

Abstract
De novo synthesis of cytoskeleton-regulatory proteins triggered by the MKL (megakaryoblastic leukemia)/SRF (serum response factor) transcriptional system in response to pro-angiogenic growth factors lies at the heart of endothelial cell (EC) migration (a critical element of angiogenesis) and neovascularization. This study explores whether pharmacological intervention of MKL/SRF signaling axis by CCG-1423 is able to suppress angiogenesis. Our studies show that CCG-1423 inhibits migration and cord morphogenesis of EC in vitro and sprouting angiogenesis ex vivo and in vivo, suggesting CCG-1423 could be a novel anti-angiogenic agent. Kymography analyses of membrane dynamics of EC revealed that CCG-1423 treatment causes a major defect in membrane protrusion. CCG-1423 treatment led to attenuated expression of several actin-binding proteins that are important for driving membrane protrusion including ArpC2, VASP and profilin1 (Pfn1) with the most drastic effect seen on the expression of Pfn1. Finally, depletion of Pfn1 alone is also sufficient for a dramatic decrease in sprouting angiogenesis of EC in vitro and ex vivo, further suggesting that Pfn1 depletion may be one of the mechanisms of the anti-angiogenic action of CCG-1423.
Keywords: CCG-1423, angiogenesis, profilin-1, ArpC2, VASP, endothelial cells, cell migration, conditional knockout
INTRODUCTION
Angiogenesis is an important physiological process that occurs during expansion of the vascular network during development. However, excessive angiogenesis promotes a wide range of human pathologies such as diabetic retinopathy, cancer, atherosclerosis, and arthritis, thus providing justification of anti-angiogenic therapies in various disease contexts. Endothelial cell (EC) migration is a fundamental aspect of angiogenesis. EC activation by proangiogenic cues allows a selective population of EC (known as tip cells) to extend F-actin-rich filopodial protrusions, migrate toward guidance cues and initiate the vessel sprouting, while EC trailing behind the tip cells (known as stalk cells) proliferate to elongate the sprouts. A proper coordination of continued migration of tip cells and proliferation of stalk cells is crucial for angiogenesis [1]. Dynamic remodeling of the actin cytoskeleton in response to angiogenic stimuli lies at the heart of tip cell migration, which, in part, is thought to rely on de novo synthesis of important structural and regulatory components of the actin cytoskeletal system by the action of transcription factor SRF [serum response factor]. SRF target genes include SRF itself and those involved in regulating actin cytoskeletal, adhesion and contractility functions (e.g. actin, myosin, vinculin, filamin, integrin, calponin, etc.) [2–6]. Myocardin family transcriptional co-activators (include myocardin (and myocardin-related transcription factors (MRTF; also known as MKL (megakaryoblastic leukemia))) play an important role in stimulating transcriptional activity of SRF. EC-selective disruption of either SRF or MKL genes (MKL1 and MKL2) when conditionally induced in neonatal and adult mice, causes prominent reduction in developmental (physiological) and tumor (pathological) angiogenesis. In line with these in vivo findings, knockdown of either SRF or MKL inhibits migration and angiogenic ability of both human and rodent ECs in vitro [7–11]. While these findings provide compelling genetic evidence of an essential role of MKL/SRF signaling in 3 EC migration and angiogenesis, pharmacological targeting of MKL/SRF pathway as a potential anti-angiogenic strategy has not been explored to date.
A small molecule screen of inhibitors of Rho-induced SRF-mediated transcription initially identified CCG-1423 as an inhibitor of MKL/SRF signaling [12]. The molecular targets of CCG-1423 have been more recently uncovered. It has been shown that CCG-1423 binds to the nuclear localization signal (NLS) region of RPEL-domain containing proteins, such as those belonging to the MKL family of proteins [13]. By virtue of this interaction, CCG-1423 prevents MKL’s interaction with importin-, causing inhibition of nuclear import of MKL and MKL/SRF-mediated gene transcription. CCG-1423 can also indirectly reduce nuclear accumulation of MKL and suppress SRF activation by targeting MICAL2 (an atypical actin-binding protein) and regulating MICAL2-catalyzed redox-modification and depolymerization of nuclear actin (an inhibitor of MKL:SRF interaction) [14]. Although these studies suggest that CCG-1423 has more than one cellular target, attenuation of MKL/SRF signaling is clearly one of the main downstream effects of CCG-1423.
We herein demonstrate that pharmacological intervention of MKL/SRF axis by CCG-1423 dramatically inhibits EC migration and angiogenesis in vitro, ex vivo and in vivo. Our data suggest cellular depletion of actin-binding protein profilin1 (Pfn1), an important regulator of actin cytoskeletal dynamics, may be one of the mechanisms of the anti-angiogenic action of CCG-1423. These findings set the stage for further preclinical evaluation of CCG-1423 as a potentially novel anti-angiogenic agent.
Materials and Methods
Cell culture and transfection
HmVEC-1, a widely used human dermal microvascular EC line (source: ATCC; Manassas, VA; CRL-3243 – referred to as HmVEC hereafter), were cultured in MCDB-131 (Life Technologies; Carlsbad, CA) growth medium [10% (v/v) fetal bovine serum, 100U/mL Penicillin, 100μg/mL Streptomycin, 10ng/mL EGF, 1μg/mL Hydrocortisone, 10mM L-Glutamine]. For silencing of genes, MKL1 siRNA (Santa Cruz, Dallas, TX, USA, sc-43944), SRF siRNA (Santa Cruz, sc-36563), or Pfn1 siRNA (GE Dharmacon, Lafayette, CO, M-012003-01-0005), all at 100 nM, were transfected using Transfection Reagent 1 (GE Dharmacon) following the manufacturer’s instructions. HmVEC were treated with CCG-1423 (Santa Cruz) for 24 hours under serum-starved conditions prior to extraction. Cell viability was assessed by a live-dead staining kit (Molecular Probes) according to the manufacturer’s instruction.
