

Abstract
Background
Massively parallel, or next-generation, sequencing is a powerful technique for the assessment of somatic genomic alterations in cancer samples. Numerous gene targets can be interrogated simultaneously with a high degree of sensitivity. The clinical standard of care for many advanced solid and hematologic malignancies currently requires mutation analysis of several genes in the front-line setting, making focused next generation sequencing (NGS) assays an effective tool for clinical molecular diagnostic laboratories.
Methods
We have utilized an integrated microfluidics circuit (IFC) technology for multiplex PCR-based library preparation coupled with a bioinformatic method designed to enhance indel detection. A parallel low input PCR-based library preparation method was developed for challenging specimens with low DNA yield. Computational data filters were written to optimize analytic sensitivity and specificity for clinically relevant variants.
Results
Minimum sequencing coverage and precision of variant calls were the two primary criteria used to establish minimum DNA mass input onto the IFC. Wet-bench and bioinformatics protocols were modified based on data from the optimization and familiarization process to improve assay performance. The NGS platform was then clinically validated for single nucleotide and indel (up to 93 base pair) variant detection with overall analytic accuracy of 98% (97% sensitivity; 100% specificity) using as little as 3 ng of formalin-fixed, paraffin-embedded DNA or 0.3 ng of unfixed DNA.
Conclusions
We created a targeted clinical NGS assay for common solid and hematologic cancers with high sensitivity, high specificity, and the flexibility to test very limited tissue samples often encountered in routine clinical practice.
Keywords: Molecular sequence data, molecular targeted therapy, medical oncology, pathology, computational biology
Introduction
Knowledge of somatic genomic alterations in human cancers has accumulated rapidly with the advent of high-throughput technologies including massively parallel (next generation) sequencing (NGS). Characterization of these alterations through international consortia has demonstrated the breadth and complexity of mutational patterns within anatomically defined tumor types (1,2) and further solidified the importance of frequently occurring alterations within key biologic pathways (3). Coordinately, the number of clinically useful predictive and/or prognostic associations with specific genomic alterations has increased, shifting the clinical diagnostic paradigm away from single gene/single alteration testing towards multiplexed approaches.
Clinically effective NGS assays should be able detect mutations that impact patient management (4,5) with a high degree of analytic accuracy (6). These diagnostic procedures must be cost-effective, timely, and applicable to the range of normal specimen types routinely collected to be clinically effective (7). These considerations create challenges to the successful development and implementation of CLIA-validated clinical NGS tests for cancer diagnostics.
Selection of (I) instrumentation, (II) library preparation methods, and (III) bioinformatics software critically define the performance characteristics of a NGS-based clinical test that determine analytic sensitivity and specificity, determine minimum nucleic acid input requirements, turn-around time (TAT), and cost. Optimization of these parameters is necessary to successfully analyze a broad range of clinical samples. A significant challenge in NGS analysis of cancer is biologic heterogeneity due to the mixture of neoplastic and non-neoplastic cell types as well as possible subclonal alterations within the neoplastic cell population. Therefore, NGS assays for diagnostic oncology must have sufficient limit of detection (LOD) sensitivity to identify mutations at low variant allele fractions (VAFs) with a high level of specificity that distinguishes from artefactual false positive calls (6,8).
To meet these challenges, we have optimized and validated a targeted NGS assay focused on clinically actionable alterations in both solid and hematologic malignancies using a microfluidics PCR-based library preparation method. Previously (9) we have described an NGS data processing method with enhanced capability to detect insertion-deletion (indel) mutations. We developed additional bioinformatics strategies on top of this core pipeline coupled with modified wet-bench procedures to optimize coverage, data processing time, and analytic accuracy for mutations with low VAF. A low DNA input PCR-based library preparation method was also developed for a subset of the genes that is capable of accurately analyzing samples with very low DNA concentrations (0.1–1 ng/µL) to extend the range of limited-tissue samples we could accurately test. These design features created a NGS platform with robust analytic performance and the flexibility to test the majority of submitted clinical samples for actionable alterations.
Methods
Sample preparation
Formalin fixed, paraffin embedded (FFPE) tissue samples are macro-enriched as necessary to a minimum of 15–20% estimated tumor cellularity from 10-micron unstained sections (5–10 sections depending on tumor area) using a sterile scalpel technique with a guide hematoxylin and eosin stained slide marked up by a board certified anatomic pathologist. DNA is extracted from FFPE samples using the QIAmp DNA FFPE tissue kit per manufacturer’s instructions (Qiagen, Hilden, Germany). Extraction of unfixed samples is performed with the DNeasy blood and tissue kit [peripheral blood (PB) and bone marrow (BM)] or the AllPrep DNA/RNA mini kit [fine needle aspiration (FNA) samples] per manufacturer’s instructions (Qiagen, Hilden, Germany). DNA is quantitated using Qubit fluorometric analysis (ThermoFisher, Waltham, MA, USA). A positive control DNA sample was created by pooling DNA extracts from 5 independent cell lines at equal m/m ratios: CRL-2868 (EGFR p. E746_A750del), CCL-228 (KRAS p.G12V), WC00048 (BRAF p.V600E), CRL-9591 (FLT3 p. p.D600_L601ins10), and GM06141 (JAK2 p.V617F). CRL-5908 (positive for EGFR p.T790M and p.L858R) was used independently. Orthogonal data for KRAS codons 12/13 (Sanger sequencing), BRAF V600E (c.1799 allele-specific qPCR), FLT3-ITD (PCR sizing via capillary electrophoresis) and JAK2 V617F (allele-specific PCR with capillary electrophoresis) was generated from internal CLIA-validated assays. Orthogonal confirmation for additional mutations was obtained from send-outs to other clinical laboratories.
Assay design and Fluidigm access array [integrated microfluidics circuit (IFC)] library preparation
A target bed file was generated by a committee of anatomic and clinical pathologists at our institution based on review (in 2014) of professional guidelines (including National Comprehensive Cancer Network, American Society for Clinical Oncology, College of American Pathologists, Association for Molecular Pathology), published literature, and clinical trial information (clinicaltrials.gov). Primer design, assay multiplexing, and synthesis was performed by Fluidigm Inc. (South San Francisco, CA, USA). The design consists of 92 amplicons across regions of 21 genes (http://atm.amegroups.com/public/system/atm/supp-atm.2018.05.07-1.pdf), covering ~13.6 kb of genomic sequence with an average amplicon length of 191 bp. Sequencing libraries are prepared on the Access Array Integrated Fluidics Circuit system using IFC control modules and the Biomark instrument per manufacturer’s multiplex amplicon tagging protocols (Fluidigm, South San Francicsco, CA, USA) with several modifications. Initial experiments were performed with 1 µL sample volume per protocol specifications; subsequent experiments and validation was performed with 2 µL inputs for the sample pre-mix while maintaining a 4 µL input onto the IFC. Initial experiments utilized a single reaction inlet per sample; subsequent experiments utilized two IFC reaction inlets per sample from which harvest products were pooled together for the subsequent PCR barcoding reaction as a single final sample for sequencing. For validation experiments, 15 ng/µL minimum fixed DNA concentrations and 10 ng/µL unfixed DNA concentrations were used.
