

Abstract
Autophagy is a conserved catabolic pathway that involves the engulfment of cytoplasmic components such as large protein aggregates and organelles that are delivered to the lysosome for degradation. This process is important in maintaining neuronal function and raises the possibility of a role for autophagy in neurodegenerative diseases. Alzheimer's disease (AD) is the most prevalent form of these diseases and is characterized by the accumulation of amyloid plaques in the brain which arise due to the misfolding and aggregation of toxic peptides, including amyloid beta (Aβ). There is substantial evidence from both AD patients and animal models that autophagy is dysregulated in this disease. However, it remains to be determined whether this is protective or pathogenic as there is evidence that autophagy can act to promote the degradation as well as function in the generation of toxic Aβ peptides. Understanding the molecular details of the extensive crosstalk that occurs between the autophagic and endolysosomal cellular pathways is essential for identifying the molecular details of amyloid toxicity. Drosophila models that express the toxic proteins that aggregate in AD have been generated and have been shown to recapitulate hallmarks of the disease. Here we focus on what is known about the role of autophagy in amyloid toxicity in AD from mammalian models and how Drosophila models can be used to further investigate AD pathogenesis.
1. Introduction
Alzheimer's disease (AD) is the most prevalent form of neurodegenerative disease characterized by deficiency in memory and cognitive functions. The predominant pathological changes of AD are the development of amyloid beta (Aβ) plaque deposits in specific brain areas and neurofibrillary tangles (NFTs) within neuronal cells, leading to the progressive loss of synapses, neuronal death, and cognitive decline [1–3]. The extracellular Aβ plaques are derived from cleavage of the amyloid precursor protein (APP). The NFTs consist of intracellular aggregates of the hyperphosphorylated microtubule-associated protein tau, mutant forms of which are also found in other neurodegenerative diseases termed tauopathies. This review will focus on the role of APP and the products arising from its proteolysis (which includes Aβ42) in AD.
While the primary mechanisms responsible for AD pathology remain to be established, there is increasing evidence for a role of the autophagy pathway in AD. Macroautophagy (referred to here as autophagy) is a conserved catabolic pathway that sequesters cytoplasmic material in a double-membrane vesicle (of nonlysosomal/vacuolar origin), the autophagosome, for delivery to the lysosome. Autophagy is induced in response to cellular stress and protects cells by eliminating dysfunctional organelles and toxic protein aggregates. Aberrant regulation of autophagy has significant adverse consequences to normal cellular functions and is associated with numerous human pathologies, including neurodegenerative diseases [4]. This review will describe the pathogenesis of AD, the conservation of components of the autophagy machinery, and their known roles in neurodegeneration. We will discuss evidence of autophagy perturbation in AD and focus on Drosophila as an ideal model for understanding the molecular mechanisms by which autophagy contributes to AD.
2. The Genetics of Alzheimer's Disease (AD)
There are two types of AD based on genetic inheritance and age of onset. Familial AD is rare, affecting approximately 1–5% of individuals that are under 65 years of age. Autosomal dominant mutations have been identified in amyloid precursor protein (APP) as well as presenilin-1 (PS1) and presenilin-2 (PS2) genes that encode the catalytic subunit of γ-secretase complex that cleaves APP to promote the generation of Aβ peptides as causative agents for familial AD [13, 14]. Sporadic, late-onset AD accounts for more than 95% of cases with both genetic and environmental factors contributing to the pathogenesis. While the genetic contribution in these patients is not fully defined, genome-wide association studies have identified several loci associated with increased AD risk in genes involved in various biological pathways including cholesterol/sterol metabolism (APOE-ε4), innate immunity (CR1, CD33, and TREM2), and endolysosomal and autophagy pathways (BIN1, PICALM, and CD2AP) [15, 16]. In addition, recent studies in mammalian cells further support the role of abnormal trafficking in the endolysosomal and autophagy pathways contributing to AD [17, 18].
3. Proteolysis of Amyloid Precursor Protein
APP is a transmembrane protein that undergoes sequential cleavage by one of two pathways (Figure 1). The initial proteolytic cleavage of APP by either α-secretase (nonamyloidogenic processing) or β-secretase (amyloidogenic processing) produces APP-carboxy-terminal fragments (CTFs) as well as secreted APP peptides. In the nonamyloidogenic pathway, α-secretase (ADAM10) cleavage occurs within the Aβ region generating α-carboxy-terminal fragments (α-CTFs) and thus prevents the formation of toxic Aβ [19, 20]. The α-CTF is further cleaved by γ-secretase complex to release P3 peptide as well as an APP intracellular domain (AICD) [21]. In the amyloidogenic pathway, APP is initially cleaved by β-secretase 1 (beta-site amyloid precursor protein cleaving enzyme 1, BACE1) to produce β-carboxy-terminal fragments (β-CTFs). Subsequent cleavage of β-CTF by γ-secretase complex releases toxic amyloid-β (Aβ) peptides (Figure 1). While the processing of APP by α-secretase is predominantly localized to the cell surface, amyloidogenic cleavage occurs in endosomes, lysosomes, and autophagic vacuoles [22–24].
Figure 1.
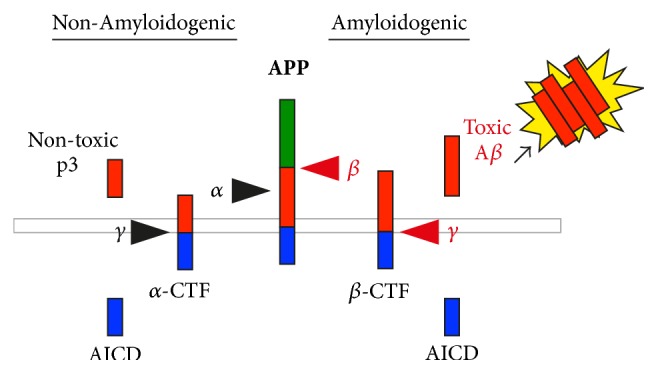
Proteolytic processing of amyloid precursor protein. In the nonamyloidogenic pathway, transmembrane APP is cleaved by α-secretase followed by γ-secretase generating a nontoxic P3 fragment and α C-terminal fragments (α-CTFs), thus preventing the generation of toxic Aβ. Alternatively, the amyloidogenic pathway involves sequential cleavages of APP by β-secretase followed by γ-secretase complex, thus generating toxic Aβ peptides in addition to the β-CTFs and amyloid precursor protein intracellular domain (AICD). The accumulation of Aβ peptides promotes oligomerisation and formation of insoluble plaques.
The amyloidogenic processing of APP increases the generation of Aβ that is susceptible to aggregation with other Aβ peptides accumulating into fibrils. This is commonly found in amyloid plaques in the brain (where Aβ42 aggregates are considered to be toxic) and is one of the hallmarks of AD. In addition to Aβ toxicity, the β-CTFs may also contribute to the pathogenesis of AD through multiple pathways [25, 26]. The AICDs of both cleavage pathways can translocate to the nucleus and induce nuclear signalling [27–29]. However, the principle physiological functions of APP remain largely undetermined. The proposed role for APP acting as a cell surface receptor or as a ligand, such as transcriptional regulation and/or synaptic functioning, requires further in vivo characterization [30]. While the generation of extracellular Aβ plaques is central to the hypothesis of amyloid as the causative agent in AD [31], additional factors have been identified which may contribute to the onset and/or progression of AD with dysregulation of autophagy thought to be an early event. Despite advances in the understanding of AD pathogenesis, further studies are required to understand the molecular mechanism by which autophagy contributes to disease pathogenesis. In addition, the consequence of Aβ as well as other products from APP processing on other cellular processes including autophagy needs further investigation.
4. Autophagy Molecular Machinery
Autophagy is a highly conserved catabolic pathway that degrades/recycles cytoplasmic material such as large protein aggregates and organelles. The cytoplasmic components are engulfed by a double-membrane vesicle, the autophagosome, for delivery to the lysosomes for degradation (Figure 2). Autophagy has essential functions in normal development, cell growth, metabolism, cell death, infection, and immunity [32–34]. It also acts to protect cells by eliminating toxic protein aggregates, unwanted cellular contents, dysfunctional organelles, and invading pathogens. Under growth-promoting conditions, low basal rates of autophagy are required to maintain cellular homeostasis. In response to extracellular and intracellular stresses, such as nutrient limitation, intracellular metabolic stress, organelle damage, and infection, high levels of autophagy are induced to recycle cytoplasmic material to maintain vital cellular processes [35]. The tightly coordinated multistep process of autophagy is regulated by a number of distinct autophagy-related (ATG) gene products that assemble into specific complexes [36]. Many of these components are evolutionarily conserved from yeast to mammals, including in Drosophila (Table 1). The autophagy process/complexes can be functionally divided into (i) initiation, (ii) autophagosome nucleation, (iii) expansion and vesicle completion, and (iv) lysosome fusion [37–40] (Figure 2).
