

Abstract
Platelets in flowing blood are sometimes exposed to elevated shear forces caused by anastomotic stenosis at the blood vessel-vascular implant interface. The objective of this study was to determine how effective upstream shear forces are in priming platelets for downstream adhesion and activation. Flow chambers with upstream stenotic regions (shear rates of 400 – 1000 s−1) were manufactured by relief molding of polydimethylsiloxane. Downstream from the stenotic regions, microcontact printing was used to covalently immobilize three different proteins (fibrinogen, collagen, or von Willebrand factor) to serve as platelet capture agents. Anticoagulated whole blood was perfused through the flow chambers and platelet adhesion to the downstream capture region was quantified. It was found that transient exposure of platelets to increased shear forces resulted in higher platelet adhesion on all three proteins. The duration of the platelet exposure to elevated shear forces was varied by changing the length of the stenotic regions. The results indicated that, in addition to the magnitude of shear forces, the duration of exposure to these forces was also an important factor in priming platelets. The effect of upstream shear forces on platelet activation was assessed by quantifying P-selectin, integrin αIIbβ3, lysosomal glycoprotein, and phosphatidylserine exposure using flow cytometry. The results suggested that increased shear forces were capable of increasing the priming of platelets for downstream activation. This study implicates the anastomotic region(s) of vascular implants as a locus of platelet pre-activation that may lead to thrombus formation downstream.
Keywords: anastomotic stenosis, platelet adhesion and activation, microfluidics, microcontact printing, flow cytometry
Graphical abstract
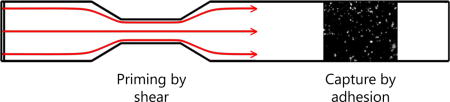
1. Introduction
Cardiovascular disease is a leading cause of death for people in the world. The disease is often treated through the surgical introduction of blood contacting devices such as stents, shunts, vascular grafts and catheters. While these devices can save the lives of patients, their surfaces are far from being truly hemocompatible. The homeostatic response to such biomaterials often necessitates that the patients remain on anticoagulation therapy [1,2]. The majority of blood compatibility studies have traditionally focused on establishing the platelet response to implanted biomaterials. These studies, when accounting only for the local platelet-biomaterial effects, often missed the dynamics of platelet adhesion and activation in circulation. In fact, it has been shown that the majority of platelet contacts at a flowing blood-biomaterial interface are transient, suggesting that upstream events may influence the downstream platelet-biomaterial interaction [3–6].
When a device such as synthetic small-diameter vascular graft is implanted into the vasculature, there are two anastomotic regions at the junctions of the graft with the native vasculature. It has been shown that vascular grafts can often become stenotic (narrow) due to intimal fibrous hyperplasia, either generally along the inside of the graft or at the anastomotic region [7,8], leading to an increased shear force on flowing platelets. Elevated shear forces have been known to lead to platelet aggregation through a variety of mechanisms [9,10]. Increased shear forces result in a motion of the red blood cells towards the middle of the flow stream, causing physical displacement of plasma and platelets towards the margination zone near the surface of the vessel [11]. Increased shear forces have also been shown to induce intracellular signaling leading to activation [12,13] as well as morphological changes in translocating platelets [14]. Elevated shear forces have also been shown to induce platelet lysis [15], resulting in the release of procoagulant factors.
A number of studies have investigated the effect of increased shear forces introduced by stenosis on platelet adhesion and activation over a wide range from as low as 270 s−1 to as high as 8.74×104 s−1 [10,12,16–21]. However, the majority of these studies investigated the platelet adhesion at the sites of stenosis while the effects of the platelet exposure to increased shear forces and subsequent downstream events were largely ignored. Transient exposure of platelets to increased shear forces is expected to pre-activate (i.e. “prime”) platelets for downstream adhesion and activation. In circulation, platelets primed by transient exposure to increased shear forces flow downstream where they may further interact with implanted biomaterials or damaged blood vessel walls. For example, if a vascular graft material is not perfectly hemocompatible, such upstream priming will increase the probability of platelet adhesion to and full activation on surface platelet binding proteins adsorbed downstream. Therefore, it is important to understand how the platelets primed by a transient passage through an upstream stenotic region respond to imperfect vascular implant surfaces downstream.
To investigate the effect of upstream shear force-dependent platelet priming on downstream adhesion and activation, we have designed experiments in which blood is perfused through a flow chamber with an upstream stenotic region and a downstream “capture” region consisting of one of three platelet binding proteins (fibrinogen, collagen or van Willebrand factor, V) covalently attached to the flow chamber wall. The flow chambers were prepared through relief molding of a silicone elastomer with a narrow upstream region to mimic the vascular graft anastomosis encountered by circulating platelets. Microcontact printing (μCP) was used to covalently attach one platelet binding protein to the surface downstream of the stenotic region. Each of the three platelet binding proteins also served the purpose of investigating which platelet receptors were activated by the upstream shear force. To further characterize the upstream platelet activation, four activation markers (P-selectin, integrin αIIbβ3, lysosomal glycoprotein, and phosphatidylserine) were quantified on platelets perfused through a flow chamber in the absence of a capture region using flow cytometry.
2. Materials and Methods
2.1. Flow chamber design and fabrication
Each flow cell consisted of a parallel array of three flow chambers designed to fit on a standard 25 × 75 mm microscope slide (Figure 1). Each flow chamber contained an upstream stenotic priming region and a downstream capture region. Flow chamber dimensions were 1 mm wide, 0.1 mm in height, and 70 mm long. Stenotic priming regions varied from 5 to 15 mm in length. Width of stenotic priming regions was varied from 0.8 mm to 0.4 mm to create 20 %, 40 % and 60 % stenosis that represented three different wall shear rates (500, 667 and 1000 s−1, respectively). Wall shear rate, γ, was calculated at a constant volumetric flow rate (Q) according to
where h was the flow chamber height and w was the chamber width. The percent of stenosis was defined as where winlet was the width of inlet of the flow chamber and wstenosis was the width of the stenotic regions. Stenotic regions were created 5 mm downstream from the inlet to allow for the development of laminar flow. Control flow chambers did not contain any upstream stenotic region (defined as 0 % stenosis) with a shear rate corresponding to 400s−1. Binding protein capture regions were placed 25 mm downstream from the end of the upstream priming regions.
Figure 1.
