profiles in LMS, we sought to increase the number of molecular markers queried in the cfDNA by including copy number alterations (CNAs) as an additional class of genomic alterations. We decided to study CNAs in the same group of patients because genomic instability resulting in complex chromosomal aberrations has been previously described in LMS (13). The analysis of 72 LMS cases within the AACR Genie project identified 15 recurrent copy number alterations in this entity (GENIE cBioPortal data accessed on June 3, 2017). This roughly classifies LMS as having intermediate levels of CNAs, compared to the pan-cancer analysis of genomic aberrations published previously by Ciriello et al. (24). Copy number calls from 22 LMS tumor specimens from 7 patients were filtered for the presence of large segmental CNAs (covered by ≥ 200 probes) with a copy number gain and loss defined as log2 ratio > 0.25 and <−0.25, respectively. With these criteria, we identified extensive CNAs across all 22 tumor samples (median of 80 CNAs per tumor, range 25 – 198) (Figure 2E, Supplementary Table S7, Supplementary Figure S3). Consistent with previous LMS studies, the most frequent CNAs included loss of chromosome 13q with RB1 locus and gain of chromosome 17p encompassing the MYOCD gene (12, 21). Copy number alterations in LMS were highly heterogeneous and the vast majority of CNAs called with the above-described criteria appear to be subclonal events (Supplementary Figure S3). The only consistently truncal CNA, found across all tumors specimens analyzed in patients LMS2, LMS3, LMS4, LMS5 and LMS7, was a 3.5Mb copy number loss on chromosome 11q24.3-q25 (Supplementary Figure S3). This genomic region includes 24 genes (Supplementary Figure S4), including ADAMTS8 gene (ADAM metallopeptidase with thrombospondin type 1 motif 8, also known as METH2) that has been previously described as a tumor suppressor in multiple types of cancer (25).
GRP was performed at a median depth of 0.21× across the whole genome on 27 plasma specimens, all of which had been profiled also by CAPP-Seq (Supplementary Table S5). We also analyzed GRP data obtained from 378 healthy donors (189 males and 189 females) to calibrate the analysis algorithm, and the data from additional 50 healthy donors was used to set the genome-wide z-score cut-off at +/− 5.44 that allowed for 98% specificity. Tumor-matched CNAs were detected in plasma specimens of 71% (5/7) of patients, with the overall sensitivity of 44% (12/27) across all samples (Table 1, Supplementary Figure S1). Based on ctDNA quantification by CAPP-Seq, the indicative limit of ctDNA detection by GRP was as low as 0.23% VAF (sample LMS3-C3). GRP libraries were prepared with 4.25 – 87.5ng of input cfDNA (Supplementary Table S6) but the detection of ctDNA by GRP was again independent of the amount of input cfDNA (two-tailed Fisher’s exact test p = 0.25).
Combination of CAPP-Seq and GRP increases the number of reporters that can be monitored in ctDNA
Individually, CAPP-Seq demonstrated a higher sensitivity for ctDNA detection than GRP, but both methods showed highly concordant ctDNA profiles in LMS patients. Interestingly, there was a high correlation between ctDNA levels detected by CAPP-Seq (quantified as VAF) and GRP (represented as percent of the whole genome affected by CNAs) across all samples (R2=0.91, p < 0.0001; Supplementary Figure S5) indicating the utility of both approaches for ctDNA monitoring in LMS. In patients LMS3 and LMS7, all of the plasma samples were concordantly positive or negative for ctDNA by both methods (Table 1, Figure 3C and F, Supplementary Figure S1). However, in patients LMS1, LMS2, LMS4 and LMS5 we demonstrated that combining CAPP-Seq and GRP might prevent false negative results in ctDNA monitoring (Figure 3A, B, D, E). Overall, 9 out of 28 plasma samples were ctDNA-positive by only one of the methods (8 samples positive only by CAPP-Seq and one sample positive only by GRP).
Figure 3.