Figure 2.

Schematic representation of the autophagy pathway and the regulatory machinery. The multiple steps of autophagy can be divided into initiation, nucleation, expansion, vesicle completion, and lysosome fusion. Several ATG proteins form distinct complexes that function in different stages of autophagy. The ULK1/ATG1 complex (consisting of ULK1, ATG13, ATG17, and ATG101) is responsible for the initiation of autophagy. The class III phosphatidylinositol 3-kinase (PI3K) complex (BECN1, VPS34, VPS15, and ATG14), ATG9, and ATG2-WIPI complex nucleate and assemble the membrane to form the double-membrane phagophore. The LC3 and ATG12 conjugation systems can be involved the formation of the autophagosome. Once completed, the autophagosome fuses with the lysosome where the enclosed components are degraded by lysosomal enzymes.
Table 1.
Conserved Autophagy-related genes in Drosophila, neurodegenerative phenotypes, and/or modification of AD models.
| Human gene | Drosophila gene | Neurodegenerative phenotype in Drosophila | Modification of AD model in Drosophila | |
|---|---|---|---|---|
| Initiation | ULK1/ATG1 | Atg1 | Decreased lifespan and climbing defect [6] | Deficiency line with decreased Atg1 reduced lifespan of Aβ42 expressing flies [5] |
| ULK2 | ||||
| ATG13 | Atg13 | |||
| FIP200/RB1CC1 | Atg17 | |||
| ATG101 | Atg101 | |||
|
| ||||
| Nucleation | BECN1 | Atg6 | ||
| ATG14 | Atg14 | |||
| PIK3R4/VPS15 | Vps15/ird1 | |||
| PIK3C3/VPS34 | Vps34/Pi3K59F | |||
|
| ||||
| Conjugation systems | ATG3 | Atg3/Aut1 | ||
|
| ||||
| Conjugation systems | ATG4A | Atg4a | ||
| ATG4B | ||||
| ATG4C | Atg4b | |||
| ATG4D | ||||
|
| ||||
| Conjugation systems | ATG5 | Atg5 | Climbing defect [7] | Decreased Atg5 reduced Aβ42 accumulation [8] |
|
| ||||
| Conjugation systems | ATG7 | Atg7 | Decreased lifespan and climbing defect [9] | |
|
| ||||
| Conjugation systems | MAP1LC3A | Atg8a | Reduced lifespan [10] | |
| MAP1LC3B | ||||
| MAP1LC3C | ||||
| GABARAP | ||||
| GABARAPL1 | Atg8b | |||
| GABARAPL2 | ||||
|
| ||||
| Conjugation systems | ATG10 | Atg10 | ||
|
| ||||
| Conjugation systems | ATG12 | Atg12 | Decreased Atg12 reduced Aβ42 accumulation [8] | |
|
| ||||
| Conjugation systems | ATG16L1 | Atg16 | Decreased lifespan and climbing defect [11] | |
|
| ||||
| ATG9 trafficking system | ATG9A | Atg9 | ||
| ATG9B | ||||
|
| ||||
| ATG9 trafficking system | ATG2A | Atg2 | ||
| ATG2B | ||||
|
| ||||
| ATG9 trafficking system | WIPI1 | |||
| WIPI2 | Atg18a | Deficiency line with decreased Atg18a reduced lifespan of Aβ42 expressing flies [5] | ||
| WDR45B/ | ||||
| WIPI3 | Atg18b | |||
| WDR45/WIPI4 | ||||
4.1. Initiation and Nucleation
Autophagy is initiated by the formation of a double-membrane structure called the phagophore (also called the isolation membrane) that further expands to encapsulate part of the cytoplasm into the autophagosome [41]. A key early step in autophagy induction requires the activity of the ULK1 (Atg1 in Drosophila) kinase complex, comprising ULK1/Atg1, ATG13, FIP200/ATG17, and ATG101. The activity of this complex is regulated in response to stress signals [42]. Nucleation from the phagophore (isolation membrane) requires active ULK1 kinase complexes for the recruitment of class III phosphatidylinositol 3-kinase (PI3K) complex. This complex consists of VPS34, VPS15, ATG14, and Beclin 1 (Atg6 in Drosophila) to generate phosphatidylinositol 3-phosphate (PI3P) required for vesicle nucleation.
4.2. Expansion and Vesicle Completion
The expansion and completion to form the autophagosome requires two ubiquitin-like conjugation systems: the Atg8/LC3-lipid phosphatidylethanolamine (PE) and the ATG12-ATG5 systems [37, 43, 44]. There are six Atg8 family members in mammals, including LC3A, LC3B, LC3C, and GABARAP proteins [45] and two in Drosophila with Atg8a shown to be essential for autophagy [46]. Prior to lipidation, LC3/Atg8 is cleaved to produce a C-terminal glycine residue (LC3-I form) by the cysteine protease, ATG4. This enables the conjugation of LC3 to PE mediated by ATG7 (E1-like enzyme) and ATG3 (E2-like enzyme). In the ATG12-ATG5 conjugation system, ATG7 and ATG10 (E1- and E2-like enzymes, respectively) mediate the conjugation of ATG12 to ATG5, which associates with ATG16. To enable phagophore expansion a supply of lipid bilayers is required and is thought to involve the transmembrane protein ATG9; however its exact function remains unclear. Membrane closure is thought to involve ATG2, in combination with WIPI1/Atg18, to regulate autophagosome formation [47].
4.3. Lysosomal Fusion
The final step in autophagy is fusion of the autophagosome with the lysosome to form an autolysosome [37]. Lysosomes are specialized organelles that function to break down extracellular materials and recycle cellular components from the secretory, endocytic, autophagic, and phagocytic pathways [48]. The lysosome contains hydrolytic enzymes required for degradation, including cathepsin proteases that are activated by the acidic pH within the lysosome generated by proton-pumping vacuolar H+ ATPase (v-ATPase) [49]. The fusion between autophagosome and lysosome requires the soluble N-ethylmaleimide-sensitive factor activating protein receptor (SNARE) complex consisting of syntaxin 17 (STX17) and synaptosomal-associated protein 29 (SNAP29). This complex forms on autophagosomes to promote tethering with vesicle-associated membrane protein 8 (VAMP8) on lysosomes, resulting in fusion to form autolysosomes [50].
4.4. Amphisome Formation
Instead of fusing with a lysosome, an autophagosome can also fuse with a late endosome/multivesicular body to form an amphisome [51]. Amphisomes contain markers of both autophagosomes (lipidated LC3) and endosomes (RAB5, RAB7, and RAB11) [52, 53]. Members of the endosomal sorting complex required for transport (ESCRT) complex are required during endocytosis as well as for later endosomal maturation and amphisome formation during autophagy [54].
4.5. Selective Autophagy
The targeting of cytoplasmic material to the autophagosome can also occur in a specific manner, by recognizing selective substrates. These can include, for example, damaged mitochondria (mitophagy), excess peroxisomes (pexophagy), and aggregate-prone proteins, including those causing many neurodegenerative conditions (aggrephagy) [38]. The selection of autophagic cargo can be determined by cargo receptors that interact with LC3 family member proteins on the membrane [55]. The multifunctional scaffold protein SQSTM1/p62 (known as Ref(2)P in Drosophila) binds ubiquitinated proteins and acts as a cargo receptor by binding LC3/Atg8 targeting ubiquitinated proteins for degradation by autophagy [56]. The type of ubiquitin linkages on the substrate can lead to different functional outcomes. The most common ubiquitin linkage tags proteins for degradation of the ubiquitin-proteasome system, whereas other linkages can direct nonproteasomal fates. There are a growing number of identified cargo receptors that bind specific substrates that are tagged with ubiquitin chains. Thus, the receptors serve as a link between ubiquitinated cargo and the autophagy pathway to enable the selective incorporation of the cargo into autophagosomes.
5. Role for Autophagy in Maintaining Neuronal Homeostasis
Multiple upstream signalling pathways regulate autophagy induction with nutrient deprivation, one of the most well characterized signals. The target of rapamycin (TOR) kinase is a central mediator in regulating the response to nutrients and growth signalling and forms a multisubunit complex, mTORC1 [57]. In the presence of growth signals, mTORC1 is activated, thus preventing autophagy by inhibiting ULK1/Atg1 kinase. Under growth-limiting conditions, mTORC1 is no longer active in enabling autophagy induction by activation of ULK1/Atg1 [58, 59]. Numerous studies link alterations of mTOR pathway to age-dependent cognitive decline and to pathogenesis of Alzheimer disease (AD) [60], highlighting the importance of maintaining physiological levels of autophagy to promote neuronal health.