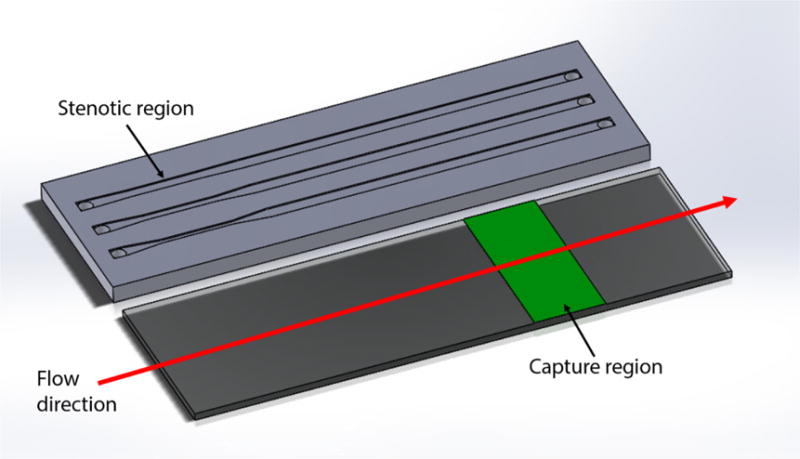
Schematic diagram of a parallel flow assay. The upstream stenotic priming region was created by controlling the width of flow cells. Proteins were immobilized downstream on a reactive glass substrate by μCP to serve as platelet capture regions.
Flow cells were manufactured through the relief molding of polydimethylsiloxane (PDMS) (Sylgard 184, Dow Corning, Midland, MI, USA). Relief for the flow chamber features was created by a polymeric tape that was patterned on a laser cutter (VLS3.60, Universal Laser Systems, Scottsdale, AZ, USA). Patterns were then transferred to the bottom of a mold, and PDMS was cast into the mold at a weight ratio of 15:1 polymer to cross-linker. The cast PDMS was degassed by placing the mold under vacuum for 30 min. The PDMS was allowed to cure, then released from the mold and cut to final dimensions (25 × 75 mm). After release from the mold, fluid vias were bored from the top of the PDMS into the flow cells to provide access for the inlet and outlet of blood.
2.2. Preparation of downstream capture regions
The downstream capture regions (10 mm in length) served to capture primed platelets. Fibrinogen and von Willebrand factor (vWF, Haematologic Technologies, Essex Junction, VT, USA), and collagen (type I, Ibidi, Fitchburg, WI, USA) were each covalently immobilized at 100 % surface coverage in the capture regions by μCP as previously described [22]. PDMS stamps coated with protein was brought into contact with the surface of Nexterion-H slides (Schott, Tempe, AZ, USA) covered with a poly(ethylene oxide)-based polymer containing reactive N-hydroxysuccinimide (NHS) esters, thus providing means for covalent protein immobilization by protein amino groups [23]. The protein coated stamps were left in contact with the Nexterion-H slide surface for 1 hr. After protein immobilization, the capture regions of the slides were rinsed with deionized water and dried under a stream of nitrogen gas.
2.3. Flow chamber assembly
The flow chambers were assembled by inverting the PDMS molded flow chamber onto the stamped Nexterion-H glass slide. Reversible sealing held the PDMS in place strongly enough to prevent leakage for the duration of the experiments. The flow chambers were connected to a syringe pump (Kent Scientific, Torrington, CT, USA) using polytetrafluorethylene tubing (shear rate 39 s−1, PTFE, Cole-Parmer, Vernon Hills, IL, USA,). PTFE tubing was kept as short as possible to minimize any effects it might have on platelet activation. After assembly, a solution of human serum albumin (HSA, 1 mg/mL in PBS, Sigma Aldrich, St. Louis, MO, USA) was perfused through each flow cell and was allowed to incubate at room temperature for 1 hr. Covalently attached HSA served to passivate the unreacted regions of the glass slide, the walls of each flow chamber and PTFE tubing by physical adsorption.
2.4. Flow chamber operation
Fresh whole human blood was drawn from healthy human donors under protocols approved by the University of Utah Institutional Review Board. Blood was drawn into buffered 3.2% (0.105 M) sodium citrate and was treated with Phe-Pro-Arg-chloromethylketone (PPACK, 80 μM, Haematologic Technologies, Essex Junction, VT, USA) within 5 min of draw to prevent thrombin-induced coagulation. Blood samples were kept at 37 °C in a water bath until use. All samples were used within 90 min of draw from donors. Blood samples were incubated with calcein AM metabolic dye (0.1 mg/mL in PBS, Fisher Scientific, Waltham, MA, USA) for 1 hr at 37 °C to fluorescently label the platelets. Blood was drawn through the flow chambers at a constant flow rate of 2.4 mL/hr in all experiments. Flow was sustained for 5 min, after which the flow chambers were rinsed with pre-warmed Tyrode’s buffer (37 °C, pH 7.4) to remove any nonattached cells. Attached cells were fixed in 4 % paraformaldehyde and imaged using a fluorescence microscope (Diaphot 200, Nikon, Tokyo, Japan). Finally, adhered platelet surface coverages were analyzed using ImageJ (NIH, USA).
2.5. Flow cytometry
Flow cytometry was used to measure the expression levels of CD62P (P-selectin), integrin αIIbβ3, CD63 (lysosomal glycoprotein), and phosphatidylserine. Following blood perfusion through an albumin coated flow chamber in the absence of a capture region, a 5 μL aliquot of blood supernatant was collected and incubated for 20 minutes with anti-human CD62P, PAC-1, anti-human CD63, or annexin V (BD Biosciences, San Jose, CA, USA) to label P-selectin, active αIIbβ3, lysosomal glycoprotein, or phosphatidylserine, respectively. Two 5 μL blood aliquots were labeled prior to perfusion. One aliquot was stimulated by the addition of thrombin immediately prior to labeling (c = 0.1 units/mL, EMD Millipore, Billerica, MA, USA) and the other was left unstimulated to serve as positive and negative controls, respectively. A 5 μL aliquot was also labeled with anti-human CD41b (BD Biosciences, San Jose, CA, USA), which binds to the αIIb subunit of integrin αIIbβ3 regardless of the activation state of the receptor, to locate platelets during flow cytometry analysis. Following labeling, platelets were fixed in 1 % paraformaldehyde and stored at 4 °C. Analysis of 100,000 events was conducted on a FACScanto analyzer (BD Biosciences, San Jose, CA, USA).