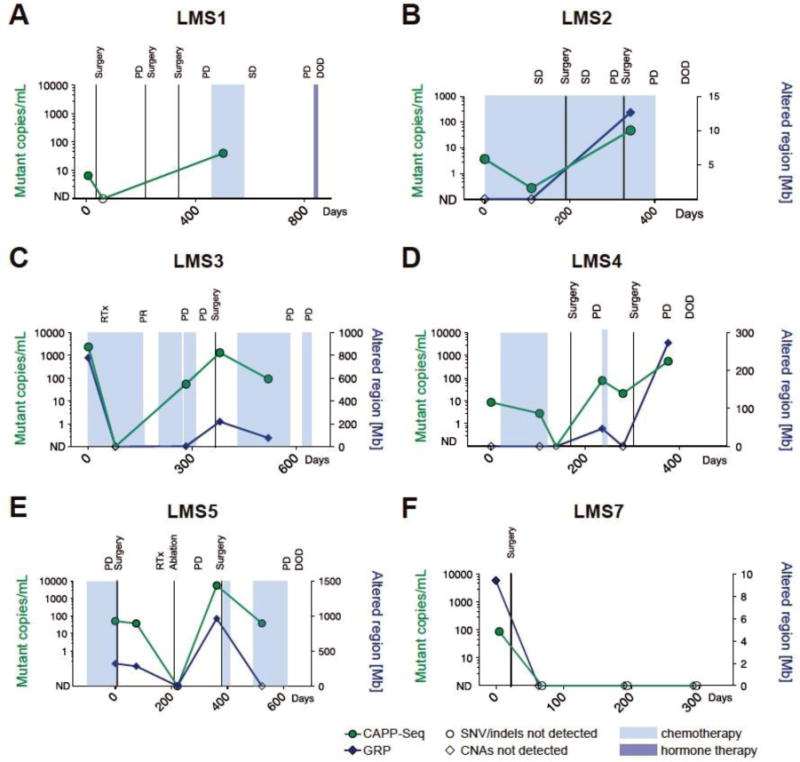
Patient-specific ctDNA profiles characterized by CAPP-Seq and GRP. The levels of ctDNA decreased in selected patients in response to surgery (LMS1 and LMS7) or chemotherapy (LMS2, LMS3, LMS4). In patients LMS1, LMS2, LMS3, LMS4 and LMS5, the increasing levels of ctDNA correlated with progression of disease throughout the treatment. (SD – stable disease; PD – progressive disease; DOD – dead of disease; Mb - megabases)
The number of SNVs/indels detected in tumors of LMS patients enrolled in this study was lower than expected (a median of 1 detected mutation vs. 3 expected mutations per tumor). This contrasted with prior CAPP-Seq studies in lung cancer and lymphoma where it was possible to track multiple markers per patient due to more recurrently mutated genomic regions in these diseases (5, 14). Counting each tumor-matched CNA detected in plasma as an additional reporter that confirms the presence of ctDNA, the average number of reporters detected per plasma sample increased from 1 to 17 (range 0 - 195). These results show that the combination approach to liquid biopsy in LMS patients substantially increases the number of molecular markers that can be monitored in plasma. Both approaches cross-validate each other’s results, which increases the confidence of detecting tumor-specific aberrations in plasma of LMS patients. Combination of CAPP-Seq and GRP resulted in the overall sensitivity of ctDNA detection of 71% (20/28) across all plasma samples.
Clinical utility of ctDNA monitoring in LMS
Our results demonstrate the feasibility of a ctDNA-based assessment of treatment response in LMS patients. In two patients, LMS1 and LMS7, the ctDNA signal became undetectable after surgery for a primary extremity tumor and a metastatic liver tumor, respectively (Figure 3A, F). ctDNA levels in three patients (LMS2, 3 and 4) reflected the initial response to chemo- and radiotherapy (Figure 3B–D). In LMS2, we observed a decrease of ctDNA level from 3.66 to 0.27 mutant copies per mL of plasma after the first 110 days of temozolomide treatment. In patient LMS3, ctDNA signal dropped to undetectable levels by CAPP-Seq and GRP after the first two cycles of gemcitabine and docetaxel treatment combined with radiation therapy. In patient LMS4, we showed a gradual decrease of ctDNA levels in the first three plasma samples collected before, during and after completion of aldoxorubicin treatment. All patients analyzed in this study eventually progressed based on imaging studies, which was reflected by the increasing levels of ctDNA in all but one patient (Figure 3). The single patient without evidence of ctDNA during progression (patient LMS7) had metastases in lymph nodes and lung but no metastases in soft tissue. Interestingly, we found a highly significant correlation between presence of ctDNA and the location of metastatic tumors in LMS patients. There was a lower rate of ctDNA-positive plasma samples in patients with metastases located in lymph nodes or lung only, compared to patients with metastatic tumors present also in soft tissues, liver and bone (Table 2, p < 0.0001, two-tailed Fisher’s exact test). Considering all the plasma samples analyzed from the 7 patients in this cohort, 12.5% (1/8) of the samples collected at time points when patients had only lymph node and/or lung metastases had detectable ctDNA, compared to 95% (18/19) of samples collected at time points when patients soft tissues, liver and/or bone metastases. These results suggest that lymph node and lung nodules in LMS patients may shed less DNA into circulation than metastases present in other sites. While the reason underlying this observation is unclear based on the results from the present study, we hypothesize that it is most likely due the differences in aggregate tumor size. All nodules found in lymph nodes and lung were small and growing slowly, which may indicate their relatively low activity. Larger studies are required to determine whether the size and/or site of metastatic deposits might have an effect on the ctDNA burden in LMS patients.