Several nervous system-specific conditional knockout mouse models of autophagy pathway genes have highlighted the importance of autophagy in maintaining the normal functions and homeostasis of the nervous system. The conditional deletion of Atg5 and Atg7 in neuronal precursor cells results in autophagy deficiency, accompanied by the accumulation of intraneuronal aggregates in neurons resulting in neuronal loss and neurodegeneration [61–63]. The accumulation of these aggregates in otherwise normal mice suggests that autophagy plays a key role in removing aggregate-prone proteins. Other mouse models of autophagy deficiency, including conditional knockout for FIP200 and Wipi4, as well as Ulk1/2 double knockout [64–66], similarly show reduced survival and early-onset, progressive neurodegeneration across broad areas of the brain. However, each model presents variations in the pathology observed which may be due to the specific stage of autophagy that is disrupted, as well as any potential autophagy independent gene functions.
Reduced function of conserved autophagy genes in Drosophila also results in neurodegenerative phenotypes (Table 1). Initiation of autophagy requires Atg17/Fip200, and reduced expression in adult flies resulted in a climbing defect as well as decreased survival [6]. Atg5 null flies displayed mobility defects [7], and decreased Atg16 resulted in climbing defects and decreased survival [11]. Atg7 mutants show a shortened lifespan as well as accumulation of aggregated ubiquitin-positive lesions in neuronal cells [9] while Atg8a mutants that are viable show decreased lifespan [10]. Taken together, these studies demonstrate the critical and conserved role of autophagy in neuronal homeostasis with the impaired clearance by autophagy likely to be a key factor in the accumulation of toxic peptides in the neurons.
6. Autophagy in Alzheimer's Disease
A hallmark of AD and other neurodegenerative diseases is the accumulation of large protein aggregates/inclusions and defective organelles. Autophagy is an essential degradation pathway involved in the clearance of abnormal protein aggregates as well as maintaining protein homeostasis in neuronal cells [67]. There is substantial evidence from both AD patients as well as animal models for the dysregulation of autophagy in this disease. Current findings suggest that impairment of the autophagy pathway leads to defects in the clearance of protein aggregates which is likely to occur early in the pathogenic process, before plaque formation or NFTs deposition [68–70]. However the role of autophagy in AD (in particular which stage is affected) and its alteration during disease progression in neurons is complex and remains largely unclear. Alterations to autophagy have also been identified in other neurodegenerative diseases, including Parkinson's disease and Huntington's disease [71]. There is also evidence for mitophagy in these diseases; however, that is outside the focus of this review and has been reviewed elsewhere [72]. In healthy neurons, autophagy is constitutively active and highly efficient, with low levels of autophagosomes detected [73]. Early observations revealed the accumulation of abnormal subcellular vesicles in the dystrophic or swollen neurites in AD patient brains [74]. Further evidence for disruption to autophagy flux in AD was revealed by the identification of autophagosomes and other immature autophagic vesicles that accumulated in dystrophic neurites in AD brains [68]. While clinical data has identified defects in autophagosomal biogenesis, whether this is pathogenic or a consequence of earlier defects is still controversial. Also, there is evidence that autophagy may not only act to promote the degradation of Aβ but may also be involved in its generation [8, 75].
To aid in understanding the role of autophagy in AD, animal models have provided a tool for in vivo studies. A number of transgenic mouse models have been generated based on the genetic pathways disrupted in AD [76]. In an APP/PS1 transgenic mouse model that contains human transgenes for APP and PS1, both of which are carrying human disease mutations, neuronal autophagy is detected in the brain before the appearance of Aβ plaques [22]. Consequently, autophagosomes and late autophagic vacuoles/intracellular trafficking vesicles accumulate in dystrophic dendrites, suggesting impaired maturation of autophagosomes to lysosomes [22]. Similarly, in another study young (4- to 6-month-old) APP/PS1 mice accumulated abnormal immature autophagosomes in axons of hippocampus neurons before neuronal loss [77]. The localization of both APP and PS1 to autophagic vacuoles suggests that Aβ may be generated during autophagy [22, 78]. This indicates that accumulation of autophagic vacuoles/intracellular trafficking vesicles may be a source of Aβ production contributing to AD progression.
Altering the level of autophagy has also been examined in AD models. APP transgenic mice with Atg7 deletion showed a reduction in Aβ extracellular secretion and plaque formation [79, 80]. This block in Aβ secretion resulted in an accumulation of intracellular Aβ and enhanced neurodegeneration was observed. An increase in the level of autophagy by rapamycin inhibition of mTOR in APP transgenic mice reduced Aβ levels and prevented AD-like cognitive deficits [81]. These findings suggest that autophagy may function in either degradation or secretion in Aβ and supports a role for autophagy in limiting the accumulation of toxic Aβ.
There is further evidence from animal models that basal autophagy is beneficial for decreasing the pathology in AD. In the APP mouse model of AD, heterozygous deletion of BECN1 decreases neuronal autophagy and increases the accumulation of both intraneuronal and extracellular Aβ deposits followed by neurodegeneration [82]. In support of this, reduced levels of Beclin 1/BECN1 have been detected in the brains of patients with severe AD [82]. Consistent with this, a mouse knockin of a Beclin 1 gain of function mutation resulted in constitutively active autophagy and, when combined with an AD mouse model, showed reduced Aβ accumulation, prevented cognitive decline, and restored survival [83]. This suggests that in AD, BECN1 induced autophagy contributes to reduction in levels of Aβ peptides/aggregates. In an alternative approach, aged (7-month-old) APP/PS1 transgenic mice were transfected with miR-124 lentiviral vector that downregulates BACE1 [84]. These mice also showed increased Beclin 1 with alleviation of AD pathology but surprisingly they had decreased expression of other autophagy markers. This suggests that Beclin 1 may not be acting via the autophagic pathway in this system and may have other roles such as in the PtdIns 3-kinase complex (Rubicon-UVRAG-Beclin 1-hVps34-hVps15) that localizes to the late endosome/lysosome and inhibits autophagy [85]. Together these data highlight the need for comprehensive in vivo analyses to dissect the role of individual autophagy genes in AD pathogenesis.
7. Crosstalk between Autophagy and the Endolysosomal System in AD
The subcellular distribution of APP plays a key role in Aβ production and occurs within the autophagy and endolysosomal systems [86, 87]. The early endosome is the site of colocalization of APP and BACE1 promoting the proteolytic cleavage of APP [88, 89]. Indeed, endosomal pathology is one of the earliest defects observed in AD [90, 91]. Altered levels of the endosomal small GTPase, Rab5, precede Aβ deposition [91], and Aβ colocalizes in Rab5 endosomes in neurons from AD brain [23]. More recently, expression of a dominant negative Rab5 mutant was shown to reduce APP-induced axonal blockages in both cultured neurons and an in vivo Drosophila model [92]. Genome-wide association studies identified mutations in endosomal genes including BIN1, CD2AP, and PICALM, which supports the involvement of the endosomal network in processing and trafficking of APP proteolytic fragments [15]. Drosophila homologues of these genes show interactions with increased tau expression [17, 93, 94] but they have not been tested with respect to amyloid pathology.
The metabolism of APP in endolysosomal and autophagy networks is consistent with crosstalk between these pathways. Autophagic and lysosomal genes are coordinately regulated by a complex transcriptional program mediated by Transcription Factor EB (TFEB) [95]. TFEB levels have been found to be decreased in brains of Alzheimer's patients [96] while an increase in TFEB expression has been shown to be protective for Aβ-induced pathogenesis [97]. Similarly, in an APP/PS1 mouse model, the overexpression of TFEB increases lysosome biogenesis and reduces Aβ levels [98]. In Drosophila there is a single TFEB orthologue, Mitf, which has been shown to have a role in regulation of the v-ATPase proton pump as well as other components of the lysosomal-autophagic pathway to promote clearance of protein aggregates [99, 100].