Low input PCR library preparation
The low input method consists of a 28 amplicon subset of the full assay covering 4.2 kb of sequence across six genes (Table S1). These amplicons were separated into four multiplex reactions with final individual primer concentrations of 30 nM. The PCR master mix (Roche, Basel, Switzerland) used is the same as for the IFC library method described above; sample input volume is 3 µL at minimum concentrations of 0.1 or 1 ng/µL for unfixed and formalin fixed samples respectively into a final PCR reaction volume of 15 µL. The PCR cycling parameters are identical to those used for the IFC protocol, except for removal of a 20’ preliminary thermal mixing step which is specific to the microfluidics chip. Following target specific amplicon generation, the four individual reactions per sample are pooled volumetrically into the same barcoding PCR workflow with IFC library samples.
Table S1. Off chip amplicon bed file.
| Chromosome | Start | Stop | Name |
|---|---|---|---|
| chr1 | 115256420 | 115256612 | NRAS_3_3 |
| chr1 | 115256461 | 115256647 | NRAS_3_2 |
| chr1 | 115258655 | 115258842 | NRAS_2_2 |
| chr11 | 533799 | 533998 | HRAS_3_2 |
| chr11 | 534178 | 534369 | HRAS_2_2 |
| chr12 | 25380197 | 25380388 | KRAS_3_2 |
| chr12 | 25398166 | 25398346 | KRAS_2_2 |
| chr3 | 178916767 | 178916937 | PIK3CA_targeted_2_1 |
| chr3 | 178916833 | 178917032 | PIK3CA_targeted_2_2 |
| chr3 | 178921353 | 178921552 | PIK3CA_targeted_5_1 |
| chr3 | 178921429 | 178921599 | PIK3CA_targeted_5_2 |
| chr3 | 178921528 | 178921706 | PIK3CA_targeted_5_3 |
| chr3 | 178928151 | 178928345 | PIK3CA_9_1 |
| chr3 | 178928256 | 178928426 | PIK3CA_9_2 |
| chr3 | 178935915 | 178936068 | PIK3CA_10_1 |
| chr3 | 178936035 | 178936196 | PIK3CA_10_4 |
| chr3 | 178947897 | 178948091 | PIK3CA_20_1 |
| chr3 | 178948024 | 178948222 | PIK3CA_20_2 |
| chr3 | 178951921 | 178952120 | PIK3CA_targeted_21_1 |
| chr3 | 178952037 | 178952227 | PIK3CA_targeted_21_2 |
| chr7 | 55241575 | 55241751 | EGFR_18_1 |
| chr7 | 55241624 | 55241798 | EGFR_18_2 |
| chr7 | 55242380 | 55242572 | EGFR_19_2 |
| chr7 | 55248904 | 55249100 | EGFR_20_1 |
| chr7 | 55249028 | 55249220 | EGFR_20_2 |
| chr7 | 55259370 | 55259567 | EGFR_21_1 |
| chr7 | 55259407 | 55259604 | EGFR_21_2 |
| chr7 | 140453060 | 140453259 | BRAF_15_2 |
Next generation sequencing
Barcoded PCR reactions products are pooled and subjected to AMPure XP (Beckman Coulter, Brea, CA, USA) clean up following Fluidigm protocols. Quality control of individual sample target-specific products, barcoded products, and both pre- and post-clean up library pools is performed with the QIAxcel instrument (Qiagen, Hilden, Germany). The final library pool is diluted to 6 pM, combined with a 2.5% PhiX control library spike-in, and loaded onto a MiSeq V3 reagent cartridge per manufacturer’s protocols (Illumina, San Diego, CA, USA) with Fluidigm modifications for spike in of assay-specific sequencing primers. For routine sequencing runs, cycles are limited to 226 per read. Sequencing quality control targets are: cluster density =600–1,100 K/mm2; clusters passing filter =92–99%; %Q30 ≥67%; reads passing filter >12 M.
Bioinformatics analysis
Samples were run through the ScanIndel v.2.0 pipeline (https://github.com/cauyrd/ScanIndel, outlined in Figure 1) (9). Results were filtered using a custom R script to remove variants not meeting quality control criteria: amplicons are required to have 500× minimum coverage for any variants to be called; indels greater than 3 bp in length must have a VAF of at least 1%, and smaller indels must have a VAF of at least 5%; SNPs in hotspot locations must have a VAF of at least 5%, while SNPs elsewhere must have a VAF of at least 10% (outlined in Figure 2). Coverage data was compiled using bedtools (v.2.17.0) (10). Concordance was calculated for filtered variants; only identical variants (same alternate allele) were considered concordant. For preliminary concordance comparisons (Table 1), 12 fixed samples (inputs of 10, 15, and 20 ng DNA) and 12 unfixed (inputs of 10 ng) were run in replicate. Validation of the platform assessed all genes sequenced simultaneously by the IFC or low input library methods at minimum DNA concentrations specified above. Clinical implementation of the platform was offered with 10 different disease specific mini-panels (7 for solid tumors; 3 for acute myeloid leukemia or myeloproliferative neoplasms) ranging from 3–14 genes). The R script limits the filtered clinical vcf to an input gene list specific to the clinical order.
Figure 1.