Angiogenesis assays
For in vitro angiogenesis assessment, cord formation assay in matrigel (mimics the natural basement membrane matrix of EC), was performed as previous described [15]. Briefly, HmVEC were plated on top of polymerized matrigel and allowed to adhere prior to treatment with DMSO or CCG. Cord formation was assessed after 16 hours by phase-contrast microscopy. Cord-formation data was quantified by measuring the total length of all cords using the angiogenesis plugin for ImageJ. For CCG experiments, cells were allowed to adhere for 4 hours before switching to serum-free media with 30 ng/ml VEGF-A (Cell Signaling) and CCG-1423. For EC-spheroid angiogenesis assay, collagen solution [1 mg/ml collagen with 0.25% (w/v) methylcellulose diluted in MCDB-131] was pH-adjusted using 5N NaOH. EC spheroids (formed by culturing cells in ultra-low attachment plate) were mixed with chilled collagen solution and seeded into the wells of a pre-warmed 24-well plate (the seeding volume was equal to 600 μL which typically contained 15-20 spheroids). To permit complete polymerization, the gels were incubated at 37°C for 1 hour, and then overlaid with 500 μL of MCDB-131 growth medium. End-point analysis of sprouts was performed after 24 hrs of culture.
For ex vivo angiogenesis assessment, aortic ring angiogenesis assay was performed. Briefly, thoracic aortas of 9-11 week-old mice were isolated, cleaned of fatty tissues and branching vessels, and excised into ~1 mm segments. When applicable, aortic segments were transfected for 24 hrs with appropriate siRNAs in Opti-MEM (Life Technologies) media, and embedded in either type I collagen (1 mg/ml - diluted in DMEM) or growth-factor reduced matrigel. After polymerizing the matrix for 45 minutes, ex vivo culture was maintained in Opti-MEM containing 2.5% (v/v) FBS and 30 ng/ml VEGF-A for an additional 3-4 days to allow vascular sprouting to take place. For CCG experiments, DMSO or CCG-1423 was added to the media following embedding of aortic rings. For Pfn1 siRNA experiments, control of Pfn1 siRNA was added to the media following embedding of aortic rings. At the end of incubation, aortic rings were fixed with 3% (v/v) formalin for 30 minutes, stained with 0.05 mg/ml rhodamine-conjugated lectin (labels EC) for 2 hours at room temperature and further washed before acquiring images at a 4X magnification. For Pfn1 immunostaining, following fixation, aortic rings were permeabilized with two consecutive treatments of 0.25% (v/v) Triton-X-100 (Fisher Scientific) for 15 minutes. After blocking with 10% (v/v) goat serum for 1 hour, rings were stained with Pfn1 polyclonal antibody (1:200; Abcam, Cambridge, MA) overnight at 4 °C, washed extensively, labeled with FITC-conjugated Goat Anti-Rabbit (1:100; Jackson Immunoresearch) and 0.05 mg/ml rhodamine-conjugated lectin for 2 hours at room temperature and further washed several times before image acquisition.
For in vivo angiogenesis experiments, embryos were collected from Tg(kdrl:gfp)la116 zebrafish (Danio rerio), grown at 24°C until the 20 somite stage [equivalent to 19 hours post-fertilization (hpf) at 28.5°C], dechorionated, and allocated 20 per well in a six-well plate. Embryos were exposed to 1% DMSO or graded doses of CCG-1423 (0.5-50 μM) in 30% Danieau/0.003% phenylthiourea (Sigma, St. Louis, MO) for 22 hr at 28.5°C. CCG-1423 was added directly in the water. For vessel analysis, each dose was repeated at least two times. At the end of the incubation period (41 hpf), embryos were fixed in 4% paraformaldehyde and imaged via brightfield and fluorescence on an Olympus MVX10 microscope. Selected high-resolution images were obtained using a multiphoton microscope.
Generation of EC-specific conditional Pfn1 knockout mice
Pfn1flox/flox mice (described in [16]) were bred into a Tie2-Cre background (Source: Jackson Laboratory in this transgenic C57BL/6 mouse strain, Cre expression is driven under EC-specific Tie2 promoter) to generate EC-specific heterozygous Pfn1 knockout mouse (Pfn1+/− [EC]). Genomic DNA was extracted from either the tails of pups or aortic segments of adult mice (following sacrifice) using a commercial kit (Promega). For genotyping, the following primers were used: LoxP (Primer 1 [forward]: TGGAGCGGATCCAGCGAAGG; Primer 2 [reverse]:GTCCCCAGCAGTCGGGACG, Tie2-Cre (forward: GCGGTCTGGCAGTAAAAACTATC, reverse: GTGAAACAGCATTGCTGTCACTT); PCR positive control (forward: CTAGGCCACAGAATTGAAAGATCT; reverse: GTAGGTGGAAATTCTAGCATCATCC). The recombinase activity of Cre was confirmed by the generation of a 700 bp knockout band using Primer 1 (as mentioned above) and Primer 3 [reverse]: GGACACCAACCTCAGCTGGC. All animal experiments were performed in compliance with an approved protocol by the Institutional Animal Care Committee of the University of Pittsburgh.