As both endocytic and autophagic pathways lead to the lysosome, it is not surprising that aberrant lysosomal function contributes to AD pathogenesis. Defective lysosomal membrane integrity has been detected in AD patients suggesting dysfunction [101]. Increased expression levels of lysosomal proteases in the early phase of AD patients have also been reported [102]; it is likely that this increased lysosomal function is in response to increased pathogenic load. The AD-associated risk factor gene Apolipoprotein E4 (ApoE4) also affects lysosomal function. Transgenic mice that overexpress ApoE4 accumulate Aβ42 in lysosomes and there is death of neurons in the hippocampus [103]. Also in Neuro-2a cells, ApoE4 can affect lysosomal membrane permeabilization causing the release of proteolytic enzymes that can mediate cell death [104]. Further support for the function of lysosomes in AD was highlighted by the role of PS1 in the assembly of the v-ATPase pump in the lysosomal membrane, thus promoting acidification and contributing to autophagy degradation in a γ-secretase-independent way [105]. An alternative report suggested that the lysosome dysfunction resulting from loss of PS1 could be attributed to alterations in lysosomal calcium storage [106]. Increased or sustained activation of Glycogen synthase kinase-3 also affects lysosome acidification and has been shown to affect the autophagic degradation of APP [107, 108]. In addition, consideration needs to be given to the physiology of neuronal cells where retrograde transport of distally located autophagic vacuoles (mostly amphisomes) is required before any fusion can occur with lysosomes that are located in the soma [109].
These findings and others, including cell culture studies not described here, clearly establish autophagic and endolysosomal dysfunction in AD. Using model organisms to gain an understanding of the exact contribution of these pathways to the pathogenesis of AD will be a priority to enable the development of specific therapeutic interventions that do not affect other essential cellular processes.
8. Advantages of Using Drosophila to Model Amyloid Pathology
More than 77% of human disease genes listed on the OMIM database have an orthologue in Drosophila, confirming their utility as a model for human genetic diseases [110]. In addition, it is possible to avoid complications that could arise from redundancy as there is often a single gene in Drosophila compared with multiple genes in mammalian systems as is the case for APP (Table 2). Knockdown and ectopic expression constructs are readily available in Drosophila for most genes and the genetic toolkit available for analyses is constantly being developed and refined [111]. Ectopic expression via the GAL4/UAS system is used most frequently where various tissue-specific “drivers” (i.e., gene-specific promoter regions upstream of a GAL4 transcriptional activation domain) give particular patterns of expression. Driver lines most useful for studies of molecular mechanisms of AD include the endogenous APPL promoter (appl-GAL4), the eye driver (gmr-GAL4), the neuronal driver (elav-GAL4), and ones that express specifically in cholinergic neurons (cha-gal4), glial cells (repo-GAL4), or ubiquitously (da-GAL4 or actin5C-GAL4) (Figure 3(a)) [12]. Inducible expression systems are also available (e.g., GeneSwitch) which allow for studies where the timing of transgene expression can be regulated more precisely [112].
Table 2.
The human genes that function in APP proteolysis and their Drosophila orthologues.
| Human gene | Drosophila gene | Functions |
|---|---|---|
| Amyloid precursor protein (APP) | Appl | APP is an integral membrane protein containing an Aβ-like region that is cleaved by BACE1. |
| Amyloid precursor-like proteins (APLP1 and APLP2) | Sequence divergence at the internal Aβ site of APLP1 and APLP2 prevents cleavage by BACE1. The principal functions of APLP1 and APLP2 remain unknown. | |
|
| ||
| Presenilin 1 | Presenilin | The catalytic subunit of the γ-secretase enzyme complex, also required for lysosomal acidification. |
| Presenilin 2 | Component of γ-secretase complex. | |
|
| ||
| ADAM 10 | Kuzbanian | A neuronal α-secretase that cleaves APP at the plasma membrane via nonamyloidogenic processing. |
|
| ||
| BACE1 | Bace | β-secretase enzyme activity cleaves APP in early endosome and promotes amyloidogenic processing with Aβ production. |
| BACE2 | β-secretase related to BACE1 that is thought to contribute to Alzheimer's disease. | |
Figure 3.

Drosophila models of AD and assays for neurodegeneration. (a) Drosophila models of AD. The GAL4/UAS system is routinely used in Drosophila to drive expression of a gene of interest [12]. There are ubiquitous or tissue-specific enhancers that drive expression of GAL4. By crossing lines containing the driver-GAL4 to the UAS-gene of interest, the progeny will result in ectopic expression. Eye, neuronal, glial, or ubiquitous drivers are used to express Aβ or APP and BACE1 transgenes resulting in specific phenotypes. These can be assessed for neural degeneration and dysfunction. (b) Assays to assess neuronal dysfunction. Using the eye-specific driver GMR-GAL4, APP + BACE1 can be expressed during eye development and the adult eye disruption can be observed. The degenerative eye shows disruption of ommatidial structure, reduced size, and loss of pigmentation. This is a useful system to screen for modifiers of APP + BACE1 toxicity. Lifespan analyses can be performed using neuronal, glial, or ubiquitous cell type driver lines and the effect of genetic modifiers on the longevity of APP + BACE1 flies can also be monitored. Climbing assays can be used to examine locomotor deficits that are known to degenerate with age. Flies are tapped to the bottom of a measuring cylinder and the number of flies that can climb above a certain height is recorded. Also relevant for studies in AD are assays for learning and memory such as odour preference teamed with an electrical shock treatment.
Ectopic expression of human sequences encoding full length APP (with or without BACE1) or Aβ1–42 peptides (wild-type or mutant) in transgenes under UAS control gives rise to neuronal dysfunction which can be measured as retinal degeneration, locomotor defects, decreased longevity, learning and memory defects, and alterations to various cell biological markers [113] (Figure 3). These have been used as the basis for genetic and/or pharmacological screening [114–117]. Other novel approaches to ameliorating AD symptoms in Drosophila models include immunotherapy and photodynamics [118, 119]. In addition, the contribution of nonneuronal cell types to AD disease progression is well established. Glial cells have been shown to clear neurotoxic Aβ peptides in the adult Drosophila brain through a Draper/STAT92E/JNK cascade that may be coupled to protein clearance pathways such as autophagy [120]. The genetic systems available in Drosophila also allow for elegant approaches for understanding the complex interactions that occur between neurons and glia that could contribute to AD [121].
9. Drosophila Models for Amyloid Toxicity
Components of APP proteolysis are conserved in Drosophila (see Table 2). Although there is limited sequence conservation across the Aβ42 region, it has been shown that neuronal dBACE like enzyme activity can lead to cleavage of the APP-like (APPL) protein in Drosophila where the resultant peptide gives rise to neurodegenerative phenotypes that are accompanied by Aβ-like deposits [122]. Processing of APPL gives rise to the same types of cleavage fragments shown for human APP in Figure 1 including small membrane bound intracellular CTFs and neurotoxic Aβ-like peptides, and these have been shown to be expressed throughout the nervous system during development [123]. Given that APPL is conserved throughout evolution suggests that it does have important functions, some of which have been uncovered including its role in neuronal outgrowth and synapse formation, regulation of the circadian clock, and providing neuroprotection in models for AD as well as other neurodegenerative diseases [123–125].
Despite the conservation of endogenous APP processing and function in Drosophila, disease models have predominantly been generated based on ectopic expression of human counterparts based on the mutations identified in genetic pathways involved in AD (Table 2) (Figure 3). Various model systems have been developed whereby the human gene products are ectopically expressed in Drosophila. Many studies have determined the effects of expressing the Aβ42 toxic peptide directly and it has been shown to give rise to age-dependent neurodegenerative phenotypes that are accompanied by significant disruption to the correct functioning of the autophagic-lysosomal system [126]. It was shown that Aβ42 carrying the “Arctic” APP human disease mutation (E22G) has more severe effects as it is thought to increase the rate of Aβ42 aggregation [127, 128]. However these Aβ42 expression constructs require the inclusion of signal sequences from unrelated genes to ensure their secretion and it has been shown that, at least in some cases, these can give alternative effects [129]. Nonetheless, Aβ42 is localized within endosomes and has been proposed to be the cellular source of pathogenic Aβ42 [8]. The presence of Aβ40 was also observed but found not to correlate with toxicity. Similarly comparison of Aβ40 with Aβ42 by others has also shown differential effects in memory testing [130]. In addition ectopic expression of Aβ43 was tested separately and found to be neurotoxic, potentially by acting to prime the formation of amyloid aggregates [131].
Ectopic expression of the full length (695 amino acid) APP is also used in Drosophila models for AD where again both wild-type and disease associated mutations have been investigated. Wild-type human APP expressed in combination with ectopic human BACE1 enzyme gives effective processing of APP and leads to neuropathology [132]. Synaptic abnormalities have also been reported when APP and BACE1 are coexpressed specifically in neuronal cells [133]. Interestingly, it has been shown that equivalent amounts of Aβ42 peptide produced from processing of APP (when it is expressed together with BACE1) give stronger effects in vivo than Aβ42 peptide expressed directly as the secreted form [134]. This suggests that incorporating the findings from APP and BACE1 ectopic expression models will contribute significantly to the understanding of the molecular pathogenic mechanisms of the proteolytic products of APP.