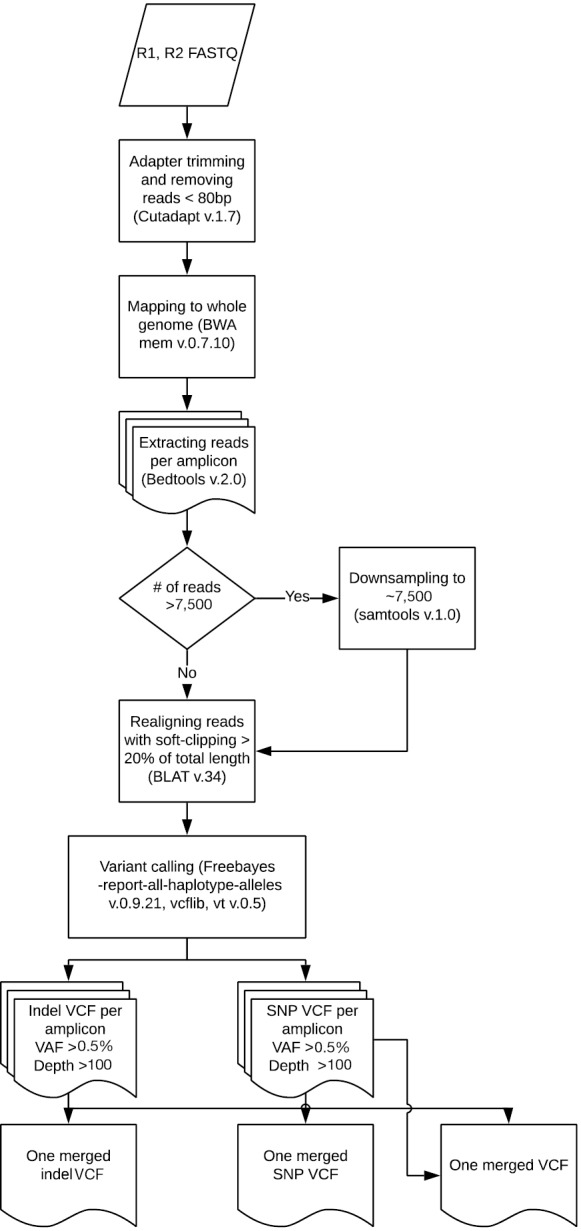
Step-wise ScanIndel v2.0 Method. Following read mapping (BWA-MEM), bedtools is used to assign each read to an amplicon of origin; samtools is then used to limit individual amplicon coverage to a target of 7,500×.
Figure 2.
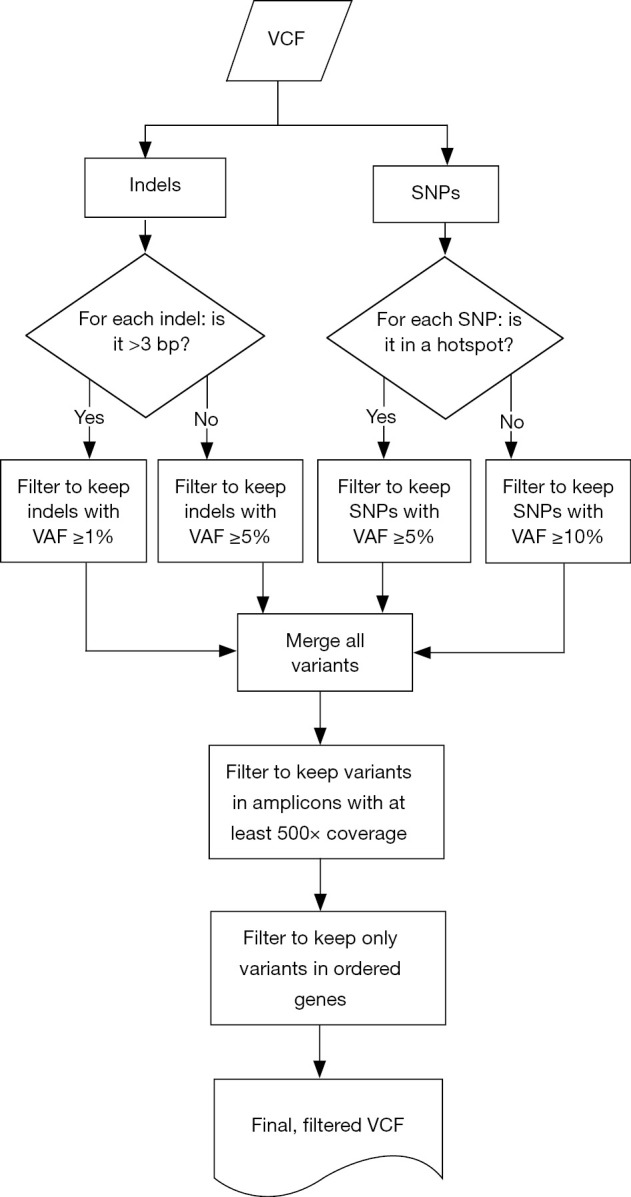
Filtering rules applied to combined vcf output from ScanIndel v2.0 to establish variants list meeting all quality control criteria for clinical reporting in ordered genes.
Table 1. Concordance of variant calls by sample and variant type at three minimum variant allele frequency thresholds.
| Sample type | Variant type | VAF >0.005, n [%] | VAF >0.01, n [%] | VAF >0.05, n [%] | VAF >0.1, n [%] |
|---|---|---|---|---|---|
| Unfixed | Indel | 1,030 [43] | 379 [60] | 99 [81] | – |
| Fixed | Indel | 774 [41] | 280 [53] | 69 [72] | – |
| Unfixed | SNV | 478 [50] | – | 218 [78] | 141 [92] |
| Fixed | SNV | 676 [30] | – | 315 [44] | 136 [76] |
n = total variants. SNV, single nucleotide variants; VAF, variant allele fractions.
Results
Characterization of coverage and development of amplicon-based data mapping
Establishment of minimum DNA input is critical to ensure reproducible results and maximize the number of clinical samples that can be successfully analyzed. We tested a broad range of DNA input levels from both (formalin) fixed and unfixed samples to define the range of minimum sequencing coverage achieved on the IFC assay (Figure 3A). These data showed substantially increased variance for fixed samples input at <15 vs. >15 ng/µL (9.5×106 vs. 2.2×106; standard deviation 3,086.0 vs. 1,495.5 respectively). Unfixed samples input at <10 vs. >10 ng/µL also showed increased variance, although the magnitude of changes was smaller (4.3×106 vs. 3.7×106; standard deviation 2,069.8 vs. 1,927.1 respectively). These parameters suggested initial minimum DNA inputs for each sample type. Further, we noted a broad distribution of mean coverage depth by amplicon from approximately 500× to 18,000× on the IFC assay (Figure 3B); over half of the amplicons within the assay demonstrated a mean coverage of >8,000×.
Figure 3.

