

Abstract
Myotonic dystrophy type 1 (DM1) and 2 (DM2) are autosomal dominant degenerative neuromuscular disorders characterized by progressive skeletal muscle weakness, atrophy, and myotonia with progeroid features. Although both DM1 and DM2 are characterized by skeletal muscle dysfunction and also share other clinical features, the diseases differ in the muscle groups that are affected. In DM1, distal muscles are mainly affected, whereas in DM2 problems are mostly found in proximal muscles. In addition, manifestation in DM1 is generally more severe, with possible congenital or childhood-onset of disease and prominent CNS involvement. DM1 and DM2 are caused by expansion of (CTG•CAG)n and (CCTG•CAGG)n repeats in the 3′ non-coding region of DMPK and in intron 1 of CNBP, respectively, and in overlapping antisense genes. This critical review will focus on the pleiotropic problems that occur during development, growth, regeneration, and aging of skeletal muscle in patients who inherited these expansions. The current best-accepted idea is that most muscle symptoms can be explained by pathomechanistic effects of repeat expansion on RNA-mediated pathways. However, aberrations in DNA replication and transcription of the DM loci or in protein translation and proteome homeostasis could also affect the control of proliferation and differentiation of muscle progenitor cells or the maintenance and physiological integrity of muscle fibers during a patient’s lifetime. Here, we will discuss these molecular and cellular processes and summarize current knowledge about the role of embryonic and adult muscle-resident stem cells in growth, homeostasis, regeneration, and premature aging of healthy and diseased muscle tissue. Of particular interest is that also progenitor cells from extramuscular sources, such as pericytes and mesoangioblasts, can participate in myogenic differentiation. We will examine the potential of all these types of cells in the application of regenerative medicine for muscular dystrophies and evaluate new possibilities for their use in future therapy of DM.
Keywords: myotonic dystrophy, myogenesis, mesoangioblast, myoblast, muscle stem cell, pericyte, proteotoxicity, RNA toxicity
Introduction
Skeletal muscle formation, growth, and maintenance in vertebrates are dynamic processes in terms of tissue differentiation, remodeling, repair, and regeneration. During the different phases of life, muscle may suffer due to injury or disease, causing weakness, pain, or paralysis, which may be even fatal. Muscle problems may be acute or short-lived, like during an infection, or be long-lasting, as in chronic disorders. Patients with inherited myopathy or muscular dystrophy, a heterogeneous group of disorders for which disease etiology is rooted in the genetically abnormal pathways that control formation and physiological integrity of skeletal muscle, commonly experience progressive muscle weakness and atrophy (i.e., loss of muscle mass). As a result, physical strength and independence are lost, which causes substantial morbidity over decades. For the development of novel therapies to halt or reverse progression of muscle problems, validated classification criteria for differential clinical diagnosis and detailed preclinical knowledge about what is going wrong at the molecular and genetic level are a prerequisite. Unfortunately, the current states of clinical and fundamental understanding—and hence the prospects for treatment—vary enormously between individual myopathies and dystrophies.
This review is meant to bring new background knowledge for myotonic dystrophy (DM). DM is one of the most prevalent and probably also one of the most difficult to understand genetic disorders, due to its heterogeneity and its highly complex and variable clinical manifestation and molecular etiology. DM is the collective name for a disease with two genetic subtypes, DM1 (OMIM #160900) and DM2 (OMIM #602668). In fact, the classification as a skeletal muscle dystrophy is only partially correct, as the disease also has neuromuscular character and cardiac, CNS and endocrine problems are commonly involved as well (1–3). Here, we will only briefly recapitulate the history of clinical and molecular research in DM as multiple comprehensive reviews have been published on this subject (1, 2, 4, 5). The focus here is on a (re)examination of studies related to the molecular and histomorphological problems that occur during growth, maintenance, and aging of skeletal muscles in patients with DM. Findings in animal model studies are included only if they faithfully reflect the muscular pathophysiology in DM patients (6–8).
The main waves of myogenesis occur during embryonic development and growth, when myoblasts undergo cell cycle arrest and fuse to form the multinucleated myotubes that ultimately become the mature myofibers (9–11). Later, regenerative myogenesis serves in muscle turnover and to replace damaged or diseased muscle (10). Relevance of embryonic and adult stem cells for each of the distinct phases of myogenesis for the manifestation of DM will be examined. We will also describe so-called non-somite skeletal myogenesis through involvement of mesoangioblasts (MABs) and pericytes (PCs) as muscle progenitor cells, and speculate about the importance of this process for DM. Finally, we will discuss possibilities to use these progenitor cells in future therapeutic strategies.
Myotonic Dystrophy
Clinical Features and Genetic Causes
A number of clinical and molecular characteristics are shared between DM1 and DM2, but the differences prevail and render them distinct disorders.
Myotonic Dystrophy Type 1
Myotonic dystrophy type 1, or Steinert’s disease, shows the highest prevalence, ranging between 0.5 and 18 cases per 100,000 individuals among different ethnic populations (12–14). Progressive muscle weakness and atrophy of the distal muscles together with myotonia are consistent features. Multiple other organs in the body can also be affected, causing combinations of symptoms. For example, heart failure due to conduction problems, insulin resistance, excessive sleepiness, intellectual disability or mental problems, and cognitive deficits are common symptoms (15–18). Anticipation is typical for DM1, which means that disease problems become more severe and occur earlier in successive generations in families. Nowadays, five partially overlapping clinical subtypes of DM1 are recognized, based on the occurrence and onset of the main symptoms: congenital (cDM), infantile, juvenile, adult, and late-onset/asymptomatic DM1 (19). This classification is not only important for patient care but also for the design of clinical trials (2). For a fair interpretation of the literature cited in this review, it is important to note that in studies that appeared before the recent redefinition and refinement of disease classes, authors mostly only discriminated between cDM and adult-onset DM1.
The sole known molecular cause of DM1 is the expansion of a (CTG•CAG)n sequence on chromosome 19q13 in the last exon of DMPK (20, 21) (Figure 1). In DM1 families, when expanded to a length above (CTG)37, the repeat is unstable and has a tendency to grow somatically and intergenerationally (22, 23). Thus, repeat expansion forms the basis for the anticipation phenotype, whereby a longer repeat correlates with more severe symptoms and an earlier disease onset. An expanded DMPK repeat is mostly an uninterrupted (CTG)n sequence of variable length. However, additional sequence variations such as CCG and CGG triplets in the 3′ end or immediate flanking DNA, or non-CTG replacements within the repeat have been found. These alterations are generally associated with milder disease manifestation and symptomatic variation in families or seem to occur somatically in certain tissues (24–26).
Figure 1.

Distinct molecular mechanisms contribute to pathology in myotonic dystrophy type 1 (DM1) and myotonic dystrophy type 2 (DM2). (1) Expanded (CTG)n and (CCTG)n repeats in DMPK and CNBP, or the complementary repeats in the antisense genes (not shown), can cause cellular stress by (1) promoting DNA replication fork stalling and R-loop formation. Expression of repeat-containing sense and antisense RNAs results in (2) sequestration of members of the MBNL protein family, leading to mRNA missplicing, alternative polyadenylation, microRNA (miRNA) deregulation, and transcription deregulation. In addition, (3) CELF1 gets hyperphosphorylated and stabilized, resulting in mRNA missplicing and dysregulation of mRNA stability and translation. (4) Formation of abnormal RNA-protein condensates by repeat RNA and RNA-binding proteins (RBPs) may alter the intracellular distribution fate and biological activity of RBPs. (5) Repeat-associated non-ATG (RAN) translation of the repeats may result in the production of toxic polymeric polypeptides, which perturb cellular proteostasis.
