

Abstract
Cancers of the gastrointestinal tract have limited available treatments and are often associated with a poor prognosis. Clinical trials and translational work associated with these trials provide the opportunity to increase understanding of the mechanisms of sensitivity and resistance to cytotoxic chemotherapy and targeted therapy in these diseases. In this review we discuss the rationale for intensive translational work within the context of academic clinical trials and the successes and challenges which have been associated with translational work at our institution over the past number of years. We reflect on tissue, plasma and radiological biomarker work including a novel patient derived organoid programme and discuss the iterative application of previous results to next generation trial design.
Keywords: Gastrointestinal cancer, gastric cancer, colorectal cancer (CRC), clinical trials, translational, next generation sequencing (NGS), ctDNA, liquid biopsy
Introduction
Most tumours of the gastrointestinal tract are associated with a poor prognosis. Whilst patients with metastatic colorectal cancer (CRC) can live for up to three years when treated with appropriate sequential chemotherapy and biological agents, patients with advanced gastroesophageal and pancreatic cancer can expect to live less than one year when treated using standard cytotoxic chemotherapy (1-3). Despite the growing body of knowledge on the molecular aberrations which underpin these cancers, with the exception of anti-EGFR therapy in RAS wildtype CRC and trastuzumab in HER2 overexpressing gastroesophageal cancer, the biology of these cancers does not yet inform many treatment choices (4-6). Clinical trials and associated translational work on clinical trials provide the opportunity to further the understanding of aspects of tumour biology which affect outcomes following standard chemotherapy as well as targeted drugs and immunotherapy. In order for this research to be successful, it must be focused and iterative. This review aims to summarise the integrative approach to translational research and clinical trials we have adopted at the Royal Marsden over the past few years, and seeks to understand the potential for success and the challenges of this approach.
Retrospective translational analysis of older clinical trials (MAGIC, REAL3)
Prior to approaching prospective trial design, rigorous evaluation and translational assessment of the available material from older trials from which material has been already been collected is warranted. The purpose of this approach is to develop hypotheses for further research in prospective studies, and to avoid hypothesis free analysis of prospectively collected trial tissue. For example, the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) trial was a large phase III randomised trial conducted in the United Kingdom which compared peri-operative epirubicin, cisplatin and 5-fluourouracil (ECF) chemotherapy before and after surgery to surgery alone for patients with resectable gastroesophageal cancer (7). The trial resulted in an improvement in overall survival for patients treated with peri-operative chemotherapy (HR 0.75, 95% CI, 0.60 to 0.93; P=0.009); and resulted in the adoption of ECF as a standard of care for this patient population for a decade from 2007–2017 (8-10). Although ECF has recently been replaced as a standard by the non-anthracycline containing FLOT (5-fluororuacil, leucovorin, oxaliplatin and docetaxel) the lessons learned from the retrospective analysis of tissue from the MAGIC trial are transferrable to trial design with novel chemotherapy combinations and provide proof of concept for the utility of this approach (11).
Previously, there was significant debate regarding the prognostic value of pathological assessment of tumour regression grading (TRG) in gastroesophageal cancer following peri-operative chemotherapy. Although several large non-randomised series existed in the literature suggesting that the presence of pathological tumour regression was independently associated with survival following chemotherapy plus surgery, other datasets conflicted on this point (12-14). Determining the value of TRG was thought to be important as this was frequently used as an endpoint in clinical trials, and was considered likely to be a valid, short term surrogate for overall survival. However, this could only be definitively shown in a randomised dataset. Therefore we evaluated whether the presence of tumour regression using the Mandard grading system was associated with survival outcomes in the MAGIC trial (15). Tissue was collected centrally and scanned using an Aperio scanner at high resolution, and the images were viewed by two pathologists blinded to the study arms. Additional material was obtained if the original slides were inadequate to assess regression grade. We demonstrated that although significant tumour regression (Mandard grade 1–2) was associated with better survival in chemotherapy treated patients, that this was not independently prognostic when evaluated alongside the presence of lymph node metastases. We went on to show that patients who did not have lymph node metastases had excellent survival even in the absence of significant tumour regression. These findings were considered important because they identified that the presence of lymph node metastases and not tumour regression grade should be the focus of further research in patients with operable gastroesophageal cancer. In particular, patients who have positive lymph nodes in the resection specimen following surgery are identified as a high-risk group who may warrant further intervention. Following these findings, the EORTC GI Trials group has initiated a clinical trial (VESTIGE GITCG-1707) in which patients who are at high risk of recurrence following peri-operative chemotherapy plus surgery (R1 resection or node positive) are randomised to continue with standard of care chemotherapy or switch to combination immunotherapy with nivolumab plus ipilimumab. The primary endpoint for this trial is 1-year disease free survival, as most recurrences were identified within this timeframe in the MAGIC dataset. Thus retrospective translational research informed both a high risk patient group for treatment in a novel clinical trial and also the endpoint for that trial.
