

Abstract
Background
Sustained β-adrenergic receptor (β-AR) stimulation causes pathophysiologic changes during heart failure (HF), including inhibition of the slow component of the delayed rectifier current, IKs. Aberrant calcium handling, including increased activation of calcium/calmodulin-dependent protein kinase II (CaMKII), contributes to arrhythmia development during HF.
Objective
To investigate CaMKII regulation of KCNQ1 (pore-forming subunit of IKs) during sustained β-AR stimulation and associated functional implications on IKs.
Methods
KCNQ1 phosphorylation was assessed using LCMS/MS following sustained β-AR stimulation with isoproterenol (ISO). Peptide fragments corresponding to KCNQ1 residues were synthesized to identify CaMKII phosphorylation at the identified sites. Dephosphorylated (alanine) and phosphorylated (aspartic acid) mimics were introduced at identified residues. Whole-cell, voltage44 clamp experiments were performed in HEK 293 cells co-expressing wild-type (WT) or mutant KCNQ1 and KCNE1 (auxiliary subunit) during ISO treatment or lentiviral δCaMKII overexpression.
Results
Novel KCNQ1 carboxyl terminus sites were identified with enhanced phosphorylation during sustained β-AR stimulation at T482 and S484. S484 peptides demonstrated the strongest δCaMKII phosphorylation. Sustained β-AR stimulation reduced IKs activation (p=0.02 versus control) similar to phosphorylated mimic (p=0.62 versus sustained β-AR). Individual phosphorylated mimics at S484 (p=0.04) but not T482 (p=0.17) reduced IKs function. Treatment with CN21 (CaMKII inhibitor) reversed the reductions in IKs versus CN21-Alanine control (p<0.01). δCaMKII overexpression reduced IKs similar to ISO treatment in WT (p<0.01) but not in the dephosphorylated S484 mimic (p=0.99).
Conclusion
CaMKII regulates KCNQ1 at S484 during sustained β-AR stimulation to inhibit IKs. The ability of CaMKII to inhibit IKs may contribute to arrhythmogenicity during HF.
Keywords: KCNQ1, CaMKII, Calcium/Calmodulin-Dependent Protein Kinase II, heart failure, beta-adrenergic receptor, delayed rectifier, Iks
INTRODUCTION
Sustained elevations in beta-adrenergic receptor (β-AR) stimulation, a hallmark pathophysiologic finding in heart failure (HF), prolong the cardiac action potential and thereby increase the risk for ventricular arrhythmogenesis and sudden cardiac death. Past investigations have demonstrated functional deficits in repolarizing potassium currents during HF, including those in the slow component of the delayed rectifier potassium current, IKs. 1-5
IKs is mediated by interaction of the pore-forming subunit, KCNQ1, with the auxiliary subunit, KCNE1. During acute or intermittent β-AR stimulation, IKs function is enhanced via KCNQ1 phosphorylation by a protein kinase A (PKA)-dependent signaling complex to stabilize cardiac conduction during rapid heart rates.6 Although acute β-AR signaling enhances IKs function to stabilize cardiac conduction, sustained β-AR stimulation has been suggested to pathologically inhibit IKs function.2, 7 Functional reductions in IKs have been demonstrated to prolong action potential duration (APD) and increase arrhythmogenesis in both HF models and in patients carrying Long QT Phenotype 1 (LQT1) mutations in the KCNQ1 gene.8, 9
Several investigations have characterized mechanisms by which LQT1 mutations disrupt IKs function. In particular, the intracellular carboxy terminus of KCNQ1 (residues 352-676) has been shown to be involved in channel gating, membrane trafficking, interaction with KCNE1, and subunit tetramerization.10-12 Though previous investigations have elucidated the functional significance of various regions of the KCNQ1 carboxy terminal, the potential for changes in carboxy terminal phosphorylation in response to sustained β-AR stimulation, and the associated functional impact on IKs, have not been well characterized.
Calcium/calmodulin-dependent protein kinase II (CaMKII) has been demonstrated to pathologically regulate many ion channels, including cardiac potassium channels, resulting in prolonged APD and increased arrhythmia development.13 In particular, CaMKII has been established as a mediator of arrhythmogenesis during sustained β-AR stimulation and HF, wherein the expression and activity of CaMKII are increased.14 The objective of this study was to assess the potential for CaMKII to regulate IKs during sustained β-AR stimulation via KCNQ1 phosphorylation.
METHODS
Cell Culture
Human endothelial kidney (HEK 293) cells were purchased from ATCC® and maintained in Modified Essential Medium with 10% Fetal Bovine Serum and 1% Pen-Strep (5,000 Units/mL Penicillin; 5,000 μg/mL Streptomycin) at 37°C and 5% CO2. To achieve β-AR stimulation, isoproterenol 100 nM (ISO; β1, β2-adrenergic receptor agonist) was added directly to culture media. IKs responsiveness to stimulation of endogenous β-ARs has been previously demonstrated in HEK 293 cells.15 The following reagents were commercially purchased: ISO (Sigma-Aldrich®); myristolated PKI (EMD Millipore®); tat-CN21, CN21, tat-CN21-Alanine, CN21-Alanine (Biopeptide Co., Inc.®); KN-92, KN-93 (Santa Cruz Biotech®).
cDNA Transfection
Complementary DNA encoding for human (h) KCNQ1 and hKCNE1 in pCDNA3.1 were used. Point mutations to alanine (A) and aspartic acid (D) were generated in hKCNQ1 by site-directed mutagenesis. hKCNQ1 (WT and mutants), hKCNE1, and enhanced green fluorescent protein (GFP) were co-98 transfected using Lipofectamine 2000 (ThermoFisher®) at the ratio: 750 ng KCNE1, 500 ng KCNQ1, 100 ng GFP.