Table 2.
Correlation between ctDNA signal detected by CAPP-Seq/GRP and the location of metastatic sites in LMS patients.
| Metastatic sites | ||
|---|---|---|
| Lymph node/lung only | Soft tissue/liver/bone/lung | |
| ctDNA positive | 1 | 18 |
| ctDNA negative | 7 | 1 |
Two-tailed Fisher’s exact test p < 0.0001
LMS tumor heterogeneity reflected by ctDNA
Overcoming tumor heterogeneity is a major challenge for the personalized treatment of cancer (26). Cancers are known to vary in their mutational profile at different sites within an individual lesion, as well as across different tumor deposits in metastatic patients (27–29). A major advantage of ctDNA compared to tissue biopsies is that ctDNA analysis simultaneously integrates contributions from multiple tumor deposits, enabling a more comprehensive analysis of tumor heterogeneity (15, 30–32). Thus, we sought to explore the utility of our approach for the study of tumor heterogeneity in LMS. We profiled multiple tumor specimens per patient (median of 3, range 1 - 7) and found evidence for intra-patient heterogeneity of mutations across different lesions in 2/5 of the patients in whom more than 1 tumor sample was sequenced (Figure 2D). However, these subclonal SNVs were not detected in ctDNA, likely due to the low overall ctDNA levels in these patients. On the other hand, we have observed substantial intra-patient tumor heterogeneity by profiling CNAs (Supplementary Figure S3). Multiple aberrations were present only in a single sample or in a single region of the same tumor, and some of these subclonal CNAs were detectable in ctDNA. For example, among the 5 plasma specimens from one of these patients (LMS5) we detected a copy number gain of chromosome 1q21.3-q42.12 in the 1st and the 4th plasma samples. We did not detect this aberration in the metastatic tumor removed shortly after the 1st blood sample collection, as we examined only one region of the multifocal abdominal recurrence. But 1/6 subsequent metastatic tumors removed shortly after the 4th blood sample collection harbored the identical chromosomal gain as detected in ctDNA (Figure 4). As has previously been shown for lymphoma and lung cancer (14, 15, 23), the analysis of ctDNA enables detection of subclonal alterations also in LMS.
Figure 4.