Cell migration and kymography assay
HmVEC were sparsely plated in a 24-well plate coated with type I collagen (Millipore) and allowed to attach prior to switching to serum-free media overnight with DMSO or CCG. Prior to taking images, cells were stimulated with 50 ng/ml of VEGF. Time-lapse images of randomly migrating EC were collected using a 10x objective for 120 minutes at a 1 minute time interval using MetaMorph (Universal Imaging). The centroid of the cell nucleus was tracked using ImageJ. For kymography analyses, HmVEC were plated and treated as before but imaged using a 20x objective at 20 second intervals for 20 minutes, as per procedure described before [17].
Protein extraction and immunoblotting
Cell lysates were prepared by a modified RIPA buffer (25 mM Tris–HCl—pH 7.5, 150 mM NaCl, 1% (v/v) NP-40, 5% (v/v) glycerol), 1 mM EDTA, 50 mM NaF, 1 mM sodium pervanadate, and protease inhibitors supplemented with 6x sample buffer diluted to 1x with the final SDS concentration in the lysis buffer was equivalent to 2%. Conditions for the various antibodies were: monoclonal Pfn1 (Abcam; 1:3000), monoclonal GAPDH (Biorad, Hercules, CA, 1:3000), monoclonal VASP (BD Biosciences, Frankin Lakes, NJ; 1:2000), polyclonal MKL1 (Santa Cruz, 1:500), polyclonal SRF (Santa Cruz, 1:500) and polyclonal p34-ArpC2 (Arp2/3) (Upstate Biotechnology, Lake Placid, NY; 1:1000).
Statistics
Statistical tests were performed with either one-way ANOVA followed by Tukey’s post-hoc test or student’s T-test when appropriate, and p < 0.05 was considered to be statistically significant.
Results
Pharmacological intervention of MKL/SRF signaling by CCG-1423 inhibits angiogenesis
First, to determine whether CCG-1423 affects the angiogenic potential of EC in vitro, we performed matrigel-induced cord formation assay with HmVEC at different concentrations (0.5, 1, 2.5, and 5 M) of CCG-1423 or DMSO (vehicle control). In these experiments, CCG-1423 significantly reduced the cord forming ability of HmVEC starting at a 1 M concentration. We found a dose-dependent inhibition in cord forming ability of HmVEC (as judged by the measurements of total cord length) with mean inhibition equaling to 45% and 80%, at 1 and 2.5 M concentrations, respectively. At the 5 M concentration of CCG-1423, the cord-forming ability of HmVEC was almost completely abolished (Fig 1A-B). By live-dead staining, we confirmed no significant cytotoxicity effect of CCG-1423 on HmVEC (supplementary Fig S1). Immunoblot analyses of parallel culture showed that CCG-1423 not only caused a prominent reduction the expression level of SRF (this is expected since inhibition of nuclear import of MKL by CCG-1423 should reduce SRF activation thus diminishing transcription of SRF itself), but interestingly, it also downregulated the expression level of MKL1. CCG-1423-induced inhibition of cord formation of HmVEC was also consistent with similar effects elicited by treatment of these cells with siRNAs targeting either MKL1 or SRF (Fig 1C-D).
Figure 1. CCG-1423 inhibits angiogenesis both in vitro and ex vivo.

A) Representative images of HmVEC cord formation after treatment with either DMSO or CCG-1423 at increasing concentrations. B) Quantification of angiogenesis readouts (measured by total cord length) of HmVEC in CCG-1423 vs DMSO treatment groups summarized from 3 independent experiments with 3 replicates/experiment (** p < 0.01) C) Immunoblots of HmVEC extracts prepared at the same time-point as the end-point of cord-formation assays showing the effect of increasing doses of CCG-1423 on the expression level of MKL1 and SRF (GAPDH - loading control). D-E) Representative images (panel D) and quantification (panel E) showing the effect of either MKL1 or SRF knockdown on the cord forming ability of HmVEC (data summarized from 3 independent experiments with 3 replicates/experiment; ** p < 0.01). F-G) Representative images (panel F; magnified regions of the lower right corner of the rings below show sprouts [arrows]) and quantification (panel G) of endothelial sprouts from aortic segments in collagen-I matrix under either DMSO or CCG-1423 treatment (sprouts were identified by FITC lectin staining; data summarized from aortic segments pooled from 4 mice; **: p<0.01).
Although cord formation assay is widely used to study the morphogenetic ability (and angiogenic potential) of EC, it fails to recapitulate the sprouting behavior of EC from the preexisting blood vessels and represent the complexities of multicellular interactions that occur doing angiogenesis in vivo. Therefore, we next assessed EC sprouting from mouse aortic rings explanted in ECM in response to either 5 M CCG-1423 or DMSO (vehicle control). Consistent with its effect on human EC line in vitro, CCG-1423 at 5 M concentration nearly completely abrogated EC sprouting from the mouse aortic segments ex vivo (Fig 1E-F), further confirming the anti-angiogenic effect of CCG-1423 ex vivo.