CaMKII Protein Immunoblot
Protein was loaded and run on 10% SDS-polyacrylamide gel, transferred to PVDF membranes (ThermoFisher®), and blocked with 5% BSA in TBST. Membranes were exposed to primary CaMKII (1:1000, Boster Biological Technology®) or CaMKII P287 (1:1000, Novus Biologicals®) antibodies, primary GAPDH (1:2000, Santa Cruz Biotech®) and secondary anti-mouse (1:5000, Santa Cruz Biotech®) antibodies, and developed using Pierce ECL Western Blotting Substrate (ThermoFisher®) and a Chemi-Doc Imager (Bio-Rad®).
Viral Transduction
Constitutively active human δCaMKII was inserted into a packaging plasmid with an amino terminal fused yellow fluorescent protein (YFP) tag. The packaging plasmid (20 μg) was co-transfected into HEK 293T cells along with viral gene plasmids [pRRE (10 μg), pRSV-Rev (5 μg), pCMV-VSV-G (6 μg)] using polyethylenimine (50 μg) in Opti-MEM (ThermoFisher®). Cells were maintained in virus114 containing filtered media (0.45 μm) along with polybrene (Sigma-Aldrich®; 8 μg: 1 mL viral media) for 6 hours. Effective transduction was confirmed by YFP expression at 2 days following infection.
Mass Spectrometry
HEK293 cells expressing hKCNQ1 / hKCNE1 were incubated with ISO 100 nM. Cells were lysed and enriched membrane preparations (100 μg) were reduced, alkylated and digested with trypsin.16 Digested samples were analyzed using a Thermo-Finnigan linear ion-trap (LTQ) mass spectrometer coupled with a Surveyor autosampler and MS HPLC system (ThermoFinnigan®). The complete LCMS/MS methods have been previously described in detail.16 Only proteins and peptides with protein probability ≥ 0.9000 and peptide probability ≥ 0.8000 are reported. Protein quantification was performed using a label-free quantification software package, IdentiQuantXL™.17
Electrophysiology
Functional measurements were assessed using the whole-cell, patch-clamp configuration in the voltage128 clamp mode at room temperature (~22°C). ISO and inhibitors were incubated in culture media and replaced with external (bath) solution during experiments that did not contain ISO. Activation currents were measured using a voltage step protocol using an EPC-9 amplifier and PatchMaster software (HEKA Elektronik®). The sampling rate was set to 20,000 samples/second. The voltage dependence of activation was assessed in elicited tail currents when the voltage returned to -40 mV after the activating steps. The internal patch solution was composed of (mM): K-Aspartate 110, KCl 20, MgCl2•6H2O 1, HEPES 10, Mg-ATP 5, EGTA 5, while the external patch solution was composed of (mM): NaCl 140, KCl 5.4, NaH2PO4 0.33, CaCl2•2H2O 1.8, MgCl2•6H2O 1, HEPES 5, Glucose 10. The internal solution was adjusted to a pH of 7.2 using KOH, and the external solution was adjusted to a pH of 7.4 using NaOH. Pipettes were pulled from borosilicate glass using a P-2000 Puller (Sutter Instrument®) with resistances of 2-7 MΩ. The mean measured liquid junctional potential for all experimental groups was 14.2±2.7 mV. Reported values were not corrected for the liquid junction potential. Series resistance compensation was not performed. The average cell capacitance was 18.6±0.4 (mean±SEM) pF and the average series resistance was 18.1±0.5 MΩ.
KCNQ1 Peptide Array
Immobilized peptides were synthesized on a modified cellulose membrane using a robotic peptide synthesizer (Intavis MultiPep®) with routine Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry as previously described.18, 19 Human δCaMKII (10 nM) was activated with Ca2+/CaM and Mg-ATP before the autophosphorylated kinase plus Mg-[γ-32P]ATP was added to the membrane, as described.18 In conditions evaluating peptide selectivity by PKA, catalytic subunit (10 nM) from bovine heart (Sigma #P2645) was added along with Mg-ATP (10 mM/0.1mM) and Mg-[γ-32P]ATP to label substrates, as described for CaMKII. Phosphorylated peptides were visualized with a phosphoimager (Fuji®) and quantified using MultiGauge® (Version 3.0).
Data Analysis
Analysis was performed using FitMaster (Version 2×73.1; HEKA®) and GraphPad Prism (Version 6.03). A Boltzmann distribution was used to fit normalized activation curves with the equation:
| (Eq. 1) |
where I/Imax is the normalized current, V is membrane voltage, V1/2 is the voltage of half-maximal activation, and k is the slope factor. Mono-exponential curves were fit to activation and tail currents to estimate rate constants of activation and deactivation at voltages that elicited measurable channel activation (0 to +60 mV). Protein abundance, phospho-stimulated luminescence, peak activation current density, V1/2, and rate constants were compared via one-way ANOVA with Tukey’s HSD post-hoc test or independent sample t-test with Welch correction. All data are expressed as mean±standard error of the mean (SEM) and alpha set to 0.05.
RESULTS
The KCNQ1 carboxy terminus is differentially phosphorylated during sustained β-AR stimulation
Basal phosphorylation of KCNQ1 was identified on five residues on the carboxy terminus (Figure 1A). The phosphorylation status at T482, S484, and S457 was decreased in the presence of KCNE1 co-169 transfection (Supplemental Figure 1). Phosphorylation was enhanced at S407, T482, S484, and T624, (p<0.02) following sustained 4-hour ISO treatment in the presence of KCNE1. The increased phosphorylation was maintained following sustained 24-hour treatment at T482 (p<0.01) and S484 (p=0.01).