2.6. Statistical methods
All tests were carried out at least in triplicate. Box and whisker plots are used to depict the distribution of platelet adhesion data as the mean, quartiles, and outliers. Bar plots present the results of platelet activation obtained by the flow cytometry analysis as mean ± standard deviation. Significant differences between the data sets were analyzed using OriginPro9.0 software (OriginLab, Northampton, MA, USA). Statistical significance was established using a paired t-test at p-values less than 0.05. The significance levels are presented as *(p < 0.05), **(p < 0.005), or ***(p < 0.0005).
3. Results and Discussion
Blood, a complex mixture of plasma and cells, circulates through the body in a closed loop carrying out myriad functions. When blood contacts an implanted device, proteins from the blood plasma such as fibrinogen and vWF may adsorb to the surface of the device [24,25]. Circulating platelets can recognize these adsorbed proteins via receptors such as glycoprotein Ib-IX-V complex and integrin αIIbβ3. After initial tethering and rolling, platelets may ultimately form stable adhesive contacts to the device surface and become fully activated. Similarly, in the case of blood vessels with damaged endothelial lining, platelets will interact with collagen present in the exposed subendothelium via their GPVI and integrin α2β1 receptors. Due to these multiple activation pathways, platelet adhesion to vascular device surfaces is the crucial step in thrombus formation that may lead to the temporary or permanent occlusion of blood vessels. We have recently showed that transient platelet contacts with upstream surface-immobilized proteins, acting as agonists, are responsible for increased platelet adhesion and activation downstream [6,26]. In the present study, we investigated how transiently increased shear forces due to upstream stenosis affect downstream platelet adhesion and activation. Fibrinogen, collagen and vWF were chosen for downstream capture regions since they are the primary adhesive ligands for platelet receptors. These surface immobilized proteins can mimic the procoagulant surface of an implanted vascular device or exposed subendothelium.
The effect of increased shear forces due to upstream stenosis on the downstream platelet adhesion was compared with the control conditions (no upstream stenotic region, shear rate of 400 s−1). Figure 2 shows the platelet surface coverage in the three downstream capture regions as a function of the shear rate in the upstream stenotic region. Figure 3 shows representative fluorescence images of adhered platelets on all three platelet binding proteins after passing through stenotic regions. In the case of capture by fibrinogen, platelet adhesion was significantly higher than the control in all three flow chambers with an upstream stenotic priming region. For capture on collagen, platelet adhesion was significantly higher than control only for the flow chambers with upstream shear rates of 667 and 1000 s−1. Similarly, for capture on vWF, platelet adhesion was only significantly higher for the flow chambers with upstream shear rates of 667 and 1000 s−1. Several studies have suggested that the interactions of surface-immobilized vWF with platelet receptor GPIb occur selectively at high shear rates (> 1000 s−1) [27,28]. In the present study, an increasing number of platelets attached to vWF capture region after being exposed to shear rates lower than 1000 s−1. However, overall platelet surface coverage on vWF was much lower than platelet adhesion to fibrinogen or collagen. At present shear rates (≤ 1000 s−1), platelet deposition on surface-immobilized vWF might have occurred independently of vWF-GPIb interaction, possibly due to interaction between vWF and αIIbβ3 integrin which is a receptor for both vWF and fibrinogen.
Figure 2.
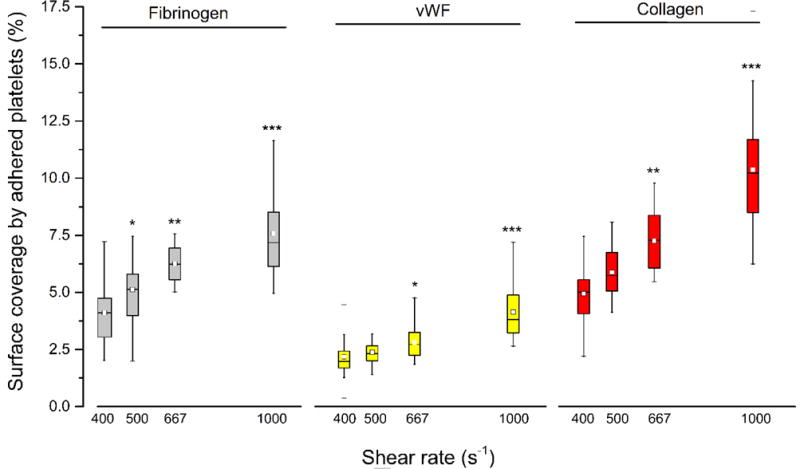
Platelet adhesion to three downstream capture proteins as a function of shear forces in the upstream stenosis compared with control sample containing no stenotic region (shear rate of 400 s−1). Statistical significance for each downstream protein was obtained using paired t-test (n = 30, *p < 0.05, **p < 0.005 and ***p < 0.0005).
Figure 3.
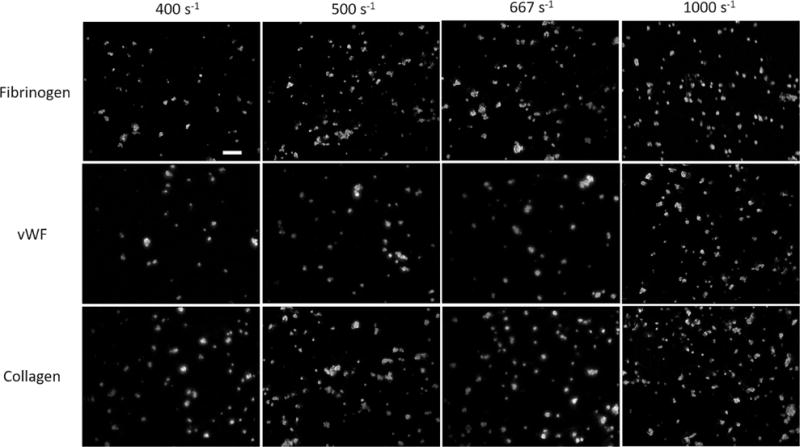
Representative fluorescence images showing platelet adhered to the three downstream proteins after passing through the upstream stenotic region or the chamber containing no stenotic region (shear rate of 400 s−1). Scale bar represents 20 μm.
For all three binding proteins, platelet adhesion to the downstream capture region increased as the shear rate in the upstream priming region was increased, suggesting that transient exposure to higher shear forces is capable of elevating the priming of platelets for downstream adhesion. Overall platelet adhesion was found to be significantly higher when circulating platelets were exposed to upstream shear rates of 667 and 1000 s−1.
