

Abstract
Background
As human longevity increases and Alzheimer’s disease (AD) increasingly becomes a significant societal burden, finding pathways or protective factors that facilitate exceptional brain aging without AD pathophysiologies (ADP) will be critical. The goal of this viewpoint is two-fold: 1) to present evidence for “exceptional brain aging” without ADP; and 2) to bring together ideas and observations from the literature and present them as testable hypotheses for biomarker studies to discover protective factors for “exceptional brain aging” without ADP and AD dementia.
Discovering pathways to exceptional aging
There are three testable hypotheses. First, discovering and quantifying links between risk factor(s) and early ADP changes in midlife using longitudinal biomarker studies will be fundamental to understanding why the majority of individuals deviate from normal aging to the AD pathway. Second, a risk factor may have quantifiably greater impact as a trigger and/or accelerator on a specific component of the biomarker cascade (amyloid, tau, neurodegeneration). Finally, and most importantly, while each risk factor may have a different mechanism of action on AD biomarkers, “exceptional aging” and protection against AD dementia will come from “net sum” protection against all components of the biomarker cascade. The knowledge of the mechanism of action of risk factor(s) from hypotheses 1 and 2 will aid in better characterization of their effect on outcomes, identification of subpopulations that would benefit, and the timing at which the risk factor(s) would have the maximal impact. Additionally, hypothesis 3 highlights the importance of multifactorial or multi-domain approaches to “exceptional aging” as well as prevention of AD dementia.
Conclusion
While important strides have been made in identifying risk factors for AD dementia incidence, further efforts are needed to translate these into effective preventive strategies. Using biomarker studies for understanding the mechanism of action, effect size estimation, selection of appropriate end-points, and better subject recruitment based on subpopulation effects are fundamental for better design and success of prevention trials.
Keywords: Exceptional Aging, AD prevention, Biomarker cascade
Background
The two primary histopathological changes to the brain due to Alzheimer’s disease (AD) are the deposition of amyloid and tau [1]. These two AD-related brain changes are the primary underlying causes of neurodegeneration and cognitive dysfunction which ultimately leads to dementia. As human longevity increases, and AD dementia increasingly becomes a major societal burden, finding pathways that lead to brain aging without AD pathologies (ADP) are critical. Currently, much of the research has been focused on resilience or cognitive reserve [2], wherein the focus has been on discovering how and why individuals are able to remain clinically unimpaired or cognitively normal despite ADP. However, it is important to investigate, using surrogates of amyloid and tau pathologies via cerebrospinal fluid (CSF) and positron emission tomography (PET), why majority of individuals develop ADP as they age and how some oldest old individuals are able to age without significant ADP. The latter individuals are called “exceptional agers” without ADP. While the absence of ADP can be defined using various thresholds, we refer to the absence of ADP as not reaching the neuropathological definition of AD in pathology studies and the imaging cutoffs of amyloid and tau positivity in imaging studies. Amyloid and tau PET scans of an exceptional ager in comparison to a clinically unimpaired individual and an AD dementia individual are shown in Fig. 1.
Fig. 1.