From the normal and mutant DMPK alleles multiple alternatively spliced transcripts are produced, all of which contain the (CUG)n repeat sequence in their 3′ untranslated region (UTR) (27). In addition, there is a partial overlap with an antisense-oriented gene, named DM1-AS, which encodes variant (CAG)n transcripts with characteristics of long non-coding RNA (lncRNA) (28).
Myotonic Dystrophy Type 2
Formerly known as proximal myotonic myopathy and proximal myotonic dystrophy, DM2 was discovered in a group of patients with clinical features that were slightly different from those in DM1 (29, 30). Prevalence for DM2 varies strongly by population, but is less well known than for DM1, since the mild DM2 phenotype often goes undiagnosed (5). As mutations have been predominantly identified in Caucasians in Northern Europe and this population also has the most registered DM2 patients (31, 32), prevalence of DM2 and DM1 may be quite similar in countries in this region (33). Although the myotonic dystrophies share a number of clinical symptoms, there are distinct differences (34, 35) (Table 1). For DM2 no congenital manifestation is known and diagnosis is always late, when patients have reached adult age. Myotonia is less evident and myotonia of grip often has a jerky quality (36). Proximal muscles are most prominently affected in DM2 and weakness and wasting of facial muscles and limbs is generally mild (29, 30, 36).
Table 1.
Similarities and differences in genetic, clinical, and histopathological features of myotonic dystrophy type 1 (DM1) and myotonic dystrophy type 2 (DM2).
| DM1 | DM2 | Reference | |
|---|---|---|---|
| Main features | |||
| Affected gene, chromosome | DMPK; 19q13.3 | CNBP; 3q21 | (20, 32) |
| Repeat expansion | (CTG)n | (CCTG)n | (20, 37) |
| Anticipation | Always present | Exceptional | (38) |
| Age of onset | Any age | Adulthood | (19) |
| Congenital form | Yes | No | (19) |
| Muscle symptoms | |||
| Predominant muscle weakness | Distal | Proximal | (39) |
| Predominantly affected muscle fibers | Type 1 | Type 2 | (40–42) |
| Histopathological findings | |||
| Fiber atrophy | Type 1 fibers (not always present) | Subgroup of highly atrophic type 2 fibers (always present) | (30) |
| Nuclear clump fibers | In end stage only | Scattered at early stage | (43) |
| Sarcoplasmic masses | Frequent in distal muscles | Extremely rare | (43) |
| Ring fibers | Frequent | May occur | (43) |
| Internal nuclei | Massive in distal muscle | Variable, mainly in type 2 fibers | (43) |
Similar to DM1, only one underlying cause of disease has been identified for DM2: all patients carry an expansion of a (CCTG)n repeat in intron 1 of CNBP (previously known as ZNF9) on chromosome 3q21 (29, 30) (Figure 1). The repeat is part of a complex (TG)n(TCTG)n(CCTG)n motif in which the (CCTG)n repeat is often interrupted and consists of up to 26 units in healthy individuals. In patients the (CCTG)n repeat is usually uninterrupted and contains 75–11,000 quadruplets (36). The DM2 repeat is extremely unstable and has a tendency to expand somatically, causing length increase and cell-to-cell heterogeneity during a patient’s life. Interestingly, by contrast to the behavior of the (CTG•CAG)n repeat in DM1, the (CCTG•CAGG)n repeat has the tendency to contract intergenerationally (44). The correlation between repeat length and disease severity is less strong than in DM1 patients and anticipation is less evident (1, 38).
Molecular Mechanisms Involved in the Etiology of DM1 and DM2
Several molecular mechanisms are thought to contribute to the muscular pathogenesis of DM throughout all phases of development and maintenance (Figure 1). Similarities with other neurological disorders that are caused by microsatellite expansions have already been comprehensively reviewed (4, 8, 18, 45, 46). Here, we aim to accentuate the relationships between the molecular and cellular levels at which problems caused by the repeat expansions may occur. The emphasis is biased toward the pathobiology of DM1, based on a longer history of study, its seemingly bigger variability and complexity of manifestation, and the broader availability of patient materials, and cell and animal models.
Problems at the Chromatin Level
The first level at which repeat expansion may contribute to disease is at the chromatin level. The (CTG•CAG)n repeat in DM1 is situated within the 3′ UTR of DMPK, within the overlapping antisense DM1-AS gene and in the promoter of SIX5 (formerly known as DMAHP). These genes lie in the center of a gene-rich region of chromosome 19, spanning also DMWD (47, 48), RSHL1, and SYMPLEKIN, within a chromatin loop that is flanked by nuclear matrix attachment regions (49). Two binding sites for the transcriptional repressor CTCF with an insulator role in regulation of transcription and chromatin architecture are within this loop, flanking the repeat area. Already soon after the discovery of the repeat, Otten and Tapscott demonstrated that long (CTG•CAG)n repeats are strong nucleosome positioning elements (50). Extreme repeat expansion as in cDM leads to the occlusion of adjacent DNase hypersensitive sites and concomitant changes in local DNA methylation in the surrounding CG-rich region (51–53), rendering the chromatin more heterochromatic and inaccessible. In turn, this process has cis-effects on gene activity in the immediate vicinity, including DMPK, DM1-AS, and SIX5 and perhaps other neighboring genes. To our knowledge, no similar studies of epigenetic changes after repeat expansion in CNBP (DM2) exist. Clearly, more work is needed to understand the biological effects that DNA methylation, histone modification and other chromatin changes due to repeat expansion in the DM1 locus have on muscle progenitor cells.
Problems at the DNA Level: Stalled Replication Forks and R-Loops
Numerous studies have addressed DNA instability of expanded (CTG•CAG)n and (CCTG•CAGG)n repeats. The influence of oxidative damage and mismatch-repair and recombination pathways for DNA repair on repeat instability have already been thoroughly discussed (54–56). Less attention has been focused on the types of cell stress that large repeats may have at the DNA level and their consequences for loss of cell viability.
DNA polymerase stalling and replication fork arrest seem to be frequent events when unusually large repeat sequences in the genome have to be replicated in S-phase (57). Cells have adequate repair systems to resolve problems with DNA replication fork processivity, either directly when proceeding through the cell cycle or later when they arrive at so-called DNA replication checkpoints (58). Different rescue systems exist in which Chk1 and γH2AX phosphorylation and p53 activation are crucial for the on-site response (58). Stalling at sites in eu- and heterochromatin may even require differential composition of the repair machinery that is recruited. For transcribed repeats, as in the DM1 and DM2 loci, there is an additional complication. Here the threat comes from the formation of so-called R-loops (59). R-loops are triple-stranded RNA-DNA structures formed by duplex formation between the template strand and the transcribed RNA, leaving the non-template strand unpaired. R-loop formation may influence DNA methylation and transcriptional activity in its immediate vicinity. Persistent presence of unresolved R-loops or structures wherein stalled DNA forks and R-loops coincide may affect cellular fitness and arrest the cell cycle. The associated stress may even cause cell death.