Figure 1.
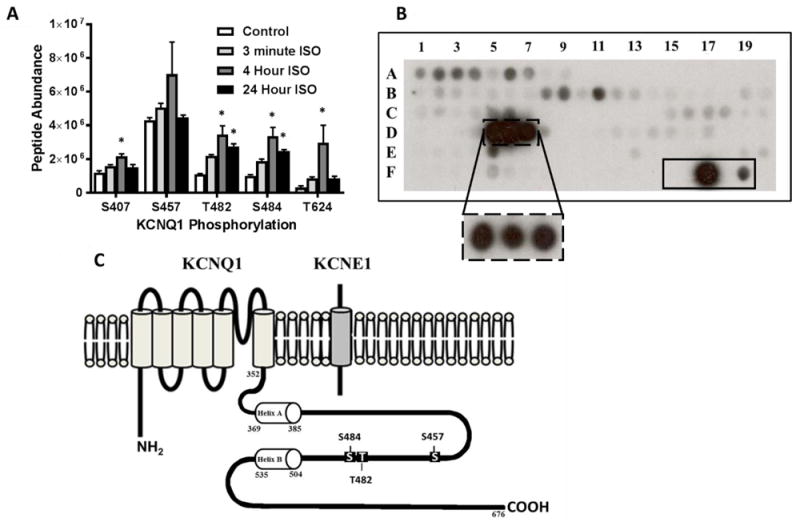
(A) Phosphorylation status of KCNQ1 carboxy terminus in HEK 293 cells (co-expressing KCNQ1 and KCNE1) following treatment with 100 nM ISO for 3 minutes, 4 hours, and 24 hours via LCMS/MS analysis. (B) Peptide fragments corresponding to the intracellular regions of KCNQ1 were exposed to activated δCaMKII for 4 minutes and 30 seconds. Each peptide was 15 amino acids in length, tiled by two residues for 13 overlapping residues per consecutive peptide. Peptide fragments containing residues T482 and S484 (solid box at D5-D7) were the strongest substrates for CaMKII phosphorylation. The dashed box at D5-D7 is following 30 second exposure of activated δCaMKII. The solid box (F15-F19) contains a autocamtide-2 negative control (T→A mutation; F15), WT autocamtide-2 positive control (F17), and kemptide control (classical PKA substrate; F19). Full peptide sequences are in Supplemental Table 1. (C) Schematic of KCNQ1 and KCNE1 subunits showing carboxy terminal sites of potential CaMKII regulation investigated. *p<0.05 vs. control
Sustained β-AR stimulation pathologically activates CaMKII which is an important mediator of arrhythmogenesis.20 Therefore, the ability of activated δCaMKII to phosphorylate intracellular regions of KCNQ1 was assessed using a peptide array. Luminescent signals corresponding to CaMKII phosphorylation were visible on peptides containing T482 and S484 (Figure 1B; complete peptide sequences in Supplemental Table 1). ScanSite3 (http://scansite3.mit.edu/#home) predicted S457 and S484 as sites of CaMKII phosphorylation. Therefore, residues S457, T482, and S484, which reside within the region connecting calmodulin-binding domains in KCNQ1 helices A and B, were assessed for CaMKII-mediated regulation (Figure 1C).
Sustained β-AR stimulation inhibits IKs
The impact of sustained β-AR stimulation on IKs function was assessed following treatment with 100 nM ISO for 12-24 hours. IKs function was similar following 4-6 hour and 12-24 hour ISO treatment (Supplemental Figure 2). As displayed in Figure 2A/B, sustained ISO treatment reduced peak corrected currents at +60 mV (voltage of maximum activation) from 45.6±7.9 pA/pF (n=20) to 21.5±4.0 (n=14), p=0.01. ISO treatment also resulted in a depolarizing shift in the voltage dependence of activation with a V1/2 of 20.6±0.8, n=19 with vehicle vs. 26.1±0.8 mV, n=12 following ISO treatment, p<0.01; Figure 2C and Supplemental Table 2. Rate constants were not statistically different (Figures 2D and 2E).
Figure 2.