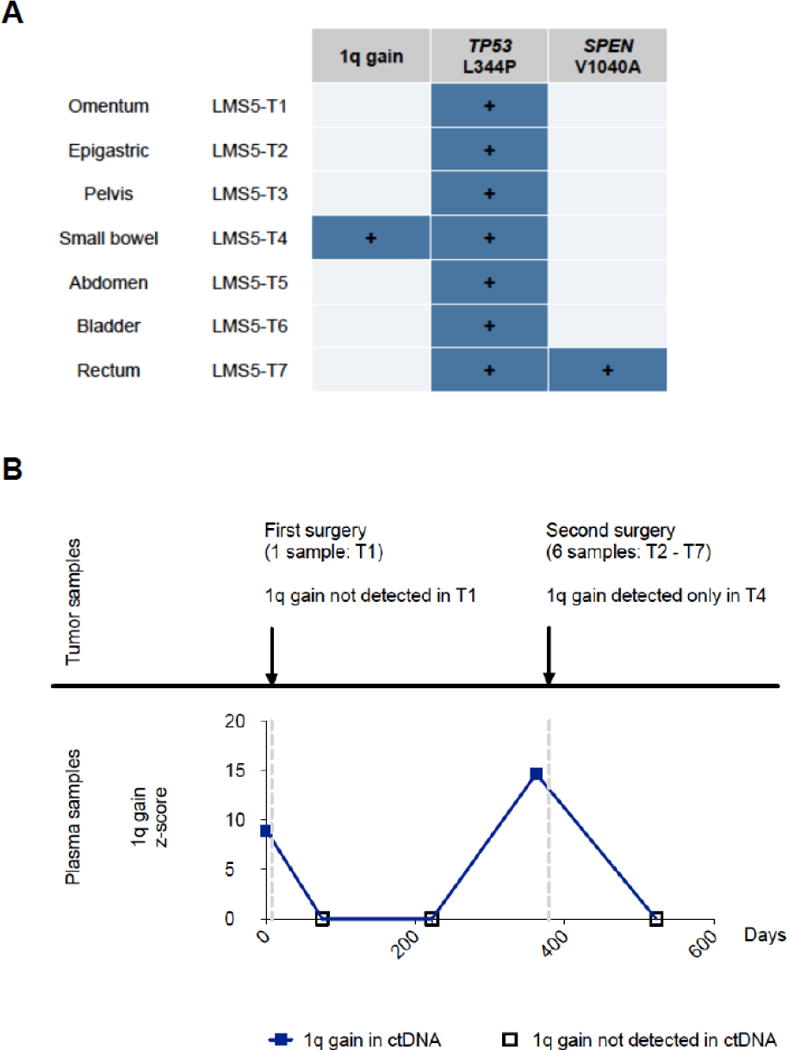
Tumor heterogeneity detected by ctDNA in patient LMS5.
A) CNA and SNV profiles of tumor specimens from patient LMS5. Copy number gain spanning genomic region 1q21.3 – 1q42.12 has been identified by SNP arrays only in specimen T4 from small bowel. Point mutation in TP53 is a truncal event detected across all recurrent and metastatic tumor specimens, while SPEN mutation was a subclonal event detected only in a single metastatic tumor specimen.
B) GRP-based ctDNA profile of region 1q21.3 – 1q42.12 across 5 longitudinal plasma samples from patient LMS5. Copy number gains in this region have been detected in ctDNA plasma sample #1 (collected 7 days before the excision of tumor T1) and plasma sample #4 (collected 17 days before the second surgery of tumors T2-T7).
DISCUSSION
ctDNA monitoring offers a great promise for improved diagnosis, prognosis, treatment selection and surveillance of cancer patients. A personalized approach through tumor-specific phylogenetic ctDNA profiling has been demonstrated to predict resistance to therapy and development of recurrent tumors in lung cancer patients (4). Clinical utility of CAPP-Seq has been demonstrated for improved prognostication in lymphomas (14), and for studying heterogeneity of resistance mechanisms to EGFR inhibitors in lung cancer patients (15). Plasma genotyping for EGFR-sensitizing mutations is already approved by the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) for selection of patients for treatments with gefitinib, erlotinib and osimetrinib when tumor genotyping is not feasible (33, 34). In prostate cancer patients, androgen receptor copy number gain in ctDNA was associated with worse overall survival in response to abiraterone (35). These examples show that ctDNA analysis may provide clinically useful information. However, given a wide variety of DNA abnormalities in different entities, ctDNA testing needs to be tailored to the genomic profiles of specific types of tumors to achieve clinically relevant sensitivity and specificity.
Here we present a combinatorial approach to ctDNA monitoring, using LMS as a disease with an intermediate level of both point mutations and copy number aberrations. We describe the application of two previously validated next generation sequencing-based methods for ctDNA detection in a group of seven patients with LMS for whom sequential blood samples were collected. The first method, CAPP-Seq, relies on deep sequencing of a tumor type-specific panel covering the most recurrent SNVs and indels in that disease. CAPP-Seq is an ultrasensitive method for ctDNA detection that is most effective in tumor types with a relatively high rate of recurrent mutations that can be covered by a capture panel of a limited size to ensure cost-effectiveness of deep sequencing, as has been previously demonstrated in lung cancer and lymphoma (5, 14, 15). The second method, GRP, is routinely used for a non-invasive prenatal screening and relies on a low pass whole genome sequencing of cfDNA. GRP has been reported to detect ctDNA in asymptomatic patients with ovarian carcinoma, Hodgkin lymphoma and follicular lymphoma (9).
