

Abstract
Background/Aims
We found recently that increasing renal epoxyeicosatrienoic acids (EETs) levels by blocking soluble epoxide hydrolase (sEH), an enzyme responsible for EETs degradation, shows renoprotective actions and retards the progression of chronic kidney disease (CKD) in Ren-2 transgenic hypertensive rats (TGR) after 5/6 renal ablation (5/6 NX). This prompted us to examine if additional protection is provided when sEH inhibitor is added to the standard renin-angiotensin system (RAS) blockade, specifically in rats with established CKD.
Methods
For RAS blockade, an angiotensin-converting enzyme inhibitor along with an angiotensin II type receptor blocker was used. RAS blockade was compared to sEH inhibition added to the RAS blockade. Treatments were initiated 6 weeks after 5/6 NX in TGR and the follow-up period was 60 weeks.
Results
Combined RAS and sEH blockade exhibited additional positive impact on the rat survival rate, further reduced albuminuria, further reduced glomerular and tubulointerstitial injury, and attenuated the decline in creatinine clearance when compared to 5/6 NX TGR subjected to RAS blockade alone. These additional beneficial actions were associated with normalization of the intrarenal EETs deficient and a further reduction of urinary angiotensinogen excretion.
Conclusion
This study provides evidence that addition of pharmacological inhibition of sEH to RAS blockade in 5/6 NX TGR enhances renoprotection and retards progression of CKD, notably, when applied at an advanced stage.
Keywords: Chronic kidney disease, 5/6 nephrectomy, Hypertension, Renin-angiotensin system, Epoxyeicosatrienoic acids, Soluble epoxide hydrolase
Introduction
Progression of chronic kidney disease (CKD) to end-stage renal disease (ESRD) is independent of the initial insult and the underlying mechanism(s) are common for all renal disorders [1–5]. However, these mechanisms remain unclear and therapeutic approaches aimed to delay the progression of CKD are debated [3–5]. Hypertension and inappropriately increased intrarenal activity of the renin-angiotensin system (RAS) are thought to be critically important for progression of CKD to ESRD [1–19], hence the use of antihypertensive regimes involving inhibition of the RAS to provide “renoprotection”. It is postulated that renoprotection provided by RAS blockers, a gold standard in CKD treatment, cannot be solely explained by their blood pressure (BP)-lowering actions: RAS blockers also exhibit BP-independent organ-protective effects [3–5, 20–24]. It was even claimed that RAS blockade could reverse kidney damage during established CKD, e.g. induce regression of glomerulosclerosis [25–27].
Nevertheless, there are recent reports that indicate that the cardio- and renoprotective effects of RAS blockade can be entirely attributed to BP reduction, [14, 28–30] and advanced CKD regression of renal damage is limited [12, 13, 15, 17, 31–33]. This points to the need for more multifaceted pharmacological strategies that target neurohormonal systems other than RAS and thereby bring additional renoprotective effects, especially in advanced CKD. However, no new drug for the treatment of CKD has been registered since 2001 [4]. In this context, considerable attention has been focused on epoxyeicosatrienoic acids (EET), cytochrome P-450 (CYP)-dependent metabolites of arachidonic acid and growing evidence indicates that increased EETs bioavailability is an important antihypertensive and organ-protective factor [34–40].
Experimental studies with 5/6 renal mass reduction (5/6 NX), a commonly used model of CKD, provided most our knowledge regarding the pathophysiology of CKD progression. Findings demonstrate that besides reduction of renal mass, hypertension and augmented intrarenal RAS activity are the major determinants for the rate of CKD progression [1, 2,9–20, 41]. The Ren-2 transgenic rat (TGR) presents a unique angiotensin II (ANG II)-dependent model of hypertension resulting from a single gene alteration and activation of the RAS [42]. Evidently, 5/6 NX TGR encompasses all risk factors for the progression of CKD and therefore this animal model is well-suited for evaluating new approaches for the treatment of CKD. We found recently that in 5/6 NX TGR increasing renal EETs by blocking soluble epoxide hydrolase (sEH), an enzyme which degrades EETs to inactive dihydroxyeicosatrienoic acids (DHETEs), a substantially improved rat survival rates, prevented BP increases, and exhibited organ-protective actions [43]. This suggests that pharmacologically-induced increases of EETs could be a novel tool to treat CKD. However, we did not examine if addition of sEH inhibition to the standard RAS blockade would exhibit additive renoprotective effects. Furthermore, the treatment was started immediately after 5/6 NX i.e. before any organ-damage could develop. It is unknown if these renoprotective effects would occur in individuals with established CKD. Considering these limitations, we examined whether combined sEH and RAS inhibition will have enhanced renoprotective effects compared with those achieved with RAS blockade alone. Importantly, we studied the TGR that were left untreated until 6 weeks after 5/6 NX (late treatment regime).
Materials and Methods
General methodological procedures
Ethical approval and animals
The studies followed the guidelines and practices established by the Animal Care and Use Committee of the Institute for Clinical and Experimental Medicine, and of the 2nd Faculty of Medicine, Charles University, Prague, which accord with the national law, the European Union policy (EEC Council Directive 86/609, OJL 358-1, December 1987) and with American Physiological Society guiding principles for the care and use of vertebrate animals in research and training. Heterozygous TGR were used in the present study and Hannover-Sprague Dawley (HanSD) rats served as transgene-negative normotensive controls. Heterozygous TGR were generated by breeding male homozygous TGR with female homozygous rats as described in the original study [42], age-matched HanSD rats served as transgene-negative normotensive controls. The animals were kept on a 12-hour/12-hour light/dark cycle. Throughout the experiments rats were fed a normal salt, normal protein diet (0.45% NaCl, 19-21% protein) produced by SEMED (Prague, Czech Republic) and had free access to tap water.
Pharmacological therapeutic regimes
The activity of RAS can be pharmacologically altered at various levels. We and others have demonstrated that the pharmacological blockade using combination of ACEi and AT1 blocker at high doses provides a cardio- and renoprotection superior to that achieved with routine dosage [14, 15, 20]. Thus, similarly as in our recent studies [14, 15], for the “RAS blockade” a combination of trandolapril (6 mg/L drinking water, Gopten; Abbot, Prague, Czech Republic), and of losartan (Lozap, 100 mg/L drinking water, Zentiva, Prague, Czech Republic), was used.
The pharmacological blockade of sEH was achieved by employing the sEH inhibitor, cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB), was prepared freshly and given in drinking water at 3 mg/L. The dose of c-AUCB was selected based on our recent studies where it elicited substantial increases in tissue concentrations of EETs without altering RAS activity [43]. We purposely chose the dose of c-AUCB that only blocks sEH activity without altering plasma and tissue ANG II levels, because the major aim of this treatment regime was to evaluate whether pharmacologically-induced EETs concentrations will exhibit additional renoprotective effects as compared with RAS blockade alone in 5/6 NX TGR.
BP measurements
Similarly as in our previous studies and in accordance with the recommendation for BP measurements in experimental animals, we used radiotelemetry system for direct BP measurements [14, 15, 17, 18, 37, 44, 45]. At week labeled -4 (i.e. 4 weeks after 5/6 NX or after sham-operation), rats were anesthetized with a combination of tiletamine, zolazepam (Zoletil, Virbac SA, Carros Cedex, France; 8 mg.kg−1), and xylazine (Rometar, Spofa, Czech Republic; 4 mg.kg−1) intramuscularly, and TA11PA-C40 radiotelemetric probes (Data Sciences International, St. Paul, MN, USA) were implanted into abdominal aorta for direct BP measurements as described previously [14, 15]. Rats were allowed 7 days to recover before basal BP was recorded and only animals with stable BP records at the end of this recovery period were used for experiments.
Evaluation of indices of kidney injury and creatinine clearance
At the end of the appropriate followup period, animals were killed, the kidneys were used to assess renal glomerular damage and kidney tubulointerstial injury. The kidneys were fixed in 4% formaldehyde, dehydrated and embedded in paraffin. The sections stained with hematoxylin–eosin and PAS (periodic acid, for Schiff reaction) were examined and evaluated in a blind-test fashion. Fifty glomeruli in each kidney were examined on a semi-quantitative scale as described previously [14, 15]: grade 0, all glomeruli normal; grade 1, sclerotic area up to 25% (minimal sclerosis); grade 2, sclerotic area 25 to 50% (moderate sclerosis); grade 3, sclerotic area 50 to 75% (moderate-to-severe sclerosis); grade 4, sclerotic area 75 to 100% (severe sclerosis). The glomerulosclerosis index (GSI) was calculated using the following formula: GSI = [ (1 × n1) + (2 × n2) + (3 × n3) + (4 × n4) ]/(n0 + n1 + n2 + n3 + n4), where nx is the number of glomeruli in each grade of glomerulosclerosis.
Renal cortical tubulointerstitial injury was evaluated as defined by Nakano et al. [46] and as used in our recent studies [14, 15], to determine inflammatory cell infiltration, tubular dilatation, atrophy, or interstitial fibrosis. The injury was graded semi-quantitatively using the following scale of lesions: grade 0, no abnormal findings; 1, mild (<25 % of the cortex); 2, moderate (25 – 50 % of the cortex); 3, severe (>50 % of the cortex). The lesions were assessed in at least 30 random and non-overlapping fields in the renal cortex.
Urinary rat albumin in urine was measured by a quantitative sandwich enzyme immunoassay technique, using the commercially available ELISA kit (ERA3201-1, AssayPro, MO, USA).
Plasma creatinine was measured by FUJI DRI-CHEM analyzer using appropriate slides for creatinine CRE-P III (FUJIFILM Corp., Toyo, Japan).
Urine creatinine was determined using Liquick Cor-CREATININE kit that is based on modificated Jaffe’s method, without deprotenization (PZ CORMAY S.A., Poland). In alkaline solution picrate reacts with creatinine to form a yellow-red 2, 4,6-trinitrocyclohexadienate. The color intensity measured by photometer at 500 nm is proportional to the creatinine concentration. Clearance of creatinine was calculated by using standard formula and was normalized per body weight of evaluated animals.
Evaluation of indices of the RAS activity
The measurement of plasma and tissue ANG II concentrations: For sampling, conscious rats were decapitated, because it is now recognized that plasma and tissue concentrations of ANG II are higher than those obtained from anesthetized rats [7, 47].