An elegant study indeed showed that transcription of a (CTG•CAG)n repeat, as in the DM1 locus, may cause convergent repeat instability and apoptosis (60). Against this background, it is tempting to speculate that proliferating cells in which DMPK and/or DM1-AS are expressed are vulnerable to the danger of formation of stalled replication forks and R-loops. Specifically, this holds for all mesodermal derivatives and embryonic and adult muscle stem cells [muscle-resident stem cells (MuSCs); see below]. An identical pathogenic cascade may be possible in DM2, since CNBP is most highly expressed in muscle (61). There is evidence for bidirectional transcription across the locus (62) and unpaired (CCT/UG)n or (CAGG)n repeats may form abnormal hairpin structures (63).
Misregulation of RNA Processing and Translation
By far the most intensely studied aspects of DM’s etiology are the pleiotropic problems caused by the production of repeat-expanded transcripts. Intranuclear residence of repeat transcripts causes trans effects, which culminate in abnormal processing of many other RNAs in the cell’s transcriptome (64).
Probably right after transcription, the repeats in RNAs of DMPK and CNBP (and the corresponding antisense genes) form stable hairpins that alter activities of two antagonistic protein families, the MBNL (Muscleblind) and CELF proteins. MBNL proteins bind anomalously across the repeat hairpin, leading them to become sequestered in nuclear aggregates, which are visualized as so-called foci under the microscope (65–71). Various other RNA-binding proteins (RBPs) such as hnRNP F, H, DDX5, -6, -17, and Staufen, some of which have intrinsically unstructured domains, are engaged in the nuclear aggregates as well (71–74). CELF1, formerly called CUGBP1, binds at the base of the hairpin and becomes hyperphosphorylated.
Altogether, these events result in an imbalance in cellular ribostasis and proteostasis, associated with depletion and a shift in the distribution of MBNL family members and an increase and redistribution of CELF1 protein. The end result is a cell type and cell state dependent whole-transcriptome effect on alternative splicing (75–78), alternative polyadenylation (79, 80), and nucleocytoplasmic transport of other transcripts for which MBNL1-3 or CELF1 play a role in RNA processing. Changes in mRNA half-life may also occur, as CELF1 has been identified as a key regulator of RNA decay or translational silencing in muscle cells (81). In turn, the changes in the transcriptome have widespread trans-acting effects on the production and makeup of multiple proteins (82–86). Some cell-stage effects of MBNL1-3, CELF1, and other ribonucleoprotein (RNP) anomalies will be discussed in more detail below, in the context of embryonic or regenerative myogenesis.
Missplicing may have the most obvious links with the myopathy in DM. For instance, abnormal splicing of ClC1 is sufficient to cause myotonia (87). Missplicing of the muscle-specific genes BIN1, TNNT3, RYR1, TTN, LDB3, and SERCA1 is linked to impaired muscle function (88). Aberrant splicing of the insulin receptor, highly expressed in skeletal muscle, results in reduced responsiveness to insulin, another contributing factor to skeletal muscle dysfunction (89–91). Furthermore, alternative splicing of CACNA1S, a calcium channel that controls skeletal muscle excitation-contraction coupling, is markedly repressed in DM1 and DM2 (92). Combined with splicing alterations in the machineries for voltage-induced Ca2+ release and for release and uptake of Ca2+ in the ER/SR store (RyR1 and SERCA1), this may lead to chronic Ca2+ overload, activate ER stress (93), or become a cause of excitotoxicity. These long-term physiological abnormalities may induce premature senescence and contribute to muscle degeneration in DM.
Not all splicing abnormalities are congruent in DM1 and DM2 muscles. For instance, TNNT3 is more often misspliced in DM2 than in DM1, and NCAM1 missplicing can be found more in nuclear clump fibers of DM2 patients (1, 94, 95). Furthermore, in muscle tissue of DM2 patients, NEDD4 was found to be disrupted. NEDD4 is an E3 ubiquitin ligase for PTEN, an important regulator of the AKT signaling pathway for protection against cellular stress. The PTEN protein level is upregulated in DM2 muscle tissue and PTEN accumulations can be found in nuclear clump 2a fibers in DM2 muscle (96).
For DM2, there may be also a direct effect on ribostasis and proteostasis. Repeat expansion in CNBP may cause pausing of transcription or retardation of splicing of its pre-mRNA, resulting in a reduction of mature CNBP mRNA and the CNBP protein product. Initial studies on this topic yielded conflicting results, as some groups found unaltered levels of CNBP RNA and protein levels in cells and tissues from DM2 patients, whereas later studies showed a clear inhibitory effect of an expanded (CCTG)n repeat (97). Studies on heterozygous knockout mice for CNBP brought further support for the idea that haploinsufficiency may be involved in myopathy in DM2 (98). The CNBP protein has a role in the regulation of translation through binding to the 5′ UTRs of terminal oligopyrimidine tract mRNAs. For example, the production of RPS17, poly(A)-binding protein 1, and elongation factors eEF1A and eEF2 are controlled by this mechanism (99).
Also other types of problems at the translational level may play a role in the distinct manifestation of DM1 and DM2. Differential involvement of CELF1 may herein be a key issue. CELF1 can act by relieving secondary structures on a subset of target RNAs that exhibit G-rich sequence stretches with a high-degree of secondary structure, thereby promoting their translatability. Furthermore, if (hyper)phosphorylated, CELF1 may form a multisubunit complex with eukaryotic initiation factor eIF2 and other translation initiation factors, promoting the translation of protein products from alternative start codons in mRNAs that bear an IRES motif (100, 101). Importantly, the different effects of CELF1 on the translation of target mRNAs depend on its phosphorylation status and on the overall level of available protein, which is controlled in accordance with the stage of myogenic differentiation. Although there is no consensus about the fate of CELF1 in DM1 and DM2 muscles, evidence points to a situation in which the available level and thus binding of CELF1 to mRNAs is reduced in DM2. By contrast to the situation in DM1, its phosphorylation status appears unaltered in DM2. When taken combined, these studies support the idea that, superimposed on aberrancies in RNA splicing and polyadenylation, aberrancies in protein translation might have distinct roles in eliciting muscle dysfunction in both forms of DM (99, 102).
RNP Condensates: Is Phase Separation of Repeat RNA Causing Cell Stress?
Revolutionary work on polymer physical properties of macromolecular assemblies that undergo liquid-to-gel phase transition and concentration into microscale structures have led to the idea that formation of abnormal condensates by repeat transcripts and RBPs may also be involved in repeat RNA toxicity in DM (103–105). Jain and Vale have recently provided evidence that poly-CUG RNA and also poly-CAG RNA, which both can engage in multivalent intra- and intermolecular reactions, can undergo phase separation in vitro (106). They also showed that (CUG)n RNA forms small phase-separated gel inclusions in cells.