Characterization of sequencing depth by sample type, input amount, and across individual PCR amplicons. (A) Box plots demonstrate the distribution of maximum (red) and minimum (blue) read depth for both formalin fixed (upper) and unfixed (lower) specimens at indicated DNA mass inputs using the integrated fluidics circuit (IFC) library preparation; (B) mean coverage by amplicon for fixed samples with 15 ng of DNA input on the IFC.
The analytic LOD of a NGS-based assay to identify low allele fraction mutations is dependent on the minimum depth of coverage achieved and on the artifact rate of the library preparation method. Conversely, high maximum coverage depths potentially achieved with PCR amplicon libraries do not significantly improve the LOD, as biologically unique reads cannot be distinguished from PCR duplicates without more sophisticated methods (11). Furthermore, we empirically observed that samples with skewed maximum coverage depths required increased computational processing times (up to 700% increase for individual outliers). Therefore, we developed a modified version of the ScanIndel method (9) (Figure 1) to assign all individual reads to an amplicon of origin following mapping. This allowed us to downsample reads within each sample on an amplicon-by-amplicon basis, which maintains adequate minimum coverage for less efficient amplicons but limits maximum coverage for highly efficient amplicons to ensure consistent computational processing times across all samples. We performed a pilot experiment by downsampling individual amplicons within each sample to a maximum depth of 7,500×. VAF for 11 mutations (including both single nucleotide variants and indels; VAF range from 0.05–0.86) were stable across 8 samples (Table S2). The average computational processing time with downsampling decreased to 1.1 hours per sample from 2.8 hours previously (~60% decrease; average of 1.5 M raw reads per sample), and no outliers were observed.
Table S2. Accuracy of VAF results with downsampling by amplicon to maximum 7,500× coverage.
| Sample type | Variant (VAF) without downsampling | VAF downsampling cutoff =7,500 |
|---|---|---|
| Fixed | BRAF V600R (0.86; 2 bp indel) | 0.87 |
| Unfixed | CALR L367Tfs*46 (0.44) | 0.47 |
| Fixed | KRAS G12G (0.22) | 0.24 |
| Unfixed | FLT3 ITD (0.05; 90 bp ins); NPM1 Y288Cfs (0.39) | 0.06; 0.40 |
| Fixed | EGFR p.L747_T751del (0.39) | 0.37 |
| Fixed | KIT Q556_K558delinsH (0.29) | 0.30 |
| Fixed | NRAS Q61L (0.41) | 0.37 |
| Unfixed | BRAF V600E (0.14); EGFR E746_A750del (0.75); KRAS G12V (0.38) | 0.17; 0.75; 0.38 |
Characterization of variant calls by DNA input
We next assessed the impact of DNA input on the distribution of variant calls in 115 replicates from 30 unique samples (52 fixed and 63 unfixed sample replicates). Figure 4 displays variation in the total number of variant calls for each DNA input level tested (range 1–50 ng total) at relaxed minimum quality filters (minimum VAF 0.005; minimum coverage depth >100×). Replicates below 20 ng of total DNA input demonstrated broad variation in the total number of variants called. Additionally, most variant calls had VAFs below 0.05–0.1 (Table 2).
Figure 4.

Distribution of the total number of variants called per sample with IFC library preparation at various DNA inputs. Box plots show the distribution in the total number of single nucleotide variants (upper) and indel (lower) variants called for both fixed (red) and unfixed (blue) specimens at the indicated DNA inputs. A total of 115 replicates from 30 biologically unique samples are represented. IFC, integrated fluidics circuit.
Table 2. Percentage of low variant allele fractions (VAFs) indel and single nucleotide variant calls by sample type.
| Sample type | % SNV calls <0.1 VAF | % Indel calls <0.05 VAF |
|---|---|---|
| Unfixed | 65.60 | 87.70 |
| Fixed | 67.60 | 89.10 |
A subset of 24 sample pairs was analyzed for variant concordance. The precision of called variants was compared at three different minimum VAF thresholds based on both variant and sample type (Table 1), as the rate of artifacts varies by these characteristics. For both single nucleotide variant (SNV) and indels, replicate concordance was higher in unfixed vs. fixed samples at all tested minimum VAF thresholds. This concordance was calculated across all sequenced positions, irrespective of intronic or exonic location. Significant causes of discrepancies included stutter artifacts related to homopolymer or trinucleotide sequences and presumed deamination artifacts in fixed samples. We noted a subset of outlier cases with low concordance for both variant types at all minimum VAFs and identified that these samples failed to achieve a minimum 500× coverage in greater than 3% of total amplicons in the assay. This observation suggested that the minimum coverage performance across the entire assay was indicative of the quality of the target specific enrichment and variant data, regardless of the specific coverage level of the amplicon in which a variant call was made. Therefore, we instituted a QC metric that all samples needed a minimum of 500× coverage in at least 97% of amplicons before variant information from individual samples can be interpreted.
Optimization of microfluidics workflow and bioinformatics processing for data precision
These preliminary results indicated that optimization of the workflow was required to balance sensitivity for low-fraction variant alleles at clinically relevant positions with stringent specificity requirements for clinical use. A primary challenge of the integrated fluidics circuit is a volumetric limitation on sample input (1 µL by standard protocol), thus requiring concentrated DNA extracts (50 ng/µL by manufacturer’s recommendation) which are difficult to obtain from limited clinical samples such as core needle biopsies and fine needle aspirates. We modified the standard wet-bench protocol in two ways to maximize the amount of input DNA. First we altered the sample:master mix ratio to increase the total DNA mass added onto each assay lane of the IFC by ~65%. Second, we loaded each sample into two reaction inlets per IFC which were then pooled for a single barcoding reaction per sample. This strategy further increased the number of individual starting DNA molecules subjected to each target-specific amplicon reaction on the IFC and we hypothesized that pooling of these independent reactions would minimize PCR artifacts and improve the analytic precision of results.
We used custom vcf-processing scripts in R software for data-filtering to improve the specificity of variant calls and achieve higher levels of sensitivity for clinically actionable mutations (Figure 2). We extended the utility of amplicon assignment following read mapping by also calling sequence variant calls on an amplicon by amplicon basis. These amplicon-assigned variant calls were then subjected to a tiered-system of filtering based on allele fraction thresholds for specific variant types and genomic coordinates. For SNVs, a list of 152 missense variants with NCCN guidelines or significant published evidence for clinical actionability was created (online: http://atm.amegroups.com/public/system/atm/supp-atm.2018.05.07-2.pdf) which were called at a minimum VAF threshold of 0.05; all remaining SNVs were called at a minimum VAF threshold of 0.1. Indels were processed based on the length of the variant: insertions or deletions of three base pairs or less were filtered at 0.05 while larger indels (>3 base pairs) were filtered at 0.01. These strategies maximized sensitivity for variants with the highest levels of clinical actionability.