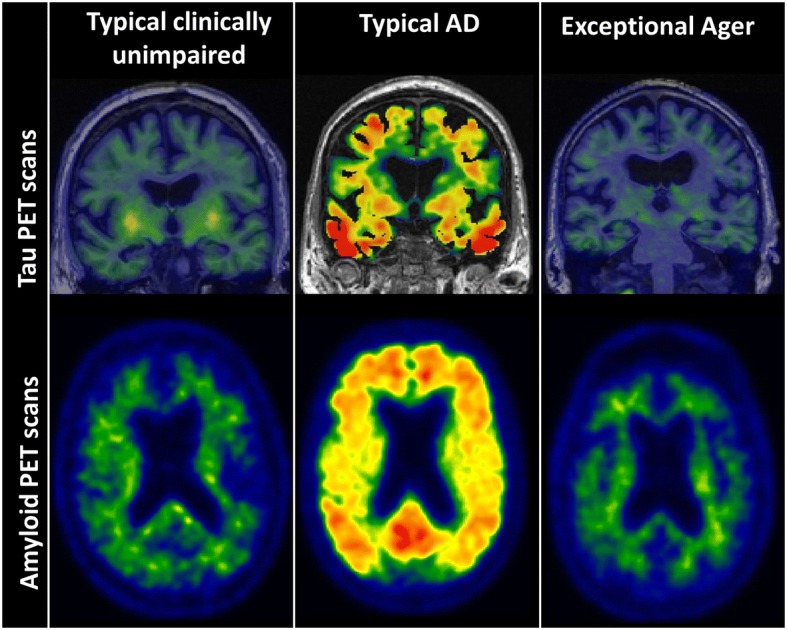
Tau and amyloid positron emission tomography (PET) scans in a typical clinically unimpaired, typical AD, and an exceptional ager (> 85-year-old APOE4 carrier)
In this view point, the main goal is to bring together ideas and observations from the literature and present them as testable hypotheses or frameworks that can be employed in biomarker studies to discover protective factors or pathways to “exceptional brain aging”. In the context of the terminology we recently proposed, for hypotheses 1 and 2 the focus is on “resistance to ADP” [3] and for hypothesis 3 the focus is on both resistance to ADP and prevention of AD dementia.
These concepts are presented in the context of the primary AD pathophysiological processes in the biomarker cascade (amyloid, tau, and neurodegeneration due to AD pathologies). The focus is on primary prevention in midlife, designing effective trials by understanding the mechanisms of action on the biomarker cascade, and looking at the net sum protection against all components of the biomarker cascade. Although additional AD processes are not explicitly addressed, such as inflammation, synaptic and microglial dysfunction that are relevant to aging and AD dementia, the concepts here can also be extended to other measurable biomarkers that are mechanistically relevant to AD.
“Exceptional brain aging” without ADP: is it really possible?
Several pathology and observational studies have provided evidence for aging without ADP [4, 5] and have focused on optimal or successful aging without cognitive decline [6–8] in the oldest old. In addition, specific evidence for “exceptional brain aging” without ADP comes from these three different lines of investigation.
Prevalence of AD pathologies
Nelson et al. [9] published an amalgamation of neuropathological literature showing that each added year of life does not lead to an increased prevalence of AD pathologies, unlike hippocampal sclerosis and cerebrovascular disease. Neuroimaging studies in the Mayo Clinic Study of Aging (MCSA) have also found non-monotonicity in the frequency of amyloid positivity in clinically unimpaired individuals [10, 11]. The data from our previous work [11] were consolidated to plot the prevalence of elevated amyloid versus excess cerebrovascular disease burden in clinically unimpaired individuals (Fig. 2a). These curves are reminiscent of two types of growth curve models in population ecology: exponential, or J-shaped, and logistic, or S-shaped, models. While exponential models have uninhibited growth in numbers, logistic growth models exhibit a slowing in growth as the population reaches its carrying capacity. Vascular pathologies show a steady increase in the prevalence or rate of growth representing an exponential model over an age range of 50–100 years. On the contrary, the amyloid elevation curves exhibit a slow saturation alluding to the fact that there may be a proportion of the population that will never develop elevated levels of amyloid, supported by evidence from Khachaturian et al. [12]. Amyloid data collected from 55 studies by Jansen et al. also showed that a logistic model was the best fit for amyloid prevalence [13].
Fig. 2.

a Prevalence of elevated amyloid levels (A+) versus vascular disease (V+) in clinically unimpaired individuals based on data from Vemuri and Knopman [11]. Vascular pathologies show a steady increase in the prevalence (exponential growth curve models) but the amyloid positivity curves exhibit a slow saturation similar to logistic growth curve models. b Data from our previous study [25] illustrates the slow longitudinal cognitive decline seen in a clinically unimpaired 80-year-old male without amyloid and cerebrovascular pathologies (in blue) in comparison with significantly greater decline seen in a clinically unimpaired individual of the same age with both elevated amyloid and cerebrovascular pathologies (in red)
Declining AD incidence and amyloid levels
Recent evidence of age-specific decline in both incidence of dementia [14, 15] and amyloid levels [16] in aging brains provides compelling evidence for the possibility of aging without AD pathologies. With the strong possibility that better medical care and increasing education levels may have contributed to these declining trends [17], investigation into the underlying mechanisms may lead us closer to understanding the differences between normal aging and developing ADP.