More basic studies into the thermodynamics of phase transition have revealed that the threshold concentration at which nano-sized biomolecular RNP condensates are formed are determined by various parameters, including the type, stoichiometry and local concentration of available RNA, and protein constituents and their folding or solubility properties. Most of these studies have been focused on phase transition under conditions with high concentrations of RNA and protein. Future research must thus reveal the requirements for RNA-protein condensate assembly and phase transition in patient cells with endogenous levels of expanded RNAs. Most importantly, the question must be answered whether the occurrence of abnormal repeat RNP gel inclusions containing DMPK, DM1-AS, or CNBP mRNA with abnormal repeat length could by itself be a trigger for stress.
Repeat-Associated Non-ATG (RAN) Translation
Since its discovery in 2011, RAN translation has been linked to proteome abnormalities in multiple repeat-expansion disorders (107). RAN translation of expanded triplet or quadruplet repeats can occur in all reading frames, resulting in the production of homopolymeric (DM1) or poly-tetrapeptide (DM2) proteins (62, 108, 109). In DM1, polyglutamine nuclear aggregates have been identified in myoblasts, skeletal muscle and peripheral blood leukocytes of patients, and in DM1 mouse tissue (108). In DM2, RAN translation across the (CCUG)n and antisense (CAGG)n repeats produces toxic poly-LPAC in neurons, astrocytes, and glia cells, while poly-QAGR proteins accumulate in white matter (62). Whether these findings can be extrapolated to DM2 muscle is an open question.
Many other unanswered questions remain about the production and relevance of RAN products in DM. How does an intronic RNA segment that is normally retained in the nucleoplasm and—without repeat gets quickly degraded—become accessible for the ribosome machinery? A similar question can be asked for DM1, since also expanded DMPK and DM1-AS RNAs are mainly retained within the nucleus, unavailable for assembly of ribosomes and subsequent translation (28). Nuclear translation is a process that has been demonstrated to occur (110, 111), but at this moment we do not know whether this could be involved. Another possibility is that the initiation of RAN translation occurs only after the onset of prometaphase in cycling cells, so when ribosome subunits are accessible because nucleoplasm and cytosol can mix. Indeed, at mitotic entry, cap-independent translation acquires a dominant role in expression regulation (112). Once polymeric proteins have been produced by RAN translation, they may—alike prion proteins—have a seeding effect in triggering abnormal protein aggregation and condensation and cause imbalance in the cellular proteome (62, 107). This may come at a considerable fitness cost for the cell in which it occurs.
Cellular Mechanisms Involved in the Etiology of DM1 and DM2
Quantitative and Qualitative Aspects Do Matter
Any of the molecular disease pathways discussed above could contribute to the myopathy during the different phases of life of patients with DM (Figure 2). However, one should realize that their involvement at the cellular level may differ dramatically with the stage of development and with the type of myofiber that is formed during muscle growth, regeneration, and aging. For example, stalling of replication forks at the (CTG•CAG)n and (CCTG•CAGG)n repeats may not be major threats in quiescent cells, but danger may increase once cells start proliferating. Similarly, reciprocal coupling does exist between the stage and type of differentiation and the mode and extend of alternative splicing in individual muscle progenitor cells or myofibers. The level of DMPK and CNBP transcripts, splicing factors, or their mRNA targets do, however, vary during muscle differentiation and maturation. So in muscle cells from cDM, DM1, or DM2 patients the complex changes in stoichiometric ratios between MBNL1-3, CELF1, and other RBPs, and the DMPK or CNBP RNA molecules that take place during natural development are superimposed by variable toxic changes caused by abnormal RBP-repeat RNA interactions (Figure 1).
Figure 2.

Abnormalities in skeletal muscle myogenesis in myotonic dystrophy (DM). For myotonic dystrophy type 1 (DM1), five clinical subtypes have been identified (19), while for myotonic dystrophy type 2 (DM2) only the adult-onset manifestation is known. The myogenic process in skeletal muscle is divided in two prenatal and four postnatal stages. The graphic summarizes which stages of the pre- and postnatal myogenic process are affected in each clinical DM (sub)type. A discontinuous bar indicates decreased life expectancy.
New supportive evidence for a mutual relationship between differentiation abnormalities and repeat expansion effects was obtained by our group in a study of isogenic CRISPR/Cas9-edited DM1 muscle cells with and without (CTG•CAG)2600 repeat (113). Monitoring of the molecular causes and cellular effect at the individual cell level, during in vitro myocyte–myotube differentiation and maturation in culture should thus become possible. Answering the chicken-egg question whether the impaired differentiation and regeneration events or the RNA processing abnormalities and associated cell stress were first in initiating the pathology in DM muscle tissue is not easy. Heterogeneity in cell type composition and developmental stage in the muscle cell population is here the confounding factor. In the next sections, we will try to provide background information on aspects of normal myogenesis and the cellular pathology and histopathology of DM muscle, to come closer to the root of this problem.
Muscle Fiber Type and Developmental-Stage Dependent Manifestation of Disease
Within human skeletal muscle there are different categories of fiber types, defined by myosin heavy chain (MyHC) isoform expression and metabolic activity (114). Individual fibers are characterized as one type of slow-twitch fiber (type 1) and three types of fast-twitch fibers [type 2a, 2c, and 2x/d (also referred to as 2b)] (115). Type 1 and 2a fibers are oxidative, whereas type 2c and 2x/d fibers are primarily glycolytic. Type 2 fibers generally produce higher forces and fatigue more quickly than type 1 fibers (116). Walled off from the main part of the muscle in the muscle spindle, highly specialized fibers, known as intrafusal fibers, can be found. These fibers serve as specialized stretch receptors that allow the perception and coordination of limb movement.
Most muscles in the human body are built as a mixture of type 1 and 2 fibers, but between individuals there are marked differences in muscle composition and size. Fiber type content and distribution is thereby coupled to aspects of physical performance, such as endurance and strength. Hence, there is also differential association with disease risk or states between individuals, as skeletal muscle fiber subtypes respond differently to (patho)physiological signals, which include atrophy signals. The ratio of type 1 and 2 fibers within a muscle is altered in muscular disorders when atrophy of one of the two types occurs. Several signaling pathways for muscle atrophy are known, mostly related to abnormalities in protein degradation (117). However, the selectivity of fiber type atrophy remains an unresolved issue (118, 119). For DM, the fiber type specificity of manifestation is a topic that deserves new attention, especially since revolutionary methodologies for transcriptome, proteome, and microscopy analyses at the single cell level have become available.
Skeletal muscles from all DM patients have a distinct histopathological phenotype, but biopsies show conspicuous differences between DM1 and DM2 patients (Table 1). The distal muscles mainly affected in adult DM1 show predominant loss of type 1 fibers (120), whereas the predominantly affected proximal muscles in DM2 show mostly type 2 fiber atrophy (39). Furthermore, an increased variation of fiber diameter and prominent central nuclei with chromatin clumps are present in DM1, normally observed in constantly regenerating muscle with immature fibers (39, 43, 121, 122). Another differential observation is the higher frequency of nuclear clump fibers in DM2. Nuclear clump fibers are typically observed in denervated muscles and have been termed “denervation-like” when observed in DM2 muscle, since other neuropathic alterations were not detected (123). Generally, the alterations seen in muscle of DM2 patients are rather mild and have a heterogeneous character (122). Muscle pathology in DM1 patients has a more typical appearance. However, histological reports, especially of older DM1 studies, may sometimes have a misleading message as researchers usually only draw a distinction between muscles of individuals with cDM and the adult-onset form of disease. Details about graded differences in pathology between muscles from patients with childhood, juvenile, adult, and late-onset/asymptomatic DM1 are not well known.