Preparation of kidney samples
Immediately after decapitation, kidneys were removed, dried, weighed and 0.5 g of the tissue was homogenized in 3 ml precooled methanol. The tube with homogenate was kept on ice and then centrifuged at 4 °C and 3000 g for 10 min. The supernatant was evaporated using Savant SpeedVac vacuum centrifuge,. Dried samples were stored at −20 °C or lower until solid-phase extraction and the assay. Kidney homogenates were purified by solid-phase extraction. Dried kidney samples were reconstituted with 4 ml of 50 mM sodium phosphate buffer (pH 7.4) containing 267 mg bovine serum albumin/l and kept on ice. Phenyl-bonded solid phase extraction columns (SPE) (Bond-Elut® PH, Agilent)were preconditioned with methanol (3 ml), followed by distilled-water (2× 3 ml). Thereafter, reconstituted kidney samples were applied to pre-washed columns. The columns were sequentially washed with distilled-water (3 ml), hexane (3 ml) and chloroform (3 ml); water removes salts and other polar substances from the columns. Hexane and chloroform elute contaminating lipids and hydrophobic material from the columns but do not affect angiotensin peptides recovery. At the end, angiotensin peptides were eluted from SPE columns using 2× 1 ml flush of methanol. The eluates were evaporated to dryness using a vacuum centrifuge. Dried samples were stored at −20 °C or lower until assayed. ANG II levels were measured by a competitive radioimmunoassay, using the commercially available RIA kit (ED29051, IBL Int., Hamburg, Germany).
The measurement of tissue angiotensin 1-7 (ANG 1-7) concentrations: Kidney ANG 1-7 levels were measured by a competitive radioimmunoassay using the custom-made RIA kit (BeckmanCoulter, Prague, Czech Republic).
Preparation of tissue samples was similar as for the determination of ANG II, with a few exceptions. Kidney samples were purified by SPE, however, a different protocol of SPE was used. Dried kidney samples were reconstituted with 4 ml of 50 mM sodium phosphate buffer (pH 7.4) containing 267 mg BSA/l and kept on ice. C18-bonded SPE columns (Bond-Elut® C18, Agilent) were preconditioned with mixture of ethanol +distilled-water + 4 % acetic acid (83:13:4 by volume; 5 ml), methanol (5 ml), distilled-water (5 ml) and with 4 % acetic acid (5 ml). Thereafter, reconstituted samples were applied to pre-washed columns. The columns were sequentially washed with distilled-water (5 ml) and acetone (5 ml). At the end, ANG peptides were eluted from SPE columns with 2× 1ml + 1× 1.5 ml of mixture of ethanol + distilled-water + 4 % acetic acid (83:13:4 by volume). The eluates were evaporated to dryness using a vacuum centrifuge. Dried samples were stored at −20 °C or lower until assayed.
The measurement of urine angiotensinogen
Rat total angiotensinogen concentrations were measured in urine samples, by a solid phase sandwich Enzyme-linked Immunosorbent Assay, using the commercially available ELISA kit (JP27414, IBL Int., Hamburg, Germany).
Evaluation of kidney tissue concentrations of CYP-dependent metabolites of arachidonic acid
The levels of arachidonic acid metabolites: EET, specifically 5, 6-EET, 8, 9-EET, 11, 12-EET and 14-15-EET, and DHETEs, which are the biologically active and inactive, respectively, products of CYP epoxygenase enzymatic pathway were measured in the kidney cortex. For the analysis, 20 – 40 mg of tissue was used. Homogenized tissue samples were subjected to alkaline hydrolysis and solid-phase extraction was performed as described by Rivera et al. [48]. After that, samples were analyzed using high performance liquid chromatography (HPLC), Agilent 1200SL with tandem mass spectroscopy (MS), Agilent 6460 for quantification. The setup of HPLC setting (Agilent 1200SL) was prepared as follows:
Separation was performed on a Phenomenex Kinentex column (150 × 2.1mm, 2.6 μm, 40 °C) using an ammonium acetate (solvent B)/acetonitrile (solvent A) gradient at pH 6.8.
Chromatography was carried out under the following gradient: 0 min 95% for solvent B; 1 min; 1 min 95% for solvent B; 2 min 70% for solvent B; 16 min 33% for solvent B; 17 min 5% for solvent B. The flow rate was 0.4 ml/min. The injection volume was 7.5 μl.
The setup of Triple Quad MS/MS setting (Agilent 6460) was as follows: the electrospray ionization source was used. Drying gas was adjusted at 250 °C/10 L.min−1, sheath gas 400 °C/10 L.min−1. Nebulizer pressure was adjusted to 30 psi. Capillary and nozzle voltage were optimized at 4500 V and 300 V, respectively. Analysis of CYP-dependent metabolites was performed with dynamic multiple reaction monitoring in negative mode: precursor/product ion 319.2/289.1.
The measurement of kidney gene expression
Total RNA was extracted from kidney tissue using RNAzol® RT (Molecular Research Center, Inc., Cincinnati, USA) according to the manufacturer’s directions. RNA purity and concentration were assessed using microvolume spectrophotometer (DeNovix Inc., Wilmington, USA). Total RNA was reverse transcribed and amplified using One Step SYBR® PrimeScript™ RT-PCR Kit II (TAKARA BIO INC, Shiga, Japan) in total volume 20 μl. All samples were analyzed in triplicates. The primers were designed by Primer3 software (version 4.0.0) and purchased from Generi Biotech Ltd. (Hradec Králové, Czech Republic). Primers sequences were:
- CYP2C23:
- forward 5′-GAT GCT GTC TTC CGT CAT GC-3′
- reverse 5′-GTA ATA GGC TTG ATG TCA AG-3′
- sEH:
- forward 5′-AAG CCT GTG GAG CCA GTC TA-3′
- reverse 5′-CCA GTT GTT GAC AAT GC-3′
- β-actin:
- forward 5′-TGA CTG ACT ACC TCA TGA AGA-3′
- reverse 5′-CAC GTC ACA CTT CAT GAT TG-3′
- α-smooth muscle actin (α-SMA):
- forward 5′-ATAGAACACGGCATCATCACC-3′
- reverse 5′-GGTCTCAAACATAATCTGGGTCA-3′
- E-cadherin:
- forward 5′-TGCTGCCACCAGATGACGATAC-3′
- revers 5′-TGTGCAGCTGGCTCAAATCA-3′
- monocyte chemoattractant protein-1 (MCP-1):
- forward 5′-CTGTGCTGACCCCAATAAGGAAT-3′
- reverse 5′-AGGTGGTTGTGGAAAAGAGAGTG-3′
- fibronectin:
- forward 5′-GACCATCAGCCCGGATGTCA-3′
- reverse 5′-ATCAATGGCCGTGGAGGCAT-3′
- transforming growth factor-β1 (TGF-β1):
- forward 5′-CTTTGTACAACAGCACCCGC-3′
- reverse 5′-TAGATTGCGTTGTTGCGGTC-3′
- collagen I:
- forward 5′-GAGCGGAGAGTACTGGATCGA -3′
- reverse 5′-CTGACCTGTCTCCATGTTGCA-3′
- collagen III:
- forward 5′-TGCCATTGCTGGAGTTGGA-3′
- reverse 5′-GAAGACATGATCTCCTCAGTGTTGA-3′
PCR amplifications were performed using the ViiA™ 7 Real-time PCR system (Applied Biosystems, USA) following the reaction parameters recommended by the manufacturer, using 100 ng RNA per reaction. β-actin was used as an endogenous control gene and negative controls contained water instead of cDNA. In all experiments, relative gene expression was calculated by the 2-ΔΔCt method, the most frequently used method for relative quantification in qPCR experiments [49]. The Ct (threshold cycle) is the cycle at which the fluorescence level reaches a certain amount (the threshold). This method directly uses the Ct information generated from a qPCR system to calculate relative gene expression in target and reference samples, using a reference gene as the normalizer. As normalizers housekeeping genes are used, such as β-actin, or GAPDH or 18S rRNA because their expression levels remain relatively stable in response to any treatment [49, 50]. The resultant mRNA level was normalized to a calibrator; in each case, the calibrator chosen was the basal sample. In our experiments, mRNA of CYP2C23, sEH, α-SMA, E-cadherin and collagen I and III and TGF-β1 were normalized to a group of untreated sham-operated HanSD rats. Final results were expressed as the n-fold difference in gene expression between target mRNA and calibrator mRNA as follows:
n-fold = 2− (ΔCt sample−ΔCt calibrator), where ΔCt values of the sample and calibrator were determined by subtracting average Ct value of β-actin mRNA from the average Ct value of target gene.
Western blot analysis for quantification of kidney protein expression
The kidney protein expression of CYP2C23, sEH, α-SMA, E-cadherin and collagen I and III and TGF-β1were methodically performed as described in detail in our previous studies [43, 51, 52]. Briefly, kidney cortex was homogenized 1:3 wt:vol in ice–cold RIPA lysis buffer containing 50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% deoxycholic acid, 1 mM EDTA; supplemented with protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA), kept on ice for 30 min and centrifuged twice at 10.000g for 10 min at 4 °C. Protein concentration in the supernatant was measured using Pierce BCA protein assay (Thermo Scientific, Waltham, MA, USA). Totally 40 μg of protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto the polyvinyl difluoride (PVDF) membrane in transfer buffer at 100 V for 1.5 hours. Membranes were blocked with 5% non-fat dry milk in TRIS buffered saline with Tween20 (TBS-T) overnight at 4 °C. After washing with TBS-T, the membranes were incubated with primary antibodies overnight at 4 °C. The antibody dilutions were as follows:
anti-CYP2C23, 1:2000, manufactured and provided by professor Imig’s lab (Department of Pharmacology and Toxicology, Medical College of Wisconsin, USA);
anti-sEH, 1:2000, Santa Cruz Biotechnology, Inc. (Texas, USA);
anti-α-SMA, 1:200, Abcam (Cambridge, UK);
anti-E-cadherin, 1:1000, Cell Signaling Technology, Inc. (MA, USA);
anti-collagen I, 1:1000, Acris Antibodies (Herford, Germany);
anti-collagen III, 1:1000, Acris Antibodies (Herford, Germany);
anti-TGF-β1, 1:500, Acris Antibodies (Herford, Germany);
anti-β-actin, 1:7500, Sigma Aldrich (MO, USA).