APOE4 carriers without AD dementia and AD pathologies in the oldest old
Age and the apolipoprotein (APO)E4 genotype are the two well-established risk factors for AD [13]. Therefore, one would expect that, as people age, the odds of an APOE4 individual developing AD dementia would increase with age. However, there have been several observations showing that the association between APOE4 genotype and development of AD dementia is weak in the oldest old, i.e., there are some APOE4 carriers who live into their 90s without AD dementia [12, 18–20]. While these studies have proven the presence of very old APOE4 carriers without AD dementia, one may argue that protection against AD dementia primarily comes from “resilience to ADP, i.e., coping with pathology”. However, the presence of amyloid-negative APOE4 cognitively normal individuals at 85 years of age (~ 25%) in a large meta-analysis [13] supports the idea of “resistance to ADP” in the oldest old APOE4 carriers.
While the observed evidence can be attributed to excess mortality early in life in those at risk (for example, for APOE4 carriers), it is important to study and understand how some individuals are able to age without ADP.
Discovering pathways to “exceptional aging”
Given the possibility of “exceptional aging”, how does one discover the important protective factors. Three inter-related ideas or hypotheses are presented here that, when taken together, can aid in discovering protective pathways and help design effective preventive strategies.
Hypothesis 1 (primary prevention in midlife)
Discovering and quantifying links between risk factors and early ADP changes in midlife using longitudinal biomarker studies is fundamental to understanding why the majority of individuals deviate from normal aging to the AD pathway.
Normal aging versus pathological aging
Aging acts through a number of biological mechanisms at the cellular or tissue level that lead to loss of reserve and function [21]. Prominent aging-related changes occur in the brain during midlife, and more so in the sixth to seventh decades. Midlife also represents the time during which (neurodegenerative and cerebrovascular) pathologies are observed in brain autopsies [9]. Even in the absence of pathologies, individuals suffer from age-related neural structure alterations [22, 23] and alterations in gene expression [24] starting in midlife. However, in the presence of neurodegenerative and cerebrovascular pathologies, the structural and functional deterioration of the brain has been observed to be greater. This accelerated decline in brain health due to neurodegenerative and cerebrovascular pathologies is the primary observed cause of dementia. By age 80, > 60% of clinically unimpaired individuals have either ADP or cerebrovascular disease. Figure 2b based on data from our previous study [25] illustrates the slow longitudinal cognitive decline seen in a clinically unimpaired 80-year-old male without amyloid and cerebrovascular pathologies (in blue) in comparison with a significantly greater decline in a clinically unimpaired individual of the same age with both amyloid and cerebrovascular pathologies (in red). There is also consensus about the significant heterogeneity in the cognitive aging process [7]. All these studies taken together provide evidence that normal aging is different from pathological aging and late midlife represents a critical time period during which we observe noticeable divergence of these two pathways. Given that slowing of age-related changes in midlife can be observed with better lifestyle factors such as physical activity and ideal levels of cardiovascular health [26–28], our focus should be on primary prevention during midlife and early adulthood.
There is well-established literature supporting that midlife conditions have a significant impact on late-life dementia, especially cardiorespiratory fitness [29] and vascular risk factors [30]. The relationship between several risk factors (obesity, hypertension, dyslipidemia) and dementia incidence has been observed to be U-shaped in nature with the greatest association during midlife [31–33]. Additionally, the prevalence of amyloid curves (as mentioned above) follows a logistic growth curve model with the greatest rate of amyloid accumulation in the population during late midlife. The first hypothesis proposes that greater focus needs to be placed on longitudinal biomarker studies that can discover and quantify links between risk factors in midlife and increased ADP accumulation in late midlife to understand why individuals deviate from the normal aging process.