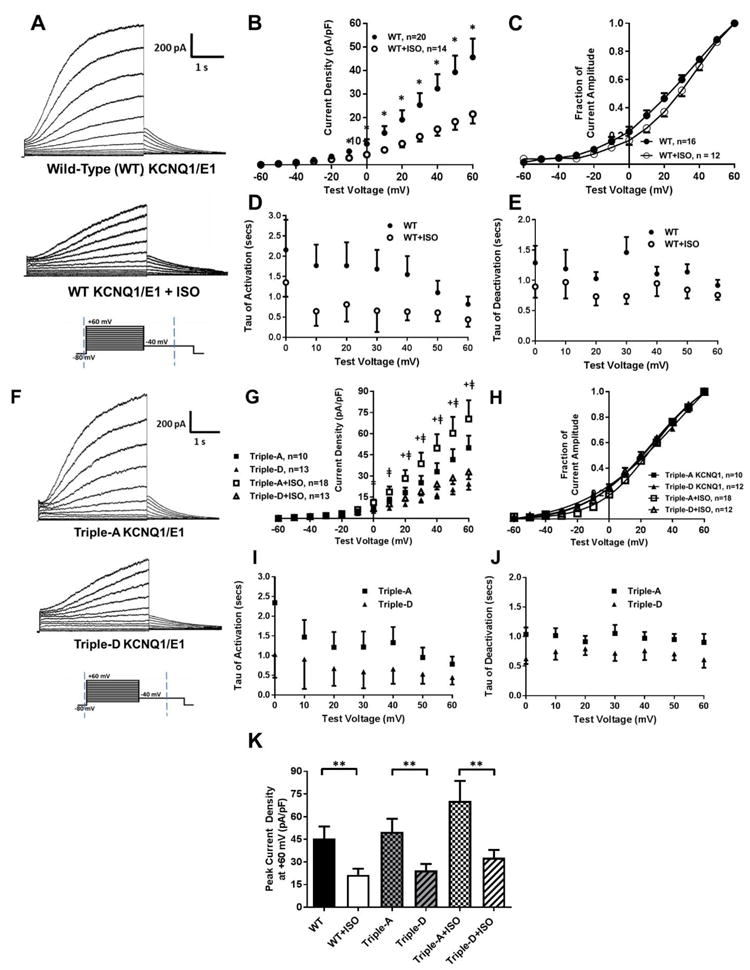
(A) Representative traces of IKs activation currents from WT KCNQ1/KCNE1 following 100 nM ISO or vehicle for 12-24 hours. (B) I-V plots, (C) activation curves (normalized to the voltage of maximum activation), and (D) rate constants of activation and (E) deactivation following treatment with ISO or vehicle. (F) Representative traces of IKs from KCNQ1 combination mimics of dephosphorylation (Triple-A) and phosphorylation (Triple-D) co-expressed with KCNE1. (G) I-V plots, (H) activation curves, and (I) rate constants of activation and (J) deactivation for Triple-A and Triple-D KCNQ1. (K) Peak current density at +60 mV for WT KCNQ1 and combination mimics following ISO or vehicle. *p<0.05 vs. WT+ISO, + p<0.05 for Triple-A vs. Triple-D, ǂp<0.05 for Triple-A+ISO vs. Triple-D+ISO, **p<0.05 for comparison indicated
Phosphorylation at S457, T482, and S484 in combination inhibit IKs activation currents
The functional implications of phosphorylation at the identified residues were investigated in HEK 293 cells co-expressing KCNE1 and KCNQ1 mutants conferring mimics of dephosphorylation (A) or phosphorylation (D) at S457, T482, and S484 in combination: triple-alanine KCNQ1 (Triple-A; dephosphorylated mimic) and triple-aspartic acid KCNQ1 (Triple-D; phosphorylated mimic). As displayed in Figure 2G, Triple-A mutants increased mean peak corrected activation currents (50.0±8.7 pA/pF, n=10 at +60 mV) relative to Triple-D mutants (24.5±4.2, n=13, p=0.02). Sustained treatment with ISO did not decrease IKs with Triple-A (50.0±8.7, n=10 with vehicle vs. 70.5±13.3, n=18 following ISO, p=0.21) or Triple-D mutants (24.5±4.2, n=13 with vehicle vs. 32.9±5.1, n=13 following ISO, p=0.22). The V1/2 was not different between Triple-A and Triple-D mutants (19.5±0.9 mV, n=10 vs. 20.2±1.1, n=13, respectively; p=0.66), and sustained ISO treatment did not alter V1/2 in Triple-A (19.5±0.9, n=10 with vehicle vs. 22.3±0.6, n=18 following ISO, p=0.25) or Triple-D mutants (V1/2 of 20.2±1.1 mV, n=13 with vehicle vs. 19.2±0.6, n=12 following ISO, p=0.44; Figure 2H). Rate constants of activation or deactivation were not different between mutants (Figure 2I and 2J). Relative to Triple- A, Triple-D mutants demonstrate reduced IKs currents similar to WT with sustained ISO (Figure 2K).
Phosphorylation at S457 and S484, but not at T482, inhibit IKs
Mimics of dephosphorylation (A) or phosphorylation (D) were individually introduced at S457, T482, and S484. IKs activation was decreased with S457D KCNQ1 relative to S457A with a mean of 70.7±12.3 (n=16) in S457A and 40.4±4.1 pA/pF (n=19) in S457D at +60 mV, p=0.02; (V1/2 of 28.6±2.5 mV, n=15 for S457D vs. 21.6±1.7, n=12 for S457A, p=0.03) Figure 3A. Conversely, IKs activation current density were not different between T482 mimics (64.3±11.4, n=22 in T482D vs. 73.8±11.3, n=22 in T482A at +60 mV, p=0.53 and V1/2 of 24.8±4.2 mV, n=14 for T482D vs. 24.7±2.2, n=16 for T482A, p=0.99; Figure 3B). Mimics of phosphorylation at S484 decreased IKs current density and right shifted the voltage dependence of activation relative to dephosphorylation mimics (61.9±10.8, n=20 in S484D vs. 100.4±13.7, n=22 in S484A at +60 mV, p=0.04 and V1/2 of 28.3±3.1 mV, n=15 for S484D vs. 19.6±1.8, n=11 for S484A, p=0.02; Figure 3C). Reduced IKs function with S484D versus S484A was only observed during co-expression with KCNE1 (Figure 3C). KCNQ1-related current was not different between S484A and S484D in the absence of KCNE1 expression (Supplemental Figure 3).
Figure 3.