10. Role for Autophagy in Drosophila Models for AD
Similar to mammalian systems there is accumulating evidence for a role of autophagy in the pathogenesis of Drosophila models for AD. Amyloid toxicity models tested to date have concentrated on those ectopically expressing the Aβ42 peptide (Table 2). Increased basal autophagy by various methods in these models suppresses ectopic Aβ42 induced phenotypes [135, 136]. Specific components of the autophagy pathway have also been investigated by genetic modification analyses in these Aβ42 models. Decreased expression of Atg1 or Atg18 was found to enhance the neurotoxic effect in flies expressing Aβ42, also supporting a protective role for autophagy [5]. However, contrary to this, the knockdown of Atg5 or Atg12 was shown to decrease accumulation of Aβ42 [8]. These findings suggest that there is a complex role for components of the autophagic pathway in AD which may be attributed to the particular stage of the process and/or correlate with timing of disease progression. Together they highlight the need for a comprehensive genetic dissection of the autophagy pathway to determine its contribution to AD.
11. Ageing, Autophagy, and AD
Age is the most prominent risk factor in the development of AD. Age-related dysfunction of autophagy may play a causative role in the onset and progression of AD. It has been suggested that the neuronal autophagy-lysosomal system may shift from a functional and protective state to a pathological and deleterious state either during brain ageing or via Aβ42 neurotoxicity [137]. In support of this there is also an age-related decline in clearance of Aβ42 via the X-box protein 1 [138]. An aged onset model has been developed in Drosophila where human APP and human BACE1 are expressed at low levels during development followed by increased expression throughout adulthood [139]. This type of model will enable in vivo studies in Drosophila to more closely represent disease progression as it occurs in humans. Drosophila is also an excellent model to dissect the molecular mechanisms of ageing that are relevant for AD related neuronal dysfunction [140].
12. Conclusions and Future Directions
The contribution of autophagy to AD has been controversial. In particular, it remains to be determined whether autophagy plays a causative or a protective role in AD or whether autophagy defects are a consequence of disease progression. The detection of aberrant autophagy alone is not sufficient to support a causative role, and further detailed molecular analysis is required. However, there is clear evidence to suggest that autophagy is involved in AD pathophysiology. With therapeutic intervention based on modulating autophagy, it will be critical to understand the role of autophagy in the different stages of the disease as well as defining the molecular mechanisms underlying autophagy dysfunction in AD. While the strongest evidence for the contribution of dysfunctional autophagy to AD comes from in vivo studies, in vitro cell studies have contributed to the understanding of autophagy defects in AD.
Disruption to autophagy could occur at different steps in the pathway from initiation, elongation, cargo selection, lysosomal fusion, and degradation. This may result in altered autophagic flux, with accumulation of autophagosomes, autolysosomes and/or amphisomes, and lysosomal defects that may present as different pathological outcomes. In addition, there is a tissue-specific requirement for distinct components of the autophagic machinery as well as autophagy independent functions of a number of Atg genes [46, 141]. Given the controversy as to the protective and/or pathogenic role of autophagy in AD, using Drosophila models to dissect out the contribution of the different steps will provide important information about the origin of dysfunctional autophagic processes in AD.
Alzheimer's disease pathology is remarkably complex and human genetic mutations have highlighted alterations to amyloid processing as a primary event that gives rise to neuronal toxicity. Autophagy as part of a cellular clearance mechanism has been shown to play a prominent role in disease progression but its functional contribution to neurotoxicity and/or neuroprotection has not been fully defined. In addition some clues have emerged as to the role of nonneuronal cells, in particular glial cells and their interactions with neuronal cells that can affect neuronal function. Using the genetic platform provided by Drosophila, these pathways can be fully dissected and cellular mechanisms of neuronal dysfunction identified. This could include a multigenic approach where more than one candidate can be tested for their effects on APP processing and disease progression. In addition, given that ageing is the most prominent risk factor in AD, the time-frame that would be required for determining efficacies of drugs in humans is not feasible. With the development of technology that can detect amyloid in the blood as an early biomarker for Alzheimer's disease [142, 143], this now provides the opportunity for early intervention and there is a pressing need for identifying new therapeutic compounds. By understanding the role of autophagy in progression/prognosis, this will provide potential novel ways to treat AD and/or provide prognostic biomarkers of disease. Again Drosophila presents as an ideal system where specific autophagic mechanisms could be targeted for the development of novel therapies for early intervention in AD.
Acknowledgments
The research involving autophagy in DD laboratory was supported by the National Health and Medical Research Council of Australia Project Grant (1124490).
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this paper.
References
- 1.Holtzman D. M., Morris J. C., Goate A. M. Alzheimer's disease: the challenge of the second century. Science Translational Medicine. 2011;3(77) doi: 10.1126/scitranslmed.3002369.77sr1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang Y., Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148(6):1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingelsson M., Fukumoto H., Newell K. L., et al. Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62(6):925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 4.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Omata Y., et al. Age-induced reduction of autophagy-related gene expression is associated with onset of Alzheimer's disease. American Journal of Neurodegenerative Disease. 2014;3(3):134–142. [PMC free article] [PubMed] [Google Scholar]
- 6.Kim M., Park H. L., Park H.-W., et al. Drosophila Fip200 is an essential regulator of autophagy that attenuates both growth and aging. Autophagy. 2013;9(8):1201–1213. doi: 10.4161/auto.24811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim M., Sandford E., Gatica D., et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife. 2016;5 doi: 10.7554/eLife.12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling D., Magallanes M., Salvaterra P. M. Accumulation of amyloid-like Aβ1-42 in AEL (autophagy-endosomal-lysosomal) vesicles: potential implications for plaque biogenesis. ASN Neuro. 2014;6(2):95–109. doi: 10.1042/AN20130044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Juhász G., Érdi B., Sass M., Neufeld T. P. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes & Development. 2007;21(23):3061–3066. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simonsen A., Cumming R. C., Brech A., Isakson P., Schubert D. R., Finley K. D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4(2):176–184. doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- 11.Varga K., Nagy P., Arsikin Csordás K., Kovács A. L., Hegedü K., Juhász G. Loss of Atg16 delays the alcohol-induced sedation response via regulation of Corazonin neuropeptide production in Drosophila. Scientific Reports. 2016;6 doi: 10.1038/srep34641.34641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brand A. H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118(2):401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 13.Karch C. M., Cruchaga C., Goate A. M. Alzheimer's disease genetics: from the bench to the clinic. Neuron. 2014;83(1):11–26. doi: 10.1016/j.neuron.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bekris L. M., Yu C.-E., Bird T. D., Tsuang D. W. Review article: genetics of alzheimer disease. Journal of Geriatric Psychiatry and Neurology. 2010;23(4):213–227. doi: 10.1177/0891988710383571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lambert J. C., Ibrahim-Verbaas C. A., Harold D., et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nature Genetics. 2013;45(12):1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ridge P. G., Mukherjee S., Crane P. K., Kauwe J. S. K. Alzheimer's disease: analyzing the missing heritability. PLoS ONE. 2013;8(11) doi: 10.1371/journal.pone.0079771.e79771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moreau K., Fleming A., Imarisio S., et al. PICALM modulates autophagy activity and tau accumulation. Nature Communications. 2014;5, article 4998 doi: 10.1038/ncomms5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyagawa T., Ebinuma I., Morohashi Y., et al. BIN1 regulates BACE1 intracellular trafficking and amyloid-β production. Human Molecular Genetics. 2016;25(14):2948–2958. doi: 10.1093/hmg/ddw146. [DOI] [PubMed] [Google Scholar]
- 19.Kuhn P.-H., Wang H., Dislich B., et al. ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO Journal. 2010;29(17):3020–3032. doi: 10.1038/emboj.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sisodia S. S., Koo E. H., Beyreuther K., Unterbeck A., Price D. L. Evidence that β-amyloid protein in Alzheimer's disease is not derived by normal processing. Science. 1990;248(4954):492–495. doi: 10.1126/science.1691865. [DOI] [PubMed] [Google Scholar]