The MAGIC dataset has also been used to demonstrate the value or not of peri-operative chemotherapy in biologically defined patient subsets which may, in the future, inform trial design and clinical care paradigms. For example, although microsatellite instability as a result of mismatch repair deficiency had been identified as a positive prognostic marker in patients with resected gastroesophageal adenocarcinoma in multiple retrospective datasets, this did not impact clinical practice (16) In MAGIC, as expected, it was demonstrated that for patients treated with surgery alone, the presence of microsatellite instability was associated with a good prognosis (17). However, surprisingly, it was also demonstrated that patients who had mismatch repair deficient cancers did not appear to benefit from peri-operative chemotherapy, in contrast to patients with mismatch repair proficient cancers. These findings were subsequently validated in an analysis of the Asian CLASSIC trial, in which patients with resected gastric cancer were randomised to adjuvant chemotherapy or no further treatment (18,19). As the results from these retrospective analyses of practice defining trials are filtered through the oncology community, trials are currently being designed in which patients with mismatch repair deficient tumours are not treated with peri-operative chemotherapy but with immune checkpoint blockade. As mismatch repair deficient tumours are intrinsically immunogenic, this may provide superior survival benefit than cytotoxic chemotherapy alone (20,21).
The previous two examples of the benefits of translational research based on the MAGIC trial dataset provide proof of concept for the value of retrospective translational research on trial datasets, and how this can inform future trial design and deliver better outcomes for cancer patients. However, lessons were learned from the challenges of working with this dataset. Firstly, relatively limited tumour material was collected; this did not allow for complete evaluation of the tumour bed and limited the scope of the TRG systems which could be used (15). Secondly, tissue was not collected for all patients on this study, which limited the power of analyses, in particular in biomarker identified subgroups. Had tissue been collected prospectively it is likely that it would have been available from a larger proportion of the population, and had the tissue requirements been identified a priori a larger volume of material might have been collected which could have facilitated a more extensive translational analysis. These issues were both addressed by the prospective consent for and collection of material in the subsequent UK Medical Research Council ST03 study (22).
In patients with metastatic cancer in whom tumour material is less freely available, informative translational research is still possible. This can even be useful in the context of a negative clinical trial. For example, in the REAL 3 trial patients with treatment naïve metastatic or locally advanced unresectable gastroesophageal cancer were treated with either epirubicin, oxaliplatin and capecitabine or the same plus the anti-EGFR monoclonal antibody panitumumab (23). The trial was stopped early following an interim analysis which demonstrated inferior survival for patients treated with EOX-P (HR 1.37, 95% CI, 1.07–1.76; P=0.013). Despite these negative findings, there have been several potentially useful clinical findings from this dataset. As poly (ADP-ribose) polymerase (PARP) inhibitors had recently been licenced in ovarian cancer, we initially chose to focus on whether a potentially PARP sensitive population could be identified in gastroesophageal cancer patients. In particular, as genomic instability quantified using an algorithmic approach on a standard NGS panel as an LOH-high (loss of heterozygosity-high) signature was associated with a benefit from PARP inhibition in non-BRCA mutant ovarian cancer, we attempted to identify a similar population in the REAL3 dataset (24,25). We hypothesised that such a population would have enhanced sensitivity to platinum chemotherapy and prolonged survival as a result. In collaboration with Clovis, the manufacturer of rucaparib, we analysed diagnostic biopsy material using the Foundation Medicine One panel (26). Unfortunately, previous translational projects had exhausted much of the material and DNA of sufficient quality and quantity for LOH analysis was only available for a limited number of patients from the trial (27). Nonetheless, we identified a group of patients with high levels of genomic instability (LOH-high, 14% of the assessed population), who had longer progression free and overall survival than patients who were LOH-low HR for overall survival (OS) in patients who did not undergo surgery 18.3 vs. 10.2 months (HR 0.43, 95% CI, 0.20–0.92, P=0.02 for LOH-high vs. low); PFS was 10.5 vs. 7.2 months (HR 0.55, 95% CI, 0.26–1.17 for LOH-high vs. low respectively). Concurrently with this translational research we set up the PLATFORM trial (NCT02678182) which evaluates various maintenance therapies in patients who have demonstrated sensitivity to first line chemotherapy for advanced gastroesophageal cancer. One arm of the study will evaluate the efficacy of rucaparib in this setting and attempt to validate the predictive value of the LOH biomarker. In this way, translational research from a negative trial has informed future development of unrelated compounds.