We next examined the effect of CCG-1423 on sprouting angiogenesis in developing zebrafish Tg(kdrl:gfp)la116 embryos. These zebrafish express GFP driven by the EC-specific kdrl/flk1/vegfr2 promoter, enabling visualization of blood vessels by fluorescence microscopy [18]. Embryos were exposed to CCG-1423 at 19 hpf (20 somites), just prior to the onset of intersegmental vessel (ISV) sprouting from the dorsal aorta, which initiates at 20 hpf [19]. We initially performed a qualitative assessment of the effect of various doses of CCG-1423 ranging from 0.5 to 50 M on overall development and vascular sprouting, the results of which are displayed in a tabular form in Fig 2A. At concentrations 10 M and higher, the embryos exhibited a complete developmental arrest. Although vascular sprouting was dramatically inhibited in the concentration range of 2.5 - 5 M CCG-1423 (data not shown), there was also a prominent developmental delay of the embryos, and therefore we excluded these concentrations for the ISV analyses. Since no gross developmental anomaly was apparent within 0.5 to1.0 M CCG-1423 (Fig 2B), we evaluated ISV development specifically at these two doses of the compound. With DMSO treatment, ISVs sprouted normally from the dorsal aorta, and rostral and caudal branches from these primary ISVs interconnected to form the dorsal longitudinal anastomotic vessel (DLAV) (Fig 2C-E). These features were not adversely affected at 0.5 M CCG-1423. However, at 1 M CCG-1423, ~70% embryos exhibited defects in ISV sprouting which was marked by lower number of initiated sprouts and/or incomplete extension of ISVs. In many of those cases, DLAV formation was not apparent likely resulting from the initiation and/or extension defects). On average, there was a 25% reduction in the number of ISV and 41% decrease in the total length of ISV in response to 1.0 M CCG-1423 relative to the vehicle control, and this difference was statistically significant (p<0.01; Fig 2F-G). Taken together with foregoing in vitro and ex vivo angiogenesis data, these in vivo data demonstrate anti-angiogenic effect of CCG-1423.
Figure 2. CCG-1423 inhibits developmental angiogenesis in zebrafish embryos.
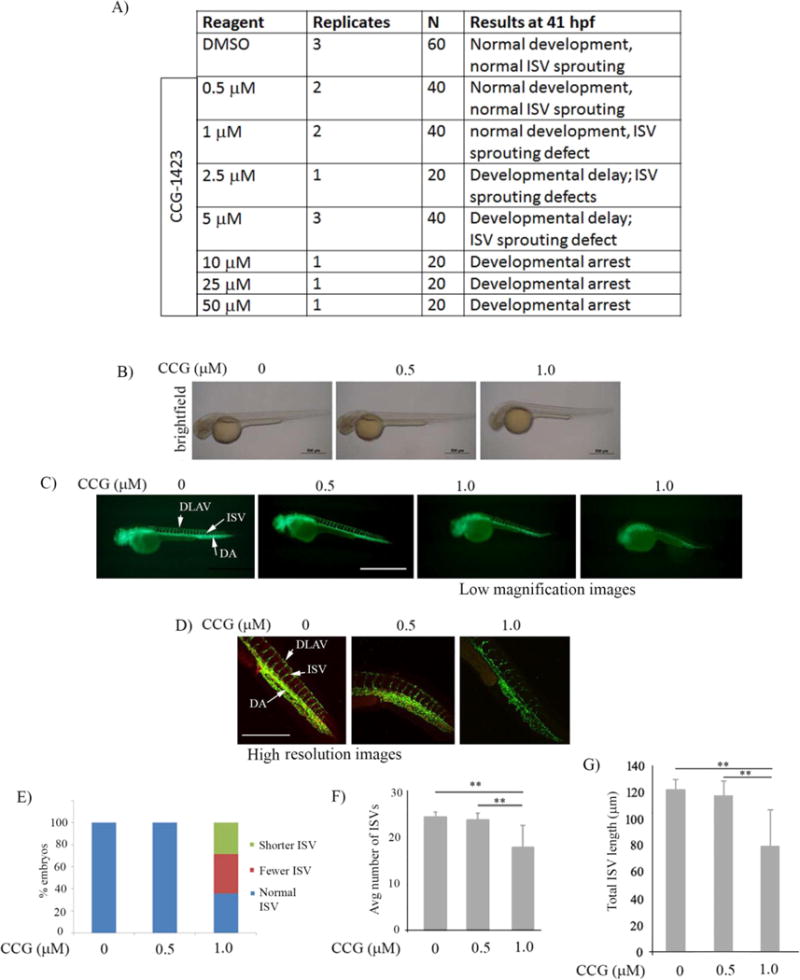
A) Summary of phenotypes of Tg(flk1:gfp)lal16 zebrafish embryos at 22 hr after treatment with the indicated doses of either CCG-1423 or DMSO (control); N indicates the number of embryos in each treatment group. B-D) Brightfield (panel B; scale bar – 500 m) and fluorescence images (panels C [scale bar – 500 m] and D; panel D shows multiphoton images [scale bar – 50 m] of zebrafish embryos at the indicated doses of CCG-1423 relative to DMSO (ISV: intersegmental vessel; DA – dorsal aorta, DLAV – dorsal longitudinal anastomotic vessel). E-G) Bar graph summarizing the ISV phenotypes (panel E), average number of ISV (panel F) and the total length of ISV (panel G) per embryo at 41 hpf under DMSO vs low concentration CCG-1423 treatment (** p < .01).
CCG-1423 inhibits membrane protrusion and impedes EC migration
Since cell migration is an important event in sprouting angiogenesis, we next assessed the effect of CCG-1423 on EC migration. Specifically, we analyzed random migration of HmVEC in serum-starved vs VEGF-stimulated conditions without or with 5 M CCG-1423 treatment (Fig 3A). In the absence of CCG-1423, VEGF stimulation increased the average speed of migration of HmVEC by approximately 2-fold. CCG-1423 treatment not only reduced both basal (i.e. in serum-starved state) and VEGF-induced migration dramatically, but also abrogated VEGF-induced motility response, demonstrating that CCG-1423 has a prominent anti-migratory effect in EC.