In the previous CAPP-Seq studies in lung cancer, a selector designed to detect a median of 8 mutations per tumor yielded a median 6 mutations per tumor when applied to an independent group of patients (6). This number of expected reporters tracked in plasma was high enough to reliably quantify ctDNA with a very high specificity and sensitivity. In the present study, we applied a similar approach to design an LMS-specific CAPP-Seq selector based on TCGA data from 77 LMS cases as described previously for lung cancer (5). Our LMS-specific capture panel was expected to detect 3 mutations per LMS tumor. Similar to the lung cancer CAPP-Seq study, we detected a lower than expected number of mutations also in LMS patients (a median of 1 mutation per tumor). The discrepancy between the expected and detected number of SNV/indels in LMS patients is most likely a combination of 1) the selector design being over-fit due to the relatively small number of LMS exomes used to design the panel, 2) the small size of our pilot cohort, 3) the heterogeneity of somatic mutations in this disease, and 4) more stringent analysis criteria applied in CAPP-Seq analysis as opposed to TCGA dataset. Because of the lower than expected number of SNV/indels detected in LMS tumors, we employed a combination approach to simultaneously detect CNAs in tumor and plasma specimens. Separate sequencing of libraries for SNV/indel and CNA analyses may not be practical in the clinical setting. We propose that this strategy could be further improved by developing more robust experimental and bioinformatics methods that would combine targeted detection of SNV/indels with a genome-wide interrogation of copy number changes in a single experiment.
To obtain the highest level of confidence in the measurement of LMS ctDNA in plasma samples, we required each aberration to be present in patient-matched tumor and plasma specimens, and required each somatic SNV/indel to have duplex support. Our data show that ctDNA could be measured in 86% (6/7) of LMS patients with active disease at >98% specificity, and suggest that the levels of ctDNA correlate with the response to treatment. We demonstrate that the orthogonal analysis of ctDNA using two separate methods increased the number of molecular markers that can be monitored in plasma. By CAPP-Seq, we measured the VAF of each mutation, which can be translated into mutant copies of ctDNA per mL of plasma, an absolute quantification of ctDNA levels. By GRP, it is not possible to perform an absolute measurement of the abundance of ctDNA. Therefore, to represent the GRP data we used the extent of ctDNA with tumor-matched CNAs measured as Mb or the fraction of genome altered by CNAs. While detection of copy number alterations in ctDNA is in principle limited by the mutant allele fraction and the original profile of CNAs in the tumor, GRP provided a good proxy for ctDNA load compared to CAPP-Seq results (Supplementary Figure S5). We propose that a similar strategy may also prove beneficial in other types of tumors that are characterized by moderate levels of both SNVs/indels and CNAs, such as ovarian, breast or head and neck squamous cell carcinomas (24). Cohen et al. have recently demonstrated a different combinatorial strategy for an improved sensitivity of liquid biopsy testing in early stage pancreatic cancer patients by integrated detection of genomic alterations and measurement of protein biomarkers, including CEA (carcinoembryonic antigen) and CA 19-9 (carbohydrate antigen 19-9) in plasma specimens (36). This approach increased sensitivity because a large portion of patients was positive for only a single marker.
This proof-of-principle study shows for the first time the feasibility of ctDNA monitoring in LMS patients. Our report provides a rationale for prospective studies in a larger cohort of LMS patients to evaluate whether ctDNA testing may complement current imaging-based regimens for evaluation of LMS patients and drive better-informed clinical decisions on the post-operative treatment, allowing for early detection of residual disease or disease recurrence in asymptomatic patients. In the future, ctDNA-based assessment of treatment response in LMS patients could also lead to a significant improvement of the efficiency of clinical trials. We also hypothesize that ctDNA testing may also be useful for a non-invasive distinction between LMS and the much more common benign leiomyomas in the uterus. Treatment of sarcomas poses a significant clinical challenge because imaging-based methods may not accurately assess tumor response to treatment. We hypothesize that a ctDNA-based assay for evaluation of response to therapy may allow for more economical monitoring and could spare patients an exposure to radiation. The cost of next generation sequencing is steadily decreasing as the technology matures and we envision that ctDNA assay may, in the future, become complimentary to standard imaging.