- 21.Haass C., Schlossmacher M. G., Hung A. Y., et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359(6393):322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 22.Yu W. H., Cuervo A. M., Kumar A., et al. Macroautophagy—a novel β-amyloid peptide-generating pathway activated in Alzheimer's disease. The Journal of Cell Biology. 2005;171(1):87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cataldo A. M., Petanceska S., Terio N. B., et al. Aβ localization in abnormal endosomes: Association with earliest Aβ elevations in AD and Down syndrome. Neurobiology of Aging. 2004;25(10):1263–1272. doi: 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 24.Liu R.-Q., Zhou Q.-H., Ji S.-R., et al. Membrane localization of β-amyloid 1-42 in lysosomes: a possible mechanism for lysosome labilization. The Journal of Biological Chemistry. 2010;285(26):19986–19996. doi: 10.1074/jbc.M109.036798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lauritzen I., Pardossi-Piquard R., Bourgeois A., et al. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathologica. 2016;132(2):257–276. doi: 10.1007/s00401-016-1577-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pera M., Larrea D., Guardia-Laguarta C., et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO Journal. 2017;36(22):3356–3371. doi: 10.15252/embj.201796797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao X., Sudhof T. C. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293(5527):115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 28.Walsh D. M., Fadeeva J. V., LaVoie M. J., et al. γ-secretase cleavage and binding to FE65 regulate the nuclear translocation of the intracellular C-terminal domain (ICD) of the APP family of proteins. Biochemistry. 2003;42(22):6664–6673. doi: 10.1021/bi027375c. [DOI] [PubMed] [Google Scholar]
- 29.Goodger Z. V., Rajendran L., Trutzel A., Kohli B. M., Nitsch R. M., Konietzko U. Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. Journal of Cell Science. 2009;122(20):3703–3714. doi: 10.1242/jcs.048090. [DOI] [PubMed] [Google Scholar]
- 30.Müller U. C., Deller T., Korte M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nature Reviews Neuroscience. 2017;18(5):281–298. doi: 10.1038/nrn.2017.29. [DOI] [PubMed] [Google Scholar]
- 31.Selkoe D. J., Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Molecular Medicine. 2016;8(6):595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denton D., Xu T., Kumar S. Autophagy as a pro-death pathway. Immunology & Cell Biology. 2015;93(1):35–42. doi: 10.1038/icb.2014.85. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H., Baehrecke E. H. Eaten alive: novel insights into autophagy from multicellular model systems. Trends in Cell Biology. 2015;25(7):376–387. doi: 10.1016/j.tcb.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marino G., Madeo F., Kroemer G. Autophagy for tissue homeostasis and neuroprotection. Current Opinion in Cell Biology. 2011;23(2):198–206. doi: 10.1016/j.ceb.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 35.Kroemer G., Marino G., Levine B. Autophagy and the integrated stress response. Molecular Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klionsky D. J., Codogno P., Cuervo A. M., et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy. 2010;6(4):438–448. doi: 10.4161/auto.6.4.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antonioli M., Di Rienzo M., Piacentini M., Fimia G. M. Emerging mechanisms in initiating and terminating autophagy. Trends in Biochemical Sciences. 2017;42(1):28–41. doi: 10.1016/j.tibs.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Galluzzi L., Baehrecke E. H., Ballabio A., et al. Molecular definitions of autophagy and related processes. EMBO Journal. 2017;36(13):1811–1836. doi: 10.15252/embj.201796697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu L., Chen Y., Tooze S. A. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2017 doi: 10.1080/15548627.2017.1378838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bento C. F., Renna M., Ghislat G., et al. Mammalian autophagy: how does it work? Annual Review of Biochemistry. 2016;85:685–713. doi: 10.1146/annurev-biochem-060815-014556. [DOI] [PubMed] [Google Scholar]
- 41.Lamb C. A., Yoshimori T., Tooze S. A. The autophagosome: origins unknown, biogenesis complex. Nature Reviews Molecular Cell Biology. 2013;14(12):759–774. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- 42.Lin M. G., Hurley J. H. Structure and function of the ULK1 complex in autophagy. Current Opinion in Cell Biology. 2016;39:61–68. doi: 10.1016/j.ceb.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 44.Nakatogawa H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays in Biochemistry. 2013;55(1):39–50. doi: 10.1042/BSE0550039. [DOI] [PubMed] [Google Scholar]
- 45.Shpilka T., Weidberg H., Pietrokovski S., Elazar Z. Atg8: an autophagy-related ubiquitin-like protein family. Genome Biology. 2011;12(7, article 226) doi: 10.1186/gb-2011-12-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu T., Nicolson S., Denton D., Kumar S. Distinct requirements of Autophagy-related genes in programmed cell death. Cell Death & Differentiation. 2015;22(11):1792–1802. doi: 10.1038/cdd.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Velikkakath A. K. G., Nishimura T., Oita E., Ishihara N., Mizushima N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Molecular Biology of the Cell (MBoC) 2012;23(5):896–909. doi: 10.1091/mbc.E11-09-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perera R. M., Zoncu R. The Lysosome as a Regulatory Hub. Annual Review of Cell and Developmental Biology. 2016;32:223–253. doi: 10.1146/annurev-cellbio-111315-125125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mindell J. A. Lysosomal acidification mechanisms. Annual Review of Physiology. 2012;74:69–86. doi: 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- 50.Nakamura S., Yoshimori T. New insights into autophagosome-lysosome fusion. Journal of Cell Science. 2017;130(7):1209–1216. doi: 10.1242/jcs.196352. [DOI] [PubMed] [Google Scholar]
- 51.Gordon P. B., Seglen P. O. Prelysosomal convergence of autophagic and endocytic pathways. Biochemical and Biophysical Research Communications. 1988;151(1):40–47. doi: 10.1016/0006-291X(88)90556-6. [DOI] [PubMed] [Google Scholar]
- 52.Chandra P., Ghanwat S., Matta S. K., et al. Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages. Scientific Reports. 2015;5 doi: 10.1038/srep16320.16320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fader C. M., Sánchez D., Furlán M., Colombo M. I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in K562 cells. Traffic. 2008;9(2):230–250. doi: 10.1111/j.1600-0854.2007.00677.x. [DOI] [PubMed] [Google Scholar]
- 54.Lefebvre C., Legouis R., Culetto E. ESCRT and autophagies: endosomal functions and beyond. Seminars in Cell and Developmental Biology. 2018;74:21–28. doi: 10.1016/j.semcdb.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 55.Birgisdottir A. B., Lamark T., Johansen T. The LIR motif - crucial for selective autophagy. Journal of Cell Science. 2013;126(15):3237–3247. doi: 10.1242/jcs.126128. [DOI] [PubMed] [Google Scholar]
- 56.Pankiv S., Clausen T. H., Lamark T., et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. The Journal of Biological Chemistry. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 57.Sengupta S., Peterson T. R., Sabatini D. M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Molecular Cell. 2010;40(2):310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jung C. H., Jun C. B., Ro S.-H., et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Molecular Biology of the Cell (MBoC) 2009;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dunlop E. A., Tee A. R. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Seminars in Cell & Developmental Biology. 2014;36:121–129. doi: 10.1016/j.semcdb.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 60.Perluigi M., Di Domenico F., Butterfield D. A. mTOR signaling in aging and neurodegeneration: at the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiology of Disease. 2015;84:39–49. doi: 10.1016/j.nbd.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 61.Hara T., Nakamura K., Matsui M., et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 62.Komatsu M., Waguri S., Chiba T., et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 63.Komatsu M., Qing J. W., Holstein G. R., et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proceedings of the National Acadamy of Sciences of the United States of America. 2007;104(36):14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Joo J. H., Wang B., Frankel E., et al. The noncanonical role of ULK/ATG1 in ER-to-Golgi trafficking is essential for cellular homeostasis. Molecular Cell. 2016;62(6):p. 982. doi: 10.1016/j.molcel.2016.05.030. [DOI] [PubMed] [Google Scholar]
- 65.Liang C.-C., Wang C., Peng X., Gan B., Guan J.-L. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. The Journal of Biological Chemistry. 2010;285(5):3499–3509. doi: 10.1074/jbc.M109.072389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao Y. G., Sun L., Miao G., et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy. 2015;11(6):881–890. doi: 10.1080/15548627.2015.1047127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nixon R. A., Wegiel J., Kumar A., et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. Journal of Neuropathology & Experimental Neurology. 2005;64(2):113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 69.Nixon R. A., Yang D.-S. Autophagy failure in Alzheimer's disease-locating the primary defect. Neurobiology of Disease. 2011;43(1):38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Correia S. C., Resende R., Moreira P. I., Pereira C. M. Alzheimer's disease-related misfolded proteins and dysfunctional organelles on autophagy menu. DNA and Cell Biology. 2015;34(4):261–273. doi: 10.1089/dna.2014.2757. [DOI] [PubMed] [Google Scholar]