After variant calling and filtering by these parameters, a final merged vcf is created. Amplicon-based variant calling (Figure 2) and the overlapping amplicon design of the assay is then leveraged to further increase specificity. The pipeline labels each called variant with the amplicon(s) in which it was called and with the amplicon(s) in which that genomic coordinate is sequenced. Both deamination and PCR artifacts generated during target enrichment are not equally amplified in distinct but overlapping amplicons; thus, a variant not identified in all expected amplicons is flagged as a probable artifact for pathologist quality control (QC) review. Figure 5A illustrates the effectiveness of this strategy: a G>A variant was detected in exon 3 of NRAS near codon 61 that was present with a VAF of 0.19 in amplicon 3_2 but not detected by amplicon 3_3. Additionally, sanger sequencing with both primer sets did not identify this variant (data not shown), confirming this as an artifact.
Figure 5.

Advantages of amplicon-based variant calling. (A) Example of deamination artifact (G>A/C>T) in mapped sequencing reads which was captured in early rounds of PCR by only one of two overlapping amplicons. True variants (including low abundance alleles near assay VAF thresholds) do not demonstrate this finding. (B) Example of allele drop out due to a clinically relevant indel affecting primer binding in one of two overlapping amplicons. Amplicon-based variant calling more accurately estimates variant allele fraction by computing the AO/RO ratio only for reads from amplicon 9_1 (most 5’ amplicon displayed).
Amplicon based calling also improves the detection accuracy and VAF estimation for variants affecting primer binding sites. Indel sequence alterations impacting primer binding may cause allele-specific amplicon drop out, thus under-estimating the allele fraction or entirely missing the variant. Figure 5B demonstrates a 52 base pair exon 9 CALR deletion that is accurately detected by amplicon 9_1 with a VAF =0.22, but this deletion partially overlaps with the 5’ primer binding sequence of amplicon 9_2, leading to allele dropout. Thus, the variant fraction is significantly under-estimated (~10%) in the composite data based on coverage at the affected genomic coordinates from both amplicons. In this situation, pathologist review of the pileups clearly indicates that allele drop out is the cause of the QC flag, and the call is approved as a true positive with an appropriate VAF from amplicon 9_1. These examples demonstrate how amplicon-based variant calling improves assay specificity, sensitivity, and accuracy in variant calling and VAF estimation.
Development and optimization of low input target enrichment assay
Even with enhanced sample DNA loading procedures for the IFC (described above), we concluded that concentrations of 10–15 ng/µL would still be required for accurate target enrichment on the microfluidics assay for unfixed and fixed samples respectively. This would lead to unacceptably high rates of quantity not sufficient (QNS) designations in our clinical laboratory, particularly for advanced solid tumors assessed only by FNA, core needle, or endoscopic biopsies. Therefore, we developed a 4.2 kb assay consisting of 6 key genes required for clinical management in these disease areas (Table S1). Four primer-limited multiplex pools and modified cycling parameters were developed (see methods) and an unfixed, cell-line derived control was used to characterize assay performance at 0.1, 0.5, 1, and 5 ng/µL of DNA input. Mean coverage ranged from 4,000×–5,700× (Figure 6) and all input levels achieved minimum coverage of 500× in >90% of amplicons. VAF for three known mutations were consistent across all input levels (Table 3). Empiric testing of a small cohort of independent FFPE samples demonstrated similar coverage characteristics at 1 ng/uL inputs (~4,000× mean coverage and >90% of amplicons with minimum 500× coverage at all positions; data not shown). Therefore, these sample input parameters (0.1 ng/µL for unfixed and 1 ng/µL for fixed samples) and assay conditions were set for the final phase of clinical validation of the low input library preparation.
Figure 6.
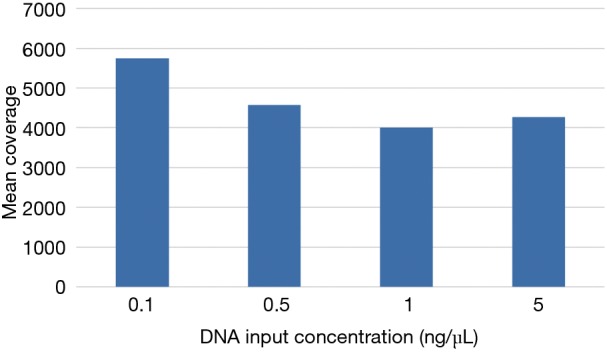
Mean coverage of low input library preparation by DNA input. A primer-limited PCR library preparation method for 6 genes was developed for tissue-limited clinical samples. Average coverage was stable across a range of input concentrations from 0.1–5 ng/µL of DNA.
Table 3. Stability of variant allele fractions across DNA input levels for the low input library preparation.
| Gene | Mutation.AA | 0.1 ng/ìL | 0.5 ng/ìL | 1 ng/ìL | 5 ng/ìL |
|---|---|---|---|---|---|
| EGFR | p.E746_A750del | 0.78 | 0.81 | 0.83 | 0.84 |
| BRAF | p.V600E | 0.11 | 0.10 | 0.09 | 0.10 |
| KRAS | p.G12V | 0.64 | 0.66 | 0.64 | 0.54 |
Optimization of read length
The average and maximum amplicon size for this assay design were 192 and 200 bp respectively; therefore, read lengths greater than 200 bp are potentially unnecessary and extend sequencing time. However, we hypothesized that longer read lengths could improve accuracy for the detection of larger insertions (particularly FLT3 internal tandem duplications). To test this, we created pilot data at 300 bp read lengths for a series of samples with 13 biologically independent insertion mutations in FLT3, NPM1, NRAS, and KIT (range, 4–93 bp). FASTQ files were then in silico trimmed to 250, 225, and 200 bp lengths. Variant calls made from the 250 and 225 bp read length data sets detected 100% of these variants, but the 2 largest FLT3 insertions (93 bp each) were missed in the 200 bp read length data (Table 4). Otherwise, VAFs were similar for each mutation across each read length assessed, indicating stability of the variant calling algorithm at the different read lengths. Based on this data a read length of 225 bp was chosen for clinical validation.