In summary, we report that ctDNA analysis may have significant potential to improve monitoring of LMS patients. While CAPP-Seq outperformed GRP in the number of samples that were called positive for the presence of ctDNA, we show that integrated detection of multiple classes of genomic alterations substantially increased the number of reporters that can be tracked in ctDNA of LMS patients. We propose that such combination approach to liquid biopsies may be applicable also to many other types of tumors that harbor moderate levels of different types of genomic aberrations.
Supplementary Material
Statement of translational relevance.
The detection of circulating tumor DNA (ctDNA) has potential to improve prognostication, molecular profiling and surveillance, especially in cancer types with highly recurrent genomic alterations. Leiomyosarcoma (LMS) represents a type of tumor that harbors a wide spectrum of heterogeneous genomic abnormalities; thus, targeting a narrow genomic region for ctDNA monitoring may not be practical. In this study, we demonstrate a combination approach that integrates different sequencing protocols for the orthogonal detection of multiple types of alterations in ctDNA. This strategy substantially increases the number of molecular markers that can be tracked in plasma, and improves the confidence of ctDNA detection in LMS patients and could be applied to a wide range of tumors characterized by genomic profiles of comparable complexity.
Acknowledgments
We would like to thank Sharon Anderson and Shohreh Monshipouri for invaluable assistance with the enrollment of the patients.
Financial support
Research supported by the 2016 QuadW Foundation-AACR Fellowship for Clinical/Translational Sarcoma Research Grant Number 16-40-37-PRZY (J.P.), The Leiomyosarcoma Direct Research Foundation (M.vdR.), the National Science Foundation (J.J.C.; DGE-114747), the Virginia and D.K. Ludwig Fund for Cancer Research (M.D., A.A.A. and M.vdR.), US National Institutes of Health Director’s New Innovator Award Program (M.D.; 1-DP2-CA186569), the CRK Faculty Scholar Fund (M.D.), K.U. Leuven Concentrated Action Grant number 2011/010 (M.D.R.), and R00 CA187192 NCI grant (A.M.N.).
Footnotes
Conflict of interest
A.M.N., A.A.A., and M.D. are co-inventors on patent applications related to CAPP-Seq. A.M.N., A.A.A., and M.D. are consultants for Roche Molecular Systems. A.A.A. has served as a consultant for Genentech, Gilead, and Celgene. M.D. has served as a consultant for Novartis and Quanticel Pharmaceuticals. M.D. has received research funding from Varian Medical Systems.
References
- 1.Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96:9236–41. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leary RJ, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med. 2010;2:20ra14. doi: 10.1126/scitranslmed.3000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014;20:430–5. doi: 10.1038/nm.3511. [DOI] [PubMed] [Google Scholar]
- 4.Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545:446–51. doi: 10.1038/nature22364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548–54. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547–55. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bayindir B, Dehaspe L, Brison N, Brady P, Ardui S, Kammoun M, et al. Noninvasive prenatal testing using a novel analysis pipeline to screen for all autosomal fetal aneuploidies improves pregnancy management. Eur J Hum Genet. 2015;23:1286–93. doi: 10.1038/ejhg.2014.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amant F, Verheecke M, Wlodarska I, Dehaspe L, Brady P, Brison N, et al. Presymptomatic Identification of Cancers in Pregnant Women During Noninvasive Prenatal Testing. JAMA Oncol. 2015;1:814–9. doi: 10.1001/jamaoncol.2015.1883. [DOI] [PubMed] [Google Scholar]
- 10.Vandenberghe P, Wlodarska I, Tousseyn T, Dehaspe L, Dierickx D, Verheecke M, et al. Non-invasive detection of genomic imbalances in Hodgkin/Reed-Sternberg cells in early and advanced stage Hodgkin’s lymphoma by sequencing of circulating cell-free DNA: a technical proof-of-principle study. Lancet Haematol. 2015;2:e55–65. doi: 10.1016/S2352-3026(14)00039-8. [DOI] [PubMed] [Google Scholar]
- 11.Heitzer E, Ulz P, Belic J, Gutschi S, Quehenberger F, Fischereder K, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013;5:30. doi: 10.1186/gm434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazar A, Evans HL, Shipley J. Smooth-muscle tumors Leiomyoma of deep soft tissue Leiomyosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO Classification of Tumours of Soft Tissue and Bone. Lyon: IARC; 2013. pp. 111–3. [Google Scholar]