We simulated flow in the four perfusion chambers using COMSOL Multiphysics (COMSOL, Inc., Burlington, MA, USA) to examine the impact of upstream stenosis on blood flow. We utilized continuity and Navier-Stokes equations to derive the velocity profiles (Figure 4). The computational results indicated that the blood flow velocity is highly affected by upstream narrowing, especially for 40 % and 60 % stenosis. The flow chambers with upstream stenosis exhibited steeper velocity gradients towards the walls and consequently higher wall shear rates. Increasing wall shear rates have been shown to increase the number of red blood cells in the central portion of flowing blood and thus promote platelet margination towards the vessel wall [29,30]. Such enhanced platelet margination at higher shear rates acts as an important priming factor for platelet adhesion to surface-immobilized binding proteins downstream.
Figure 4.
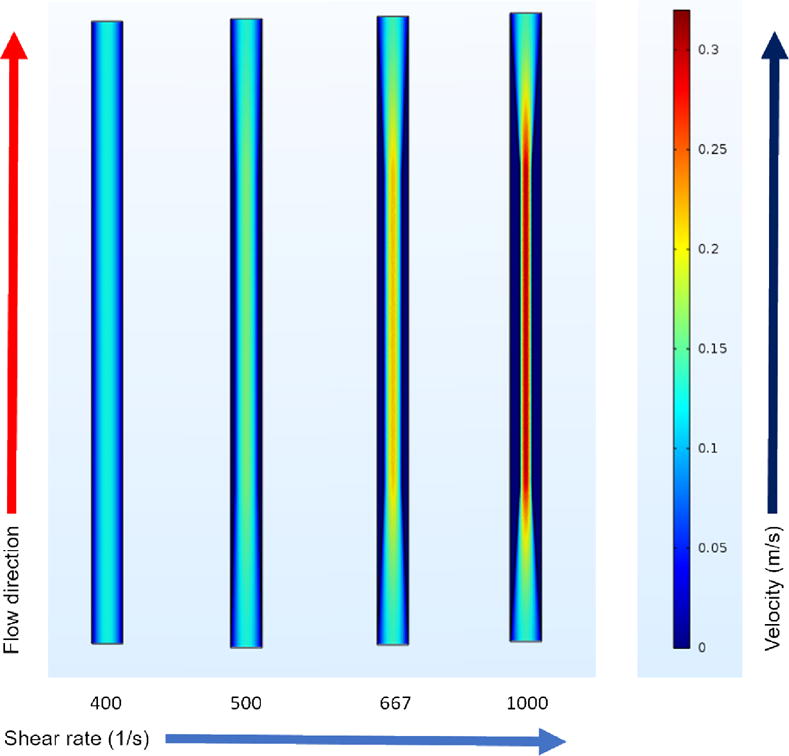
Velocity profiles in four different flow chambers. The first 20 mm of each experimental flow chamber was simulated using COMSOL Multiphysics to visualize the velocity profiles in the anastomotic regions. The profiles show the velocity cross-section at the middle height of the channel (at the depth of 0.05 mm).
Elevated shear forces may activate platelets via intracellular signaling cascades or cause platelet lysis. For shear-induced intracellular signaling, elevated shear forces have been shown to initially promote the interaction of plasma vWF with the GPIb-IX-V receptor complex [31]. This interaction starts an intercellular signaling cascade characterized by Ca2+ mobilization [32,33], actin polymerization and cytoskeletal rearrangements [13], protein kinase C expression [34], and αIIbβ3 expression/activation [33,35]. These events are coupled with the further release of soluble vWF from platelet α-granules. Surface-immobilized platelet binding proteins work synergistically with plasma proteins to form stable adhesive contacts [27]. Mechanical damage due to increased shear forces on the platelets can cause platelet lysis [15], resulting in the release of procoagulant factors and subsequent activation of surrounding platelets.
To investigate the effect of the time duration of platelet exposure to increased shear forces, we investigated how three different lengths (5, 10 and 15 mm) of upstream stenotic regions influence the downstream adhesion. Both lower and higher shear rates (500 and 1000 s−1) were used. Figure 5 shows that the extent of platelet adhesion downstream correlated with both the magnitude and duration of upstream shear forces. At the lower shear rate of 500 s−1, even though average platelet adhesion to all three binding proteins increased for the 10 mm long stenotic region compared to the 5 mm length, the increase was not statistically significant (p > 0.05). For the 15 mm stenotic length, platelet adhesion to fibrinogen and vWF at 500 s−1 was significantly higher than for the 5 mm length, but the increases were not significant when compared to the 10 mm length (p > 0.05). For collagen, platelet adhesion for the 15 mm stenotic length was significantly higher compared to both lengths of 5 and 10 mm (Fig 5A).
Figure 5.
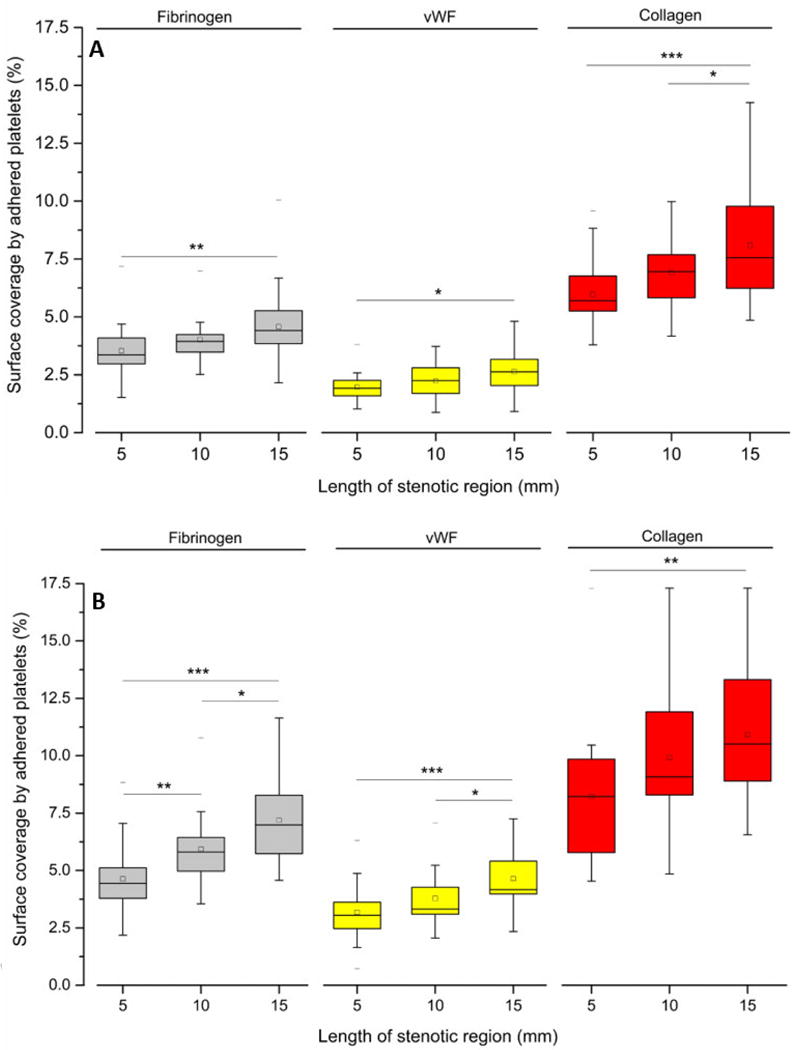
Platelet adhesion to three downstream capture proteins at upstream shear rates of (A) 500 s−1 and (B) 1000 s−1 for three different lengths of stenotic regions. Statistical significance was obtained using paired t-test (n = 30, *p < 0.05, **p < 0.005 and ***p < 0.0005).