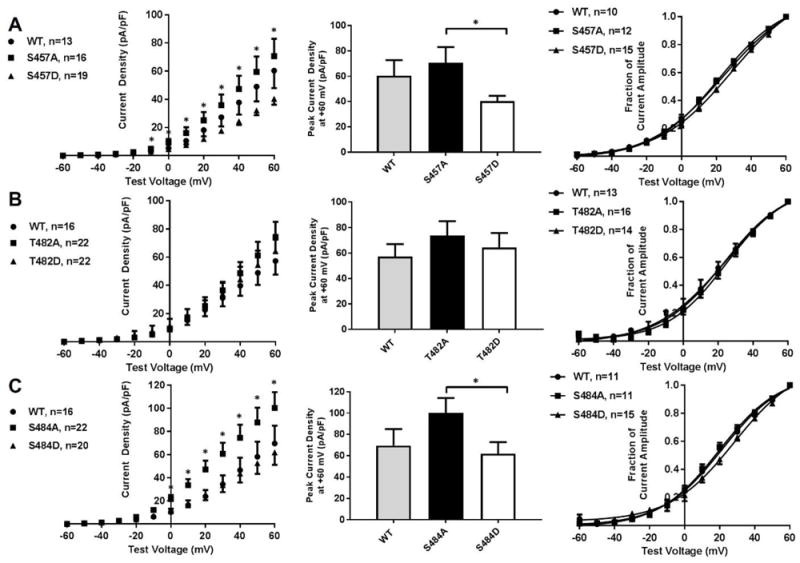
(A) I-V plots, peak current density, and activation curves for WT, S457A, and S457D KCNQ1/KCNE1 (B) I-V plots, peak current density, and activation curves for WT, T482A, and T482D KCNQ1/KCNE1. (C) I-V plots, peak current density, and activation curves for WT, S484A, and S484D KCNQ1/KCNE1. *p<0.05
CaMKII mediates functional inhibition of IKs during sustained β-AR stimulation
IKs currents were assessed during sustained ISO and co-treatment with the CaMKII peptide inhibitor CN21 or its inactive analogue, CN21-Alanine (10 μM tat-CN21 or tat-CN21-Ala in culture media for 4 hours and 1 μM CN-21 or CN21-Ala in pipette solution). CN21 reversed sustained ISO induced reductions in IKs activation (71.9±5.1 pA/pF, n=10 with CN21 vs. 47.2±5.9, n=12 with CN21-Ala, p<0.01; Figure 4A/B). The sustained ISO induced depolarizing shift in the voltage dependence of activation was reversed with CN21 (V1/2 of 13.8±1.3 mV, n=6 with CN21 vs. 20.2±2.1, n=6 with CN21- Ala, p<0.01; Figure 4C). Co-incubation with the PKA peptide inhibitor myristoylated-PKI (1 μM in culture media for 4 hours and in pipette solution) did not attenuate ISO-induced changes in IKs (Figure 4A-C). The chemical CaMKII inhibitor KN-93 also attenuated ISO-induced changes in IKs relative to KN-92 control (Supplemental Figure 4C-E).
Figure 4.
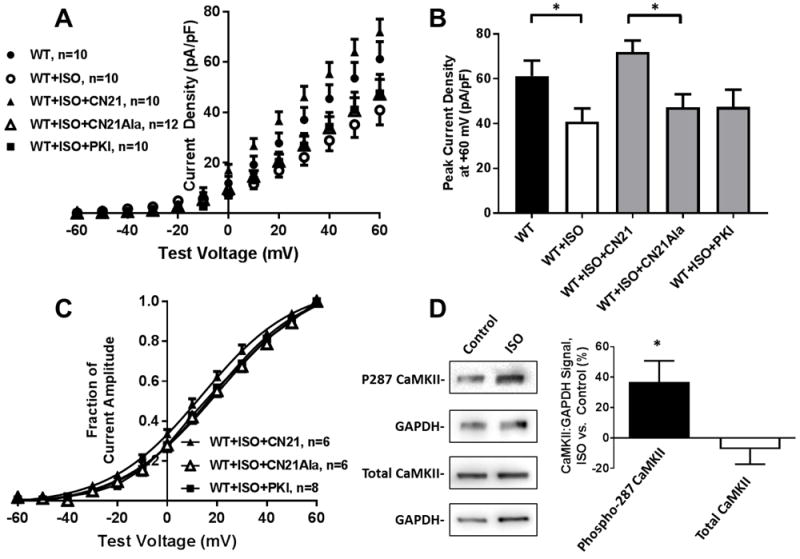
(A) I-V plots and (B) peak current density for WT KCNQ1/KCNE1 following treatment with ISO (100 nM for 12-24 hours) and CN21, CN21-Ala, or myr-PKI. (C) Normalized activation curves following ISO with CN21, CN21-Ala, or myr-PKI. (D) Immunoblots and percent changes in CaMKII and CaMKII T287 phosphorylation following 100 nM ISO for 24 hours. *p<0.05
CaMKII activity was assessed following sustained ISO treatment via phosphorylation at T287, a residue at which CaMKII is autophosphorylated to confer constitutive kinase activity.21 T287 phosphorylation relative to GAPDH was increased by 36.8% during ISO treatment versus control (p<0.05) while CaMKII expression relative to GAPDH was not changed following ISO treatment; p=0.62 (Figure 4D).
CaMKII inhibits IKs through phosphorylation at S484
A peptide array was used to assess site-specific δCaMKII phosphorylation of the KCNQ1 carboxy terminal residues identified by LCMS/MS. The strongest δCaMKII phosphorylation signals were detected in peptides containing T482 and S484 (Figure 5). By individually mutating T482 or S484 to alanine, strong phosphorylation signals were detected in all peptide fragments containing WT S484, including the T482KO (Spot 3). Peptides containing the T482 residue as the lone potential phosphorylation site (S484A, Spot 5) displayed negligible signals relative to peptides containing S484 alone (40.7± 1.9 for S484A vs. 1605.3±67.8 for the T482KO, p<0.01). Peptides containing S407, S457, T482, S484, and T624 were not phosphorylated when exposed to activated PKA (<2.5% of positive control signal, Supplemental Figure 5). Peptide sequences are defined in Supplemental Table 3, and the full membranes displayed in Supplemental Figure 6.
Figure 5.