One may argue that there has been extensive literature already supporting the hypothesis that midlife risk factors such as vascular risk factors increase late life dementia incidence. However, the results from intervention studies based on a reduction of vascular risk factors [34] highlights the need for longitudinal biomarker studies in midlife that focus on understanding the mechanisms of action of the suggested risk factors as early ADP changes evolve. This is especially important for risk or protective factors that are highly debated in the literature [35–37]. Understanding how the risk factors or combination of risk factors impact early ADP changes (whether it is amyloid, tau, or neurodegeneration) using longitudinal studies will facilitate a better understanding of how protective factors can be employed for primary prevention [38, 39]. While significant focus has been placed on amyloid imaging since it has been available from the mid-2000s, the same concepts can be extended to tau-related studies as longitudinal tau data become available [40].
Hypothesis 2 (designing effective trials)
A specific risk factor may have quantifiably greater impact as a trigger and/or accelerator on a specific component of the biomarker cascade (amyloid, tau, or neurodegeneration).
The biomarker cascade framework and quantifying the impact of each risk/protective factor
Although amyloid and tau deposition can be initiated independently, there is sufficient recent evidence supporting the hypothesis that amyloid deposition accelerates tau deposition which, in turn, is closely associated with cognitive decline [41–44]. Autosomal dominant AD studies that represent younger-onset pure AD cases have confirmed the sequence of amyloid followed by tau, followed by cognitive decline [45, 46]. The biomarker model presented and refined based on the literature by Jack et al. [43] synthesized AD processes into a set of testable hypotheses. Amyloid, tau, neurodegeneration, and cognitive decline form the biomarker cascade and this framework has helped significantly improve our understanding of disease onset and progression [41, 47–49].
The presence of suspected non-AD pathophysiology (SNAP; neurodegeneration in the absence of amyloid) [50] and primary age-related tauopathy (PART) in the absence of amyloid [51] illustrate the heterogeneity in the age-related neurodegenerative processes and share some pathophysiological aspects (neurodegeneration or tau) of the AD biomarker cascade. Since each of these pathophysiologies plays a role in the development of AD dementia, as discussed further in hypothesis 3 below, studying independent triggers and accelerators for each component of the AD biomarker cascade will be important. In the second hypothesis, it is proposed that looking at each individual component of the biomarker cascade (amyloid, tau, neurodegeneration) to explore the impact of the risk factor of interest will aid in understanding the mechanisms through which the specific risk factor impacts AD processes.
Importance of knowing the mechanisms
Although a vast amount of literature has provided evidence for the impact of risk factors on dementia incidence, less has been published on the impact of each individual risk factor on the primary disease mechanisms. Discerning the disease stage at which the reduction of a specific risk factor would be helpful will be important for designing effective preventive strategies. A recent example was the failure of the TOMORROW trial, which targeted diabetes medications for reduction of dementia [38]. While there has been substantial evidence that diabetes is associated with AD dementia incidence, the primary mechanism of action may be through neurodegeneration (discussed further below) [52]. Therefore, with diabetes as a preventive strategy, the focus should be on measuring the reduction in neurodegeneration and not on reduction in amyloid deposition. Another example is that of sleep as a preventive strategy. While poor sleep has been shown to impact amyloid deposition through poor clearance of amyloid [53, 54], and thus could mechanistically be linked to greater dementia incidence [55] and brain atrophy [56], improving sleep quality as a preventive strategy for AD dementia may fail in individuals who have high levels of amyloid. Therefore, quantifying the effect size of risk factors on each component of the biomarker cascade will aid in choosing appropriate outcomes and the sample sizes required. In addition, determining the effect modifiers (main biological and disease-related factors that may influence the treatment response such as additional interactions of the risk factors with age and APOE4 status) will aid in better enrichment strategies and intervention optimization.
Figure 3 illustrates well-established triggers and accelerators for some of the components of the biomarker cascade. A specific example of vascular health and neurodegeneration is discussed here. Poor vascular health and vascular risk factors are clearly related to higher incidence of dementia [57] as well as causing significant brain changes independent of amyloid and tau [58]. While there has been no doubt that vascular risk factors, specifically diabetes and hypertension, increase neurodegeneration (cortical thinning and hippocampal atrophy), there has been considerable debate about the impact of vascular risk on amyloid deposition. In a recent study, we found that the impact of vascular health was quantifiably greater on neurodegeneration than on amyloid deposition supporting the second hypothesis [52]. If one were to consider that vascular risk factors cause significantly greater neurodegeneration and cognitive decline compared with their effect on early amyloid deposition, it strongly supports the epidemiological findings that vascular risk factors lower the threshold of dementia detection and are related to a higher incidence of dementia [57].