After O/N incubation, the membranes were washed again and incubated with horseradish peroxidaseconjugated secondary antibody for 1 hour at room temperature. After last washing, the immunoblots were exposed to SuperSignal West Dura Substrate (Thermo Scientific, Rockford, IL, USA) for chemiluminiscent detection. Relative densitometry was determined using ImageJ software (NIH, Bethesda, MD, USA). All protein data was normalized to the housekeeping protein β-actin.
Specific experimental design
Series 1: Effects of RAS blockade alone and combined RAS and sEH blockade on survival rate, albuminuria, clearance of endogenous creatinine in animals with established CKD
In the week labeled -6, at the age of 6 weeks, male HanSD rats and TGR from several litters were exposed to 5/6 NX under general anesthesia (tiletamine + zolazepam, Virbac SA, Carros Cedex, France, 8 mg/kg; and xylasine, Spofa, Czech Republic, 4 mg/kg intramuscularly) as described previously [36, 37, 44]. After 24 hours’ recovery, animals were randomly assigned to experimental groups and were left with no treatment. Six weeks later (in the week 0), an appropriate treatment regime was initiated or rats were left further with no treatment. The following experimental groups were investigated:
Sham-operated HanSD rats + water (initial n = 8)
Sham-operated TGR + water (n = 8)
5/6 NX TGR + water (n = 26)
5/6 NX TGR + RAS blockade (n = 24)
5/6 NX TGR + RAS blockade + sEH blockade (n = 25)
The follow-up period was until week +60. In the weeks labeled 2, 4, 14, 22, 30, 38, 46, 54 and 60 and after appropriate habituation training, the animals were placed in individual metabolic cages and their 24-hour urine was collected for determination of daily albuminuria and urinary creatinine excretion. In addition, in the weeks 2, 4, 14, 22 and 60 daily urinary excretion of angiotensinogen was determined.
Series 2: Effects of RAS blockade alone and combined RAS and sEH blockade on the development of renal glomerular damage and renal cortical tubulointerstitial injury
Animals were again exposed to the same procedure (i.e. 5/6 NX or sham-operation) as in series 1. In the week 0 (6 weeks after 5/6 NX or sham operation) the following groups were studied:
Sham-operated HanSD rats + water (n = 6)
Sham-operated TGR + water (n = 6)
5/6 NX TGR + water (n = 10)
In the weeks labeled 2, 4, 6, 8 the following separate groups were investigated (n represents always the number of animals at appropriate time point):
Sham-operated HanSD rats + water (n = 6)
Sham-operated TGR + water (n = 6)
5/6 NX TGR + water (n = 10)
5/6 NX TGR + RAS blockade (n = 10)
5/6 NX TGR + RAS blockade + sEH blockade (n = 10)
At the end of the appropriate follow-up period, animals were killed, and renal glomerular damage and kidney tubulointerstial injury were assessed. The aim was a) to evaluate the course of renal damage in untreated and treated animals with established CKD throughout the critical periods of the experiment; b) to investigate whether the addition of sEH inhibitor to standard RAS blockade will exhibit some beneficial effects on renal damage in addition to standard RAS blockade alone.
Series 3: Effects of RAS blockade alone and combined RAS and sEH blockade on BP (radiotelemetry) and cardiac hypertrophy in animals with established CKD
Animals were exposed to 5/6 NX or sham-operation as in series 1. In accordance with the recommendation for BP measurement in experimental animals, we employed a radiotelemetry system for direct BP measurements [45]. Starting on the day labeled -7, BP recording was initiated, on day 0 an appropriate treatment regime was initiated or rats were left with no treatment (i.e. 6 weeks after 5/6 NX, the same design as in series 1) and BP was recorded for additional 21 days.
The following experimental groups were investigated:
Sham-operated HanSD rats + water (n = 6)
Sham-operated TGR + water (n = 6)
5/6 NX TGR + water (initial n = 9)
5/6 NX TGR + RAS blockade (initial n = 9)
5/6 NX TGR + RAS blockade + sEH blockade (initial n = 9)
The aim of this series was to evaluate whether the addition of sEH inhibitor to standard RAS blockade will exhibit additional BP-lowering effects. The ratio of left ventricle weight (LVW) to tibial length (TL), LVW/TL, was employed to determine the degree of cardiac hypertrophy.
Series 4: Effects of RAS blockade alone and combined RAS and sEH blockade on kidney ANG II, angiotensin-(1-7) (ANG 1-7), EETs and DHETEs concentrations and gene and protein expression of CYP2C23 and sEH enzyme, and gene expression associated with renal fibrosis in animals with established CKD
Animals were prepared as in series 1 and the same experimental groups were formed (n = 14 in each group, one-half of samples were used for measurements of biologically active agents and the second half for gene and protein expression). In the week labeled 0 appropriate treatment regimes were started or rats were left with no treatment. Two weeks later (eight weeks after 5/6 NX), the rats were decapitated and kidney ANG II, ANG 1-7, EETs and DHETES levels were measured, and kidney gene and protein expression of CYP2C23 and sEH enzyme were evaluated. In addition, the kidney gene expression of α-smooth muscle actin (α-SMA), monocyte chemoattractant protein-1 (MCP-1), fibronectin, transforming growth factor-β1 (TGF-β1), collagen I and III, and E-cadherin were estimated. The aim of this series was to evaluate the degree of the activation of the RAS and the rate of CYP-dependent epoxygenase pathway activity and the degree of activation of markers involved in the process of renal fibrosis. The time point: eight weeks after 5/6 NX, was chosen because at this time the animals of all experimental groups were alive, and untreated 5/6 NX TGR were just before beginning to die.
Statistical Analyses
All values are expressed as mean ± SEM. Statistical analysis was done by Student’s t-test, Wilcoxon’s signed-rank test for unpaired data, or one-way analysis of variance (ANOVA) when appropriate, using the Graph-Pad Prism software (Graph Pad Software, San Diego, CA, USA), ANOVA for repeated measurements, followed by Student-Newman-Keuls test performed for the analysis within groups, when appropriate. Comparison of survival curves was performed by log-rank (Mantel-Cox) test followed by Gehan-Breslow-Wilcoxon test. The values exceeding 95% probability limits (p<0.05) were considered statistically significant.
Results
Series 1: Effects of RAS blockade alone and combined RAS and sEH blockade on survival rate, albuminuria, and creatinine clearance in animals with established CKD
All sham-operated TGR and HanSD rats survived until the end of experiment. As shown in Fig. 1A, untreated 5/6 NX TGR began to die in the week labeled 2 (8 weeks after 5/6 NX) and by week 14 no animal survived. Both therapeutic regimes substantially improved the survival rate, however, the efficiency of RAS blockade considerably decreased from week 12 (20 weeks after 5/6 NX) and the final survival rate was 23 %. In contrast, the combined RAS and sEH blockade was more effective throughout the study and the final survival rate was 42 %, which was significantly higher compared with RAS blockade alone.
Fig. 1.
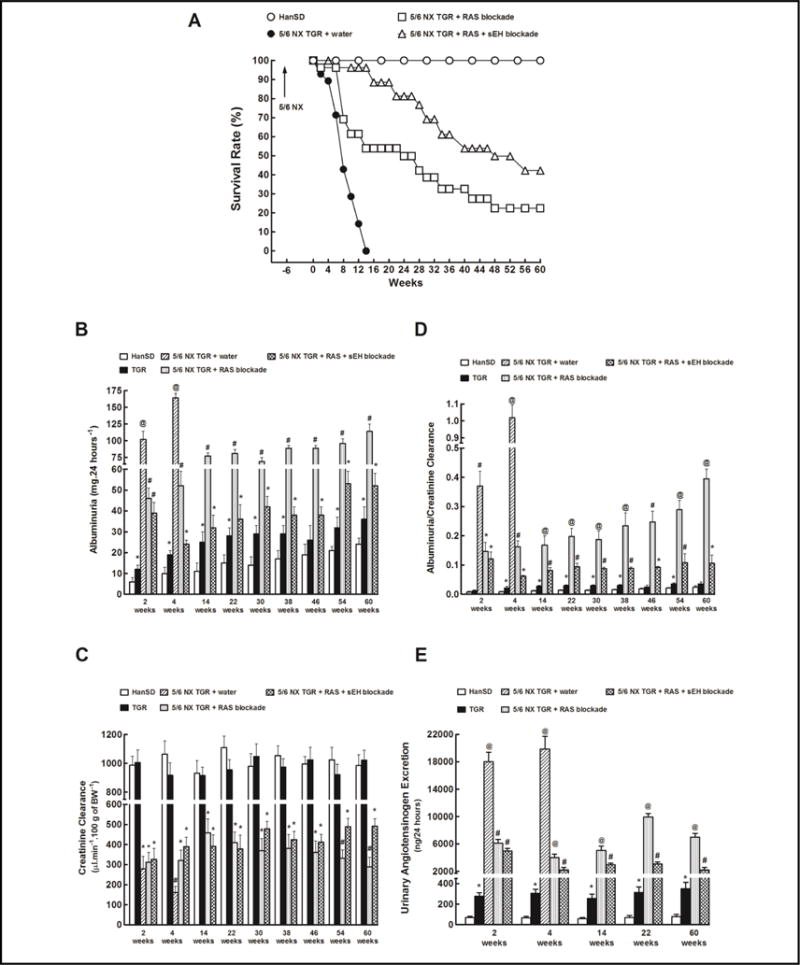
Survival rate (A), albuminuria (B), creatinine clearance (C), ratio of albuminuria to creatinine clearance (D) and urinary angiotensinogen excretion (E) in sham-operated Hannover Sprague-Dawley (HanSD, transgene-negative) rats and in heterozygous Ren-2 transgenic rats (TGR), and in 5/6 nephrectomized (5/6 NX) TGR, untreated (water) or receiving renin-angiotensin system (RAS) blockade (an angiotensin converting enzyme inhibitor and an angiotensin II receptor blocker). Alternatively, the above two-drug RAS blockade was combined with the soluble epoxide hydrolase (sEH) inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid (c-AUCB). *P<0.05 compared with sham-operated HanSD rats at the same time point. #P<0.05 compared with all the other groups at the same time point. @ P<0.05 compared with # marked values at the same time point. 5/6 NX TGR + water exhibited the worst survival rate curve among all experimental groups. 5/6 NX TGR + RAS + sEH blockade revealed better survival rate curve than 5/6 NX TGR + RAS blockade according to log-rank Mantel-Cox test followed by Gehan-Breslow-Wilcoxon test.