Table 4. Optimization of read length vs. indel call accuracy by in silico trimming.
| Gene_bp_indel | Read length | |||
|---|---|---|---|---|
| 300_bp | 250_bp | 225_bp | 200_bp | |
| FLT3_30_ins | 0.16 | 0.17 | 0.17 | 0.15 |
| FLT3_30_ins | 0.13 | 0.15 | 0.15 | 0.12 |
| FLT3_36_ins | 0.36 | 0.37 | 0.37 | 0.30 |
| FLT3_54_ins | 0.63 | 0.68 | 0.64 | 0.59 |
| FLT3_78_ins | 0.01 | 0.01 | 0.02 | 0.01 |
| FLT3_93_ins | 0.02 | 0.02 | 0.02 | Not detected |
| FLT3_93_ins | 0.05 | 0.07 | 0.10 | Not detected |
| KIT_6_delins | 0.31 | 0.40 | 0.40 | 0.40 |
| NPM_4_ins | 0.45 | 0.45 | 0.45 | 0.45 |
| NPM_4_ins | 0.46 | 0.46 | 0.46 | 0.46 |
| NPM_4_ins | 0.44 | 0.45 | 0.45 | 0.45 |
| NPM_4_ins | 0.4 | 0.40 | 0.40 | 0.40 |
| NRAS_30_ins | 0.43 | 0.34 | 0.34 | 0.34 |
Clinical validation of the optimized next generation sequencing assay
Following optimization of the IFC and low input library preparation methods and bioinformatics pipeline, we devised a three-part clinical validation strategy for the parallel IFC and low-input workflows. First, linearity and LOD was established with serial dilutions of a cell line control sample. Six mutations were assessed from the IFC assay (4 SNV, 2 indel) and 3 from the low-input assay (2 SNV, 1 deletion); all mutations were detected as predicted above minimum clinical VAF thresholds (0.05 for SNV and 0.01 for indels >4 bp; Tables 5,6). Further, mutant allele reads were detected in the mapped read files to variant fractions less than 1%, well below the minimum VAF thresholds of the assay defined to ensure specificity. Linearity of detection for all SNVs and the FLT3 insertion demonstrated r2>0.95 across the dilution series; the EGFR deletion instead demonstrated an exponential trend line with r2=0.99931 (Figure 7). This phenomenon is due to the increased replication efficiency of the shorter (deleted) amplicon which causes relative over-representation compared to the wildtype allele. Linearity was nearly identical for the low input assay (Figure S1). Collectively, these data demonstrated LOD sensitivity above specificity thresholds and consistent linearity for both the IFC and low input library preparation methods.
Table 5. Limit of detection for integrated microfluidics circuit (IFC) library preparation method.
| IFC | BRAF_V600E | KRAS_G12V | EGFR E746_A750del | FLT3 D600_L601ins10 | JAK2_V617F | TP53_R273H | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Predicted | Observed | Predicted | Observed | Predicted | Observed | Predicted | Observed | Predicted | Observed | Predicted | Observed | ||||||
| Straight | – | 0.150 | – | 0.571 | – | 0.757 | – | 0.115 | – | 0.533 | – | 0.372 | |||||
| 1:4 | 0.037 | 0.026 | 0.143 | 0.269 | 0.189 | 0.549 | 0.029 | 0.041 | 0.133 | 0.217 | 0.093 | 0.140 | |||||
| 1:16 | 0.009 | 0.009 | 0.036 | 0.085 | 0.047 | 0.327 | 0.007 | 0.004 | 0.033 | 0.095 | 0.023 | 0.048 | |||||
| 1:64 | 0.002 | 0.010 | 0.009 | 0.020 | 0.012 | 0.093 | 0.002 | 0.004 | 0.008 | 0.022 | 0.006 | 0.021 | |||||
Table 6. Limit of detection for low input library preparation method.
| Low Input | BRAF_V600E | KRAS_G12V | EGFR E746_A750del | |||||
|---|---|---|---|---|---|---|---|---|
| Predicted | Observed | Predicted | Observed | Predicted | Observed | |||
| Straight | – | 0.087 | – | 0.651 | – | 0.795 | ||
| 1:4 | 0.022 | 0.032 | 0.163 | 0.262 | 0.199 | 0.576 | ||
| 1:16 | 0.005 | 0.014 | 0.041 | 0.101 | 0.050 | 0.277 | ||
| 1:64 | 0.001 | 0.002 | 0.010 | 0.021 | 0.012 | 0.104 | ||
Figure 7.

Linearity of variant allele fractions across serial dilutions. Serial dilutions were made of a cell line DNA mixture with 6 characterized mutations into negative control DNA. Four single nucleotide variants (SNVs) (BRAF p.V600E, KRAS p.G12V, JAK2 p.V617F, and TP53 p.R273H), 1 insertion (FLT3 p.D600_L601ins10) and 1 deletion (EGFR p.E746_A750del) were assessed. VAF, variant allele fractions.
Figure S1.

Linearity of variant allele fractions across serial dilutions prepared by the low input library preparation method. Two single nucleotide variants (SNVs) (BRAF p.V600E, KRAS p.G12V) and 1 deletion (EGFR p.E746_A750del) were assessed. Linearity was similar to results generated with the integrated microfluidics circuit (IFC) library preparation method (see Figure 7 of primary manuscript).
Second, we performed precision analyses of both library preparation methods. Concordance of variant calls between sample replicates was assessed for missense SNVs, SNVs affecting canonical splice positions, and all indels above minimum VAF thresholds. Nineteen replicate pairs were compared including BM, PB, FNA, and formalin fixed paraffin embedded (FFPE) samples. After exclusion of artifacts by pathologist review, precision was 99% (Table 7) including both intra- and inter-run comparisons for both library preparation methods. Data breakdown by each library preparation workflow and intra- vs. inter-run comparisons are presented in Tables S3,S4. Overall, there were 75 concordant variants called between replicates and 1 true discrepancy due to a low VAF mutation called above the 5% SNV threshold in one replicate but present below threshold in the other. In addition, 19 discrepant calls were excluded by molecular pathologist review due to intronic homopolymer sizing artifact [18] or indel mapping discrepancy [1]. This highlights the importance of quality control review and interpretation by a molecular pathologist to discriminate artifacts from potentially relevant calls, particularly with indels. There are clinically relevant indel mutations that span intron-exon boundaries or are located within intronic sequence (including MET, KIT, and FLT3) (12-14). We were concerned that highly stringent bioinformatics filtering rules which eliminate all artifacts could also exclude true indel mutations. Thus, we chose direct pathologist review of all indel calls as a quality control step to prevent potential false negative results.