Prospective translational trial design (FGFR)
Translational research can also inform the development of prospective translational clinical trials which can be amended iteratively pending the results which are observed. In a national Royal Marsden sponsored clinical trial, we hypothesised that patients with FGFR amplified cancers would respond to treatment with the orally available tyrosine kinase inhibitor AZD4547 (28). Initially, three patient cohorts were screened, each with a known prevalence of FGFR amplification; these were breast, gastroesophageal and non-small cell lung cancer. Initially, patients were screened centrally using a FISH assay developed by the study funder. The initial iteration of the trial had a pharmacodynamic endpoint based on dual biopsies using a phosphoprotein assay, however following testing dual biopsies of the first cohort of patients it became apparent that this was not feasible as a primary endpoint and therefore a more traditional endpoint of objective response rate was adopted. As the study progressed, due to limited responses observed in breast and NSCLC FGFR amplified patients, these cohorts were closed to recruitment. However, in patients with FGFR2 amplified gastroesophageal cancers, three excellent responses were observed in the first nine amplified patients treated (29). Focusing on the intensive translational programme that was associated with the trial, we found that only patients with high levels of FGFR2 ctDNA in plasma had responded to AZD4547, thus identifying a novel predictive biomarker (28). We also demonstrated that within the patient population of FGFR2 amplified gastroesophageal cancers that only patients with high level and homogenous FGFR2 amplification were associated with high levels of FGFR2 ctDNA in plasma. Following this analysis, we went on to develop a screening tool based on FGFR2 ctDNA using ddPCR which was used nationally to screen for the trial. In doing so, we were amongst the first groups to screen for a biomarker using a ctDNA assay. Unfortunately, following this, recruitment to the study was inadequate and due to a paucity of further evidence regarding the lack of efficacy of AZD4547, the study was closed to recruitment. This is unfortunate, as several groups have now demonstrated frequencies of FGFR2 amplification in plasma of up to 8% using NGS technologies in large patient cohorts (30). The low prevalence of this biomarker may mean that targeting FGFR amplified patients in future may require international collaboration.
Biomarkers from radiology (PROSPECT-R)
The concomitant use of translational tools and novel imaging techniques can lead to innovative research and novel biomarker discovery. Regorafenib, a multitargeted anti-angiogenic tyrosine kinase inhibitor, is licenced in patients with advanced colorectal cancer refractory to standard chemotherapy (31,32). Treatment with regorafenib is associated with radiological response rates of less than 5% and also non-trivial toxicities such as fatigue, hypertension, gastrointestinal perforation and palmar plantar erythrodysesthesia. Despite extensive research, no molecular biomarker exists to predict for response to regorafenib (33-35). As colorectal xenograft models suggested that regorafenib leads to decreased tumour vascularity when quantified by dynamic contrast enhanced (DCE)-magnetic resonance imaging (MRI), we established a translational phase II clinical trial using regorafenib in chemorefractory RAS mutant colorectal cancer patients with a view to understanding whether DCE-MRI dynamics could act as a radiological biomarker to predict outcomes in patients treated with regorafenib therapy (36,37). In the PROSPECT-R trial all patients were treated with standard dose of regorafenib, and patients on the study were mandated to have pretreatment biopsies (six cores targeted towards the MRI identified index lesion), another biopsy at 2 months (if response or stable disease by RECIST V.1.1 criteria) and at the time of progression (38). Participants who had metastatic lesions which were at least 2 cm in diameter and could safely undergo MRI underwent matched DCE and diffusion weighted (DW)- MRI at pretreatment and at day 15 on treatment. A significant drop in most DCE MRI parameters at two weeks supported the anti-angiogenic effect of regorafenib; in particular we focused on the KEF measurement [product of (Ktrans) and enhancing fraction (EF) (summarised median values of Ktrans × EF)]; which is a composite endpoint which corrects for therapy induced tumour volume changes (39). Using a ROC curve analysis, we defined a KEF drop of >70% as optimal to define two patient populations with differential responses to regorafenib (Figure 1). Patients with a decrease in KEF of >70% had improved progression free survival (PFS) and OS compared to those who did not demonstrate a similar fall in KEF value (PFS HR 0.16; 95% CI, 0.04 to 0.72, P=0.02, OS HR 0.08; 95% CI, 0.01 to 0.63, P=0.02). OS at one year was 75% in the >70% KEF drop group compared with 13.8% in the <70% drop group respectively. Contemporaneous biopsies allowed confirmation of the significance of the anti-angiogenic effect of regorafenib; a lower CD31 score on biopsy at 2 months was associated with superior PFS (HR 0.13; 95% CI, 0.03 to 0.52, P=0.004) and OS (HR 0.30; 95% CI, 0.08 to 1.06, P=0.06). In contrast, neither changes on DW-MRI nor tissue-based markers of apoptosis (caspase-3) on repeat biopsy were associated with disease control rates, PFS, or OS. Thus, the beneficial effects of regorafenib on overall survival appear to be driven primarily by angiogenesis. Finally, in order to evaluate whether liquid biopsy could be equivalent or superior to DCE MRI in predicting regorafenib treatment outcomes, we performed serial analysis of plasma using digital droplet PCR for RAS mutant clones. Whilst all treated patients demonstrated a reduction in fractional abundance of RAS mutation on initial treatment, only a sustained drop following two months of treatment was predictive of improved survival. This led us to conclude that KEF decrease at day 15 has a 6-week lead time advantage over liquid biopsy in predicting regorafenib treatment outcomes. In order to further validate our findings and explore the use of KEF derived from DCE-MRI as a predictive marker, we are now planning a larger, national phase II trial which will aim to validate KEF as an early predictor of response or resistance to regorafenib. This will be a phase II study of mCRC patients who have progressed on at least two lines of systemic therapy. All eligible patients will be offered regorafenib treatment and an optional tumour tissue biopsy will be performed prior to commencement of regorafenib and again at development of resistance. Early DCE-MRI along with diffusion weighted (DW)-MRI imaging at the beginning, and on day 15±7 post treatment will be performed in all participants. On day 15 DCE-MRI median KEF will be generated; patients with ≥70% drop in KEF will continue with regorafenib until progressive disease (PD) or intolerance to treatment. Patients with <70% KEF drop will also continue with regorafenib until progression. Patients in either group will be switched to trifluridine-tipiracil and will be treated until progression, if they had no previous exposure to this therapy and are deemed clinically suitable for it at the time of progression on regorafenib. Moreover, candidate markers of resistance will be identified in tissue biopsies through genetic and other molecular analysis and parallel collection of serial blood specimens will facilitate the identification of resistance markers and the tracking of resistance evolution in circulating nucleic acids. Disease will be monitored with serial DECT/CT-CAP every eight weeks, until PD. This study will be statistically powered to validate KEF as a biomarker of response to regorafenib therapy and may potentially take us a step closer to application of precision medicine in the field of gastrointestinal cancer medicine.