Fig. 1B shows that sham-operated TGR and HanSD rats exhibited at the start only minimal albuminuria, which slightly increased with age and this increase appeared more pronounced in TGR than in HanSD rats, however, compared with animals after 5/6 NX, in sham-operated animals the albuminuria remained noticeably lower. Untreated 5/6 NX TGR showed albuminuria up to 20fold greater than in the sham-operated animals. RAS blockade alone attenuated albuminuria in 5/6 NX TGR, but it remained significantly higher than in sham-operated TGR. Remarkably, the combined RAS and sEH blockade prevented increases in albuminuria after 5/6 NX: it usually remained similar as in sham-operated TGR over the course of the study.
There were no significant differences in creatinine clearance between sham-operated TGR and HanSD rats throughout the study (Fig. 1C). Untreated 5/6 NX TGR showed a profound decline in creatinine clearance which was most obvious in the week 4, just before animals began to die. Both treatment regimes in 5/6 NX TGR attenuated these decreases, however, the creatinine clearance remained significantly lower than that observed in sham-operated TGR. Nevertheless, after week 54, creatinine clearance in 5/6 NX TGR treated with RAS blockade alone began to decline and was significantly lower than observed in 5/6 NX TGR under combined RAS and sEH blockade.
As shown in Fig. 1D, in 5/6 NX TGR treated with RAS blockade alone the albuminuria normalized for glomerular filtration rate (albuminuria-to-creatinine clearance ratio) increased progressively during the study and at the end of experiment this ratio was almost 4fold higher than observed in 5/6 NX TGR treated with the combined RAS and sEH blockade.
The urinary angiotensinogen excretion was almost 4fold higher in sham-operated TGR than in sham-operated HanSD rats (Fig. 1E). Untreated 5/6 NX TGR showed in the week 2 (8 weeks after 5/6 NX) a dramatic increase in urinary angiotensinogen excretion that was more than 250-fold higher than in sham-operated HanSD rats. Both treatment regimes markedly reduced urinary angiotensinogen excretion in 5/6 NX TGR, but the combined RAS and sEH blockade was significantly more effective.
Series 2: Effects of RAS blockade alone and combined RAS and sEH blockade on the development of renal glomerular damage and renal cortical tubulointerstitial injury
As shown in Fig. 2A, already in the week 0 (6 weeks after 5/6 NX) untreated 5/6 NX TGR showed a prominent GSI increase, then the index increased further and in the week 4 (10 weeks after 5/6 NX) reached the value of almost 3, indicating an extremely serious renal glomerular damage. RAS blockade alone attenuated the increases in GSI in 5/6 NX TGR, however its efficiency considerably decreased beginning from week 6. In contrast, the combined RAS and sEH blockade remained effective throughout the experiment.
Fig. 2.
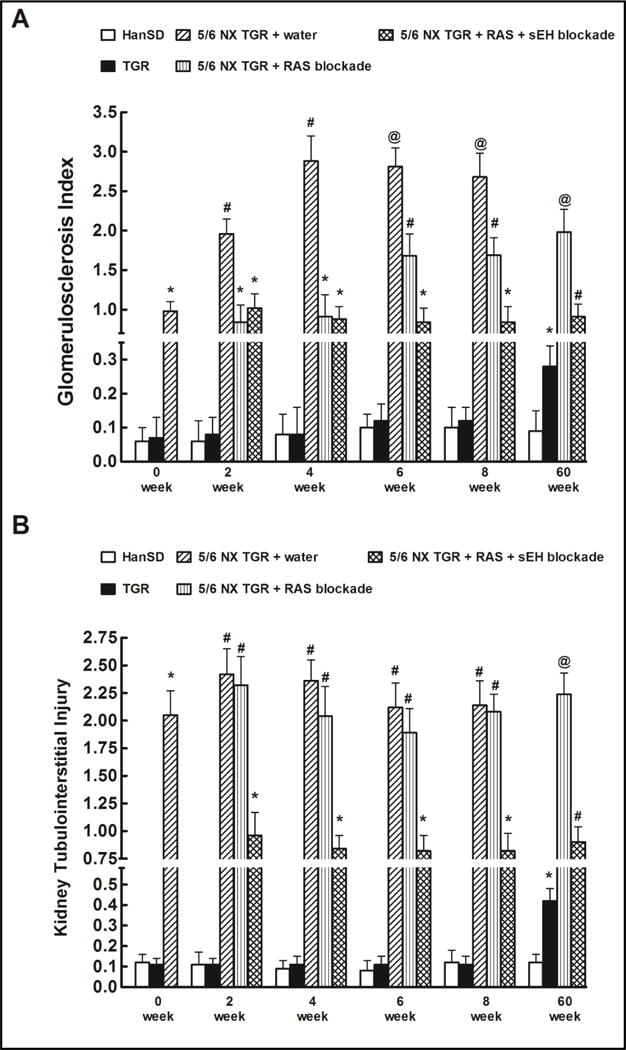
Glomerulosclerosis index (A) and kidney cortical tubulointerstitial injury (B) in shamoperated Hannover Sprague-Dawley (HanSD, transgene-negative) rats and in heterozygous Ren-2 transgenic rats (TGR), and in 5/6 nephrectomized (5/6 NX) TGR, untreated (water) or receiving renin-angiotensin system (RAS) blockade (an angiotensin converting enzyme inhibitor and an angiotensin II receptor blocker). Alternatively, the above two-drug RAS blockade was combined with the soluble epoxide hydrolase (sEH) inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy] benzoic acid (c-AUCB). *P<0.05 compared with sham-operated HanSD rats at the same time point. #P<0.05 compared with all the other groups at the same time point. @P<0.05 compared with # marked values at the same time point.
Kidney tubulointerstitial injury followed a change pattern similar as observed for GSI, however, the changes were more pronounced and RAS blockade alone did not ameliorate the kidney tubulointerstitial injury in 5/6 NX TGR. In contrast, the combined RAS and sEH blockade effectively attenuated kidney tubulointerstitial injury in 5/6 NX TGR throughout the study (Fig. 2B).
Series 3: Effects of RAS blockade alone and combined RAS and sEH blockade on BP (radiotelemetry) and cardiac hypertrophy in animals with established CKD
Fig. 3A shows that 6 weeks after 5/6 NX all TGR groups exhibited basal systolic BP (SBP) about 30 mmHg higher compared with sham-operated TGR. Either treatment regime decreased SBP in 5/6 NX TGR within 72 hours after initiation of treatment to levels that were not significantly different from the values measured in sham-operated HanSD rats.
Fig. 3.
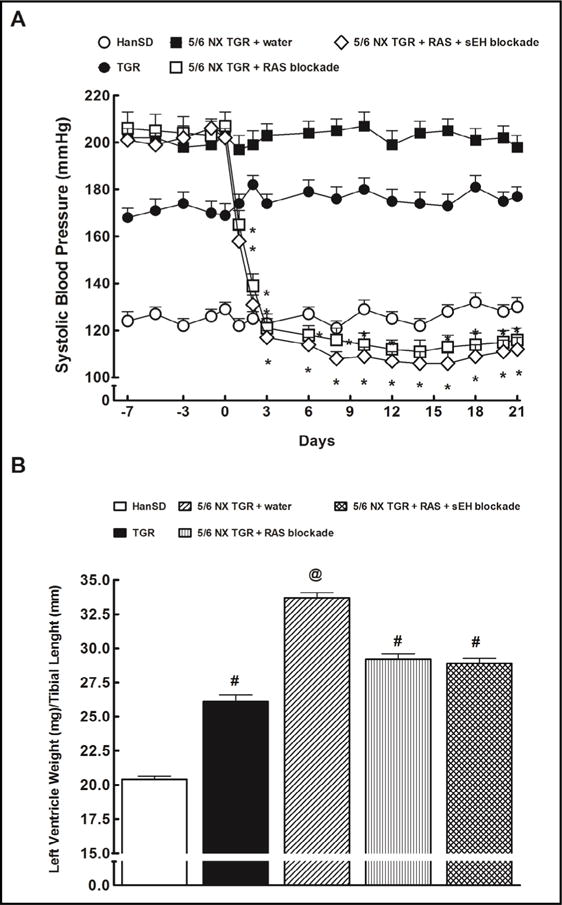
Systolic blood pressure (A) and cardiac hypertrophy (B) in sham-operated Hannover Sprague-Dawley (HanSD, transgenenegative) rats and in heterozygous Ren-2 transgenic rats (TGR), and in 5/6 nephrectomized (5/6 NX) TGR, untreated (water) or receiving renin-angiotensin system (RAS) blockade (an angiotensin converting enzyme inhibitor and an angiotensin II receptor blocker). Alternatively, the above two-drug RAS blockade was combined with the soluble epoxide hydrolase (sEH) inhibitor cis-4-[4-(3-adamantan-1-yl-ureido) cyclohexyloxy]benzoic acid. *P<0.05 compared with baseline values. #P<0.05 compared with sham-operated HanSD rats. @ P<0.05 compared with all the other groups.
The ratio of LVW/TL (an index of cardiac hypertrophy) was significantly higher in shamoperated TGR as compared with sham-operated HanSD rats (Fig. 3B). Untreated 5/6 NX TGR exhibited marked increases in this ratio as compared with sham-operated TGR. Three weeks of RAS blockade alone as well as of the combined RAS and sEH blockade resulted in significant decreases in this ratio in 5/6 NX TGR, but it still remained significantly higher than in sham-operated TGR.
Series 4: Effects of RAS blockade alone and combined RAS and sEH blockade on kidney ANG II, angiotensin-(1-7) (ANG 1-7), EETs and DHETEs concentrations and gene and protein expression of CYP2C23 and sEH enzyme, and gene expression associated with renal fibrosis in animals with established CKD
As shown in Fig. 4A, kidney ANG II concentrations were more than 2fold higher in sham-operated TGR than in sham-operated HanSD rats. Untreated 5/6 NX TGR exhibited additional increase in kidney ANG II concentrations leading to a further 2fold increase above the level seen in sham-operated TGR. Two weeks of RAS blockade alone as well as of the combined RAS and sEH blockade decreased kidney ANG II concentrations to levels not significantly different from the values observed in sham-operated HanSD rats.
Fig. 4.