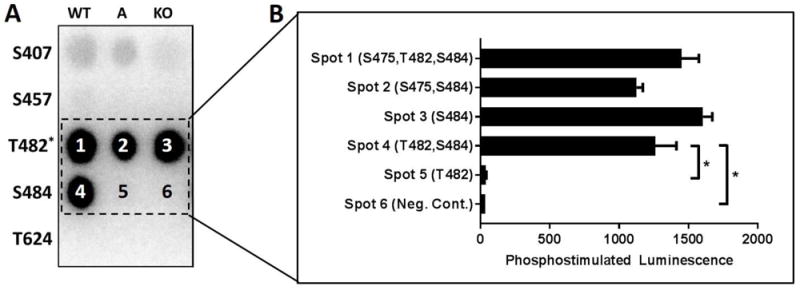
(A) KCNQ1 carboxy peptides were exposed to activated δCaMKII. Each row contains peptides corresponding to the labeled KCNQ1 residue with columns for WT, A (phospho-acceptor site mutated to alanine), or KO (all serine and threonine mutated to alanine with the exception of T482 wherein S484 was not mutated; n=5 for each condition). (B) Quantification of phosphostimulated luminescence for WT, A, and KO peptides corresponding to KCNQ1 T482 and S484 during exposure to activated δCaMKII. *p<0.05, ns = not significant
The potential for CaMKII to mediate IKs function at S484 and/or S457 was assessed in a HEK 293 cell line that stably overexpressed YFP-tagged constitutively active δCaMKII (T287D) and transiently co-expressed WT or mutant KCNQ1 with KCNE1. A lentiviral plasmid containing YFP was stably expressed as control. As shown in Figure 6A/B, IKs current density was inhibited during CaMKII overexpression relative to control (67.3±9.3 pA/pF, n=15 in control vs. 35.8±5.8, n=15 in CaMKII at +60 mV, p=0.01). The CaMKII induced IKs inhibition observed in WT KCNQ1 was reversed with S484A (60.9±9.1, n=10 for S484A mutants in CaMKII vs. 35.8±5.8, n=15 for WT KCNQ1 in CaMKII, p=0.04) but not S457A (35.6±7.0, n=10 for S457A in CaMKII vs. 35.8±5.8, n=15 for WT KCNQ1 in CaMKII, p=0.99). Relative to lentiviral control, CaMKII induced a depolarizing shift in the voltage dependence of activation similar to that of sustained ISO (V1/2 of 18.5±1.4 mV, n=11 in control vs. 22.2±0.8, n=11 in CaMKII, p=0.03; Figure 6C). Rate constants of activation were not different (Figure 6D), however, rate constants of deactivation were reduced in cells expressing WT KCNQ1 relative to S484A with CaMKII (Figure 6E).
Figure 6.
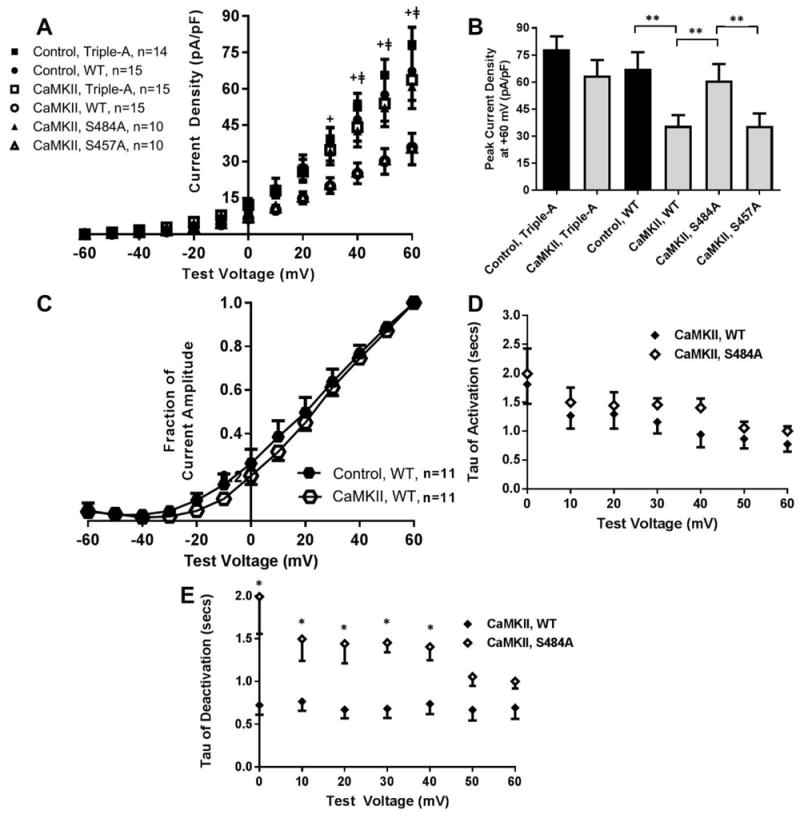
(A) I-V plots and (B) peak current density for WT or mutant KCNQ1 co-expressed with KCNE1 in cells overexpressing constitutively active δCaMKII or YFP control. (C) Normalized activation curves for WT KCNQ1 when expressed in CaMKII overexpression and control. (D) Rate constants of activation and (E) deactivation for WT KCNQ1 and S484A during CaMKII overexpression. + p<0.05 for Control, WT vs. CaMKII, WT, ǂp<0.05 for CaMKII, S484A vs. CaMKII, WT, **p<0.05 for comparison indicated, *p<0.05 for CaMKII, WT vs. CaMKII, S484A
DISCUSSION
In contrast to enhancement during acute stimulation, sustained β-AR stimulation reduces IKs function by an unclear mechanism.6, 7 Although functional deficits in IKs contribute to an increased risk of ventricular arrhythmias, the regulation of IKs during sustained β-AR stimulation is poorly defined. In this study, we identified a CaMKII-dependent reduction in IKs current density during sustained β-AR stimulation through phosphorylation at a residue (S484) in the region connecting two alpha-helical calmodulin-binding domains on KCNQ1.