Figure 1.
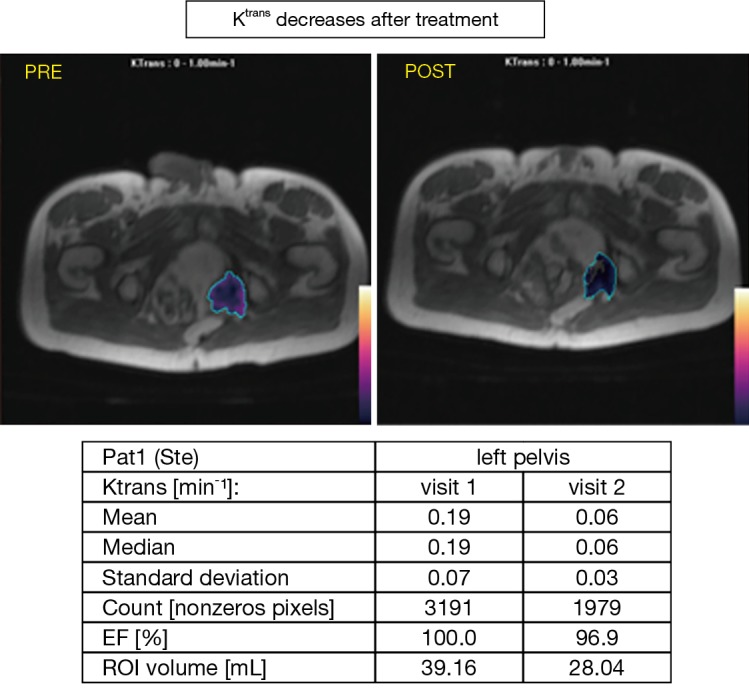
Decrease in Ktrans following regorafenib in MRI study.
Patient derived organoid models
Truly personalized medicine must reflect intra- and interpatient heterogeneity, and standard precision medicine using next generation sequencing has not yet delivered improved survival using targeted therapies for most gastrointestinal cancers. A co-clinical trial paradigm where patient response to treatment is predicted using personalized preclinical models is an attractive approach, however patient derived xenograft models are challenging to develop, expensive and unlikely to be adopted as clinical standards of care in the short to medium term. A realistic and pragmatic alternative to xenograft models are patient derived organoids, which are cultured from LRG5+ tumour stem cells propagated as epithelial organoids in vitro to provide multi-omic information and drug sensitivity testing (40). The extensive biopsy driven clinical trial portfolio we have undertaken has provided an opportunity to develop a patient derived organoid programme which has the potential to provide co-clinical trials and also predictive models for standard chemotherapy. As components of this programme we have developed patient derived organoids (PDOs) from liver, pelvic, peritoneal and nodal metastases and also from serial biopsies pre and post-treatment (Figure 2) (41). Using immunohistochemical markers, we showed that these PDOs were carried the same markers as the tissue from which they were derived (e.g., CDX-2, CK7 in colorectal). Underlying genetic aberrations such as ERBB2 and FGFR2 amplifications were also preserved. In total, an overlap of 96% between PDOs and the mutational spectrum of their parental biopsies, confirming the capability of organoids to act as surrogates for the primary tumour in drug discovery. Finally, PDOs have the capacity to capture intratumoural and temporal heterogeneity when multiple biopsies are performed, and are stable over multiple growth passages. These data suggest that co-clinical trials using organoids could be a cost-efficient and accurate method to determine individual patient sensitivity to chemo or targeted therapy.
Figure 2.