At the higher shear rate of 1000 s−1, the increase of platelet adhesion to fibrinogen was significantly higher at longer stenotic lengths. For vWF, the platelet adhesion increase was less than that of the other two binding proteins. Platelet adhesion to vWF was significantly higher for the 15 mm stenotic length compared to each of the two shorter stenotic lengths. For collagen capture, platelet adhesion at 1000 s−1 was significantly higher for the 15 mm stenotic length only when compared with the 5 mm stenotic region. Overall, these findings indicated that besides the magnitude and duration of applied upstream shear force, a threshold level of platelet priming also depends on the type of capture protein.
Using these flow experiments we found that upstream stenosis increases platelet adhesion downstream in a shear rate-proportional manner. To further differentiate the effect of upstream shear forces on platelet activation, the platelets that passed through stenotic regions were characterized using flow cytometry. Four activation markers (P-selectin, integrin αIIbβ3, lysosomal glycoprotein, and phosphatidylserine) were quantified after anticoagulated whole blood was perfused through four albumin-coated flow chambers with no capture regions. The percent of expression events out of 100,000 for each marker was recorded and compared to blood with no prior perfusion (negative control) and to blood with no prior perfusion after thrombin stimulation (positive control). Expression levels of P-selectin, integrin αIIbβ3, lysosomal glycoprotein, and phosphatidylserine are shown in Figures 6 and 7.
Figure 6.
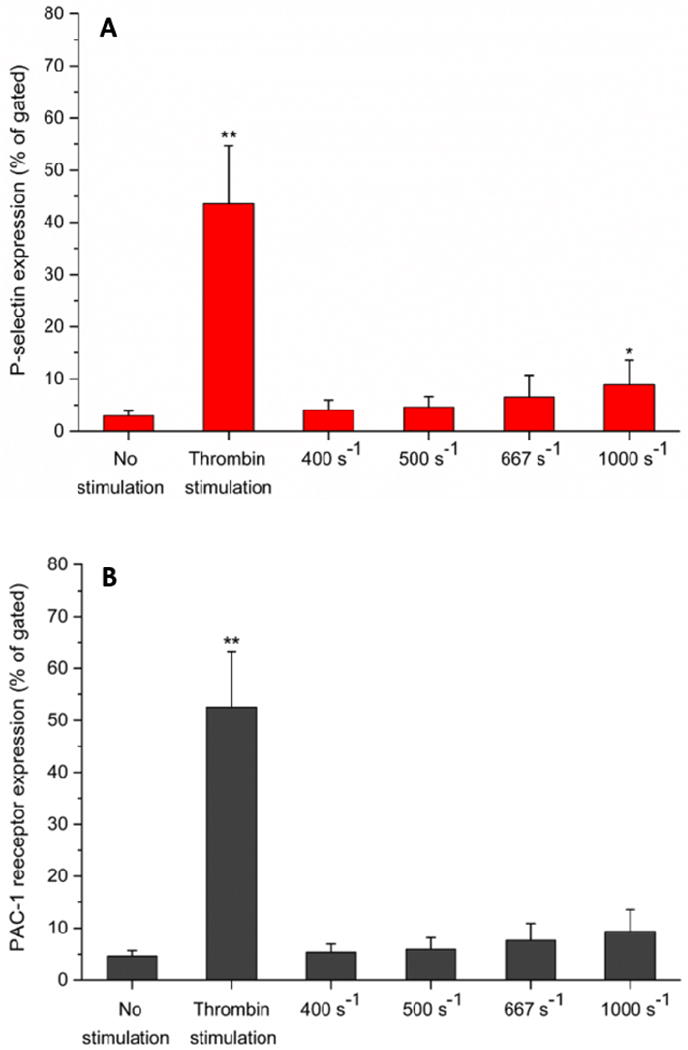
Flow cytometry analysis of platelet activation. Expression levels of (A) P-selectin and (B) PAC-1 receptor (integrin αIIbβ3) in perfused blood samples were compared with unstimulated (negative control) and thrombin stimulated (positive control) samples collected prior to perfusion. Analysis of 100,000 events for each sample was conducted, and events of platelets expressing each marker were recorded. Statistical significance was obtained using paired t-test (n = 3, *p < 0.05 and **p < 0.005 relative to “no stimulation” control).
Figure 7.