Already early on it was recognized that cDM is associated with a much broader spectrum of morpho-anatomical muscle problems, with type 1 fiber preponderance and hypotrophy and common occurrence of type 2b fiber deficiency (52, 124). Undifferentiated thin fibers and an increase in satellite cells at birth indicate immature muscle fiber growth and delayed muscle fiber differentiation (125, 126). Also, outside the body in in vitro culture the differentiation and maturation capacity of progenitor cells from embryonic muscle of cDM patients was found to be defective (127). The percentage of myoblasts fusing to form myotubes was reduced, the myotube morphology was abnormal, and only immature MyHC protein isoforms were expressed, primarily the embryonic isoform. Also conspicuous aberrancies in intrafusal fiber and muscle spindle presence or morphology were reported. These latter features and the specific fiber type effects may point to additional abnormalities in innervation, motor unit formation, or neurotrophic signaling during the later phases of embryonic development and early prenatal muscle maturation (128–130) (Figure 2).
How Healthy and DM Muscles are Built and Maintained
Myogenesis During Early Embryogenesis
The skeletal muscles of limb and torso and head muscles in vertebrates derive from the paraxial and prechordal mesoderm layers in the early embryo. The myogenic process starts when the paraxial mesoderm forms multiple somites, which then further specialize and form the dermomyotome. First, a large proportion of stem cells in the somites and later in the forming limb buds undergo frequent mitosis, under influence of factors such as IGF-1 and PDGF. The proliferating progenitor cells derived from the embryonic mesenchyme of the somite then undergo different phases of myotome development. This process starts with programmed maturation accompanied by adoption of skeletal muscle fate and withdrawal from the cell cycle, giving rise to a layer of non-proliferating myoblasts that form the primary myotome beneath the dermomyotome (131, 132). When more and more cells are progressively added and start to fuse to already committed myoblasts (myocytes) that already reside in the myotome this leads to the formation of the first myofibers and the onset of embryonic muscle growth (133). The following sections will describe the different steps on the road to maturation of skeletal muscles before and after birth.
Cell Cycle Exit During Myogenesis
During all stages of contribution to muscle formation and regeneration, myoblasts first need to stop their proliferation process by exiting the cell cycle (134). This occurs by activation of cyclin-dependent kinase (cdk) inhibitor p21 and retinoblastoma protein (Rb), a downstream target of p21. p21 is also partially responsible for the decreased Cdk1 activity observed in differentiating cells (135–137). Formation of Rb–E2F complexes is necessary for maintenance of inhibition of cell cycle progression and for cell cycle withdrawal (138). The role of CELF1 in this regulatory circuit is considered an important link to myogenic problems in DM.
Phosphorylation of CELF1 regulates its intracellular localization and activity. Normally, CELF1 is phosphorylated by AKT and cyclin D3/cdk4 at Ser28 and Ser302, respectively. This posttranslational modification is crucial for myogenic progression. Induction of AKT activity is otherwise involved in the suppression of apoptosis during myogenesis (139). In DM1 myoblasts, CELF1 appears to become hyperphosphorylated by AKT (140), whereas in myotubes, CELF1 phosphorylation by cyclin D3/cdk4 seems to be reduced (141). Alterations in the activity of GSK3β influence the activity in the cyclin D3-CDK4 phosphorylation signaling pathway from upstream. The abnormalities in phosphorylation status compromise CELF1’s role as a translational regulator of a specific population of mRNAs. As an end effect, the changes lead to an increase of cyclin D1, an important regulator of proliferation of myoblasts, and to a reduction of p21 in DM1 myotubes. Together, the changes in the AKT-CELF1-cyclin D1 and cyclin D3/cdk4-CELF1-p21 pathways affect the myogenic process in DM1 (68, 141, 142). Also the Rb–E2F repressor complex appears not to be formed, underscoring that impairment of cell cycle withdrawal may have a role in both forms of DM manifestation (68). However, because not all pathways in which CELF1 is involved are similarly abnormal in DM1 and DM2, other obstructions in myogenic programming might be at play in DM2 as well.
Myoblast Fusion
After cell cycle arrest, the fusion of competent myoblasts to form multinucleated myotubes begins. Fusion is a tightly controlled process that involves distinct mechanistic steps, including cell–cell interaction, recognition, and adhesion, followed by membrane coalescence and merging of competent myoblasts to form the multinucleated myotube (143). Extracellular signals from adjacent tissues have an important role in the initiation of several of these steps. Two waves of fusion events take place to form the muscle. Primary myofibers that determine the shape and identity of muscles are formed in the first wave. Secondary myofibers align alongside the primary myofibers and add mass to the muscles in the second wave. Distinct events govern these stages for promotion of differentiation and growth of muscle: first, individual myoblasts fuse to form nascent myotubes and then multinuclear myotubes are formed during subsequent fusion steps between myotubes and additional individual myoblasts (144–146).
The factors that trigger cell fusion (i.e., fusogens) are not precisely known, but numerous proteins that coordinate the formation of primary and secondary myotubes have been identified (144, 146–148). Myomaker, a plasma membrane, Golgi and organellar membrane embedded protein seems crucial (149, 150). Its importance is illustrated by the finding that mutations in myomaker cause a congenital myopathy, Carey-Fineman-Ziter syndrome (151, 152). Other proteins that have an essential role in the myoblast fusion process are myomixer and myomerger. Myomixer, localized to the plasma membrane, associates with myomaker. Myomixer together with myomaker are strong promoters of cell fusion, driving the formation of multinucleated cells from myoblasts (153). Myomerger is only expressed on myocytes and induces the fusogenicity, while myomaker is essential to make a cell fusion competent (148).
Rearrangements in the actin cytoskeleton are first involved in the formation of membrane protrusions between the incoming myoblast and the partner myoblast or myotube. Later they are important for pore formation and cytoarchitectural rearrangements in the resulting multinuclear cell. The entire network that controls the actin network in cells is too complex to discuss here (154, 155), but one issue related to DMPK splice variants may be important. Tentative evidence points to a role for the kinase activity of DMPK, a member of the Rho kinase family, in the regulation of myosin light chain phosphorylation. DMPK may, therefore, functionally link to plasticity of the actomyosin network (156, 157). DMPK is dispensable for myogenesis, as DMPK knockout mice are viable and make muscles with only minor abnormalities (156). However, the possibility that DMPK splicing becomes spatiotemporally deranged by presence of very long (CUG)n repeats and exerts a modulatory effect on actomyosin cytoskeleton dynamics during early and late myoblast-myotube fusion still exists. Tight regulation of DMPK isoform E during early muscle differentiation is essential for normal development (158) and alternative splicing causes downregulation of DMPK E during myoblast to myotube differentiation (159).