Figure 3. CCG-1423 attenuates expression of several actin-binding proteins, suppresses membrane protrusion and motility of HmVEC.
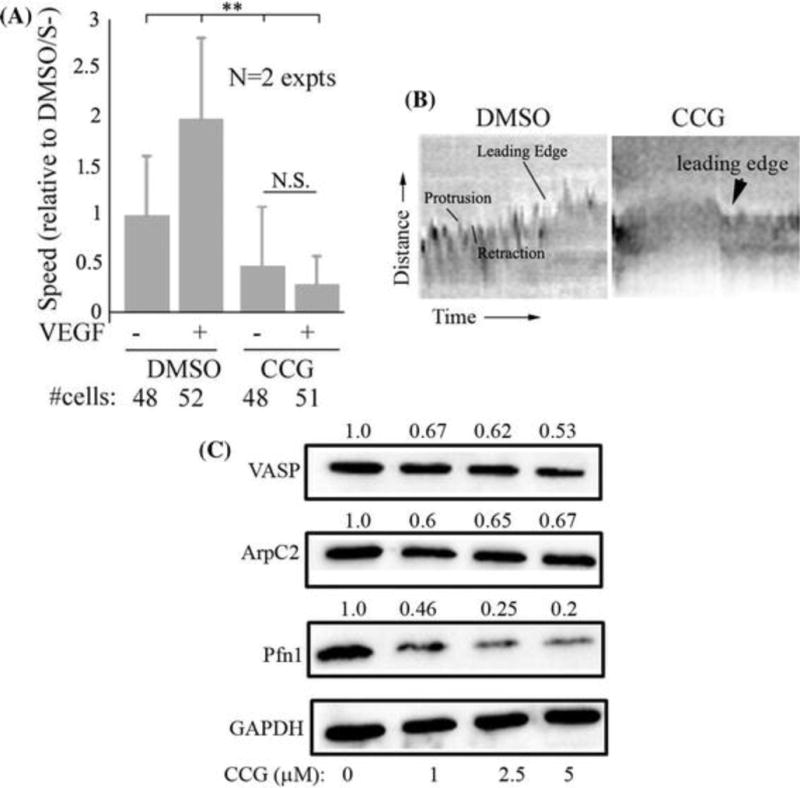
A) Quantitative analyses of the effect of 5 M CCG-1423 treatment on VEGF-induced motility of HmVEC (these data are based on time-lapse analyses of randomly migrating HmVEC cells for 2 hrs) (**: p<0.01; data summarized from 2 independent experiments; N.S: not significant; S-: serum-starved state). B) Representative images of kymograph traces of VEGF-stimulated cells under DMSO vs CCG-1423 (5 M) treatment. C) Representative immunoblots of HmVEC extracts for VASP, ArpC2, Pfn1, and GAPDH (loading control) at various concentrations of CCG-1423 treatment (the numbers indicate the expression level relative to DMSO control averaged from 2 independent experiments). HmVEC lysates were prepared at the same time-point as the end-point cord formation or cell motility assay.
Lamellipodial protrusion is the defining step of cell migration. To determine whether CCG-1423 has any effect on protrusion dynamics of EC, we performed kymography analyses of the dynamic behavior of the leading edge in migrating HmVEC. Representative kymography traces show that within the 20 min time-frame, control cells (i.e. subjected to DMSO) exhibited many cycles of membrane protrusions and retractions (marked by the saw-tooth waveform– protrusions and retractions are denoted by the ascending and descending portions of saw-tooth waveform, respectively), with an overall positive slope indicating a net protrusive behavior (Fig 3B). By contrast, the generally flatter kymograph traces of CCG-1423-treated cells suggested that membrane protrusion is dramatically suppressed by the action of CCG-1423.
CCG-1423 reduces the expressions of several key regulators of actin assembly with most dramatic effect on Pfn1
Actin polymerization at the leading edge drives membrane protrusion. The above results promoted us to probe for expression of a few key regulators of actin assembly the actions of which are critical for membrane protrusion including ArpC2 (a key component of the Arp 2/3 complex), VASP and Pfn1, in response to different doses (1-5 M) of CCG-1423. This dose range was selected because of anti-angiogenic effect of CCG-1423 in this range of concentrations. We chose these three proteins because they belong to different classes of actin-binding proteins and stimulate actin polymerization through different mechanisms of action, albeit in a coordinated fashion [20]. Specifically, the action of Arp 2/3 facilitates the nucleation step of actin assembly. VASP, a key member of the Ena/VASP family of actin-binding proteins, promotes the elongation of actin filaments. Finally, Pfn1 promotes actin assembly by a) facilitating an ADP-to-ATP nucleotide exchange on actin, and b) delivering ATP-G-actin to various F-actin elongating factors (including Ena/VASP proteins) through its direct interaction. We have also found that Pfn1 expression can be influenced post-transcriptionally by either perturbations (knockdown and overexpression) of MKL or high concentration of CCG-1423 (unpublished observation: manuscript under consideration). Whether CCG-1423 has any dose effect on Pfn1 expression in cells and is capable of affecting Pfn1 expression in a low concentration range are not known. Immunoblot analyses of HmVEC extracts showed attenuation in the expression of all three actin-binding proteins in response to CCG-1423 with the effect being most pronounced for Pfn1 (Fig 3C). The cellular level of Pfn1 was depleted by almost 80% at 5 M concentration of CCG-1423; in comparison, VASP and ArpC2 levels were reduced by 30-40%. Even at 1 M concentration of CCG-1423, reduction of Pfn1 expression was fairly dramatic (~54%). Among these three proteins, a dose-dependent trend of CCG-1423 was observed mainly for Pfn1. Attenuation of these actin assembly factors was clearly consistent with the defective protrusion phenotype of CCG-1423 treated cells. CCG-1423 induced downregulation of Pfn1 expression and concomitant defects in migration and cord morphogenesis of EC are consistent with our previously reported findings of similar phenotypes associated with loss of expression of Pfn1 in EC including HmVEC [15, 21].