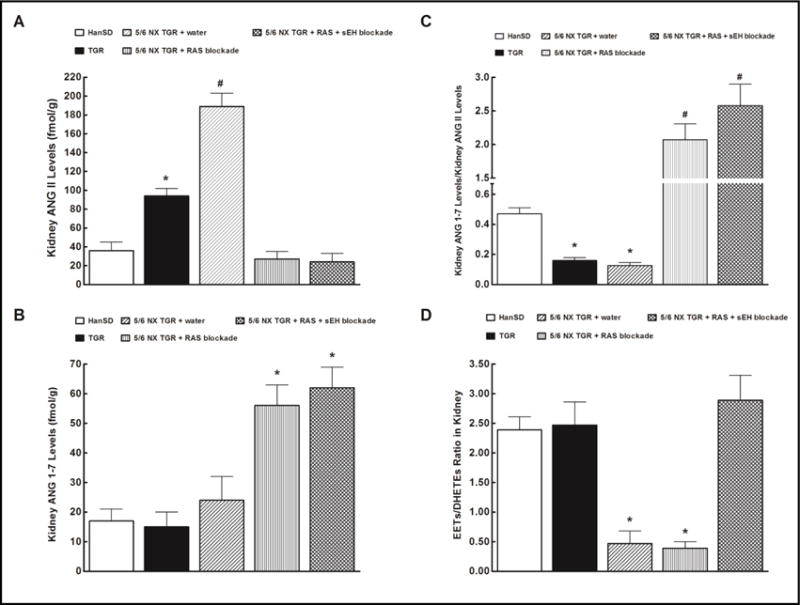
Effects of two weeks treatment with renin-angiotensin (RAS) blockade (an angiotensin converting enzyme inhibitor and an angiotensin II receptor blocker) and the two-drug RAS blockade combined with the soluble epoxide hydrolase (sEH) inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid on kidney angiotensin II (ANG II) levels (A), kidney angiotensin-1-7 (ANG 1-7) levels (B), kidney ANG 1-7 to ANG II ratio (C) and in sham-operated Hannover Sprague-Dawley (HanSD, transgene-negative) rats and kidney epoxyeicosatrienoic acids (EETs) to dihydroxyeicosatrienoic acids (DHETEs) ratio (D), in heterozygous Ren-2 transgenic rats (TGR), and in 5/6 nephrectomized (5/6 NX) TGR, untreated (water). *P<0.05 compared with sham-operated HanSD rats. #P<0.05 compared with all the other groups.
There were no significant differences in kidney ANG 1-7 concentrations between sham-operated HanSD rats, sham-operated TGR and untreated 5/6 NX TGR (Fig. 4B). RAS blockade alone as well as the combined RAS and sEH blockade caused significant increases in kidney ANG 1-7 concentrations in 5/6 NX TGR.
Fig. 4C shows the state of intrarenal balance between vasodilator and vasoconstrictor axes of the RAS expressed as the ratio of ANG 1-7 to ANG II (we and other investigators validated this ratio as a reliable marker of the activity of angiotensin-converting enzyme type 2 (ACE2)/ANG 1-7 axis of the RAS). It is seen that the ratio value in sham-operated TGR and untreated 5/6 NX TGR are only one-half of that in sham-operated HanSD rats. RAS blockade alone as well as the combined RAS and sEH blockade substantially increased this ratio in 5/6 NX TGR, to a value more than 15fold higher than in untreated 5/6 NX TGR.
As shown in Fig. 4D, the intrarenal availability of biologically active epoxygenase metabolites, expressed as the EETs/DHETEs ratio, did not significantly differ between sham-operated TGR and sham-operated HanSD rats. Untreated 5/6 NX TGR showed a profound decrease in the renal availability of epoxygenase metabolites compared with sham-operated TGR. RAS blockade alone did not change EETs/DHETEs ratio in 5/6 NX TGR. In contrast, the combined RAS and sEH blockade normalized EETs/DHETEs ratio in 5/6 NX TGR to values observed in sham-operated TGR.
As shown in Fig. 5A and 5B, kidney CYP2C23 gene and protein expressions were significantly higher in sham-operated TGR than in sham-operated HanSD rats and they remained elevated also in all 5/6 NX TGR groups.
Fig. 5.
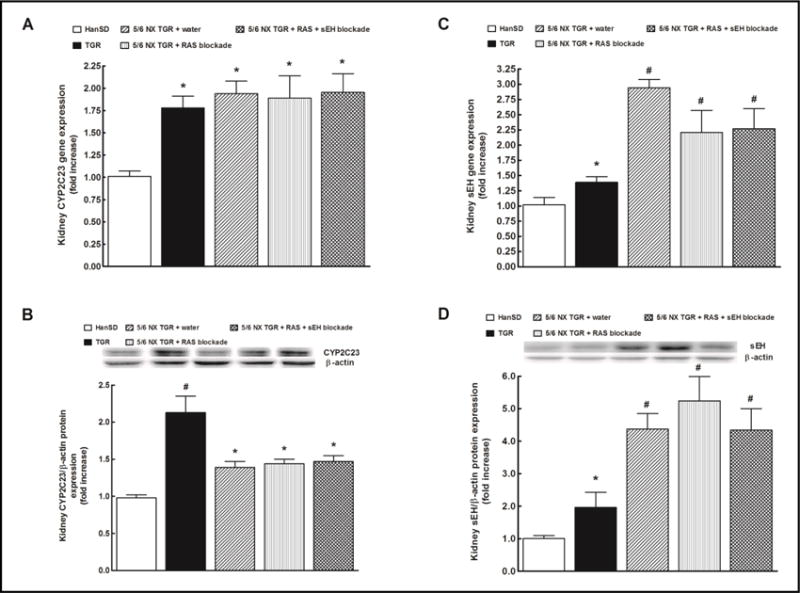
Effects of two weeks treatment with renin-angiotensin (RAS) blockade (an angiotensin converting enzyme inhibitor and an angiotensin II receptor blocker) and the two-drug RAS blockade combined with the soluble epoxide hydrolase (sEH) inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)cyclohexyloxy]benzoic acid on kidney CYP2C23 gene (A) and protein (B) and kidney sEH gene (C) and protein (D) expressions in sham-operated Hannover Sprague-Dawley (HanSD, transgene-negative), in heterozygous Ren-2 transgenic rats (TGR), and in 5/6 nephrectomized (5/6 NX) TGR, untreated (water). *P<0.05 compared with shamoperated HanSD rats. #P<0.05 compared with all the other groups.
Kidney sEH gene and protein expressions were significantly higher in sham-operated TGR as compared with sham-operated HanSD rats, and in all groups of 5/6 NX TGR the gene as well as protein expressions of sEH were further markedly enhanced (Fig. 5C and 5D).
Fig. 6A to 6E show that untreated 5/6 NX TGR exhibited markedly elevated kidney gene expressions of α-smooth muscle actin (α-SMA), monocyte chemoattractant protein-1 (MCP-1), fibronection, transforming growth factor-β1 (TGF-β1) and collagen I as compared with sham-operated HanSD rats. RAS blockade alone or the combined RAS and sEH blockade did not alter α-SMA, MCP-1 and fibronectin kidney gene expressions but, in contrast, reduced to similar extent the TGF-β1 and collagen I kidney gene expressions. The kidney collagen III gene expression showed the same pattern as did collagen I and the data are not shown.
Fig. 6.
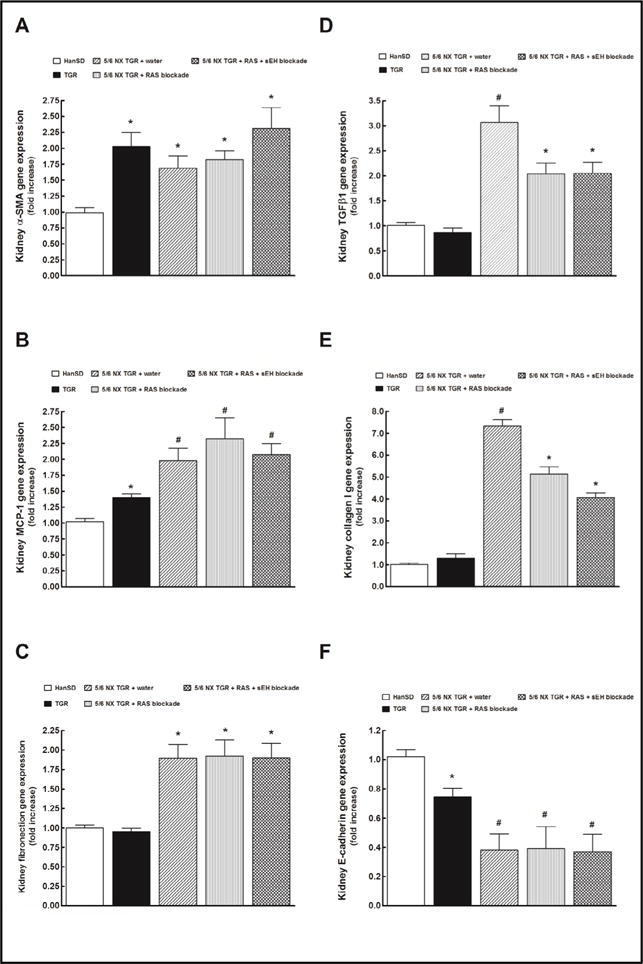
Effects of two weeks treatment with renin-angiotensin (RAS) blockade (an angiotensin converting enzyme inhibitor and an angiotensin II receptor blocker) and the two-drug RAS blockade combined with the soluble epoxide hydrolase (sEH) inhibitor cis-4-[4-(3-adamantan-1-yl-ureido) cyclohexyloxy]benzoic acid on kidney gene α-smooth muscle actin (α-SMA) (A), monocyte chemoattractant protein-1 (MCP-1) (B), fibronection (C), transforming growth factor-β1 (TGF-β1) (D), collagen I (E) and E-cadherin (F) expressions in shamoperated Hannover Sprague-Dawley (HanSD, transgene-negative), in heterozygous Ren-2 transgenic rats (TGR), and in 5/6 nephrectomized (5/6 NX) TGR, untreated (water). *P<0.05 compared with shamoperated HanSD rats. #P<0.05 compared with all the other groups.
As shown in Fig. 6F, kidney E-cadherin gene expression in untreated 5/6 NX TGR was significantly lower than in sham-operated TGR and sham-operated HanSD rats, and was not altered by RAS blockade alone or the combined RAS and sEH blockade.