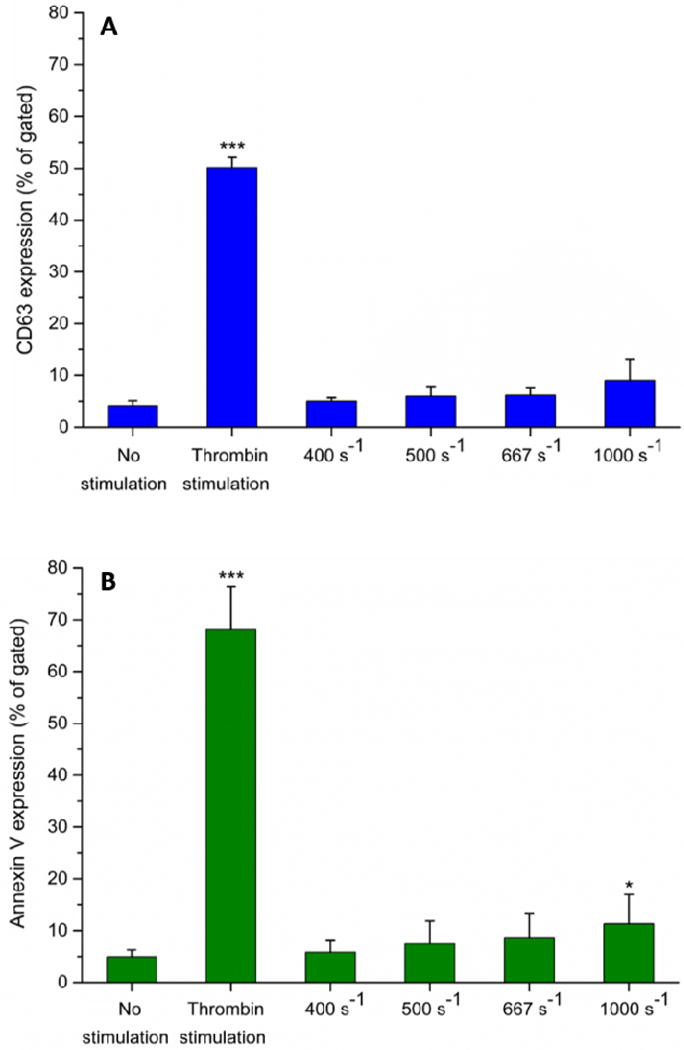
Flow cytometry analysis of platelet activation. Expression levels of (A) CD63 (lysosomal glycoprotein) and (B) phosphatidylserine (via annexin V binding) in perfused blood samples were compared with unstimulated (negative control) and thrombin stimulated (positive control) samples collected prior to perfusion. Analysis of 100,000 events for each sample was conducted, and events of platelets expressing each marker were recorded. Statistical significance was obtained using paired t-test (n = 3, *p < 0.05 and ***p < 0.0005 relative to “no stimulation” control).
It is unlikely that each shear-induced platelet priming event will end up in an adhesive event downstream. In quiescent platelets, P-selectin is located on the inner membranes of platelet granules. Following platelet activation, P-selectin is translocated from the intracellular granules to the outer membrane. It was found that P-selectin was expressed at higher levels on platelets perfused through upstream stenotic region (Fig 6A), however, a statistically different level of expression was only found at higher shear rates (1000 s−1). PAC-1 recognizes an epitope on the integrin αIIbβ3 complex of activated platelets. Like in the case of P-selectin, integrin αIIbβ3 was found to be expressed at higher levels on platelets perfused over flow cells containing upstream stenotic region but the increases were not statistically different from the negative control (Fig 6B).
In quiescent platelets, lysosomal glycoprotein CD63 is located on the membranes of dense granules and lysosomes, but relocates to the plasma membrane following platelet activation [36]. Up to 667 s−1, there was no distinguishable change in expression of CD63 compared with negative control (Figure 7A). Expression of CD63 was found higher on platelets perfused through the flow cells with the higher shear rate of 1000 s−1. Phosphatidylserine regulates the production of thrombin by enhancing the rate of activation of prothrombin to thrombin. Following platelet activation, phosphatidylserine is translocated from the cytoplasmic face of the quiescent platelet membrane to the plasma-oriented surface of discrete membrane vesicles [37]. The flow cytometry showed that expression of phosphatidylserine was higher on platelets perfused through flow cells containing upstream stenotic regions (Figure 7B). Overall, the flow cytometry results suggested that upstream increased shear forces were capable of activating platelets in a manner that could affect thrombus formation downstream. However, the increase in the expression for all four markers was significantly lower compared to thrombin stimulated positive controls.
Shear rates of 400 – 1000 s−1 used in this study were average wall shear rates, and were not uniformly distributed. In a near-wall region, there was a sharp velocity gradient compared to the flow regions far away from the wall (Figure 4). Only a fraction of platelets were exposed to near-wall regions; however, more platelets migrated toward near-wall regions upon entrance to a stenotic region. It was possible that some platelets entered a near-wall region transiently and then returned to a low shear rate region, and vice versa. We used flow cytometry to take into account both high and low shear rate flow regions. The results suggested that many more platelets were primed by increased shear rates than adhered onto downstream capture regions. Shankaran et al. [38] observed that a threshold shear stress of 80 dyne/cm2 (shear rate, approximately 2200 s−1) is necessary to induce significant platelet activation in whole blood. We observed that even moderately increased shear rates primed platelets for enhanced downstream activation. For example, a shear rate of 1000 s−1 significantly increased the activation of P-selectin and phosphatidylserine exposure (Figs 6,7). The findings support the observations reported in previous studies. Lee et al. [39] showed that platelets induced by shear rates less than 1000 s−1 tended to aggregate to a collagen-coated surface; however, maximum platelet aggregation was observed at a shear rate of 3100 s−1. Song et al. [40] showed that even 900 s−1 induced platelets for enhanced activation of CD62 (P-selectin). In these two studies [39,40] the shear forces were induced using a rotating stirrer in a micro-chamber where platelets were continuously sheared for many seconds.
Vascular implants are widely used to replace or repair damaged or occluded vasculature. Anastomotic regions of vascular implants are often characterized by higher rates of stenosis – and accordingly higher shear forces – and may also disrupt the subendothelium layer of the vasculature. While the majority of past studies involving stenosis investigated platelet aggregation at higher shear rates (> 1000 s−1) [10,17,41], the present study was focused on the effect of increased upstream shear forces on priming platelets for downstream adhesion on protein coated substrates. If not perfectly passivated against plasma protein adsorption, surfaces of vascular implants may become procoagulant and provide platelets with potential contact with platelet binding proteins adsorbed from blood plasma. A combination of higher upstream shear forces and adsorbed platelet binding proteins presents an ideal environment for platelet adhesion and activation that may have serious consequences for the blood compatibility of vascular implants. We tested such combinations of higher upstream shear rates and downstream surface-bound platelet binding proteins to better understand their role in vascular device failure. Although our study was limited to shear rates of up to 1000 s−1, the results showed that the upstream increase of the shear rates resulted in a higher platelet adhesion to downstream immobilized proteins. We have also found that an increased duration of platelet exposure to elevated shear rates also led to an increase of downstream platelet adhesion. It remains to be further investigated how even higher shear forces (> 1000 s−1), typically found in severe stenosis, prime platelets for downstream adhesion and activation.