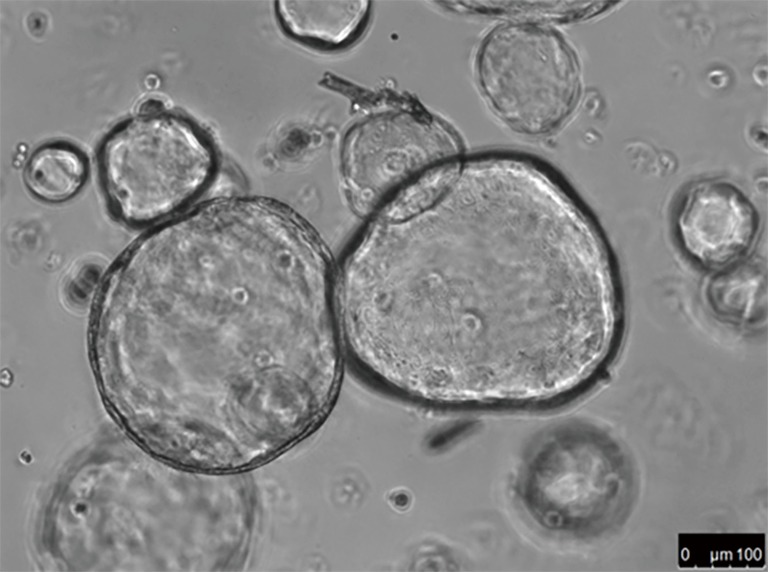
High magnification contrast phase image of PDOs (patient derived organoids) from a liver metastasis of a chemo-refractory CRC patient. CRC, colorectal cancer.
In drug screening assays using PDOs we confirmed expected sensitivities in molecularly selected PDO models, for example sensitivity to lapatinib in a ERBB2 amplified oesophago-gastric PDO and sensitivity to vemurafenib in a colorectal BRAF mutant PDO. Downstream signalling pathway analysis confirmed abrogation of MEK/ERK signalling in these models. We thus demonstrated that our PDO models can predict for response to molecularly targeted therapy. In terms of current clinical relevance, PDO may also be used to predict for response to standard cytotoxic chemotherapy. In one initially paclitaxel sensitive patient, serial biopsies of sensitive and subsequently resistant liver metastases demonstrated 4-fold lower (growth inhibitory 50) GI50 for paclitaxel in sensitive versus resistant PDO models. Furthermore, sensitivity of the PDO to paclitaxel on acquired resistance mirrored that of two PDOs acquired from patients who were primarily refractory to paclitaxel. Similar results were observed for cisplatin/5-fluorouracil sensitive and resistant models in the first line setting, emphasizing the applicability of this system over diverse settings and drug treatments.
In future, development of PDOs in a timely manner may be helpful in determining specific chemotherapy and targeted therapy regimens for gastrointestinal cancer patients. One example of the utility of this model system is exemplified by colorectal cancer treated with anti-EGFR inhibitors. In colorectal cancer although RAS mutation and possibly BRAF mutation predict for lack of cetuximab efficacy, many patients fail to respond to the anti-EGFR therapy even in the absence of these findings (5). We demonstrated in cetuximab refractory PDO models that markers that would be considered traditional predictors of response to cetuximab (e.g., EGFR amplification, high amphiregulin levels) were also found in patients with cetuximab refractory cancer. Thus in these patients PDO appeared to better predict outcome than standard molecular parameters alone. The established PDO programme also shows capacity to capture intra-patient heterogeneity in mCRC patients treated with cytotoxic chemotherapy. TAS-102 is a novel fluoropyrimidine used to treat chemorefractory CRC (42). In the same patient, GI50 from a TAS-102 responsive metastasis was 8-fold lower than the GI50 from a rapidly progressing metastasis, which also demonstrated high thymidine kinase-1 (TK1) expression (a putative resistance marker to TAS-102). Similar findings linking GI50 and TAS-102 sensitivity in a further three TAS-102 treated patients validated these findings. In total, for the PDOs that we generated, sensitivity, specificity, positive predictive and negative predictive values for predicting response to chemotherapy or targeted therapy were 100%, 93%, 88% and 100% respectively. These results strongly support integration of PDOs into clinical trials and in future, routine clinical care as predictors of response to treatment which could reduce needless toxicity and provide and allow for more effective use of resources.