Pfn1 depletion causes defects in sprouting angiogenesis
Given the drastic effect of CCG-1423 on the expression of Pfn1 and previous evidence of impaired cord morphogenesis of EC upon knockdown of Pfn1 expression, we further asked whether loss-of-function of Pfn1 alone is sufficient to impair sprouting angiogenesis, phenocopying the action of CCG-1423. To test this, we first performed spheroid-based EC sprouting assay with HmVEC following treatment with either non-targeting control or Pfn1-specific siRNA. Our experiments revealed that knockdown of Pfn1 expression (Fig 4A) dramatically inhibited EC sprouting as judged by a 55% reduction in the average number of sprouts from the EC spheroids (Fig 4B-C). To further extend the relevance of these findings ex vivo, we treated mouse aortic segments with either control or Pfn1 siRNA, and performed aortic ring angiogenesis assay. These experiments also showed a significantly lower number of endothelial sprouts from the aortic segments in Pfn1 siRNA-treated culture compare to the control culture (Fig 4D-E). Pfn1 immunostaining performed at the end of the experiment revealed a reduction but not complete loss of Pfn1 expression throughout the aortic ring and in sprouts (Supplementary Fig S2). Note that the apparent large variation in aortic sprouting can be due to several biological and experimental factors including sensitivity of EC sprouting to the age of mice the aortic segments are prepared from, zonal variation in EC sprouting even within an aortic segment and possibly unequal exposure of EC in aortic ring to siRNA in an embedded condition. Since siRNA is not likely to be targeted selectively in EC in this experimental set-up, we also examined the effects of EC-specific gene deletion of Pfn1 on vascular sprouting from mouse aortic segments ex vivo. Note that Pfn1+/− [EC] mice (Fig 4F) were born at the expected frequency (~50%); however, in three rounds of breeding yielding in a total of 21 pups, no Pfn1−/− [EC] mice were born (expected frequency: 25%) which may suggest that complete deficiency of Pfn1 in EC might be embryonically lethal. Therefore, we compared angiogenic sprouting between aortic segments derived from wild-type (WT) and Pfn1+/− [EC] mice. Although heterozygosity of Pfn1 did not result in any obvious defect in vascular sprouting in collagen-I matrix (data not shown), in matrigel Pfn1+/− [EC] aortic rings exhibited significantly lower number of sprouts than WT aortic rings at 72 hours (Fig 4G-H). Overall, these in vitro and ex vivo studies demonstrate that Pfn1 is an important molecular player for sprouting angiogenesis. Conditional knockout experiments further suggest that angiogenesis in certain ECM environment is sensitive to even a partial loss of Pfn1 in EC. Note that although Pfn1+/− [EC] mice do not exhibit any obvious phenotype, possibility of certain subtle vascularization-related developmental delay upon heterozygosity of Pfn1 cannot be absolutely ruled out which should be investigated in the future. Given that cellular depletion of Pfn1 alone is sufficient to cause a prominent defect in sprouting angiogenesis and at least qualitatively phenocopies the anti-angiogenic effect of CCG-1423, cellular attenuation of Pfn1 may be one the possible mechanisms of the anti-angiogenic action of CCG-1423. Note that the degree of inhibition of sprouting angiogenesis upon Pfn1 depletion is lower than that elicited by CCG-1423 (this is discussed later).
Figure 4. Pfn1 depletion inhibits angiogenic sprouting of EC.

A) Pfn1 and tubulin (loading control) immunoblots HmVEC extracts 5 days after transfection with control or Pfn1 siRNA (5-day represents the end time-point of the parallel EC spheroid assay). B-C) Representative images (panel B) and quantification (panel C) of EC spheroid sprouting following treatment with either control of Pfn1 siRNA after 5 days (** p < .01). D-E) Representative images (panel D) and quantification (panel E) of aortic ring sprouting angiogenesis (in collagen-I) following treatment of aortic rings with either control or Pfn1-specific siRNA after 120 hours (vessels are identified by FITC lectin staining) (** p < 0.01; N indicates the number of independent experiments; in each experiment, aortic segments prepared from 1-2 mice was randomly distributed between the two groups). F) Genotyping confirmation of Pfn1+/− [EC] mice (Tie2-Cre-mediated excision of floxed Pfn1 allele gives rise to a 700 bp knockout (KO) band in EC; EC were harvested from aortic segments). G-H) Representative images (panel G) and quantification (panel H) of sprouting angiogenesis in matrigel from WT vs Pfn1+/− [EC] aortic segments (N: number of mice for each genotype; *:p<0.05).