4. Conclusions
In this study, flow chambers were designed to mimic the physical environment of vasculature with an upstream vascular-graft anastomosis and a downstream exposed subendothelium or a procoagulant surface of an implanted vascular device. The results indicated that platelets primed by the transient exposure to increased shear force adhered downstream to the surface-immobilized proteins in a shear rate proportional manner. In addition to the magnitude of upstream shear forces, downstream platelet adhesion was also dependent on the duration of exposure to these forces. The effect of increased shear forces on platelet activation was quantified by measuring the expression levels of P-selectin, integrin αIIbβ3, lysosomal glycoprotein, and phosphatidylserine exposure. Expression of each of these four markers increased after perfusion through the flow chambers containing an upstream stenotic region. Since the anastomotic regions are often characterized by both high shear forces as well as by damage of vessel endothelium, we expect that the results of the present study could help in reevaluating vascular device designs to minimize downstream effects of elevated shear forces.
Statement of significance.
A synthetic small-diameter vascular graft can often become stenotic due to intimal fibrous hyperplasia, either generally along the inside of the graft or at the anastomotic regions, leading to an increased shear force on flowing platelets. Our lab is studying how the upstream platelet pre-activation (aka “priming”) in flowing blood affects their downstream adhesion and activation. This manuscript describes a study in which priming of platelets is achieved by upstream stenotic narrowing in a microfluidics flow chamber. Such experimental design was intended to mimic a vascular implant with stenotic upstream anastomosis and downstream exposed platelet protein agonists. Understanding how the pre-activated platelets respond to imperfect vascular implant surfaces downstream is an important factor in designing better vascular implants.
Acknowledgments
This work was supported by the National Institutes of Health (R01 HL126864). The authors would like to thank Dr. A. Fogelson for valuable discussions and Dr. A. Weyrich’s lab for providing human blood necessary to complete the experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Bertrand ME, Legrand V, Boland J, Fleck E, Bonnier J, Emmanuelson H, Vrolix M, Missault L, Chierchia S, Casaccia M, Niccoli L, Oto A, White C, Webb-Peploe M, McFadden EP. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. Circulation. 1998;98:1597–1603. doi: 10.1161/01.cir.98.16.1597. [DOI] [PubMed] [Google Scholar]
- 2.Jackson MR, Johnson WC, Williford WO, Valentine RJ, Clagett GP. The effect of anticoagulation therapy and graft selection on the ischemic consequences of femoropopliteal bypass graft occlusion: results from a multicenter randomized clinical trial. J Vasc Surg. 2002;35:292–298. doi: 10.1067/mva.2002.120383. [DOI] [PubMed] [Google Scholar]
- 3.Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand Factor. Cell. 1996;84:289–297. doi: 10.1016/s0092-8674(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 4.Moroi M, Jung SM, Shinmyozu K, Tomiyama Y, Ordinas A, Diaz-Ricart M. Analysis of platelet adhesion to a collagen-coated surface under flow conditions: the involvement of glycoprotein VI in the platelet adhesion. Blood. 1996;88:2081–2092. [PubMed] [Google Scholar]
- 5.Godo MN, Sefton MV. Characterization of transient platelet contacts on a polyvinyl alcohol hydrogel by video microscopy. Biomaterials. 1999;20:1117–1126. doi: 10.1016/s0142-9612(99)00012-5. [DOI] [PubMed] [Google Scholar]
- 6.Corum LE, Hlady V. The effect of upstream platelet–fibrinogen interactions on downstream adhesion and activation. Biomaterials. 2012;33:1255–1260. doi: 10.1016/j.biomaterials.2011.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies AH, Magee TR, Baird RN, Sheffield E, Horrocks M. Vein compliance: a preoperative indicator of vein morphology and of veins at risk of vascular graft stenosis. Br J Surg. 1992;79:1019–1021. doi: 10.1002/bjs.1800791011. [DOI] [PubMed] [Google Scholar]
- 8.Wilson YG, Davies AH, Currie IC, Morgan M. Vein graft stenosis: incidence and intervention. Eur J Vasc Endovasc Surg. 1996;11:164–169. doi: 10.1016/s1078-5884(96)80046-3. [DOI] [PubMed] [Google Scholar]
- 9.Miyazaki Y, Nomura S, Miyake T, Kagawa H, Kitada C, Taniguchi H, Komiyama Y, Fujimura Y, Ikeda Y, Fukuhara S. High shear stress can initiate both platelet aggregation and shedding of procoagulant containing microparticles. Blood. 1996;88:3456–3464. [PubMed] [Google Scholar]
- 10.Nesbitt WS, Westein E, Tovar FJ. A shear gradient–dependent platelet aggregation mechanism drives thrombus formation. Nat Med. 2009;15:665–673. doi: 10.1038/nm.1955. [DOI] [PubMed] [Google Scholar]
- 11.Bark DL, Ku DN. Platelet transport rates and binding kinetics at high shear over a thrombus. Biophys J. 2013;105:502–511. doi: 10.1016/j.bpj.2013.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holme PA, Orvim U, Hamers MJ, Solum NO, Brosstad FR, Barstad RM, Sakariassen KS. Shear-induced platelet activation and platelet microparticle formation at blood flow conditions as in arteries with a severe stenosis. Arterioscler Thromb Vasc Biol. 1997;17:646–653. doi: 10.1161/01.atv.17.4.646. [DOI] [PubMed] [Google Scholar]
- 13.Yuan Y, Kulkarni S, Ulsemer P, Cranmer SL. The von Willebrand factor-glycoprotein Ib/V/IX interaction induces actin polymerization and cytoskeletal reorganization in rolling platelets and glycoprotein Ib/V/IX-transfected cells. J Biol Chem. 1999;274:36241–36251. doi: 10.1074/jbc.274.51.36241. [DOI] [PubMed] [Google Scholar]
- 14.Maxwell MJ, Dopheide SM, Turner SJ, Jackson SP. Shear induces a unique series of morphological changes in translocating platelets: effects of morphology on translocation dynamics. Arterioscler Thromb Vasc Biol. 2006;26:663–669. doi: 10.1161/01.ATV.0000201931.16535.e1. [DOI] [PubMed] [Google Scholar]
- 15.Anderson GH, Hellums JD, Moake JL, Alfrey CP. Platelet lysis and aggregation in shear fields. Blood Cells. 1977;4:499–511. [PubMed] [Google Scholar]