- 71.Wong E., Cuervo A. M. Autophagy gone awry in neurodegenerative diseases. Nature Neuroscience. 2010;13(7):805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rodolfo C., Campello S., Cecconi F. Mitophagy in neurodegenerative diseases. Neurochemistry International. 2017 doi: 10.1016/j.neuint.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 73.Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Molecular Biology of the Cell (MBoC) 2004;15(3):1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Suzuki K., Terry R. D. Fine structural localization of acid phosphatase in senile plaques in Alzheimer's presenile dementia. Acta Neuropathologica. 1967;8(3):276–284. doi: 10.1007/BF00688828. [DOI] [PubMed] [Google Scholar]
- 75.Nilsson P., Saido T. C. Dual roles for autophagy: degradation and secretion of Alzheimer's disease Aβ peptide. BioEssays. 2014;36(6):570–578. doi: 10.1002/bies.201400002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sasaguri H., Nilsson P., Hashimoto S., et al. APP mouse models for Alzheimer's disease preclinical studies. EMBO Journal. 2017;36(17):2473–2487. doi: 10.15252/embj.201797397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanchez-Varo R., Trujillo-Estrada L., Sanchez-Mejias E., et al. Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer's mice hippocampus. Acta Neuropathologica. 2012;123(1):53–70. doi: 10.1007/s00401-011-0896-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu W. H., Kumar A., Peterhoff C., et al. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for β-amyloid peptide over-production and localization in Alzheimer's disease. The International Journal of Biochemistry & Cell Biology. 2004;36(12):2531–2540. doi: 10.1016/j.biocel.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 79.Nilsson P., Loganathan K., Sekiguchi M., et al. Aβ secretion and plaque formation depend on autophagy. Cell Reports. 2013;5(1):61–69. doi: 10.1016/j.celrep.2013.08.042. [DOI] [PubMed] [Google Scholar]
- 80.Nilsson P., Sekiguchi M., Akagi T. Autophagy-related protein 7 deficiency in amyloid beta (Abeta) precursor protein transgenic mice decreases Abeta in the multivesicular bodies and induces Abeta accumulation in the Golgi. The American Journal of Pathology. 2015;185(2):305–313. doi: 10.1016/j.ajpath.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 81.Spilman P., Podlutskaya N., Hart M. J., et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS ONE. 2010;5(4) doi: 10.1371/journal.pone.0009979.e9979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pickford F., et al. The autophagy-related protein becn1 show reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. The Journal of Clinical Investigation. 2008;118(2):2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rocchi A., Yamamoto S., Ting T., et al. A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer's disease. PLoS Genetics. 2017;13(8) doi: 10.1371/journal.pgen.1006962.e1006962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Du X., Huo X., Yang Y., et al. miR-124 downregulates BACE 1 and alters autophagy in APP/PS1 transgenic mice. Toxicology Letters. 2017;280:195–205. doi: 10.1016/j.toxlet.2017.08.082. [DOI] [PubMed] [Google Scholar]
- 85.Matsunaga K., Saitoh T., Tabata K., et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nature Cell Biology. 2009;11(4):385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 86.Whyte L. S., Lau A. A., Hemsley K. M., Hopwood J. J., Sargeant T. J. Endo-lysosomal and autophagic dysfunction: a driving factor in Alzheimer's disease? Journal of Neurochemistry. 2017;140(5):703–717. doi: 10.1111/jnc.13935. [DOI] [PubMed] [Google Scholar]
- 87.Pimplikar S. W., Nixon R. A., Robakis N. K., Shen J., Tsai L. Amyloid-independent mechanisms in Alzheimer's disease pathogenesis. The Journal of Neuroscience. 2010;30(45):14946–14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koo E. H., Squazzo S. L. Evidence that production and release of amyloid β-protein involves the endocytic pathway. The Journal of Biological Chemistry. 1994;269(26):17386–17389. [PubMed] [Google Scholar]
- 89.Soriano S., Chyung A. S. C., Chen X., Stokin G. B., Lee V. M.-Y., Koo E. H. Expression of β-amyloid precursor protein-CD3γ chimeras to demonstrate the selective generation of amyloid/α1-40 and amyloid β1-42 peptides within secretory and endocytic compartments. The Journal of Biological Chemistry. 1999;274(45):32295–32300. doi: 10.1074/jbc.274.45.32295. [DOI] [PubMed] [Google Scholar]
- 90.Cataldo A. M., Barnett J. L., Pieroni C., Nixon R. A. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: Neuropathologic evidence for a mechanism of increased β-amyloidogenesis. The Journal of Neuroscience. 1997;17(16):6142–6151. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cataldo A. M., Peterhoff C. M., Troncoso J. C., Gomez-Isla T., Hyman B. T., Nixon R. A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome. The American Journal of Pathology. 2000;157(1):277–286. doi: 10.1016/S0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu W., Fang F., Ding J., Wu C. Dysregulation of Rab5-mediated endocytic pathways in Alzheimer's disease. Traffic. 2018;19(4):253–262. doi: 10.1111/tra.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dourlen P., Fernandez-Gomez F. J., Dupont C., et al. Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of Tau pathology. Molecular Psychiatry. 2017;22(6):874–883. doi: 10.1038/mp.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chapuis J., Hansmannel F., Gistelinck M., et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Molecular Psychiatry. 2013;18(11):1225–1234. doi: 10.1038/mp.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Settembre C., di Malta C., Polito V. A., et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang H., Wang R., Xu S., Lakshmana M. K. Transcription factor EB is selectively reduced in the nuclear fractions of alzheimer’s and amyotrophic lateral sclerosis brains. Neuroscience Journal. 2016;2016:8. doi: 10.1155/2016/4732837.4732837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang Y.-D., Zhao J.-J. TFEB Participates in the Aβ-Induced Pathogenesis of Alzheimer's Disease by Regulating the Autophagy-Lysosome Pathway. DNA and Cell Biology. 2015;34(11):661–668. doi: 10.1089/dna.2014.2738. [DOI] [PubMed] [Google Scholar]
- 98.Xiao Q., Yan P., Ma X., et al. Neuronal-targeted TFEB accelerates lysosomal degradation of app, reducing Aβ generation and amyloid plaque pathogenesis. The Journal of Neuroscience. 2015;35(35):12137–12151. doi: 10.1523/JNEUROSCI.0705-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bouché V., Espinosa A. P., Leone L., Sardiello M., Ballabio A., Botas J. Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy. 2016;12(3):484–498. doi: 10.1080/15548627.2015.1134081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tognon E., Kobia F., Busi I., Fumagalli A., De Masi F., Vaccari T. Control of lysosomal biogenesis and Notch-dependent tissue patterning by components of the TFEB-V-ATPase axis in Drosophila melanogaster. Autophagy. 2016;12(3):499–514. doi: 10.1080/15548627.2015.1134080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Perez S. E., He B., Nadeem M., et al. Hippocampal endosomal, lysosomal, and autophagic dysregulation in mild cognitive impairment: correlation with Aβ and tau pathology. Journal of Neuropathology & Experimental Neurology. 2015;74(4):345–358. doi: 10.1097/NEN.0000000000000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cataldo A. M., Barnett J. L., Berman S. A., et al. Gene expression and cellular content of cathepsin D in Alzheimer's disease brain: Evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 1995;14(3):671–680. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 103.Belinson H., Lev D., Masliah E., Michaelson D. M. Activation of the amyloid cascade in apolipoprotein E4 transgenic mice induces lysosomal activation and neurodegeneration resulting in marked cognitive deficits. The Journal of Neuroscience. 2008;28(18):4690–4701. doi: 10.1523/JNEUROSCI.5633-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ji Z.-S., Müllendorff K., Cheng I. H., Miranda R. D., Huang Y., Mahley R. W. Reactivity of apolipoprotein E4 and amyloid β peptide: lysosomal stability and neurodegeneration. The Journal of Biological Chemistry. 2006;281(5):2683–2692. doi: 10.1074/jbc.M506646200. [DOI] [PubMed] [Google Scholar]