Discussion
The crucial finding of this study is that prolonged pharmacological inhibition of sEH when added to the RAS blockade provided additional renoprotection and retarded the CKD progression in 5/6 NX TGR with established phase of the disease. The rat survival rate improved, albuminuria was further reduced as was also the glomerular and tubulointerstitial injury, and the decrease in glomerular filtration rate was further attenuated. Notably, while the value of the 5/6 NX model is long established, only few studies explored the phase of established CKD [12–15, 17, 43]. Furthermore, while most studies showed beneficial effects of sEH inhibition on the progression of end-organ damage under various experimental conditions [34–40, 43, 53], it was also reported that such blockade in 5/6 NX mice provided no antihypertensive effect, no improvement in survival rate and no attenuation of renal glomerular and tubulointerstitial injury; indeed, augmentation of albuminuria suggested faster rather than slowed-down CKD progression [43, 54]. In this context, the present findings deserve special attention.
First, they strengthen the evidence that hypertension and increased activity of the RAS in the remnant kidney are two major determinants of the rate of progression of CKD. Therefore, antihypertensive therapy based on the blockade of RAS (“RAS-dependent”), especially the blockade at more than one level, inhibits the progression of CKD more efficiently, than can be achieved with “RAS-independent” therapy, and is currently accepted as the gold standard for the therapy used to delay the progression of CKD to ERSD [5, 8,16, 20–23].
Second, our results support the evidence that therapeutic RAS blockade does not revert CKD and, when applied in its advanced stage, fails to exhibit renoprotective effects. This points to the need for new therapeutic approaches [5, 12, 13, 15, 17, 31–33], which requires a sound knowledge of the underlying pathomechanisms.
What are the mechanism(s) underlying additional renoprotective effects of the combined RAS and sEH blockade in 5/6 NX TGR?
Our data show that untreated 5/6 NX TGR exhibit a reduced availability of biologically active epoxygenase products in the remnant kidney when compared to that observed in sham-operated TGR and HanSD rats. This deficiency of intrarenal EETs is clearly not the consequence of compromised endogenous EETs formation (as indicated by increased CYP2C23 gene and protein expression) but results from increased sEH-mediated conversion of EETs to DHETEs (as suggested by increased sEH gene and protein expression). Notably, measurements of the EETs/DHETEs ratio indicate that in 5/6 NX TGR normalization of the intrarenal EETs availability could be achieved only with the combined RAS and sEH blockade. Therefore, the additional renoprotective actions are very likely related to this normalization; however, the detailed mechanism(s) of the EETs-mediated renoprotective action require further analysis.
Earlier studies seriously questioned the “BP-independent” organ-protective actions of the RAS-dependent antihypertensive therapy: it was found that when equal BP-lowering effects were achieved with RAS-dependent or RAS-independent therapy, the same degree of cardio- and renoprotection in 5/6 NX animal was observed [14, 28–30]. Therefore, we first examined if addition of sEH inhibitor to the two-levels RAS blockade would have an additional BP-lowering effect: if so, additional renoprotection could be simply attributed to this effect. Such explanation is supported by findings that intrarenal deficiency of EETs is a permissive factor in the development of ANG II-dependent hypertension, and that antihypertensive effects of treatment with sEH inhibitor are associated with an increase in intrarenal EETs bioavailability [36–38, 40]. However, since our data show that either antihypertensive regime brought BP in 5/6 NX TGR to similar and clearly normotensive levels, it is evident that additional BP-lowering effect was not the reason for additional renoprotective effects of the combined RAS and sEH treatment.
Despite growing evidence indicating that, at least in 5/6 NX rats, renoprotection is predominantly BP-dependent [14, 28, 30], numerous studies support a critical role of the RAS in the pathophysiology of end-organ damage [6, 7,10, 11, 15–20, 40–44, 54–57]. Since we demonstrated recently that enhanced intrarenal tissue availability of EETs could suppress the elevated intrarenal RAS activity in experimental hypertension [58, 59], we examined if addition of sEH inhibitor to RAS blockade would cause more profound suppression of RAS activity in the remnant kidney: an effect that could be responsible for the additional renoprotective action. Our data show that two weeks’ treatment using either therapeutic regime sufficed to normalize kidney ANG II in 5/6 NX TGR to values observed in shamoperated HanSD rats. Since ANG II-mediated activation of type 1 (AT1) is responsible for the hypertensiogenic and end-organ damaging actions of the RAS [7], our data indicate that either treatment regime successfully normalized the inappropriately activated hypertensiogenic axis of the RAS. In addition, we found that either treatment regime markedly increased intrarenal ANG 1-7, the most important peptide of the vasodilator axis of the RAS which is believed to counterbalance deleterious actions of the hypertensiogenic RAS [44, 60]. Thus, either of the two treatment procedures activated also the vasodilatory and organ-protective axis of the RAS. Moreover, an increase of the ANG 1-7/ANG II ratio indicated that the intrarenal balance between the vasodilator and the hypertensiogenic axis was markedly shifted toward the prevalence of the former, both with RAS blockade alone and with the combined RAS and sEH blockade. On the whole, these data indicate that either treatment regime successfully and to similar extent suppressed inappropriately augmented hypertensiogenic axis of the RAS and concurrently activated vasodilator axis of the RAS in our 5/6 NX TGR. This suggests the additional renoprotective effects of the combined RAS and sEH blockade did not depend on different action of the two treatment regimes on the intrarenal activity of the RAS. Importantly, however, this conclusion regarding the balance of the two RAS axes is valid only for the relatively early phase of CKD, specifically for animals eight weeks after 5/6 NX and treated for two weeks.
To obtain a still better insight in the status of intrarenal RAS as affected by our treatment regimes, in the same animals we analysed urinary excretion of angiotensinogen, a very reliable indicator of this status [61, 62]. This enabled continuous follow-up of intrarenal RAS activity throughout the experiment. We found that in 5/6 NX TGR treated by either regime angiotensinogen excretion was still markedly higher than in sham-operated TGR or in HanSD rats. However, in the late phase of the study (beginning from the 28th week after 5/6 NX and 22nd week of treatment) the combined RAS and sEH blockade was distinctly more efficient in reducing urinary angiotensinogen excretion compared with RAS blockade alone. These findings clearly show that intrarenal RAS activity after 5/6 NX was markedly activated throughout the study, and this was so even with the two-levels RAS blockade (ACEi plus AT1 receptor blocker) applied using the high dosage with established efficiency [5, 20, 24, 33, 63, 64]. Therefore, it is reasonable to assume that such inappropriately augmented intrarenal RAS activity contributes to the progression of CKD to ERSD in the very late phase. However, the finding of utmost importance in this study is that addition of sEH inhibitor to the thorough RAS blockade resulted (in the advanced phase of CKD) in greater suppression of inappropriately activated intrarenal RAS. We postulate that it was the main cause for improved renoprotective action of the combined RAS and sEH blockade in 5/6 NX TGR. It is reminded that the sEH inhibitor dose employed here when given as a sole treatment did not influence systemic or kidney RAS activity or retard progression of CKD [43].
Of considerable interest are our data on the markers of the processes of renal fibrosis. In accordance with previous studies in untreated 5/6 NX TGR these markers were markedly increased, and E-cadherin, a marker of the loss of epithelial proteins during epithelial to mesenchymal transition was decreased [65, 66]. We found also that either treatment regime similarly and significantly decreased TGF-β, probably the major fibrogenic signaling molecule [67–69]. Since ANG II is the main stimulator of TGF-β expression in the kidney [19, 55, 56, 67, 68] and we found that the combined RAS and sEH blockade suppressed intrarenal RAS activity more efficiently, one would expect that addition of sEH inhibitor to the standard RAS blockade should reduce the kidney gene expressions of TGF-β relatively more. However, we did not see that, despite the fact the tubulointerstitial injury in 5/6 NX TGR treated with the combined RAS and sEH blockade was significantly lower. We have no explanation for this inconsistency; evidently, further studies are needed here.
Conclusion
This study provides evidence that addition of pharmacological inhibition of sEH to the RAS blockade brings additional renoprotective effects and further retards the progression of CKD in 5/6 NX TGR, also when the treatment is initiated at the advanced stage of established CKD. The data suggest that additive renoprotective actions of the combined RAS and sEH blockade depend on greater suppression of inappropriately activated intrarenal RAS. This information derived from our present experimental study should be considered in attempts at the development of new therapeutic approaches for the treatment of advanced stages of CKD in humans.
Acknowledgments
This study was primarily supported by the Ministry of Health of the Czech Republic grant no. 15-28671A to V.Č.Ch. All rights reserved. L.Č. was also supported by Ministry of Health of the Czech Republic within the project for the development of research organization 00023001 (IKEM) – institutional support. This work was also partially supported by a National Institute of Health grant (DK103616) to John D Imig. Partial support was supplied by NIEHS R01 ES002710 and Superfund Research Program P42 ES004699 awarded to Bruce D. Hammock.
Footnotes
Disclosure Statement
The authors declare they have no conflicts of interest regarding the publication of this article.