CaMKII is a serine/threonine kinase known to pathologically regulate ion channel function and excitation-contraction coupling in cardiomyocytes during HF.20 Furthermore, CaMKII mediates arrhythmia development in a vast range of cardiac diseases, including HF, through aberrant calcium handling.14, 22, 23 Through regulation of cardiac ion channels, including the repolarizing potassium channels Ito and IK1, CaMKII facilitates increases in both action potential duration and arrhythmogenic propensity in HF models.24, 25 While CaMKII regulation of IKs has not previously been established, investigations of LQT1 mutants in the KCNQ1 carboxy terminus demonstrate a necessary role for calmodulin in IKs function.10, 26
The KCNQ1 carboxy terminus is required for proper channel co-assembly, trafficking, and regulation.10-12 Therefore, post-translational modifications in the KCNQ1 carboxy terminus could have profound effects on IKs. While there is a paucity of data investigating the functional effects of site282 specific phosphorylation, LQT1 mutations in carboxy terminal domains have been investigated. For example, helix A mutations (R366W, A371T, S373P, and W392R) disrupt calmodulin binding and reduce IKs function by altering channel assembly, stabilizing inactivation, and decreasing current density.26 In the current study, the reduction of IKs via CaMKII occurred through phosphorylation at S484, a residue in the region connecting the calmodulin-binding domains in helices A (residues 370- 389) and B (residues 506-532). Interestingly, the co-expression of KCNE1 with KCNQ1 reduced basal phosphorylation at S484 versus KCNQ1 alone. Additionally, phosphorylation and dephosporylation mimics did not alter KCNQ1-related current in the absence of KCNE1. Together, these results suggest that reductions in IKs may involve alterations in KCNQ1 and auxiliary subunit interactions through phosphorylation but further mechanistic information is needed.
Aflaki et al. assessed the regulation of IKs during sustained β-AR stimulation in a guinea pig model.7 In accordance with our results, IKs was inhibited in response to ISO treatment for 30 hours. Additionally, they demonstrated a role for the exchange protein activated by cyclic-AMP (Epac) pathway in mediating IKs during sustained β-AR stimulation. Since CaMKII is enhanced downstream of Epac activation, it is not surprising that functional reductions in IKs were reversed with CaMKII inhibition during sustained β-AR stimulation.27, 28 Therefore, the findings from Aflaki et al. support the current findings that CaMKII regulates IKs during sustained β-AR stimulation.
In this study, the significance of the S484 residue was demonstrated by alanine substitutions that reversed the effects of CaMKII which was further supported by peptide array and biochemical data. A potential limitation of this approach is that basal phosphorylation exists on the identified residues. Therefore, it is not surprising that alanine substitutions (S484A, S4857A, and T482A alone and in combination) trended, albeit non-significantly, toward a current density increase versus WT. Aspartic acid is also not a perfect mimic of phosphorylation and, therefore, the assessment of WT versus D substitutions may not represent the full effect of phosphorylation. Additional limitations include those associated with the cellular model which lack the precise control of cell surface expression. Given potential variability between experiments, controls were assessed during the time of each experimental group and the ratio of KCNQ1 to KCNE1 was standardized to produce classical characteristics of IKs.
CONCLUSIONS
In summary, in response to sustained β-AR stimulation, CaMKII phosphorylates KCNQ1 at S484 to inhibit IKs function. Inhibition of IKs activation is consistent with the depolarizing shifts in the voltage dependence of activation during ISO treatment and CaMKII overexpression. It is also consistent with decreases in deactivation rate constants observed during CaMKII overexpression. These functional findings are consistent with our proteomic and biochemical analyses, which demonstrate that phosphorylation at S484 is enhanced during sustained β-AR stimulation and that S484 is a specific site of CaMKII phosphorylation. This study expands on previous findings to propose a molecular understanding of how CaMKII regulates IKs function during sustained β-AR stimulation. The potential for CaMKII to inhibit IKs during sustained β-AR stimulation may contribute to arrhythmogenicity during HF. Future investigations are warranted to assess the pathophysiological role of S484 regulation via CaMKII in cardiac tissue.
Supplementary Material
Acknowledgments
Supported by HL095655 (BO) from the National Heart, Lung, and Blood Institute (NHLBI) and NS078171 (AH) and NS053422 (TRC and AH) from the National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (Bethesda, MD).