- 13.Chibon F, Lagarde P, Salas S, Perot G, Brouste V, Tirode F, et al. Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat Med. 2010;16:781–7. doi: 10.1038/nm.2174. [DOI] [PubMed] [Google Scholar]
- 14.Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AF, Esfahani MS, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8:364ra155. doi: 10.1126/scitranslmed.aai8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi: 10.1038/ncomms11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 2017 doi: 10.1158/2159-8290.CD-17-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tryka KA, Hao L, Sturcke A, Jin Y, Wang ZY, Ziyabari L, et al. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2014;42:D975–9. doi: 10.1093/nar/gkt1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45:D777–d83. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makinen N, Aavikko M, Heikkinen T, Taipale M, Taipale J, Koivisto-Korander R, et al. Exome Sequencing of Uterine Leiomyosarcomas Identifies Frequent Mutations in TP53, ATRX, and MED12. PLoS Genet. 2016;12:e1005850. doi: 10.1371/journal.pgen.1005850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang CY, Liau JY, Huang WJ, Chang YT, Chang MC, Lee JC, et al. Targeted next- generation sequencing of cancer genes identified frequent TP53 and ATRX mutations in leiomyosarcoma. Am J Transl Res. 2015;7:2072–81. [PMC free article] [PubMed] [Google Scholar]
- 21.Agaram NP, Zhang L, LeLoarer F, Silk T, Sung YS, Scott SN, et al. Targeted exome sequencing profiles genetic alterations in leiomyosarcoma. Genes Chromosomes Cancer. 2016;55:124–30. doi: 10.1002/gcc.22318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–33. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi GC, Li J, Wang Y, Li L, Zhong L, Ma B, et al. The metalloprotease ADAMTS8 displays antitumor properties through antagonizing EGFR-MEK-ERK signaling and is silenced in carcinomas by CpG methylation. Mol Cancer Res. 2014;12:228–38. doi: 10.1158/1541-7786.MCR-13-0195. [DOI] [PubMed] [Google Scholar]
- 26.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 27.de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–6. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346:256–9. doi: 10.1126/science.1256930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diaz LA, Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–40. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10:472–84. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 33.Wan R, Wang Z, Lee JJ, Wang S, Li Q, Tang F, et al. Comprehensive Analysis of the Discordance of EGFR Mutation Status between Tumor Tissues and Matched Circulating Tumor DNA in Advanced Non-Small Cell Lung Cancer. J Thorac Oncol. 2017 doi: 10.1016/j.jtho.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 34.Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, et al. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2016;34:3375–82. doi: 10.1200/JCO.2016.66.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romanel A, Gasi Tandefelt D, Conteduca V, Jayaram A, Casiraghi N, Wetterskog D, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015;7:312re10. doi: 10.1126/scitranslmed.aac9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen JD, Javed AA, Thoburn C, Wong F, Tie J, Gibbs P, et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci U S A. 2017;114:10202–7. doi: 10.1073/pnas.1704961114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013 [Available from: arXiv:1303.3997v2. [Google Scholar]
- 38.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next- generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015;43:D670–81. doi: 10.1093/nar/gku1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yost SE, Smith EN, Schwab RB, Bao L, Jung H, Wang X, et al. Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens. Nucleic Acids Res. 2012;40:e107. doi: 10.1093/nar/gks299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schweiger MR, Kerick M, Timmermann B, Albrecht MW, Borodina T, Parkhomchuk D, et al. Genome-wide massively parallel sequencing of formaldehyde fixed-paraffin embedded (FFPE) tumor tissues for copy-number- and mutation- analysis. PLoS One. 2009;4:e5548. doi: 10.1371/journal.pone.0005548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smit A, Hubley R, Green P. RepeatMasker Open-4.0 2013–2015. [Available from: http://www.repeatmasker.org.
- 47.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.