DISCUSSION
Clinical efforts to develop anti-angiogenic therapies have largely focused on inhibiting the action of pro-angiogenic factor VEGF. While these therapies have shown promise in some clinical settings, in other cases therapeutic resistance (partly due to compensatory action of other pro-angiogenic factors) and/or toxicities have been encountered [22]. Therefore, there is a need to discover new anti-angiogenesis targets and therapy. Since its initial discovery as an inhibitor of Rho-induced SRF-mediated transcription, CCG-1423 has demonstrated significant promise in various preclinical disease models to date, such as an anti-fibrotic agent [23] or a possible therapeutic agent in Type II diabetes [24]. The findings from the present study further extend the utility of CCG-1423 as a novel anti-angiogenic agent.
Therefore, anti-angiogenic effect of CCG-1423 is consistent with the genetic proof-of-principle of importance of MKL and SRF in angiogenesis. The molecular links between MKL activity and EC migration/angiogenesis are still not completely understand. This insufficiency in the knowledge partly stems from the fact that MKL/SRF signaling controls the expression of a large number of genes. Several MKL/SRF-regulated genes that have been identified as pro-angiogenic (e.g. subunits of integrin, VE-cadherin and Myl9) are also required for homeostatic functions of EC and vascular integrity. Those studies either lacked detailed phenotypic comparisons between loss-of-function of MKL and the putative downstream genes, or in some cases, knockdown of those genes failed to recapitulate MKL-associated EC phenotypes, or the role of those genes as mediators of MKL-directed angiogenesis was not conclusively proven [9, 25] [7, 26],[27]. A recent study has linked MKL1 and its transcriptional targets CCN1 and CCN2 to angiogenesis-promoting effect of actin-binding protein thymosin 4 [10]; however, whether the defects in tip cell-driven angiogenesis caused by MKL deficiency is linked to alterations in CCN isoforms is not known. The present study shows that CCG-1423 treatment leads to attenuation of several key promoters of actin assembly (such as ArpC2, VASP and Pfn1) and defect in protrusive activity and migration of EC, and Pfn1 depletion phenocopies the anti-angiogenic effect of CCG-1423, at least from qualitative standpoints. These data now open up additional avenues by which suppression of MKL/SRF signaling inhibits angiogenesis. Since the degree of inhibition of sprouting angiogenesis upon Pfn1 depletion is lower than that elicited by CCG-1423, we speculate that Pfn1 depletion is one of the several mechanisms underlying the anti-migratory and anti-angiogenic actions of CCG-1423. This is not surprising given the broad nature of gene expression program elicited by MKL/SRF pathway. Interestingly, a recent microarray study performed in PC3 prostate cancer cell line showed that CCG-1423 causes downregulation in transcription of VEGF-A gene [28]. If this also occurs in EC, it can partly account for the anti-angiogenic action of CCG-1423, and furthermore, since VEGF signaling regulates VEGFR (VEGF receptor) expression, it may also partly explain why CCG-1423-treated EC fail to acquire motile phenotype in response to VEGF-stimulation. To what extent Pfn1 depletion contributes to the anti-migratory and anti-angiogenic action of CCG-1423 should be evaluated in the future by rescue experiments involving forced overexpression of Pfn1. Along this line, although we have only examined the effect of CCG-1423 on the protrusive activity of cells, it is possible that other aspects of cell migration such as adhesion and/or contractility are altered upon CCG-1423 treatment given that MKL/SRF signaling regulates transcription of genes (e.g. integrin, myosin) involved in these processes.
The exact mechanisms by which the various actin-binding proteins are coordinately downregulated by CCG-1423 are currently under investigation in our laboratory. At least in the case of Pfn1, our studies suggest that externalization control may be a key mechanism and that MKL regulates Pfn1 in an SRF-independent manner (unpublished observation - manuscript under consideration). Although CCG-1423 has been identified as an inhibitor of nuclear translocation of MKL, interestingly, we show that CCG-1423 can also substantially reduce the expression level of MKL1. Since MKL1 promoter has an SRF-binding site [29], it is possible that CCG-1423 transcriptionally downregulates MKL through suppressing SRF activation. We think it is somewhat unlikely since we have not found evidence of any appreciable change in the cellular level of MKL upon knockdown of SRF (unpublished observation). Another possibility is that alterations in MKL’s subcellular localization and/or interaction with other binding partners by CCG-1423 may somehow impact the protein stability of MKL. These questions need to be addressed in the future. Finally, a global assessment of transcriptomic and/or proteomic changes in EC will be valuable for a better understanding of pro- or anti-angiogenic pathways that are affected by CCG-1423 treatment.
Supplementary Material
Supplemental Figure S1. Live/dead staining of HmVEC after treatment with either DMSO or CCG-1423 (10 M). Green depicts live cells and red (arrow) shows dead cells.
Supplemental Figure S2. Lectin and Pfn1 immunostaining of aortic rings treated with either control or Pfn1 siRNA (note that Pfn1-siRNA treated rings show an overall weaker staining Pfn1; white and yellow arrowheads depict sprouts with undetectable and intermediate levels of Pfn1).
Acknowledgments
This work was supported by a grant from the National Institute of Health (2R01CA108607) to PR. David Gau was supported by a National Science Foundation pre-doctoral fellowship (2012139050) and an NIH Cardiovascular Bioengineering pre-doctoral training grant (2T32HL076124 to SG).
William Veon was supported by 2T32HL076124. The authors acknowledge the technical assistance from Dr .Takashi Kozai for multiphoton confocal imaging.