Fig. 3.

Framework for the second hypothesis and examples of triggers and accelerators of some of the components of the biomarker cascade. AD, Alzheimer’s disease; APOE, apolipoprotein E
Hypothesis 3 (net sum game)
“Exceptional aging” as well as protection against AD dementia will come from “net sum” protection against all the components of the AD biomarker cascade.
If protection against AD pathology in each individual were viewed as a “net sum” of effects from all triggers and accelerators (lifestyle, midlife risk factors, chronic conditions, net difference between protective and risk genes) as well as additive and interactive non-AD processes, then “exceptional aging” without ADP and ultimately without AD dementia would be possible if a large positive “net sum” were present. This hypothesis highlights the importance for multifactorial or multidomain approaches to “exceptional aging” without ADP and AD dementia.
The support for “net sum” against AD dementia primarily comes from dementia risk score studies [59, 60] that have shown that a combination of several risk factors are best at predicting dementia risk compared with individual risk factors. The large positive “net sum” against ADP was also observed in our recent study where we found that, irrespective of the impact of a risk factor on amyloid or neurodegeneration, several protective factors (absence of midlife risk factors, lower chronic conditions) had moderate effect sizes in predicting those who were greater than or equal to 85 years of age without abnormal amyloid and neurodegeneration levels compared with those who had significant amyloid and neurodegeneration [37]. In addition, greater intellectual enrichment can further aid in delaying the onset of impairment through its impact on cognition, as illustrated by Fig. 4a [61–63].
Fig. 4.

Impact of intellectual enrichment (e.g., education, occupation, cognitive activity) and Other neurodegenerative processes on the Alzheimer’s disease (AD) trajectories. Cognition curves are superimposed on the ADP curves (amyloid or tau) shown in blue. The horizontal line indicates the cognitive impairment threshold. The time at which cognitive function meets the threshold allows us to deduce the ADP levels at the same time point on the superimposed ADP curves. a Illustration of individuals with high (green curve) and low (red dashed line) intellectual enrichment and the ADP levels (shown by the circles) at which cognitive impairment would be observed in both groups of individuals. b Illustration of individuals with only AD path (green curve) and AD path in addition to other neurodegenerative pathologies (red dashed line) and the ADP levels (shown by the circles) at which cognitive impairment would be observed in both groups of individuals
The presence of non-AD processes such as cerebrovascular disease, TDP-43, Lewy bodies (often alongside AD processes) and their contribution to cognitive impairment are important to consider in this context since non-AD neurodegenerative pathologies reduce the threshold to AD dementia when present along with ADP [57, 64]. This concept can be observed in Fig. 4b, which illustrates two subsets of individuals: the first have cognitive decline or neurodegeneration only due to ADP, and the second have a greater rate of neurodegeneration or cognitive decline due to other non-AD neurodegenerative processes along with ADP. A clear difference can be observed in the levels of ADP at which the same level of cognitive impairment would be expected for both groups. The second group would need a much lower level of amyloid to experience the same level of cognitive impairment as the first group. This figure illustrates the importance of viewing protection against AD dementia as protection against all components of the AD biomarker cascade.
A major limitation of this work was limiting the scope to the three main AD biomarkers for simplicity. However the concepts illustrated in Figs. 3 and 4 can be extended after inclusion of additional measurable AD-specific processes such as inflammation as well as non-AD processes and pathologies.
Conclusions
While important strides have been made in identifying risk factors for AD dementia incidence, future efforts need to be directed towards discovering the timing and mechanism of action of each of these risk factors on AD processes. In this work, three inter-related ideas are presented that are important to consider while studying risk factors and may help us move towards developing effective preventive strategies to maneuver individuals away from the AD pathway towards the pathway of “exceptional brain aging” without ADP.