References
- 1.Brenner BM. Nephron adaptation to renal injury or ablation. Am J Physiol. 1985;249:F324–F337. doi: 10.1152/ajprenal.1985.249.3.F324. [DOI] [PubMed] [Google Scholar]
- 2.Zoja C, Abbate M, Remuzzi G. Progression of chronic kidney disease: insight from animal models. Curr Opin Nephrol Hypertens. 2006;15:250–257. doi: 10.1097/01.mnh.0000222691.53970.83. [DOI] [PubMed] [Google Scholar]
- 3.Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet. 2017;389:1238–1252. doi: 10.1016/S0140-6736(16)32064-5. [DOI] [PubMed] [Google Scholar]
- 4.Breyer M, Sustak K. The next generation of therapeutic for chronic kidney disease. Nat Rev Drug Discov. 2016;15:568–588. doi: 10.1038/nrd.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cortinovis M, Ruggenenti P, Remuzzi G. Progression, remission and regression of chronic renal diseases. Nephron. 2016;34:20–24. doi: 10.1159/000445844. [DOI] [PubMed] [Google Scholar]
- 6.Carlstrom M, Wilcox CS, Arendshorst WJ. Renal autoregulation in health and disease. Physiol Rev. 2015;95:405–511. doi: 10.1152/physrev.00042.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 8.Turner JM, Bauer C, Abramowitz MK, Melamed ML, Hostetter TH. Treatment of chronic kidney disease. Kidney Int. 2012;81:351–362. doi: 10.1038/ki.2011.380. [DOI] [PubMed] [Google Scholar]
- 9.Fukuda A, Wickman LT, Venkatareddy MP, Sato Y, Chowdhury MA, Wang SQ, Shedden KA, Dysko RC, Wiggins JE, Wiggins RC. Angiotensin II-dependent persistent podocyte loss form destabilized glomeruli causes progression of end stage kidney disease. Kidney Int. 2012;81:40–55. doi: 10.1038/ki.2011.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilbert RE, Wu LL, Kelly DJ, Cox A, Wilkinson-Berka JL, Johnston CI, Cooper ME. Pathological expression of renin and angiotensin II in the renal tubule after subtotal nephrectomy: implications of the pathogenesis of tubulointerstitial fibrosis. Am J Pathol. 1999;155:429–440. doi: 10.1016/S0002-9440(10)65139-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goncalves AR, Fujihara CK, Mattar AL, Malheiros DM, Noronha Ide L, de Nucci G, Zatz R. Renal expression of COX-2, ANG II, and AT1 receptor in remnant kidney: strong renoprotection by therapy with losartan and nonsteroidal anti-inflammatory. Am J Physiol. 2004;286:F945–F954. doi: 10.1152/ajprenal.00238.2003. [DOI] [PubMed] [Google Scholar]
- 12.Arias SC, Valente CP, Machado FG, Fanelli C, Origassa CS, de Brito T, Camara NO, Malheiros DM, Zatz R, Fujihara CK. Regression of albuminuria and hypertension and arrest of severe renal injury by a losartan-hydrochlorothiazide association in a model of very advanced nephropathy. PLos One. 2013;8:e56215. doi: 10.1371/journal.pone.0056215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arias SC, Souza RA, Malheiros DM, Fanelli C, Fujihara CK, Zatz R. An association of losartan-hydrochlothiazide, but not losartan-furosemide, completely arrests progressive injury in the remnant kidney. Am J Physiol. 2016;310:F135–F143. doi: 10.1152/ajprenal.00388.2015. [DOI] [PubMed] [Google Scholar]
- 14.Kujal P, Certikova Chabova V, Vernerova Z, Walkowska A, Kompanowska-Jezierska E, Sadowski J, Vanourkova Z, Huskova Z, Opocensky M, Skaroupkova P, Schejbalova S, Kramer HJ, Rakusan D, Maly J, Netuka I, Vaneckova I, Kopkan L, Cervenka L. Similar renoprotection after renin-angiotensin-dependent and -independent antihypertensive therapy in 5/6-nephrectomized Ren-2 transgenic rats: are there blood pressure-independent effects? Clin Exp Pharmacol Physiol. 2010;37:1159–1169. doi: 10.1111/j.1440-1681.2010.05453.x. [DOI] [PubMed] [Google Scholar]
- 15.Sedlakova L, Certikova Châbovâ V, Dolezelova S, Skaroupkova P, Kopkan L, Huskova Z, Cervenkova L, Kikerlova S, Vaneckova I, Sadowski J, Kompanovska-Jezierska E, Kujal P, Kramer HJ, Cervenka L. Renin-angiotensin system blockade alone or combined with ETA receptor blockade: effects on the course of chronic kidney disease in 5/6 nephrectomized Ren-2 transgenic hypertensive rats. Clin Exp Hypertens. 2017;39:183–195. doi: 10.1080/10641963.2016.1235184. [DOI] [PubMed] [Google Scholar]
- 16.Rüster C, Wolf G. Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol. 2006;17:2985–2991. doi: 10.1681/ASN.2006040356. [DOI] [PubMed] [Google Scholar]
- 17.Certikova Chabova V, Vernerova Z, Kujal P, Huskova Z, Skaroupkova P, Tesar V, Kramer HJ, Kompanowska-Jezierska E, Walkowska A, Sadowski J, Cervenka L, Vaneckova I. Addition of ETA receptor blockade increases renoprotection provided by renin-angiotensin system blockade in 5/6 nephrectomized Ren-2 transgenic rats. Life Sci. 2014;118:297–305. doi: 10.1016/j.lfs.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 18.Vaneckova I, Kujal P, Huskova Z, Vanourkova Z, Vernerova Z, Certikova Chabova V, Skaroupkova P, Kramer HJ, Tesar V, Cervenka L. Effects of combined endothelin A receptor and renin-angiotensin system blockade on the course of end-organ damage in 5/6 nephrectomized Ren-2 hypertensive rats. Kidney Blood Press Res. 2012;35:382–392. doi: 10.1159/000336823. [DOI] [PubMed] [Google Scholar]
- 19.Zhou L, Mo H, Miao J, Zhou D, Tan RJ, Hou FF, Liu Y. Klotho ameliorates kidney injury and fibrosis and normalizes blood pressure by targeting the renin-angiotensin system. Am J Pathol. 2015;185:3211–3223. doi: 10.1016/j.ajpath.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujihara CK, Velho M, Malheiros DM, Zatz R. An extremely high dose of losartan affords superior renoprotection in the remnant model. Kidney Int. 2005;67:1913–1924. doi: 10.1111/j.1523-1755.2005.00290.x. [DOI] [PubMed] [Google Scholar]
- 21.Ripley E. Complementary effects of angiotensin-converting enzyme inhibitors and angiotensin receptors blockers in slowing the progression of chronic kidney disease. Am Heart J. 2009;157:S7–S16. doi: 10.1016/j.ahj.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 22.Ptinopolou AG, Pikilidou MI, Lasaridis N. The effect of antihypertensive drugs on chronic kidney disease: a comprehensive review. Hypertens Res. 2013;36:91–101. doi: 10.1038/hr.2012.157. [DOI] [PubMed] [Google Scholar]
- 23.Kakinuma Y, Kawamura T, Bills T, Yoshioka T, Ichikawa I, Fogo A. Blood pressure-independent effect of angiotensin inhibition on vascular lesions of chronic renal failure. Kidney Int. 1992;42:46–55. doi: 10.1038/ki.1992.259. [DOI] [PubMed] [Google Scholar]
- 24.Berl T. Renal protection by inhibition of the renin-angiotensin-aldosterone system. J Renin Angiotensin Aldosterone Syst. 2009;10:1–8. doi: 10.1177/1470320309102747. [DOI] [PubMed] [Google Scholar]
- 25.Adamczam M, Gross ML, Krtil J, Koch A, Tyralla K, Amann K, Ritz E. Reversal of glomerulosclerosis after highdose enalapril treatment in subtotally nephrectomized rats. J Am Soc Nephrol. 2003;14:2833–2842. doi: 10.1097/01.asn.0000095248.91994.d3. [DOI] [PubMed] [Google Scholar]
- 26.Remuzi A, Gagliardinin E, Sangalli F, Bonomelli M, Piccinelli M, Benigni A, Remuzii G. ACE inhibition reduces glomerulosclerosis and regenerates glomerular tissue in a model of progressive renal disease. Kidney Int. 2006;69:1124–1130. doi: 10.1038/sj.ki.5000060. [DOI] [PubMed] [Google Scholar]
- 27.Remuzzi A, Sangalli F, Macconi D, Tomason S, Cattaneo I, Rizzo P, Bonandrini B, Bresciani E, Longaretti L, Gagliardini E, Conti S, Benigni A, Remuzzi G. Regression of renal disease by angiotensin II antagonism is caused by regeneration of kidney vasculature. J Am Soc Nephrol. 2016;27:699–705. doi: 10.1681/ASN.2014100971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bidani AK, Polichnowski AJ, Loutzenhiser R, Griffin KA. Renal microvascular dysfunction, hypertension and CKD progression. Curr Opin Nephrol Hypertens. 2013;22:1–9. doi: 10.1097/MNH.0b013e32835b36c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffin KA, Abu-Amarah I, Picken MM, Bidani AK. Renoprotection by ACE inhibition or aldosterone blockade is blood pressure-dependent. Hypertension. 2003;41:201–206. doi: 10.1161/01.hyp.0000049881.25304.73. [DOI] [PubMed] [Google Scholar]
- 30.Griffin KA, Picken MM, Bidani AK. Blood pressure lability and glomerulosclerosis after normotensive 5/6 renal mass reduction in rat. Kidney Int. 2004;65:209–218. doi: 10.1111/j.1523-1755.2004.00356.x. [DOI] [PubMed] [Google Scholar]
- 31.Perico N, Amuchastegui SC, Colosio V, Sonzogni G, Bertani T, Remuzzi G. Evidence that an angiotensinconverting enzyme inhibitor has a different effect on glomerular injury according to the different phase of the disease at which the treatment is started. J Am Soc Nephrol. 1994;5:1139–1146. doi: 10.1681/ASN.V541139. [DOI] [PubMed] [Google Scholar]
- 32.Gordon J, Kopp JB. Off the beaten renin-angiotensin-aldoserone system pathway: new perspectives on antiproteinuric therapy. Adv Chron Kidney Dis. 2011;18:300–311. doi: 10.1053/j.ackd.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Certikova Chabova V, Cervenka L. The dilemma of dual renin-angiotensin system blockade in chronic kidney disease: why beneficial in animal experiments but not in the clinic? Physiol Res. 2017;66:181–192. doi: 10.33549/physiolres.933607. [DOI] [PubMed] [Google Scholar]
- 34.Huang H, Morisseau C, Wang JF, Yang T, Falck JR, Hammock BD, Wang MH. Increasing or stabilizing renal epoxyeicosatrienoic acid production attenuates abnormal renal function and hypertension in obese rats. Am J Physiol. 2007;293:F342–F349. doi: 10.1152/ajprenal.00004.2007. [DOI] [PubMed] [Google Scholar]