Footnotes
Conflicts of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akar FG, Wu RC, Juang GJ, Tian Y, Burysek M, Disilvestre D, Xiong W, Armoundas AA, Tomaselli GF. Molecular mechanisms underlying K+ current downregulation in canine tachycardia-induced heart failure. Am J Physiol Heart Circ Physiol. 2005;288:H2887–96. doi: 10.1152/ajpheart.00320.2004. [DOI] [PubMed] [Google Scholar]
- 2.Li D, Melnyk P, Feng J, Wang Z, Petrecca K, Shrier A, Nattel S. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation. 2000;101:2631–8. doi: 10.1161/01.cir.101.22.2631. [DOI] [PubMed] [Google Scholar]
- 3.Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1031–41. doi: 10.1152/ajpheart.00105.2002. [DOI] [PubMed] [Google Scholar]
- 4.Tsuji Y, Opthof T, Kamiya K, Yasui K, Liu W, Lu Z, Kodama I. Pacing-induced heart failure causes a reduction of delayed rectifier potassium currents along with decreases in calcium and transient outward currents in rabbit ventricle. Cardiovasc Res. 2000;48:300–9. doi: 10.1016/s0008-6363(00)00180-2. [DOI] [PubMed] [Google Scholar]
- 5.Tsuji Y, Zicha S, Qi XY, Kodama I, Nattel S. Potassium channel subunit remodeling in rabbits exposed to long-term bradycardia or tachycardia: discrete arrhythmogenic consequences related to differential delayed-rectifier changes. Circulation. 2006;113:345–55. doi: 10.1161/CIRCULATIONAHA.105.552968. [DOI] [PubMed] [Google Scholar]
- 6.Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–9. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 7.Aflaki M, Qi XY, Xiao L, Ordog B, Tadevosyan A, Luo X, Maguy A, Shi Y, Tardif JC, Nattel S. Exchange protein directly activated by cAMP mediates slow delayed-rectifier current remodeling by sustained beta-adrenergic activation in guinea pig hearts. Circ Res. 2014;114:993–1003. doi: 10.1161/CIRCRESAHA.113.302982. [DOI] [PubMed] [Google Scholar]
- 8.Cheng HC, Incardona J. Models of torsades de pointes: effects of FPL64176, DPI201106, dofetilide, and chromanol 293B in isolated rabbit and guinea pig hearts. J Pharmacol Toxicol Method. 2009;60:174–84. doi: 10.1016/j.vascn.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 9.Lengyel C, Varro A, Tabori K, Papp JG, Baczko I. Combined pharmacological block of I(Kr) and I(Ks) increases short-term QT interval variability and provokes torsades de pointes. Br J Pharmacol. 2007;151:941–51. doi: 10.1038/sj.bjp.0707297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh S, Nunziato DA, Pitt GS. KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ Res. 2006;98:1048–54. doi: 10.1161/01.RES.0000218863.44140.f2. [DOI] [PubMed] [Google Scholar]
- 11.Howard RJ, Clark KA, Holton JM, Minor DL., Jr Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron. 2007;53:663–75. doi: 10.1016/j.neuron.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiener R, Haitin Y, Shamgar L, Fernandez-Alonso MC, Martos A, Chomsky-Hecht O, Rivas G, Attali B, Hirsch JA. The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J Biol Chem. 2008;283:5815–30. doi: 10.1074/jbc.M707541200. [DOI] [PubMed] [Google Scholar]
- 13.Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:180–7. doi: 10.1097/FJC.0b013e3181a25078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999;84:713–21. doi: 10.1161/01.res.84.6.713. [DOI] [PubMed] [Google Scholar]
- 15.Imredy JP, Penniman JR, Dech SJ, Irving WD, Salata JJ. Modeling of the adrenergic response of the human IKs current (hKCNQ1/hKCNE1) stably expressed in HEK-293 cells. Am J Physiol Heart Circ Physiol. 2008;295:H1867–81. doi: 10.1152/ajpheart.433.2008. [DOI] [PubMed] [Google Scholar]
- 16.Lai X. Reproducible method to enrich membrane proteins with high purity and high yield for an LC-MS/MS approach in quantitative membrane proteomics. Electrophoresis. 2013;34:809–17. doi: 10.1002/elps.201200503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai X, Wang L, Tang H, Witzmann FA. A novel alignment method and multiple filters for exclusion of unqualified peptides to enhance label-free quantification using peptide intensity in LC-MS/MS. J Proteome Res. 2011;10:4799–812. doi: 10.1021/pr2005633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, Bers DM, Hudmon A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J Biol Chem. 2012;287:19856–69. doi: 10.1074/jbc.M111.322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashpole NM, Hudmon A. Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol Cell Neurosci. 2011;46:720–30. doi: 10.1016/j.mcn.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–73. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin430 dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–22. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 23.Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. 2003;92:904–11. doi: 10.1161/01.RES.0000069685.20258.F1. [DOI] [PubMed] [Google Scholar]
- 24.Nagy N, Acsai K, Kormos A, Sebok Z, Farkas AS, Jost N, Nanasi PP, Papp JG, Varro A, Toth A. [Ca(2)(+)] i-induced augmentation of the inward rectifier potassium current (IK1) in canine and human ventricular myocardium. Pflugers Archiv. 2013;465:1621–35. doi: 10.1007/s00424-013-1309-x. [DOI] [PubMed] [Google Scholar]
- 25.Wagner S, Hacker E, Grandi E, Weber SL, Dybkova N, Sossalla S, Sowa T, Fabritz L, Kirchhof P, Bers DM, Maier LS. Ca/calmodulin kinase II differentially modulates potassium currents. Circ Arrhythm Electrophysiol. 2009;2:285–94. doi: 10.1161/CIRCEP.108.842799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shamgar L, Ma L, Schmitt N, Haitin Y, Peretz A, Wiener R, Hirsch J, Pongs O, Attali B. Calmodulin is essential for cardiac IKS channel gating and assembly: impaired function in long- QT mutations. Circ Res. 2006;98:1055–63. doi: 10.1161/01.RES.0000218979.40770.69. [DOI] [PubMed] [Google Scholar]
- 27.Metrich M, Morel E, Berthouze M, Pereira L, Charron P, Gomez AM, Lezoualc’h F. Functional characterization of the cAMP-binding proteins Epac in cardiac myocytes. Pharmacol Rep. 2009;61:146–53. doi: 10.1016/s1734-1140(09)70017-9. [DOI] [PubMed] [Google Scholar]
- 28.Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Epac and phospholipase Cepsilon regulate Ca2+ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. J Biol Chem. 2009;284:1514–22. doi: 10.1074/jbc.M806994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.