Acknowledgements
The author would like to thank David S. Knopman, MD, Eider M. Arenaza-Urquijo, PhD, and the reviewers for their excellent comments, as well as Heather Wiste and Timothy Lesnick for their help generating Fig. 2. For the images used, we would like to thank AVID Radiopharmaceuticals for the provision of AV-1451 precursor, chemistry production advice and oversight, and FDA regulatory cross-filing permission and documentation.
Funding
The author was funded by NIH grants (R01 NS097495 and R01 AG056366).
Authors’ contributions
The author read and approved the final manuscript.
Ethics approval and consent to participate
The data reported here are from Mayo Clinic Study of Aging and from publications by the author. These studies were approved by the Mayo Clinic and Olmsted Medical Center institutional review board. Informed consent was obtained from all participants or their surrogates.
Competing interests
The author declares that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 2.Stern Y. Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurol. 2012;11(11):1006–1012. doi: 10.1016/S1474-4422(12)70191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arenaza-Urquijo EM, Vemuri P. Resistance vs resilience to Alzheimer disease: clarifying terminology for preclinical studies. Neurology. 2018;90(15):695–703. doi: 10.1212/WNL.0000000000005303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett DA, et al. Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol. 2012;72(4):599–609. doi: 10.1002/ana.23654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Besser LM, et al. Late-life vascular risk factors and Alzheimer disease neuropathology in individuals with normal cognition. J Neuropathol Exp Neurol. 2016;75(10):955–962. doi: 10.1093/jnen/nlw072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goveas JS, et al. Predictors of optimal cognitive aging in 80+ women: the Women’s Health Initiative Memory Study. J Gerontol A Biol Sci Med Sci. 2016;71 Suppl 1:S62–S71. doi: 10.1093/gerona/glv055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayden KM, et al. Cognitive decline in the elderly: an analysis of population heterogeneity. Age Ageing. 2011;40(6):684–689. doi: 10.1093/ageing/afr101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogalski E, et al. Cognitive trajectories and spectrum of neuropathology in SuperAgers: the first 10 cases. Hippocampus. 2018. 10.1002/hipo.22828. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 9.Nelson PT, et al. Alzheimer’s disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011;121(5):571–587. doi: 10.1007/s00401-011-0826-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jack CR, Jr, et al. Age-specific population frequencies of cerebral beta-amyloidosis and neurodegeneration among people with normal cognitive function aged 50-89 years: a cross-sectional study. Lancet Neurol. 2014;13(10):997–1005. doi: 10.1016/S1474-4422(14)70194-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vemuri P, Knopman DS. The role of cerebrovascular disease when there is concomitant Alzheimer disease. Biochim Biophys Acta. 2016;1862(5):952–956. doi: 10.1016/j.bbadis.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khachaturian AS, et al. Apolipoprotein E epsilon4 count affects age at onset of Alzheimer disease, but not lifetime susceptibility: the Cache County Study. Arch Gen Psychiatry. 2004;61(5):518–524. doi: 10.1001/archpsyc.61.5.518. [DOI] [PubMed] [Google Scholar]
- 13.Jansen WJ, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015;313(19):1924–1938. doi: 10.1001/jama.2015.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Satizabal CL, et al. Incidence of dementia over three decades in the Framingham Heart Study. N Engl J Med. 2016;374(6):523–532. doi: 10.1056/NEJMoa1504327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matthews FE, et al. A two decade dementia incidence comparison from the Cognitive Function and Ageing Studies I and II. Nat Commun. 2016;7:11398. doi: 10.1038/ncomms11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kovari E, et al. Amyloid deposition is decreasing in aging brains: an autopsy study of 1,599 older people. Neurology. 2014;82(4):326–331. doi: 10.1212/WNL.0000000000000069. [DOI] [PubMed] [Google Scholar]
- 17.Norton S, et al. Potential for primary prevention of Alzheimer's disease: an analysis of population-based data. Lancet Neurol. 2014;13(8):788–794. doi: 10.1016/S1474-4422(14)70136-X. [DOI] [PubMed] [Google Scholar]