- 16.Grabowski EF, Rodriguez M. Platelet adhesion/aggregation in an in vitro model of coronary artery stenosis. Cathet Cardiovasc Diagn. 1993;28:65–71. doi: 10.1002/ccd.1810280113. [DOI] [PubMed] [Google Scholar]
- 17.Li M, Ku DN, Forest CR. Microfluidic system for simultaneous optical measurement of platelet aggregation at multiple shear rates in whole blood. Lab Chip. 2012;12:1355–1362. doi: 10.1039/c2lc21145a. [DOI] [PubMed] [Google Scholar]
- 18.Ha H, Lee SJ. Hemodynamic features and platelet aggregation in a stenosed microchannel. Microvasc Res. 2013;90:96–105. doi: 10.1016/j.mvr.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Westein E, van der Meer AD, Kuijpers MJE, Frimat JP, van den Berg A, Heemskerk JWM. Atherosclerotic geometries exacerbate pathological thrombus formation poststenosis in a von Willebrand factor-dependent manner. Proc Natl Acad Sci. 2013;110:1357–1362. doi: 10.1073/pnas.1209905110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fallah MA, Huck V, Niemeyer V, Desch A, Angerer JI, McKinnon TAJ, Wixforth A, Schneider SW, Schneider MF. Circulating but not immobilized N-deglycosylated von Willebrand factor increases platelet adhesion under flow conditions. Biomicrofluidics. 2013;7:044124. doi: 10.1063/1.4819746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jain A, Graveline A, Waterhouse A, Vernet A, Flaumenhaft R, Ingber D. A shear gradient-activated microfluidic device for automated monitoring of whole blood hemostasis and platelet function. Nat Commun. 2016;7:10176. doi: 10.1038/ncomms10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corum LE, Eichinger CD, Hsiao TW, Hlady V. Using microcontact printing of fibrinogen to control surface-induced platelet adhesion and activation. Langmuir. 2011;27:8316–8322. doi: 10.1021/la201064d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi H, Emoto K, Dubey M, Castner DG, Grainger DW. Imaging surface immobilization chemistry: correlation with cell patterning on non‐adhesive hydrogel thin films. Adv Funct Mater. 2008;18:2079–2088. doi: 10.1002/adfm.200800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vroman L, Adams AL. Identification of rapid changes at plasma-solid interfaces. J Biomed Mater Res. 1969;3:43–67. doi: 10.1002/jbm.820030106. [DOI] [PubMed] [Google Scholar]
- 25.Andrade JD, Hlady V. Protein adsorption and materials biocompatibility: a tutorial review and suggested hypotheses. Adv Polym Sci. 1986;79:1–63. [Google Scholar]
- 26.Eichinger CD, Hlady V. Binary agonist surface patterns prime platelets for downstream adhesion in flowing whole blood. Biointerphases. 2017;12:02C406. doi: 10.1116/1.4982596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate–receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–666. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 28.Sugimoto M, Tsuji S, Kuwahara M, Matsui H. Shear-dependent functions of the interaction between soluble von Willebrand factor and platelet glycoprotein Ib in mural thrombus formation on a collagen surface. Int J Hematol. 1999;69:48–53. [PubMed] [Google Scholar]
- 29.Aarts P, van den Broek S. Blood platelets are concentrated near the wall and red blood cells, in the center in flowing blood. Arterioscler Thromb Vasc Biol. 1988;8:819–824. doi: 10.1161/01.atv.8.6.819. [DOI] [PubMed] [Google Scholar]
- 30.Zhao R, Kameneva MV, Antaki JF. Investigation of platelet margination phenomena at elevated shear stress. Biorheology. 2007;44:161–177. [PubMed] [Google Scholar]
- 31.Deng W, Xu Y, Chen W, Paul DS, Syed AK, Dragovich MA, Liang X, Zakas P, Berndt MC, Di Paola J, Ware J, Lanza F, Doering CB, Bergmeier W, Zhang XF, Li R. Platelet clearance via shear-induced unfolding of a membrane mechanoreceptor. Nat Commun. 2016;7:12863. doi: 10.1038/ncomms12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nesbitt WS, Kulkarni S, Giuliano S, Goncalves I, Dopheide SM, Yap CL, Salem HH, Jackson SP. Distinct glycoprotein Ib/V/IX and integrin IIb 3-dependent calcium signals cooperatively regulate platelet adhesion under flow. J Biol Chem. 2001;277:2965–1972. doi: 10.1074/jbc.M110070200. [DOI] [PubMed] [Google Scholar]
- 33.Kasirer-Friede A, Cozzi MR, Mazzucato M, Marco LD, Ruggeri ZM, Shattil SJ. Signaling through GP Ib-IX-V activates αIIbβ3 independently of other receptors. Blood. 2004;103:3403–3411. doi: 10.1182/blood-2003-10-3664. [DOI] [PubMed] [Google Scholar]
- 34.Kroll MH, Hellums JD, Guo Z, Durante W, Razdan K, Hrbolich JK, Schafer AI. Protein kinase C is activated in platelets subjected to pathological shear stress. J Biol Chem. 1993;268:3520–3524. [PubMed] [Google Scholar]
- 35.Nesbitt WS, Giuliano S, Kulkarni S. Intercellular calcium communication regulates platelet aggregation and thrombus growth. J Cell Biol. 2003;160:1151–1161. doi: 10.1083/jcb.200207119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Israels SJ, McMillan-Ward EM. CD63 modulates spreading and tyrosine phosphorylation of platelets on immobilized fibrinogen. Thromb Haemost. 2005;93:311–318. doi: 10.1160/TH04-08-0503. [DOI] [PubMed] [Google Scholar]
- 37.Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res. 2003;42:423–438. doi: 10.1016/s0163-7827(03)00025-0. [DOI] [PubMed] [Google Scholar]
- 38.Shankaran H, Alexandridis P, Neelamegham S. Aspects of hydrodynamic shear regulating shear-induced platelet activation and self-association of von Willebrand factor in suspension. Blood. 2003;101:2637–2645. doi: 10.1182/blood-2002-05-1550. [DOI] [PubMed] [Google Scholar]
- 39.Lee H, Kim G, Lim C, Lee B, Shin S. A simple method for activating the platelets used in microfluidic platelet aggregation tests: stirring-induced platelet activation. Biomicrofluidics. 2016;10:064118. doi: 10.1063/1.4972077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song SH, Lim CS, Shin S. Migration distance-based platelet function analysis in a microfluidic system. Biomicrofluidics. 2013;7:064101. doi: 10.1063/1.4829095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colace TV, Diamond SL. Direct observation of von Willebrand factor elongation and fiber formation on collagen during acute whole blood exposure to pathological flow. Arterioscler Thromb Vasc Biol. 2012;33:105–113. doi: 10.1161/ATVBAHA.112.300522. [DOI] [PMC free article] [PubMed] [Google Scholar]