- 35.Lee CR, Imig JD, Edin ML, Foley J, DeGraff LM, Bradbury JA, Graves JP, Lih Fb, Clark J, Myers P, Perrow AL, Lepp AN, Kannon MA, Ronnekleiv OK, Alkayed NJ, Falck JR, Tomer KB, Zeldin DC. Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J. 2010;24:3770–3781. doi: 10.1096/fj.10-160119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neckar J, Kopkan L, Huskova Z, Kolar F, Papousek F, Kramer HJ, Hwang SH, Hammck BD, Imig JD, Maly J, Netuka I, Ostadal B, Cervenka L. Inhibition of soluble epoxide hydrolase by cis-4-[4-(3-adamantan-I-ylureido) cyclohexyl-oxy]benzoic acid exhibits antihypertensive and cardioprotective actions in transgenic rats with angiotensin II-dependent hypertension. Clin Sci. 2012;122:513–525. doi: 10.1042/CS20110622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Honetschlàgerova Z, Sporkova A, Kopkan L, Huskova Z, Hwang SH, Hammock BD, Imig JD, Kramer HJ, Kujal P, Certikova Chabova V, Tesar V, Cervenka L. Inhibition of soluble epoxide hydrolase improves the impaired pressure-natriuresis relationship and attenuates the development of hypertension and hypertension-associated end-organ damage in Cyp1a1-Ren-2 transgenic rats. J Hypertens. 2011;29:1590–1601. doi: 10.1097/HJH.0b013e328349062f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imig JD. Epoxyeicosatrienoic acids, hypertension, and kidney injury. Hypertension. 2015;65:476–482. doi: 10.1161/HYPERTENSIONAHA.114.03585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan F, Muoya Y, Roman RJ. Cytochrome P450 eicosanoids in hypertension and renal disease. Curr Opin Nephrol Hypertens. 2015;24:37–46. doi: 10.1097/MNH.0000000000000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elmarakby AA. Reno-protective mechanisms of epoxyeicosatrienoic acids in cardiovascular disease. Am J Physiol. 2012;302:R321–R330. doi: 10.1152/ajpregu.00606.2011. [DOI] [PubMed] [Google Scholar]
- 41.Shimamura T, Morrison AB. A progressive glomerulosclerosis occurring in partial five-sixths nephrectomized rats. Am J Pathol. 1975;79:95–106. [PMC free article] [PubMed] [Google Scholar]
- 42.Mullins JJ, Peters J, Ganten D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature. 1990;344:541–544. doi: 10.1038/344541a0. [DOI] [PubMed] [Google Scholar]
- 43.Kujal P, Certikova Chabova V, Skaroupkova P, Huskova Z, Vernerova Z, Kramer HJ, Walkowska A, Kompanowska- Jezierska E, Sadowski J, Kitada K, Nishiyama A, Hwang SH, Hammock BD, Imig JD, Cervenka L. Inhibition of soluble epoxide hydrolase is renoprotective in 5/6 nephrectomized Ren-2 transgenic rats. Clin Exp Pharmacol Physiol. 2014;41:227–237. doi: 10.1111/1440-1681.12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huskova Z, Kopkan L, Cervenkova L, Dolezelova S, Vanourkova Z, Skaroupkova P, Nishiyama A, Kompanowska- Jezierska E, Sadowski J, Kramer HJ, Cervenka L. Intrarenal alterations of the angiotensin-converting type 2/ angiotensin 1-7 complex of the renin-angiotensin system do not alter the course of malignant hypertension in Cyp1a1-Ren-2 transgenic rats. Clin Exp Pharmacol Physiol. 2016;43:438–449. doi: 10.1111/1440-1681.12553. [DOI] [PubMed] [Google Scholar]
- 45.Kurtz TW, Griffin KA, Bidani AK, Davisson RL, Hall JE. Recommendations for blood pressure measurements in humans and experimental animals. Part 2: Blood pressure measurements in experimental animals. Hypertension. 2005;45:299–310. doi: 10.1161/01.HYP.0000150857.39919.cb. [DOI] [PubMed] [Google Scholar]
- 46.Nakano Y, Hirano T, Uehara K, Nishibayashi S, Hattori K, Aihara M, Yamada Y. New rat model induced by anti-glomerular basement membrane antibody shows severe glomerular adhesion in early stage and quickly progress to end-stage renal failure. Pathol Int. 2008;58:361–370. doi: 10.1111/j.1440-1827.2008.02237.x. [DOI] [PubMed] [Google Scholar]
- 47.Huskova Z, Kramer HJ, Vanourkova Z, Cervenka L. Effects of changes in sodium balance on plasma and kidney angiotensin II levels in anesthetized and conscious Ren-2 trangenic rats. J Hypertens. 2006;24:517–522. doi: 10.1097/01.hjh.0000209988.51606.c7. [DOI] [PubMed] [Google Scholar]
- 48.Rivera J, Ward N, Hodgson J, Hodgson J, Puddey IB, Falck JR, Croft KD. Measurement of 20-hydroxyeicosatetraenoic acid in human urine by gas chromatography-mass spectrometry. Clin Chem. 2004;50:224–226. doi: 10.1373/clinchem.2003.025775. [DOI] [PubMed] [Google Scholar]
- 49.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 50.Bas A, Forsberg G, Hammarstrom S, Hammarstrom ML. Utility of the housekeeping genes 18S rRNA, betaactin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase-polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand J Immunol. 2004;59:566–573. doi: 10.1111/j.0300-9475.2004.01440.x. [DOI] [PubMed] [Google Scholar]
- 51.Jichova S, Dolezelova S, Kopkan L, Kompanowska-Jezierska E, Sadowski J, Cervenka L. Fenofibrate attenuates malignant hypertension by suppression of the renin-angiotensin system: a study in Cyp1a1-Ren-2 transgenic rats. Am J Med Sci. 2016;352:618–630. doi: 10.1016/j.amjms.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 52.Sporkova A, Certikova Chabova V, Dolezelova S, Jichova S, Kopkan L, Vanourkova Z, Kompanowska-Jezierska E, Sadowski J, Maxova H, Cervenka L. Fenofibrate attenuates hypertension in Goldblatt hypertensive rats: role of 20-hydroxyeicosatetraenoic acid in the nonclipped kidney. Am J Med Sci. 2017;353:568–579. doi: 10.1016/j.amjms.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 53.Zhao G, Tu L, Li X, Yang S, Chen C, Xu X, Wang P, Wang DW. Delivery of AAV2-CYP2J2 protects remnant kidney in the 5/6-nephrectomized rat via inhibition of apoptosis and fibrosis. Hum Gene Ther. 2012;23:688–699. doi: 10.1089/hum.2011.135. [DOI] [PubMed] [Google Scholar]
- 54.Jung O, Jansen F, Mieth A, Barbosa-Sicard E, Pliquett RU, Babelova A, Morisseau C, Hwang SH, Tsai C, Hammock BD, Schaefer L, Geisslinger G, Amann K, Brandes RP. Inhibition of the soluble epoxide hydrolase promotes albuminuria in mice with progressive renal disease. PloS One. 2010;5:e11979. doi: 10.1371/journal.pone.0011979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wolf G. Renal injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-β pathway. Kidney Int. 2006;70:1914–1919. doi: 10.1038/sj.ki.5001846. [DOI] [PubMed] [Google Scholar]
- 56.Wu LL, Cox A, Roe CJ, Dziadek M, Cooper ME, Gilbert RE. Transforming growth factor β1 and renal injury following subtotal nephrectomy in the rat: role of the renin-angiotensin system. Kidney Int. 1997;51:1553–1567. doi: 10.1038/ki.1997.214. [DOI] [PubMed] [Google Scholar]
- 57.Lv LL, Liu BC. Role of non-classical renin-angiotensin system axis in renal fibrosis. Front Physiol. 2015;6:117. doi: 10.3389/fphys.2015.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Varcabova S, Huskova Z, Kramer HJ, Hwang SH, Hammock BD, Imig JD, Kitada K, Cervenka L. Antihypertensive action of soluble epoxide hydrolase inhibition in Ren-2 transgenic rats is mediated by suppression of the intrarenal renin-angiotensin system. Clin Exp Pharmacol Physiol. 2012;40:273–281. doi: 10.1111/1440-1681.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jichova S, Kopkan L, Huskova Z, Dolezelova S, Neckar J, Kujal P, Vernerova Z, Kramer HJ, Sadowski J, Kompanowska-Jezierska E, Redy RN, Falck JR, Imig JD, Cervenka L. Epoxyeicosatrienoic acid analog attenuates the development of malignant hypertension, but does not reverse it once established: a study in Cyp1a1-Ren-2 transgenic rats. J Hypertens. 2016;34:2008–2025. doi: 10.1097/HJH.0000000000001029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Santos RA. Angiotensin-(1–7) Hypertension. 2014;63:1138–1147. doi: 10.1161/HYPERTENSIONAHA.113.01274. [DOI] [PubMed] [Google Scholar]
- 61.Kobori H, Nishiyama A, Harrison-Bernard LM, Navar LG. Urinary angiotensinogen as an indicator of intrarenal angiotensin status in hypertension. Hypertension. 2003;41:42–49. doi: 10.1161/01.hyp.0000050102.90932.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kobori H, Urushihara M. Augmented intrarenal and urinary angiotensinogen in hypertension and chronic kidney disease. Pflugers Arch - Eur J Physiol. 2013;465:3–12. doi: 10.1007/s00424-012-1143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cao Z, Bonnet F, Davis B, Allen TJ, Cooper ME. Additive and anti-albuminuric effects of angiotensin-converting enzyme inhibition and angiotensin receptor antagonist in diabetic spontaneously hypertensive rats. Clin Sci. 2001;100:591–599. [PubMed] [Google Scholar]
- 64.Azizi M, Ménard J. Combined blockade of the renin-angiotensin system with angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists. Circulation. 2004;109:2492–2499. doi: 10.1161/01.CIR.0000131449.94713.AD. [DOI] [PubMed] [Google Scholar]
- 65.Kim J, Imig JD, Yang J, Hammock BD, Padanilam BJ. Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am J Physiol. 2014;307:F971–F980. doi: 10.1152/ajprenal.00256.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiao Y, Liu J, Peng Y, Huang L, Yang H, Zhang J, Tao L. GSTA3 attenuates renal interstitial fibrosis by inhibiting TGF-beta-induced tubular epithelial-mesenchymal transition and fibronectin expression. PLoS One. 2016;11:e0160855. doi: 10.1371/journal.pone.0160855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gewin L, Zent R, Pozzi A. Progression of chronic kidney disease: too much cellular talk causes damage. Kidney Int. 2017;91:52–560. doi: 10.1016/j.kint.2016.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leaf IA, Duffield JS. What can target kidney fibrosis? Nephrol Dial Transplant. 2017;32:i89–i97. doi: 10.1093/ndt/gfw388. [DOI] [PubMed] [Google Scholar]
- 69.Schnaper HW. The tubulointerstitial pathophysiology of progressive kidney disease. Adv Chronic Kidney Dis. 2017;24:107–116. doi: 10.1053/j.ackd.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]