Challenges of routine genomic assessment outside clinical trials
Despite these encouraging results, there are some limitations to the applicability of tumour molecular profiling in routine clinical practice. In particular, use of routine next generation sequencing (NGS) in the absence of downstream molecular trials may be an inefficient use of resources. At the Royal Marsden hospital, a prospective study, Feasibility of a Molecular Characterisation Approach to Treatment (FOrMAT) was established to investigate the feasibility of molecular profiling to Clinical Pathology Accreditation (CPA)-certified standards in gastrointestinal malignancies in routine clinical practice (43). Two hundred and twenty two patients were recruited between February 2014 and November 2015, most of whom had metastatic cancer (96.4%). Of the 222 patients, 215 had available tumour material, however only 61.2% had ≥1 gene successfully sequenced. As expected, resection samples were more likely to yield adequate DNA for NGS compared to biopsy samples (82.4% vs. 47.6%) and high cellularity and tumour content were also associated with an increased likelihood of obtaining a successful NGS result. Considering five genes to be “actionable” (TP53, KRAS, BRAF, PIK3CA and CDKN2A), 40.5% of sequenced patients were positive for this finding. However only two patients (<1%) entered a clinical trial based on the finding of an “actionable” mutation. These results highlight the challenges and perhaps futility of providing routine sequencing for patients with advanced gastrointestinal cancers outside the context of appropriate downstream targeted trials. In more than one third of patients sequencing could not be performed, most commonly due to lack of available tissue in a biopsy or inadequate DNA yield. However, as many biopsies were processed after routine diagnostic use whereby immunohistochemistry panels, RAS mutation status (CRC) or HER2 analysis (oesophago gastric had been performed, this is not surprising. If NGS were to be used routinely, consideration of the volume of diagnostic material obtained during work up for a cancer diagnosis might be reconsidered. Additionally sample processing times should be improved, as the median time from enrollment to obtaining a result was between 14 and 28 weeks depending on the cohort. Although this could be institution dependent, in view of the urgency of treatment of patients with metastatic cancers, these metrics would require significant improvement if this technology were to be mainstreamed. In clinical trials such as NCI-MATCH, a 7-day turnaround is reported, whereas intra-institutional results at MDACC are reported in approximately 26 days (44,45). Finally, the extremely low proportion of patients who entered matched clinical trial is lower than previously observed (3-13%) (45,46). Based on these findings, we conclude that outside the context of a definitive translational question or appropriate clinical trials, routine sequencing of patients with gastrointestinal cancers is an inefficient use of resources, in particular in a universal healthcare system.
Conclusions
This review aimed to focus on the recent successes and failures relating to translational research in gastrointestinal oncology in our institution over the past number of years. The infrastructure required to deliver these studies is substantial and requires collaboration between pathologists, radiologists, basic and translational scientists. In addition to this there must be clinical trials infrastructure to support prospective studies (Figure 3). We have also made significant investment in our biological specimen co-ordinator team who have standardised protocols for tissue and plasma sample collection across separate protocols. Collaboration with the pharmaceutical industry has also been important in driving both translational research and how this informs future trial design. The limitations of processing routine diagnostic samples are readily apparent in terms of the residual volume of tumour available and also the quality of this following routine formalin fixation and paraffin embedding. This can in part be overcome through growth in patient derived organoids, however this is not a widely available or a pragmatic choice currently. Our current research goals relate primarily to intensive translational assessment of patients in clinical trials, as molecular characterisation of patients in clinical practice has been demonstrated to be unfulfilling. The next generation of clinical trials will evaluate the interface between targeted therapy and the immune system. This is particularly important in view of the relatively low response rates which are observed with immune-oncology drugs in gastrointestinal cancers (approximately 12% for gastroesophageal cancer PD-1 inhibition and negligible in colon cancer) (47,48). For example, in a trial in development assessing the value of addition of HDAC inhibitor to the anti-PD-L1 inhibitor in gastroesophageal and colorectal cancer (EMERGE), we will perform pre-treatment, post 4SC-202/pre-avelumab biopsies and a third whilst patients are on combination therapy. Liquid biopsies will also form an important component of these serial assessments. The aim of such intensive biopsy programmes is to identify biomarkers associated with treatment response at an early stage of combination drug development to provide more refined cohorts for later trials. As such the translational component of the study should be developed concurrently with the clinical question posed by the trial. This structural design will allow the best use of resources and is the most efficient way to expedite patient benefit from drug development.
Figure 3.
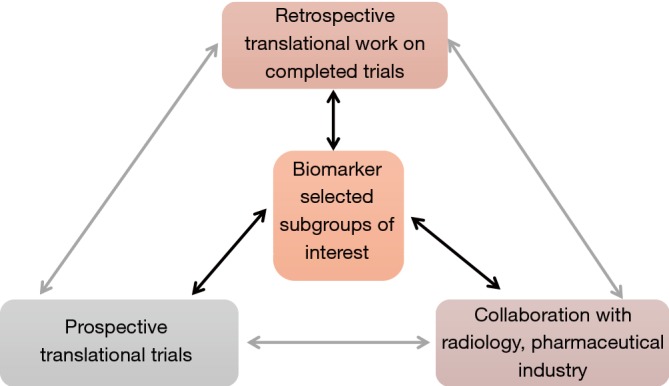
Iterative process of translational trial design.
Acknowledgements
None.