Generally, the muscle problems in adult DM patients are difficult to attribute to any of the distinct phases that determine the differentiation, fusion, or senescence or death of different types of muscle cells in vivo. In vitro studies on myoblast cultures of adult-onset DM1 with intermediate expansions or DM2 patients are scarce. New methodology was recently published for the immortalization of primary satellite cells, which stimulate in vitro studies of differentiation capacity (86). Interestingly, DM2 satellite cells with (CCTG•CAGG)4000 repeats did not have a significantly altered myogenic capacity, confirming earlier findings (66, 160). By contrast, more attention has been concentrated on the study of embryonic or early postnatally derived muscle progenitor cells from cDM muscle. These cells consistently showed impaired myogenic potential and reduced myogenic differentiation capacity during culture in vitro (66, 127, 160–164).
Transcription Factor-Induced Programming of Myogenic Lineages
To better understand pathological changes in muscle in DM patients, we will first examine the molecular processes that govern normal muscle development (165–168) and discuss these against the background of repeat expansion. The molecular cascade that directs the fate of somite-derived cells during developmental maturation is principally determined by PAX3 and PAX7. These transcription factors trigger the sequential expression of a group of highly conserved myogenic regulatory factors, collectively known as MRFs. MRFs contain a basic helix-loop-helix domain and recognize the E-box in the promoter of target genes (169). MYF5 and MYF6 (also known as MRF4) act as upstream regulators of MYOD, perhaps the best-known member of the family. Co-expression of these three factors is required for myogenic commitment. Then a fourth factor, myogenin (MYOG) activates advancement to the myocyte stage and terminal differentiation of the muscle cell (166–168, 170). In this circuit, myogenic transcription factors act in a complex feedback and feedforward network. For instance, the temporal coordination of MRF-mediated gene expression is achieved by allowing certain genes to be directly activated by an individual MRF, whereas the induction of other genes in later stages of differentiation by the same MRF requires the participation of the earlier target gene products (166). There is compelling evidence that the expression of various proteins in this MRF regulatory network, like MYOD and MYOG, is affected by the expansions in DM1 or DM2 (69). The involvement of RBPs is thereby a key event. CELF1, for example, binds and destabilizes MYOD mRNA via its GRE-motif, and an increase in CELF1 activity thus has an inhibiting effect on the progress of myogenic differentiation (72).
Members of the SIX family of homeobox genes (SIX1, SIX2, SIX4, and SIX5) are among the other upstream regulators of MRFs. In mice, Six4 and Six5 repress Myog, whereas Six1 activates it (171). Six1 and Six4 were shown to be required for Pax3 and MRF expression during myogenesis (172). Interestingly, SIX5 is immediately adjacent to DMPK and its mRNA level seems decreased in DM1 patients (173). Six5 knockout mice, however, show essentially no muscle symptoms. Hence, the role and relevance of Six5 in DM1 muscle pathology is not very well established (174–179).
Once it was realized that the coordinate action of transcriptional regulation and alternative splicing (plus other forms of RNA processing) is of key importance for myogenic development (72, 75, 180), also the role of isoforms of accessory transcription factors in impaired muscle differentiation in DM attracted further attention. First evidence for their significance came from a study of members of the MEF2 family. In vertebrates, four members of this family, MEF2A, -B, -C, and -D, are expressed. Although MEF2 members do not possess own myogenic activity, they act together with MRFs to activate and sustain the myogenic differentiation program (85, 181). As discussed earlier, MBNL1, -2, and -3 are key factors in the missplicing in DM. In their normal role, MBNL1 and -2 are positive regulators of muscle differentiation. MBNL3, on the other hand, inhibits muscle formation, by repressing adult mRNA splice isoforms (182–185). Lee et al. showed that MBNL3 influences myogenesis by disrupting MEF2D splicing, by favoring beta-exon exclusion (186). When the beta-including MEF2D isoform was expressed in a cell model, normal muscle differentiation was restored. Almost coincidentally, others reported on splicing changes for MEF2A and -C mRNAs. Dysregulation of MEF2B and -D and genes that are under transcriptional control of these factors, mainly those involved in calcium signaling, was found as well (88). Likewise, CELF1 upregulates translation of MEF2A mRNA via direct interaction with a GC-rich element in the transcript, causing a delay in myogenesis. Abnormal CELF1 upregulation thus explains the muscle maturation delay in DM1. For DM2 the involvement of coupled transcription-RNA processing abnormalities has not yet been documented.
First Appearance of Committed MuSCs
During early embryogenesis, a subselection of cells from the dermomyotome maintains proliferation and migrates directly to the myotome. These PAX3- and PAX7-positive cells do not express members of the MRF, homeobox or MEF families of transcription factors. These cells are known as the myogenic precursors that form the source of the majority of satellite cells in the adult skeletal muscle, and as such form the subject for further discussion in the next sections.
Embryonic and Prenatal Phases of Muscle Growth
Fiber Type Specification
In most vertebrates, fibers of diverse types are recognized in the embryo concomitantly with the earliest time points of muscle appearance, before innervation (187). Interestingly, slow MyHC-expressing fibers seem to form earlier than fast MyHC-expressing fibers. Hedgehog signaling is a determining mechanism required for muscle precursors to commit to the slow muscle fate. Later in development, beyond the late embryonic and fetal periods of prenatal development, the slow (type 1) fibers become less common and fast fibers (type 2) start to become the most abundant fiber type. External soluble signals, such as WNT, coming from tissues adjacent to the somites—i.e., the notochord and neural tube—plus cell–cell contacts in the embryonic niche play an important role in further growth of muscle and the specification of fiber types. Excellent reviews discuss the regulatory principles behind fiber specification (180, 187, 188).
The functional and architectural properties of fiber types that arise during embryonic and fetal muscle development are with the advancement of growth further modified by effects of physical activity, endocrine signals and muscle innervation (180, 187–189). This process continues further during postnatal life. For a better understanding of the distinct fiber type involvement in DM1 versus DM2, it is important to reiterate here that not only type 1 and 2 fate specification but also intrafusal fiber morphogenesis is under control of new combinations of transcription factors. Transcription factor EGR3, for example, is selectively expressed in sensory axon-contacted myotubes, and is a key factor for normal intrafusal fiber differentiation and spindle development (190–192). ERB2 signaling also plays an important role (193). As was specified above, intrafusal fiber and spindle morphology are clearly affected in cDM muscles.
Similar hierarchical networks determine the fast and slow fiber specification. Involvement of transcription factors PRDM1 and SOX6 has already been well documented. PRDM1 acts as a switch that activates the slow-twitch differentiation program in cells by direct repression of fast-twitch specific genes and indirect activation of slow-twitch specific genes through limiting the activity of the SOX6 transcriptional repressor (188).
During the transition from the embryonic to the fetal phase of development, a switch occurs from basic muscle patterning (primary myogenesis) to growth and maturation of the muscle masses and the onset of innervation (secondary myogenesis). These two waves of myogenesis are mediated by distinct embryonic and fetal myoblasts, respectively, each characterized by differentially expressed genes and properties. The differentiated cells that these myoblasts produce later have also distinct features. Expression of NFIX is an important prerequisite for the continuation of coordination of fiber specification in the switch to fetal muscle growth. Gradual changes in the networks for transcription regulation, alternative splicing and polyadenylation thereby jointly control the differential expression of fiber type specific protein isoforms. Single muscle fiber proteomics studies have revealed hundreds of proteins that vary in level or identity between the proteomes of different fiber types. Among these are protein isoforms involved in sarcomeric architecture, contractile activity, mitochondrial and carbohydrate metabolism, calcium handling, and protein turnover (194). Differential activation of genes for fiber type specific isoforms of myosin, troponin, tropomyosin, creatine kinase, B-enolase and glycolytic, and mitochondrial enzymes is typical in this specialization (195).