Table 7. Composite (intra- and inter-run) precision analysis by sample type (n=replicate pairs).
| Sample type | Concordant variants | Discrepant variants | Discrepant artifacts | Cause(s) of discrepancy |
|---|---|---|---|---|
| Unfixed (n=14) | 41 | 1* | 2 | *Single nucleotide variants (SNV) threshold [1]; intronic homopolymer [1]; indel mapping [1] |
| Fixed (n=16) | 34 | 0 | 17 | Intronic homopolymer [17] |
Table S3. Intra-run replicate precision.
| Sample_1 | Sample_2 | Concordant variants | Discrepant variants | Cause(s) of discrepancy |
|---|---|---|---|---|
| Integrated microfluidics circuit (IFC) | ||||
| BM1_DUP | BM1 | 8 | 0 | |
| PB1_DUP | PB1 | 8 | 0 | |
| CELL_DUP | CELL | 16 | 3 | Intronic homopolymer [1]; single nucleotide variants (SNV) threshold [1]; indel mapping [1] |
| FFPE1_DUP | FFPE1 | 6 | 5 | Intronic homopolymer [5] |
| FFPE2_DUP | FFPE2 | 6 | 1 | Intronic homopolymer [1] |
| FFPE3_DUP | FFPE3 | 7 | 6 | Intronic homopolymer [6] |
| FFPE4_DUP | FFPE4 | 6 | 5 | Intronic homopolymer [5] |
| Low input | ||||
| FFPE9_DUP | FFPE9 | 1 | 0 | |
| FFPE10_DUP | FFPE10 | 1 | 0 | |
| FFPE11_DUP | FFPE11 | 0 | 0 | |
| FFPE12_DUP | FFPE12 | 0 | 0 | |
| FFPE13_DUP | FFPE13 | 0 | 0 | |
| FFPE14_DUP | FFPE14 | 1 | 0 | |
| FNA1_DUP | FNA1 | 1 | 0 | |
| FNA2_DUP | FNA2 | 1 | 0 | |
| FNA3_DUP | FNA3 | 1 | 0 | |
| FNA4_DUP | FNA4 | 0 | 0 | |
| FNA5_DUP | FNA5 | 0 | 0 | |
| FNA6_DUP | FNA6 | 0 | 0 | |
Table S4. Inter-run/inter-tech precision.
| Sample | Mutation | Result |
|---|---|---|
| Integrated microfluidics circuit (IFC) | ||
| FFPE5 | IDH1 p.R132H | Concordant |
| FFPE6 | KRAS p.G12V | Concordant |
| FFPE7 | BRAF p.V600E | Concordant |
| FFPE8 | BRAF p.V600E | Concordant |
| BM2 | NPM1 p.860_863dup | Concordant |
| PB1 | JAK2 p.V617F | Concordant |
| PB2 | JAK2 p.V617F | Concordant |
| CRL5908 | EGFR p.T790M; p.L858R | Concordant |
| Low input | ||
| FFPE15 | KRAS p.G12S | Concordant |
| FFPE16 | BRAF p.V600K | Concordant |
| FNA1 | KRAS p.G13D | Concordant |
Third, analytic accuracy (sensitivity and specificity) of the IFC and off-chip library preparation methods was measured using a series of orthogonally characterized samples and cell line controls. Sixty-seven biologic samples were tested on the IFC assay: 21 from FFPE, 32 from BM biopsy, 12 from PB, and 2 independent cell line samples. These samples included 64 known mutations (28 SNV and 36 indels) across 13 genes plus 28 known negative genes (online: http://atm.amegroups.com/public/system/atm/supp-atm.2018.05.07-3.pdf). Sixty-two mutations were positively identified above established VAF thresholds, no false positives were identified, and two false negatives were missed. A 93-base pair FLT3 ITD was missed in a BM specimen due to a subthreshold (<1%) VAF confirmed by visual examination of the mapped sequencing data. A codon 12 KRAS SNV was not detected in a FFPE specimen at any VAF level in the mapped sequencing reads, including in 2 repeat NGS analyses. The orthogonal KRAS result was reported by an outside laboratory using a pyrosequencing method with a distinct DNA extraction from the same case; thus the discordance may have been due to pre-analytic variables or intra-tumoral heterogeneity (15).
Twenty-eight biologic samples were tested on the off-chip assay, including: 19 FFPE, 2 BM biopsies, 6 FNA, and one cell line sample. Twenty-two known mutations had been previously characterized in these samples (17 SNV and 5 indels) within 5 genes; there were 11 known wildtype genes in this sample set. Twenty-one mutations were positively identified above threshold and no false positives were called. One BRAF SNV was missed in a FNA specimen due to a subthreshold VAF (3%); an allele specific BRAF PCR assay with different LOD characteristics had provided orthogonal confirmation of this mutation.
Combined, 95 biologic samples were assessed across both library preparation pathways of this clinical NGS assay with a total of 83 true positive, 39 true negative, 3 false negative and 0 false positive results (Table 8). The analytic accuracy is 98%, with analytic sensitivity (percent positive agreement) of 97% (95% CI: 0.90–0.99) and analytic specificity of 100% (95% CI: 0.91–1). The positive predictive value is 100%.
Table 8. Composite analytic accuracy of variant calls.
| NGS | Orthogonal | |
|---|---|---|
| + | – | |
| + | 83 | 0 |
| − | 3 | 39 |
NGS, next generation sequencing.
Discussion
Implementation of a NGS assay for oncology diagnostics must balance LOD, specificity, and flexibility for handling low quality/quantity DNA specimens to maximize clinical effectiveness. In general, PCR amplicon-based library methods offer some advantages versus hybrid-capture approaches for oncology applications including lower DNA input requirements and more rapid turnaround time (16). Multiplexed PCR on microfluidics circuits is an amplicon library preparation method that enables moderate to high throughput in terms of both assay target size and number of samples analyzed per run (17).
Our work highlights solutions to consistent challenges for clinical implementation of amplicon-based NGS assays. To characterize key assay parameters, we investigated the distribution of minimum coverage and variant precision across a broad range of DNA inputs onto the integrated fluidics circuit. Plotting the variability of minimum coverage by DNA input and sample type (fixed vs. unfixed) identified a workable range of DNA input (10–20 ng) that was significantly less than the manufacturer’s protocol recommendation (50 ng). Analysis of replicate precision at several minimum VAF thresholds highlighted that quality filters based on a single variant allele threshold would not be clinically sufficient either for specificity or sensitivity. This preliminary precision analysis also identified that variability in minimum coverage across all amplicons on the integrated microfluidics circuit (IFC) (>3% of amplicons failing to achieve >500×) effectively predicted outlier samples with elevated rates of discordant variant calls, leading to important sample QC metric for downstream validation.