Footnotes
Conflicts of Interest: E Smyth discloses honoraria for advisory role from BMS, Servier, Five Prime Therapeutics, Gritstone Oncology and Celgene; K Khan discloses honoraria for advisor role from BMS; N Valeri discloses honoraria from Bayer, Merck, and Eli Lilly
References
- 1.Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 2008;358:36-46. 10.1056/NEJMoa073149 [DOI] [PubMed] [Google Scholar]
- 2.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817-25. 10.1056/NEJMoa1011923 [DOI] [PubMed] [Google Scholar]
- 3.Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol 2014;15:1065-75. 10.1016/S1470-2045(14)70330-4 [DOI] [PubMed] [Google Scholar]
- 4.Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010;376:687-97. 10.1016/S0140-6736(10)61121-X [DOI] [PubMed] [Google Scholar]
- 5.Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013;369:1023-34. 10.1056/NEJMoa1305275 [DOI] [PubMed] [Google Scholar]
- 6.Van Cutsem E, Lenz HJ, Kohne CH, et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 2015;33:692-700. 10.1200/JCO.2014.59.4812 [DOI] [PubMed] [Google Scholar]
- 7.Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med 2006;355:11-20. 10.1056/NEJMoa055531 [DOI] [PubMed] [Google Scholar]
- 8.Jackson C, Cunningham D, Oliveira J. Gastric cancer: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol 2009;20 Suppl 4:34-6. 10.1093/annonc/mdp122 [DOI] [PubMed] [Google Scholar]
- 9.Smyth EC, Verheij M, Allum W, et al. Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2016;27:v38-49. 10.1093/annonc/mdw350 [DOI] [PubMed] [Google Scholar]
- 10.Waddell T, Verheij M, Allum W, et al. Gastric cancer: ESMO-ESSO-ESTRO clinical practice guidelines for diagnosis, treatment and follow-up. Eur J Surg Oncol 2014;40:584-91. 10.1016/j.ejso.2013.09.020 [DOI] [PubMed] [Google Scholar]
- 11.Al-Batran SE, Homann N, Schmalenberg H, et al. Perioperative chemotherapy with docetaxel, oxaliplatin, and fluorouracil/leucovorin (FLOT) versus epirubicin, cisplatin, and fluorouracil or capecitabine (ECF/ECX) for resectable gastric or gastroesophageal junction (GEJ) adenocarcinoma (FLOT4-AIO): A multicenter, randomized phase 3 trial. J Clin Oncol 2017;35:abstr 4004.
- 12.Becker K, Langer R, Reim D, et al. Significance of histopathological tumor regression after neoadjuvant chemotherapy in gastric adenocarcinomas: a summary of 480 cases. Ann Surg 2011;253:934-9. 10.1097/SLA.0b013e318216f449 [DOI] [PubMed] [Google Scholar]
- 13.Noble F, Nolan L, Bateman AC, et al. Refining pathological evaluation of neoadjuvant therapy for adenocarcinoma of the esophagus. World J Gastroenterol 2013;19:9282-93. 10.3748/wjg.v19.i48.9282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mansour JC, Tang L, Shah M, et al. Does graded histologic response after neoadjuvant chemotherapy predict survival for completely resected gastric cancer? Ann Surg Oncol 2007;14:3412-8. 10.1245/s10434-007-9574-6 [DOI] [PubMed] [Google Scholar]
- 15.Smyth EC, Fassan M, Cunningham D, et al. Effect of pathologic tumor response and nodal status on survival in the medical research council adjuvant gastric infusional chemotherapy trial. J Clin Oncol 2016;34:2721-7. 10.1200/JCO.2015.65.7692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polom K, Marano L. Meta-analysis of microsatellite instability in relation to clinicopathological characteristics and overall survival in gastric cancer. Br J Surg 2018;105:159-67. 10.1002/bjs.10663 [DOI] [PubMed] [Google Scholar]
- 17.Smyth EC, Wotherspoon A, Peckitt C, et al. Mismatch repair deficiency, microsatellite instability, and survival: an exploratory analysis of the medical research council adjuvant gastric infusional chemotherapy (MAGIC) Trial. JAMA Oncol 2017;3:1197-203. 10.1001/jamaoncol.2016.6762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi YY, Kim H, Yang HK, et al. Clinical impact of microsatellite instability in patients with stage II and III gastric cancer: Results from the CLASSIC trial. J Clin Oncol 2017;35:4022. [Google Scholar]
- 19.Bang YJ, Kim YW, Yang HK, et al. Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet 2012;379:315-21. 10.1016/S0140-6736(11)61873-4 [DOI] [PubMed] [Google Scholar]
- 20.Le DT, Durham JN. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409-13. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372:2509-20. 10.1056/NEJMoa1500596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cunningham D, Stenning SP, Smyth EC, et al. Peri-operative chemotherapy with or without bevacizumab in operable oesophagogastric adenocarcinoma (UK Medical Research Council ST03): primary analysis results of a multicentre, open-label, randomised phase 2-3 trial. Lancet Oncol 2017;18:357-70. 10.1016/S1470-2045(17)30043-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waddell T, Chau I, Cunningham D, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open-label phase 3 trial. Lancet Oncol 2013;14:481-9. 10.1016/S1470-2045(13)70096-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol 2017;18:75-87. 10.1016/S1470-2045(16)30559-9 [DOI] [PubMed] [Google Scholar]
- 25.Lin K, Sun J, Maloney L, et al. 2701 Quantification of genomic loss of heterozygosity enables prospective selection of ovarian cancer patients who may derive benefit from the PARP inhibitor rucaparib. Eur J Cancer 2015;51:S531-2. 10.1016/S0959-8049(16)31469-1 [DOI] [Google Scholar]
- 26.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023-31. 10.1038/nbt.2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cafferkey C, Smyth E, Loehr A, et al. Genomic loss of heterozygosity (LOH) and survival in patients (pts) treated with epirubicin, oxaliplatin, capecitabine (EOC)±panitumumab (P) in the REAL3 trial. Ann Oncol 2016;27:207-42. 10.1093/annonc/mdw371.41 [DOI] [Google Scholar]
- 28.Pearson A, Smyth E, Babina IS, et al. High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov 2016;6:838-51. 10.1158/2159-8290.CD-15-1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smyth EC, Turner NC, Peckitt C, et al. Phase II multicenter proof of concept study of AZD4547 in FGFR amplified tumours. J Clin Oncol 2015;33:abstr 2508.