Until now, not much attention was paid to differential expression of genes whose products are linked to the RNA toxicity mechanism in DM. To the best of our knowledge no publications exist on differences in expression of MBNL1-3 or CELF1 between fast and slow fibers. Also reports on abnormalities in expression of DMPK and CNBP in individual fiber types in DM1 or DM2 are rare. In one early report, a decrease in DMPK expression in type 2a muscle fibers of DM1 patients, compared with the level in normal controls, was mentioned (196). Wheeler et al. have reported an abnormal foci count in subsynaptic nuclei and in nuclei of motor neurons at muscle-nerve junctions (67).
Muscle Progenitor Cells of Different Origin
During late fetal development the fiber composition of muscle is further defined and profound changes in the direct neighborhood of the muscle occur. Within the basal lamina formed around the muscle, the fibers are now located together with the now quiescent population of PAX3+/PAX7+ MuSCs, the satellite cells. At the end of the fetal period ~30% of myonuclei are satellite cell nuclei. The remaining ~70% are in the multinucleated fibers. Blood vessels permeate the interstitial spaces between fibers and nerve endings have established contact via neuromuscular junctions. During the transition to adulthood, the percentage of mononucleated cells located under the basal lamina at the muscle periphery declines sharply, due to recruitment for muscle growth and maintenance. In adult muscle, the population of satellite cells encompasses 2–5% of identifiable nuclei (11, 197, 198), which declines further during aging.
There is now compelling evidence that the skeletal muscle niches thus formed contain multiple types of cells, among them also cells with non-somitic origin, with myogenic capacity (Figure 3). Together with the satellite cells, the major skeletal muscle progenitor/stem cell population, these cells form the reservoir for use in skeletal muscle repair, regeneration, and maintenance (Table 2). Specifically, different interstitial populations of cells have now been characterized, referred to as PW1+ interstitial cells (PICs, that express PW1/PEG3) and β4-integrin+ cell (199). Other progenitor cells, MABs and PCs, are located in the fetal or postnatal muscle vasculature, respectively. PCs express alkaline phosphatase (ALP), but lack myogenic and endothelial markers (200). Using lineage tracing, it has been shown that most of these non-satellite cells are not derived from the somite, as do the true PAX3+/PAX7+ satellite cells. The vessel-derived progenitors can be traced back to Pax3+ progenitors of the paraxial mesoderm (201, 202). Both the muscle-resident satellite cells and the PCs contribute to muscle growth during prenatal and postnatal development.
Figure 3.

Skeletal muscle growth, maintenance, and repair by different myogenic progenitor cells. Satellite cells from the basal lamina of the myofiber are activated and undergo asymmetric and symmetric division to generate heterogeneous progeny. Some cells undergo self-renewal and return to quiescence, others become myoblast that will proliferate and differentiate to become myocytes, which fuse to myofibers, enabling repair and/or growth. Mesoangioblasts (MABs) can contribute to muscle regeneration during embryonic growth, while pericytes (PCs) are involved in postnatal muscle growth by repopulating the quiescent stem cell population or maybe by transforming into a myoblast. Participation in growth and/or repair or direct fusion with the myofiber probably occurs along the same pathways as given for satellite cells. Uncertainties in cell fate are indicated by dashed arrows. PW1+ interstitial cells (PICs) are mostly involved in perinatal growth. Expression signatures of differentiation markers in all different cell types are listed at the bottom.
Table 2.
Myogenic cell types.
| Cell type | Abbreviation | Definition |
|---|---|---|
| Muscle-resident stem cell | MuSC | Collective term for cells in (adult) skeletal muscle that can self-renew and give rise to muscle cells |
| Satellite cell | – | Muscle progenitor cell located in the adult stem cell niche under the basal lamina of the myofiber; upon muscle injury this cell can undergo symmetric or asymmetric cell division and produces cell progeny that undergo self-renewal or become myoblasts |
| Myoblast | – | General term for a mononuclear muscle progenitor cell that can proliferate or undergo terminal myogenic differentiation |
| Myocyte | – | Quiescent differentiated myoblast that can fuse to a myotube |
| Myotube | – | Multinuclear cell formed by the sequential fusion of myoblasts/myocytes, which will develop into a mature myofiber |
| Myofiber | – | Mature multinuclear muscle cell; the smallest contractile unit of a muscle |
| Induced pluripotent stem cell | iPSC | Pluripotent stem cell generated from an adult tissue cell (often a fibroblast) |
| Mesoangioblast | MAB | Cell isolated from the embryonic microvascular wall. A MAB has the potential to self-renew and generate multiple types of differentiated cells |
| Pericyte | PC | Cell isolated from the microvascular wall of postnatal tissue. A PC is capable of (trans)differentiating into other cell types when naturally or experimentally relocated to a different tissue |
We will next examine the role and fate of satellite cells in growth, renewal and regeneration of muscle. The biological significance of the other progenitor cells introduced above will be discussed further below in the context of regenerative medicine. DM pathobiology has only been studied in the satellite cell-derived myoblast population in vitro and by histological examinations in vivo. No data exist on the involvement of PICs, MABs, and PCs.
Muscle Renewal and Regenerative Myogenesis
Skeletal muscles endure a lot throughout a lifetime. First, muscle tissue has to grow in size. Then it must be constantly functionally and structurally renewed and maintained in accordance with physical demand and repaired after injury or disease. The role of the satellite cell compartment is thereby indispensable. The mechanisms by which satellite cells participate in renewal and regeneration of muscle have overt similarities to developmental myogenesis. Satellite cells follow largely the same trajectory as somite muscle cells during development, except for their start, which begins in a state of mitotic quiescence. The population of satellite cells must also be kept in check, to maintain functionality and to guarantee muscle homeostasis up to high age. This necessitates maintenance of a delicate balance between self-renewal and differentiation. In fact, evidence has accumulated showing that distinct satellite cell pools in anatomically defined muscles in the body are heterogeneous cell populations, with cells in different stages of development having different gene expression signatures (167, 199, 203–205).
Maintaining Tissue Homeostasis in Adult Muscle
In reaction to disease, injury or prolonged hypoxia, the local release of cytokines, growth factors, cell differentiation factors such as NOTCH and WNT, and other signals triggers satellite cells that are in a quiescent state. The muscle tissue itself and nearby fibroblasts and macrophages have a role in this process. The signaling starts a program of re-entry of satellite cells in cell cycle. Subsequent rounds of cell division, combined with differentiation programming, along similar lines as in embryonic development, in a subset of the satellite cells produces heterogeneity in the population. Some satellite cells retain stemness, and others become myoblasts or myocytes that undergo definite differentiation commitment (Figure 3).