- 18.Sobel E, et al. Lack of association of apolipoprotein E allele epsilon 4 with late-onset Alzheimer's disease among Finnish centenarians. Neurology. 1995;45(5):903–907. doi: 10.1212/WNL.45.5.903. [DOI] [PubMed] [Google Scholar]
- 19.Farrer LA, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278(16):1349–1356. doi: 10.1001/jama.1997.03550160069041. [DOI] [PubMed] [Google Scholar]
- 20.Bonham LW, et al. Age-dependent effects of APOE epsilon4 in preclinical Alzheimer's disease. Ann Clin Transl Neurol. 2016;3(9):668–677. doi: 10.1002/acn3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fabbri E, et al. Aging and multimorbidity: new tasks, priorities, and frontiers for integrated gerontological and clinical research. J Am Med Dir Assoc. 2015;16(8):640–647. doi: 10.1016/j.jamda.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jagust W. Vulnerable neural systems and the borderland of brain aging and neurodegeneration. Neuron. 2013;77(2):219–234. doi: 10.1016/j.neuron.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fjell AM, et al. What is normal in normal aging? Effects of aging, amyloid and Alzheimer's disease on the cerebral cortex and the hippocampus. Prog Neurobiol. 2014;117:20–40. doi: 10.1016/j.pneurobio.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berchtold NC, et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc Natl Acad Sci U S A. 2008;105(40):15605–15610. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vemuri P, et al. Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain. 2015;138(Pt 3):761–771. doi: 10.1093/brain/awu393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bancks MP, et al. Cardiovascular health in young adulthood and structural brain MRI in midlife: The CARDIA study. Neurology. 2017;89(7):680–686. doi: 10.1212/WNL.0000000000004222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arenaza-Urquijo EM, et al. Distinct effects of late adulthood cognitive and physical activities on gray matter volume. Brain Imaging Behav. 2017;11(2):346–356. doi: 10.1007/s11682-016-9617-3. [DOI] [PubMed] [Google Scholar]
- 28.Okonkwo OC, et al. Physical activity attenuates age-related biomarker alterations in preclinical AD. Neurology. 2014;83(19):1753–1760. doi: 10.1212/WNL.0000000000000964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Defina LF, et al. The association between midlife cardiorespiratory fitness levels and later-life dementia: a cohort study. Ann Intern Med. 2013;158(3):162–168. doi: 10.7326/0003-4819-158-3-201302050-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gottesman RF, et al. Associations between midlife vascular risk factors and 25-year incident dementia in the Atherosclerosis Risk in Communities (ARIC) cohort. JAMA Neurol. 2017;74(10):1246–1254. doi: 10.1001/jamaneurol.2017.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beydoun MA, Beydoun HA, Wang Y. Obesity and central obesity as risk factors for incident dementia and its subtypes: a systematic review and meta-analysis. Obes Rev. 2008;9(3):204–218. doi: 10.1111/j.1467-789X.2008.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker KA, Power MC, Gottesman RF. Defining the relationship between hypertension, cognitive decline, and dementia: a review. Curr Hypertens Rep. 2017;19(3):24. doi: 10.1007/s11906-017-0724-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wendell CR, Waldstein SR, Zonderman AB. Nonlinear longitudinal trajectories of cholesterol and neuropsychological function. Neuropsychology. 2014;28(1):106–112. doi: 10.1037/neu0000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moll van Charante EP, et al. Effectiveness of a 6-year multidomain vascular care intervention to prevent dementia (preDIVA): a cluster-randomised controlled trial. Lancet. 2016;388(10046):797–805. doi: 10.1016/S0140-6736(16)30950-3. [DOI] [PubMed] [Google Scholar]
- 35.Kemppainen N, et al. Brain amyloid load and its associations with cognition and vascular risk factors in FINGER study. Neurology. 2018;90(3):e206–e213. 10.1212/WNL.0000000000004827. [DOI] [PubMed]
- 36.Gottesman RF, et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA. 2017;317(14):1443–1450. doi: 10.1001/jama.2017.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vemuri P, et al. Evaluation of amyloid protective factors and Alzheimer disease neurodegeneration protective factors in elderly individuals. JAMA Neurol. 2017;74(6):718–726. doi: 10.1001/jamaneurol.2017.0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.There’s No Tomorrow for TOMMORROW, in https://www.alzforum.org/news/research-news/theres-no-tomorrow-tommorrow. 10 Feb 2018.