References
- 1.Stapor P, et al. Angiogenesis revisited ‐ role and therapeutic potential of targeting endothelial metabolism. J Cell Sci. 2014;127(Pt 20):4331–41. doi: 10.1242/jcs.153908. [DOI] [PubMed] [Google Scholar]
- 2.Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20(12):1545–56. doi: 10.1101/gad.1428006. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, et al. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci U S A. 2003;100(12):7129–34. doi: 10.1073/pnas.1232341100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miralles F, et al. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113(3):329–42. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 5.Mouilleron S, et al. Molecular basis for G‐actin binding to RPEL motifs from the serum response factor coactivator MAL. EMBO J. 2008;27(23):3198–208. doi: 10.1038/emboj.2008.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vartiainen MK, et al. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science. 2007;316(5832):1749–52. doi: 10.1126/science.1141084. [DOI] [PubMed] [Google Scholar]
- 7.Franco CA, et al. SRF selectively controls tip cell invasive behavior in angiogenesis. Development. 2013;140(11):2321–33. doi: 10.1242/dev.091074. [DOI] [PubMed] [Google Scholar]
- 8.Franco CA, Li Z. SRF in angiogenesis: branching the vascular system. Cell Adh Migr. 2009;3(3):264–7. doi: 10.4161/cam.3.3.8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franco CA, et al. Serum response factor is required for sprouting angiogenesis and vascular integrity. Dev Cell. 2008;15(3):448–61. doi: 10.1016/j.devcel.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 10.Hinkel R, et al. MRTF‐A controls vessel growth and maturation by increasing the expression of CCN1 and CCN2. Nat Commun. 2014;5:3970. doi: 10.1038/ncomms4970. [DOI] [PubMed] [Google Scholar]
- 11.Weinl C, et al. Endothelial SRF/MRTF ablation causes vascular disease phenotypes in murine retinae. J Clin Invest. 2013;123(5):2193–206. doi: 10.1172/JCI64201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evelyn CR, et al. CCG‐1423: a small‐molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther. 2007;6(8):2249–60. doi: 10.1158/1535-7163.MCT-06-0782. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi K, et al. RPEL proteins are the molecular targets for CCG‐1423, an inhibitor of Rho signaling. PLoS One. 2014;9(2):e89016. doi: 10.1371/journal.pone.0089016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lundquist MR, et al. Redox modification of nuclear actin by MICAL‐2 regulates SRF signaling. Cell. 2014;156(3):563–76. doi: 10.1016/j.cell.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding Z, et al. Silencing profilin‐1 inhibits endothelial cell proliferation, migration and cord morphogenesis. J Cell Sci. 2006;119(Pt 19):4127–37. doi: 10.1242/jcs.03178. [DOI] [PubMed] [Google Scholar]
- 16.Bottcher RT, et al. Profilin 1 is required for abscission during late cytokinesis of chondrocytes. Embo J. 2009 doi: 10.1038/emboj.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bae YH, et al. Loss of profilin‐1 expression enhances breast cancer cell motility by Ena/VASP proteins. J Cell Physiol. 2009;219(2):354–64. doi: 10.1002/jcp.21677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi J, et al. FoxH1 negatively modulates flk1 gene expression and vascular formation in zebrafish. Dev Biol. 2007;304(2):735–44. doi: 10.1016/j.ydbio.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isogai S, et al. Angiogenic network formation in the developing vertebrate trunk. Development. 2003;130(21):5281–90. doi: 10.1242/dev.00733. [DOI] [PubMed] [Google Scholar]
- 20.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112(4):453–65. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 21.Ding Z, et al. Both actin and polyproline interactions of profilin‐1 are required for migration, invasion and capillary morphogenesis of vascular endothelial cells. Exp Cell Res. 2009;315(17):2963–73. doi: 10.1016/j.yexcr.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergers G, Hanahan D. Modes of resistance to anti‐angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson LA, et al. Novel Rho/MRTF/SRF inhibitors block matrix‐stiffness and TGF‐beta‐induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Dis. 2014;20(1):154–65. doi: 10.1097/01.MIB.0000437615.98881.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin W, et al. Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. J Clin Invest. 2011;121(3):918–29. doi: 10.1172/JCI41940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shu XZ, et al. Histone acetyltransferase p300 promotes MRTF‐A‐mediates transactivation of VE‐cadherin gene in human umbilical vein endothelial cells. Gene. 2015;563(1):17–23. doi: 10.1016/j.gene.2015.02.076. [DOI] [PubMed] [Google Scholar]
- 26.Medjkane S, et al. Myocardin‐related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol. 2009;11(3):257–68. doi: 10.1038/ncb1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang R, et al. Rho/MRTF‐A‐Induced Integrin Expression Regulates Angiogenesis in Differentiated Multipotent Mesenchymal Stem Cells. Stem Cells Int. 2015;2015:534758. doi: 10.1155/2015/534758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evelyn CR, et al. Small‐Molecule Inhibition of Rho/MKL/SRF Transcription in Prostate Cancer Cells: Modulation of Cell Cycle, ER Stress, and Metastasis Gene Networks. Microarrays (Basel) 2016;5(2) doi: 10.3390/microarrays5020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esnault C, et al. Rho‐actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014;28(9):943–58. doi: 10.1101/gad.239327.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Live/dead staining of HmVEC after treatment with either DMSO or CCG-1423 (10 M). Green depicts live cells and red (arrow) shows dead cells.
Supplemental Figure S2. Lectin and Pfn1 immunostaining of aortic rings treated with either control or Pfn1 siRNA (note that Pfn1-siRNA treated rings show an overall weaker staining Pfn1; white and yellow arrowheads depict sprouts with undetectable and intermediate levels of Pfn1).