- 105.Lee J.-H., Yu W. H., Kumar A., et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coen K., Flannagan R. S., Baron S., et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. The Journal of Cell Biology. 2012;198(1):23–35. doi: 10.1083/jcb.201201076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Avrahami L., Farfara D., Shaham-Kol M., Vassar R., Frenkel D., Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the alzheimer disease mouse model: in vivo and in vitro studies. The Journal of Biological Chemistry. 2013;288(2):1295–1306. doi: 10.1074/jbc.m112.409250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Parr C., Carzaniga R., Gentleman S. M., Leuven F. V., Walter J., Sastre M. Correction to Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-β precursor protein. Molecular and Cellular Biology. 2012;32(21) 4410:p. 4418. doi: 10.1128/MCB.00930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tammineni P., Ye X., Feng T., Aikal D., Cai Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. eLife. 2017;6 doi: 10.7554/eLife.21776.e21776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Reiter L. T., Potocki L., Chien S., Gribskov M., Bier E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Research. 2001;11(6):1114–1125. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chow C. Y., Reiter L. T. Etiology of human genetic disease on the fly. Trends in Genetics. 2017;33(6):391–398. doi: 10.1016/j.tig.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 112.Roman G., Endo K., Zong L., Davis R. L. P[switch], a system for spatial and temporal control of gene expression in drosophila melanogaster. Proceedings of the National Acadamy of Sciences of the United States of America. 2001;98(22):12602–12607. doi: 10.1073/pnas.221303998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fernandez-Funez P., de Mena L., Rincon-Limas D. E. Modeling the complex pathology of Alzheimer's disease in Drosophila. Experimental Neurology. 2015;274:58–71. doi: 10.1016/j.expneurol.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Belfiori-Carrasco L. F., Marcora M. S., Bocai N. I., Ceriani M. F., Morelli L., Castaño E. M. A novel genetic screen identifies modifiers of age-dependent amyloid β toxicity in the Drosophila brain. Frontiers in Aging Neuroscience. 2017;9, article 61 doi: 10.3389/fnagi.2017.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Peng F., Zhao Y., Huang X., et al. Loss of Polo ameliorates APP-induced Alzheimer's disease-like symptoms in Drosophila. Scientific Reports. 2015;5 doi: 10.1038/srep16816.16816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mohaibes R. J., Fiol-deRoque M. A., Torres M., et al. The hydroxylated form of docosahexaenoic acid (DHA-H) modifies the brain lipid composition in a model of Alzheimer's disease, improving behavioral motor function and survival. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2017;1859(9):1596–1603. doi: 10.1016/j.bbamem.2017.02.020. [DOI] [PubMed] [Google Scholar]
- 117.Chouhan A. K., Guo C., Hsieh Y.-C., et al. Uncoupling neuronal death and dysfunction in Drosophila models of neurodegenerative disease. Acta Neuropathologica Communications. 2016;4(1):p. 62. doi: 10.1186/s40478-016-0333-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Martin-Peña A., Rincon-Limas D. E., Fernandez-Funez P. Anti-Aβ single-chain variable fragment antibodies restore memory acquisition in a Drosophila model of Alzheimer's disease. Scientific Reports. 2017;7(1) doi: 10.1038/s41598-017-11594-2.11268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee B. I., Suh Y. S., Chung Y. J., Yu K., Park C. B. Shedding light on alzheimer's β-amyloidosis: photosensitized methylene blue inhibits self-assembly of β-amyloid peptides and disintegrates their aggregates. Scientific Reports. 2017;7(1, article 7523) doi: 10.1038/s41598-017-07581-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ray A., Speese S. D., Logan M. A. Glial draper rescues Aβ toxicity in a Drosophila model of Alzheimer’s disease. The Journal of Neuroscience. 2017;37(49):11881–11893. doi: 10.1523/JNEUROSCI.0862-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu L., MacKenzie K. R., Putluri N., Maletic-Savatic M., Bellen H. J. The glia-neuron lactate shuttle and elevated ros promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metabolism. 2017;26(5):719–737.e6. doi: 10.1016/j.cmet.2017.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Carmine-Simmen K., Proctor T., Tschäpe J., et al. Neurotoxic effects induced by the Drosophila amyloid-β peptide suggest a conserved toxic function. Neurobiology of Disease. 2009;33(2):274–281. doi: 10.1016/j.nbd.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ramaker J. M., Cargill R. S., Swanson T. L., et al. Amyloid precursor proteins are dynamically trafficked and processed during neuronal development. Frontiers in Molecular Neuroscience. 2016;9(2016, article 130) doi: 10.3389/fnmol.2016.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Blake M. R., Holbrook S. D., Kotwica-Rolinska J., Chow E. S., Kretzschmar D., Giebultowicz J. M. Manipulations of amyloid precursor protein cleavage disrupt the circadian clock in aging Drosophila. Neurobiology of Disease. 2015;77:117–126. doi: 10.1016/j.nbd.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wentzell J. S., Bolkan B. J., Carmine-Simmen K., Swanson T. L., Musashe D. T., Kretzschmar D. Amyloid precursor proteins are protective in Drosophila models of progressive neurodegeneration. Neurobiology of Disease. 2012;46(1):78–87. doi: 10.1016/j.nbd.2011.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ling D., Salvaterra P. M. A central role for autophagy in Alzheimer-type neurodegeneration. Autophagy. 2009;5(5):738–740. doi: 10.4161/auto.5.5.8626. [DOI] [PubMed] [Google Scholar]
- 127.Nilsberth C., Westlind-Danielsson A., Eckman C. B., et al. The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nature Neuroscience. 2001;4(9):887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 128.Iijima K., Chiang H.-C., Hearn S. A., et al. Aβ42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS ONE. 2008;3(2) doi: 10.1371/journal.pone.0001703.e1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Allan K., Perez K. A., Barnham K. J., Camakaris J., Burke R. A commonly used Drosophila model of Alzheimer's disease generates an aberrant species of amyloid-β with an additional N-terminal glutamine residue. FEBS Letters. 2014;588(20):3739–3743. doi: 10.1016/j.febslet.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 130.Istrail S., Sutton G. G., Florea L. Whole-genome shotgun assembly and comparison of human genome assemblies. Proceedings of the National Acadamy of Sciences of the United States of America. 2004;101(7):6623–6628. doi: 10.1073/pnas.0307971100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Burnouf S., Gorsky M. K., Dols J., Grönke S., Partridge L. Aβ43 is neurotoxic and primes aggregation of Aβ40 in vivo. Acta Neuropathologica. 2015;130(1):35–47. doi: 10.1007/s00401-015-1419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Greeve I., Kretzschmar D., Tschäpe J.-A., et al. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. The Journal of Neuroscience. 2004;24(16):3899–3906. doi: 10.1523/jneurosci.0283-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mhatre S. D., Satyasi V., Killen M., et al. Synaptic abnormalities in a Drosophila model of Alzheimer's disease. Disease Models and Mechanisms. 2014;7(3):373–385. doi: 10.1242/dmm.012104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bergkvist L., Sandin L., Kagedal K., Brorsson A.-C. AβPP processing results in greater toxicity per amount of Aβ1-42 than individually expressed and secreted Aβ1-42 in drosophila melanogaster. Biology Open. 2016;5(8):1030–1039. doi: 10.1242/bio.017194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nandi N., Tyra L. K., Stenesen D., Krämer H. Stress-induced Cdk5 activity enhances cytoprotective basal autophagy in Drosophila melanogaster by phosphorylating acinus at serine437. eLife. 2017;6 doi: 10.7554/eLife.30760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kruppa A. J., Ott S., Chandraratna D. S., et al. Suppression of Aβ toxicity by puromycin-sensitive aminopeptidase is independent of its proteolytic activity. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2013;1832(12):2115–2126. doi: 10.1016/j.bbadis.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ling D., Salvaterra P. M. Brain aging and Aβ1-42 neurotoxicity converge via deterioration in autophagy-lysosomal system: a conditional Drosophila model linking Alzheimer's neurodegeneration with aging. Acta Neuropathologica. 2011;121(2):183–191. doi: 10.1007/s00401-010-0772-0. [DOI] [PubMed] [Google Scholar]
- 138.Marcora M. S., Belfiori-Carrasco L. F., Bocai N. I., Morelli L., Castaño E. M. Amyloid-β42 clearance and neuroprotection mediated by X-box binding protein 1 signaling decline with aging in the Drosophila brain. Neurobiology of Aging. 2017;60:57–70. doi: 10.1016/j.neurobiolaging.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 139.Mhatre S. D., Michelson S. J., Gomes J., Tabb L. P., Saunders A. J., Marenda D. R. Development and characterization of an aged onset model of Alzheimer's disease in Drosophila melanogaster. Experimental Neurology. 2014;261:772–781. doi: 10.1016/j.expneurol.2014.08.021. [DOI] [PubMed] [Google Scholar]
- 140.Piper M. D. W., Partridge L. Drosophila as a model for ageing. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2017 doi: 10.1016/j.bbadis.2017.09.016. [DOI] [PubMed] [Google Scholar]
- 141.Lindqvist L. M., Simon A. K., Baehrecke E. H. Current questions and possible controversies in autophagy. Cell Death Discovery. 2015;1(1) doi: 10.1038/cddiscovery.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fandos N., Pérez-Grijalba V., Pesini P., et al. Plasma amyloid β 42/40 ratios as biomarkers for amyloid β cerebral deposition in cognitively normal individuals. Alzheimer's and Dementia: Diagnosis, Assessment and Disease Monitoring. 2017;8:179–187. doi: 10.1016/j.dadm.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nakamura A., et al. High performance plasma amyloid-beta biomarkers for Alzheimer's disease. Nature. 2018;554(7691):249–254. doi: 10.1038/nature25456. [DOI] [PubMed] [Google Scholar]