From this preliminary analysis, we developed several innovative approaches to improve the performance of both library preparation and bioinformatics analysis. We altered sample loading and harvest product pooling procedures for the IFC protocol to maximize the amount of DNA analyzed per sample while maintaining the ability to prepare 24 samples per run. We also created a parallel low DNA input library preparation method that enabled us to effectively analyze very limited specimens, particularly for common clinical situations such as advanced lung carcinoma and metastatic colorectal carcinoma. The LOD and linearity characteristics of the IFC and low input methods were highly similar, allowing us to create an integrated workflow that analyzed both limited and routine specimens on the same sequencing run for validation.
The development of a bioinformatics amplicon assignment method for individual reads during mapping and variant calling had a significant impact on assay performance in several key ways. First, this allowed us to informatically balance read depth within a sample to achieve faster, more uniform computational processing time without compromising coverage of less efficient amplicons. Managing computational time was important to help ensure effective turnaround times for clinical reporting. This informatics strategy was cost and time effective for us because the sequencing capacity of the V3 MiSeq chemistry was in reasonable excess compared to the genomic footprint of the assay and average number of samples per run. The alternative of empirically balancing primer concentrations across the microfluidics assay would have been laborious and difficult to consistently reproduce across primer plate lots for routine clinical practice.
Second, the amplicon assignment method provided advantages to both sensitivity and specificity. The identification of amplicon specific variant calls and VAFs helps to ensure low fraction indels are not underestimated because of allele dropout and therefore possibly missed by minimum VAF thresholds based on composite coverage in regions with multiple overlapping amplicons. Further, amplicon assignment of SNV calls that QC flags variants not identified in all relevant amplicons effectively eliminates common causes of artifacts such as formalin-induced deamination and early round PCR error in target specific enrichment. The combination of amplicon assignment and tiered variant filtering (Figure 2) allowed us to optimize the clinical performance of the assay.
Of note, our formal validation of this laboratory-developed NGS oncology procedure predated the most recent professional guidelines from the Association of Molecular Pathology (6,8). Our utilization of 95 samples for validation of the integrated workflow exceeded the minimum sample recommendation (n=59) from these guidelines; however, with a total of 45 SNV and 41 indel mutations we were below the additional recommendation for 59 unique variants of each type of alteration analyzed by the assay. Our validation design for the number of orthogonally-verified positive and negative results was intended to meet or exceed a 95% confidence interval lower boundary of 0.9 for both sensitivity and specificity metrics. After clinical release of this assay, we have participated in 6 College of American Pathology proficiency tests with a 100% success rate.
Our experience describes solutions to common problems facing clinical implementation of amplicon-based NGS workflows for oncology care. This work created and validated a clinical NGS assay with strong analytic performance (97% sensitivity, 100% specificity). Further, in routine clinical use this method has an overall 98% sample adequacy rate (95% specifically for FFPE samples) and an average clinical turnaround time of 9.8 days. Although some of these solutions are specific to the microfluidics platform, our general approach and bioinformatics methods should assist other hospital-based laboratories in their efforts to deploy effective NGS platforms to support precision oncology medicine.
Acknowledgements
The authors wish to thank Dr. Kevin Silverstein for his technical guidance. This work was supported in part through funds from the Department of Laboratory Medicine and Pathology, University of Minnesota (C Henzler, AC Nelson) and the Eastern Star Scholars Fund, Minnesota Masonic Charities and University of Minnesota Masonic Cancer Center (AC Nelson).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 2017;45:D777-83. 10.1093/nar/gkw1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn) 2015;19:A68-77. 10.5114/wo.2014.47136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-74. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 4.Li MM, Datto M, Duncavage EJ, et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017;19:4-23. 10.1016/j.jmoldx.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Allen EM, Wagle N, Levy MA. Clinical analysis and interpretation of cancer genome data. J Clin Oncol 2013;31:1825-33. 10.1200/JCO.2013.48.7215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jennings LJ, Arcila ME, Corless C, et al. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J Mol Diagn 2017;19:341-65. 10.1016/j.jmoldx.2017.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yohe S, Thyagarajan B. Review of Clinical Next-Generation Sequencing. Arch Pathol Lab Med 2017;141:1544-57. 10.5858/arpa.2016-0501-RA [DOI] [PubMed] [Google Scholar]
- 8.Roy S, Coldren C, Karunamurthy A, et al. Standards and Guidelines for Validating Next-Generation Sequencing Bioinformatics Pipelines: A Joint Recommendation of the Association for Molecular Pathology and the College of American Pathologists. J Mol Diagn 2018;20:4-27. 10.1016/j.jmoldx.2017.11.003 [DOI] [PubMed] [Google Scholar]
- 9.Yang R, Nelson AC, Henzler C, et al. ScanIndel: a hybrid framework for indel detection via gapped alignment, split reads and de novo assembly. Genome Med 2015;7:127. 10.1186/s13073-015-0251-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841-2. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kinde I, Wu J, Papadopoulos N, et al. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A 2011;108:9530-5. 10.1073/pnas.1105422108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corless CL, McGreevey L, Town A, et al. KIT gene deletions at the intron 10-exon 11 boundary in GI stromal tumors. J Mol Diagn 2004;6:366-70. 10.1016/S1525-1578(10)60533-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov 2015;5:850-9. 10.1158/2159-8290.CD-15-0285 [DOI] [PubMed] [Google Scholar]
- 14.Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996;10:1911-8. [PubMed] [Google Scholar]
- 15.Nelson AC, Boone J, Cartwright D, et al. Optimal detection of clinically relevant mutations in colorectal carcinoma: sample pooling overcomes intra-tumoral heterogeneity. Mod Pathol 2018;31:343-9. 10.1038/modpathol.2017.120 [DOI] [PubMed] [Google Scholar]
- 16.Gagan J, Van Allen EM. Next-generation sequencing to guide cancer therapy. Genome Med 2015;7:80. 10.1186/s13073-015-0203-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bourgon R, Lu S, Yan Y, et al. High-throughput detection of clinically relevant mutations in archived tumor samples by multiplexed PCR and next-generation sequencing. Clin Cancer Res 2014;20:2080-91. 10.1158/1078-0432.CCR-13-3114 [DOI] [PubMed] [Google Scholar]