- 39.Vemuri P, et al. Effect of intellectual enrichment on AD biomarker trajectories: longitudinal imaging study. Neurology. 2016;86(12):1128–1135. doi: 10.1212/WNL.0000000000002490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jack CR Jr, et al. Longitudinal tau PET in ageing and Alzheimer's disease. Brain. 2018; [DOI] [PMC free article] [PubMed]
- 41.Villemagne VL, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 42.Sperling RA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Assocation workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jack CR, Jr, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knopman DS, et al. Selective worsening of brain injury biomarker abnormalities in cognitively normal elderly persons with beta-amyloidosis. JAMA Neurol. 2013;70(8):1030–8. doi: 10.1001/jamaneurol.2013.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fleisher AS, et al. Associations between biomarkers and age in the presenilin 1 E280A autosomal dominant Alzheimer disease kindred: a cross-sectional study. JAMA Neurol. 2015;72(3):316–324. doi: 10.1001/jamaneurol.2014.3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bateman RJ, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367(9):795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vos SJ, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12(10):957–965. doi: 10.1016/S1474-4422(13)70194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dubois B, et al. Preclinical Alzheimer's disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12(3):292–323. doi: 10.1016/j.jalz.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sperling RA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6(228):228fs13. doi: 10.1126/scitranslmed.3007941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jack CR, Jr, et al. Suspected non-Alzheimer disease pathophysiology—concept and controversy. Nat Rev Neurol. 2016;12(2):117–124. doi: 10.1038/nrneurol.2015.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crary JF. Primary age-related tauopathy and the amyloid cascade hypothesis: the exception that proves the rule? J Neurol Neuromedicine. 2016;1(6):53–57. doi: 10.29245/2572.942X/2016/6.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vemuri P, et al. Age, vascular health, and Alzheimer disease biomarkers in an elderly sample. Ann Neurol. 2017;82(5):706–18. 10.1002/ana.25071. [DOI] [PMC free article] [PubMed]
- 53.Xie L, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carvalho DZ, et al. Association of excessive daytime sleepiness with longitudinal beta-amyloid accumulation in elderly persons without dementia. JAMA Neurol. 2018. 10.1001/jamaneurol.2018.0049. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 55.Chen JC, et al. Sleep duration, cognitive decline, and dementia risk in older women. Alzheimers Dement. 2016;12(1):21–33. doi: 10.1016/j.jalz.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carvalho DZ, et al. Excessive daytime sleepiness and fatigue may indicate accelerated brain aging in cognitively normal late middle-aged and older adults. Sleep Med. 2017;32:236–243. doi: 10.1016/j.sleep.2016.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chui HC, et al. Vascular risk factors and Alzheimer's disease: are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence-based review. Alzheimers Res Ther. 2012;4(1):1. doi: 10.1186/alzrt98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jefferson AL, et al. Lower cardiac index levels relate to lower cerebral blood flow in older adults. Neurology. 2017;89(23):2327–2334. doi: 10.1212/WNL.0000000000004707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Exalto LG, et al. Midlife risk score for the prediction of dementia four decades later. Alzheimers Dement. 2014;10(5):562–570. doi: 10.1016/j.jalz.2013.05.1772. [DOI] [PubMed] [Google Scholar]
- 60.Kivipelto M, et al. Risk score for the prediction of dementia risk in 20 years among middle aged people: a longitudinal, population-based study. Lancet Neurol. 2006;5(9):735–741. doi: 10.1016/S1474-4422(06)70537-3. [DOI] [PubMed] [Google Scholar]
- 61.Vemuri P, et al. Effect of lifestyle activities on Alzheimer disease biomarkers and cognition. Ann Neurol. 2012;72(5):730–738. doi: 10.1002/ana.23665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vemuri P, et al. Cognitive reserve and Alzheimer's disease biomarkers are independent determinants of cognition. Brain. 2011;134(Pt 5):1479–1492. doi: 10.1093/brain/awr049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vemuri P, et al. Association of lifetime intellectual enrichment with cognitive decline in the older population. JAMA Neurol. 2014;71(8):1017–1024. doi: 10.1001/jamaneurol.2014.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schneider JA, et al. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66(2):200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]