- 30.Maron SB, Joshi SS, Lomnicki S, et al. Circulating tumor DNA (ctDNA) landscape and prognostic implications in advanced gastroesophageal adenocarcinoma (GEC). J Clin Oncol 2018;36:45. [Google Scholar]
- 31.Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381:303-12. 10.1016/S0140-6736(12)61900-X [DOI] [PubMed] [Google Scholar]
- 32.Li J, Qin S, Xu R, et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2015;16:619-29. 10.1016/S1470-2045(15)70156-7 [DOI] [PubMed] [Google Scholar]
- 33.Tabernero J, Lenz HJ, Siena S, et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol 2015;16:937-48. 10.1016/S1470-2045(15)00138-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giampieri R, Salvatore L, Del Prete M, et al. Angiogenesis genotyping and clinical outcome during regorafenib treatment in metastatic colorectal cancer patients. Sci Rep 2016;6:25195. 10.1038/srep25195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suenaga M, Mashima T, Kawata N, et al. Serum VEGF-A and CCL5 levels as candidate biomarkers for efficacy and toxicity of regorafenib in patients with metastatic colorectal cancer. Oncotarget 2016;7:34811-23. 10.18632/oncotarget.9187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cyran CC, Kazmierczak PM, Hirner H, et al. Regorafenib effects on human colon carcinoma xenografts monitored by dynamic contrast-enhanced computed tomography with immunohistochemical validation. PLoS One 2013;8:e76009. 10.1371/journal.pone.0076009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kazmierczak PM, Burian E, Eschbach R, et al. Monitoring cell death in regorafenib-treated experimental colon carcinomas using annexin-based optical fluorescence imaging validated by perfusion MRI. PLoS One 2015;10:e0138452. 10.1371/journal.pone.0138452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khan K, Rata M, Cunningham D, et al. Functional imaging and circulating biomarkers of response to regorafenib in treatment-refractory metastatic colorectal cancer patients in a prospective phase II study. Gut 2017. [Epub ahead of print]. 10.1136/gutjnl-2017-314178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferl GZ, O'Connor JP, Parker GJ, et al. Mixed-effects modeling of clinical DCE-MRI data: application to colorectal liver metastases treated with bevacizumab. J Magn Reson Imaging 2015;41:132-41. 10.1002/jmri.24514 [DOI] [PubMed] [Google Scholar]
- 40.Bredenoord AL, Clevers H, Knoblich JA. Human tissues in a dish: The research and ethical implications of organoid technology. Science 2017;355. [DOI] [PubMed] [Google Scholar]
- 41.Vlachogiannis G, Hedayat S, Vatsiou A, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018;359:920-6. 10.1126/science.aao2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 2015;372:1909-19. 10.1056/NEJMoa1414325 [DOI] [PubMed] [Google Scholar]
- 43.Moorcraft SY, Gonzalez de Castro D, Cunningham D, et al. Investigating the feasibility of tumour molecular profiling in gastrointestinal malignancies in routine clinical practice. Ann Oncol 2018;29:230-6. 10.1093/annonc/mdx631 [DOI] [PubMed] [Google Scholar]
- 44.Conley BA, Gray R, Chen A, et al. Abstract CT101: NCI-molecular analysis for therapy choice (NCI-MATCH) clinical trial: interim analysis. Cancer Res 2016;76:CT101 10.1158/1538-7445.AM2016-CT101 [DOI] [Google Scholar]
- 45.Meric-Bernstam F, Brusco L, Shaw K, et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol 2015;33:2753-62. 10.1200/JCO.2014.60.4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andre F, Bachelot T, Commo F, et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol 2014;15:267-74. 10.1016/S1470-2045(13)70611-9 [DOI] [PubMed] [Google Scholar]
- 47.Kang YK, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390:2461-71. 10.1016/S0140-6736(17)31827-5 [DOI] [PubMed] [Google Scholar]
- 48.O'Neil BH, Wallmark JM, Lorente D, et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLoS One 2017;12:e0189848. 10.1371/journal.pone.0189848 [DOI] [PMC free article] [PubMed] [Google Scholar]