Expansion in the muscle stem cell niche assures that some cells can remain associated with the extracellular matrix and to cells in the neighborhood. This promotes polarization and allows different cycles of asymmetric cell division, maintaining undifferentiated satellite cells, ready for reversal to quiescence (requiescence), and committed progeny for differentiation. Cells with highest expression of NUMB, an antagonist of NOTCH signaling, go back in quiescence for later self-renewal (206). Daughter cells in which p38α/β MAPK is asymmetrically activated by a so-called PAR complex, undergo commitment to myogenic differentiation (207), expand in number and form binuclear myotubes or fuse to existing fibers (168, 199, 203, 208).
A general repression of translation, mediated by the phosphorylation of translation initiation factor eIF2α, is also a key event in the maintenance of the quiescent state (209). The mitotic quiescent satellite cells express PAX7, MYF5, and CD34 and frequently also PAX3 (210–214). Entrance in cell cycle and progression through the myogenic lineage occurs under the control of MRFs. Activated satellite cells no longer express CD34 and start expressing MYOD. Once activated satellite cells proliferate and become myoblasts, PAX7 expression is downregulated, while MYOD and MYF5 expression remain (215). In silico modeling of RNA processing associated with human muscle development has provided strong evidence that also the expression of MBNL1, -2, and -3 varies during these transitions in cell state (85). In addition, MYOD induces the expression of p21. As mentioned earlier, p21 blocks cell cycle progression and it is involved in the switch from proliferating to differentiating myoblasts, i.e., when they become myocytes. This switch is essential for myogenic precursor cell, satellite cell, function in regenerating skeletal muscle (135, 216). During normal healthy life, this whole cascade of steps for the regulation of muscle differentiation and maintenance is orchestrated by a multitude of circulating hormones, such as IGFs, FGFs, TGFs, testosterone, thyroid hormones, cytokines, and exosome-secreted signals, which are secreted locally and appear in the muscle stem cell niches. Whether and how hormonal signaling controls viability, performance and half-life of multinucleated myofibers—i.e., the bulk of muscle mass in a healthy individual—is still poorly understood, as attention of study thus far has been mainly directed toward MuSCs (208).
Failure of Tissue Homeostasis in DM Muscle
Not much is known about the fate specification of terminally differentiated multinucleated myofibers in DM. One likely possibility is that the persistent abnormalities in alternative splicing, alternative polyadenylation, and unscheduled translation of aberrant transcripts lead to the production of excessive amounts of ectopic proteins. When combined with a bulk of proteins synthesized in normal accordance with the stage of muscle during adulthood, this will create a permanent disbalance in the assembly—and perhaps turnover—of multiprotein complexes in the fiber proteome. Production of polymeric proteins by RAN translation may further create proteome abnormality. Ultimately, such imbalance will lead to a culmination of problems and to proteotoxic stress alike UPS, ER stress, or other forms of stress mentioned in this review (217). When certain thresholds are exceeded, this may lead to senescence or apoptosis. Somatic expansion of repeat length during aging may further augment the stress level, causing loss of an increasing number of fibers with disease progression and aging.
Why pathology specifically involves type 1 fibers in distal muscles of adult-onset DM1 patients and type 2 fibers in proximal muscles in DM2 needs more study. The answers may not be found only in the mature fibers themselves. They also must be sought in differences between DM1 and DM2 in the fitness of their satellite cell pools, or in the modes of recruitment of satellite cells for the regeneration of damaged fibers. As addressed before, the relevance of the satellite cell pool becomes early apparent in cDM patients, who are born with an excessive number of satellite cells and have thin muscle fibers, typical markers for immature muscle fiber growth, diminished recruitment, and delayed differentiation (125, 126). Severe disruption of RNA processing is the key element in the diminished capacity of muscle precursor cells in muscle formation in cDM, as recently demonstrated by combining transcriptome profiling of muscle tissue from patients and mouse models (85). In adult-onset DM1, the number of satellite cells are increased in distal but not in proximal muscles (218). Late myogenic differentiation markers are not fully expressed (219). In cell culture, DM1 and DM2 myoblasts show a premature proliferative growth arrest compared with healthy myoblasts (5). Combined, these observations point to a situation in which the regenerative capacity of satellite cells induced in response to fiber dystrophy is constitutively impaired (36, 220).
To understand muscle wasting in greater detail, we first need to know whether the cellular effects of fiber dystrophy are indeed dominant over those of regeneration failure. Then, to deconvolute the complexity of DM further, molecular analyses are needed. First, we need to know whether failure in pools of satellite cells to adequately balance asymmetric and symmetric division and/or subsequent loss of regenerative potency after myogenic commitment could be involved. Underlying mechanisms and differences between DM1 and DM2 muscles therein must be analyzed. Other studies should be concentrated on the loss of functionality, stability, and viability of fibers in DM1 and DM2. Preferably cell- and lineage-tracing studies should be non-invasive and concentrated on the fate of individual myoblasts, myocytes and muscle fibers over longer periods of aging. For obvious reasons, these types of longitudinal analyses of individual cells are virtually impossible for human muscle. However, tracing of cells during development and maintenance in muscles of animal models of DM will also become challenging.
Premature Muscle Aging in DM
From a clinical perspective, various symptoms of DM1 can be seen as a manifestation of progeria or accelerated aging (221–223), while aging-like symptoms are not as apparent in DM2. The progression of dystrophy in skeletal muscle in DM1 patients shows similarities with sarcopenia, i.e., age-related loss of muscle mass, strength, and function (5). Experimental evidence is mostly indirect and based on descriptive studies, wherein histopathological features such as grouped atrophy, fiber size variability, and central nuclei were investigated in sarcopenic and DM1 muscle (224). Also compelling ultrastructural and molecular evidence was provided, showing that alterations in RNA metabolism in myonuclei from DM1 patients and in aging muscle share similarity (221, 222, 225).
The mechanisms underlying age-related muscle wasting and weakness are probably diverse and not well understood (226). A recent single-fiber proteomics approach showed that the senescence of type 1 and 2 muscle fibers during aging in healthy donors is characterized by several diverging mechanisms. Differential adaptations in cellular carbohydrate and energy metabolism and the networks for protein quality control and proteostasis were among the most conspicuous changes in slow and fast fibers (194). Earlier profiling studies had pointed to a glycolytic to oxidative shift (227) or non-specified overall changes caused by aging in whole human muscles (228). The numerical loss and the loss of functionality of MuSCs, rather than fibers, with aging have attracted until now more attention, as they provide an explanation for the regenerative failure of aged muscle. For more details on the molecular and cellular findings, we refer the reader to comprehensive reviews on this topic (229–231).
Within the networks for muscle regeneration and maintenance during aging, only a few players and processes have been identified that bear direct relevance for DM1 and DM2 pathophysiology. DNA repair is one important issue. Nuclei in resting satellite cells and in muscle fibers are highly efficient in DNA repair through non-homologous end joining, explaining why repeat expansion predominantly occurs in these cells (232). Ongoing somatic expansion of the (CTG•CAG)n and (CCTG•CAGG)n repeats due to DNA repair in quiescent cells may thus be an important factor in impaired muscle regeneration in patients (23). Whether age-induced changes in the production of mitochondrial reactive oxygen species also have an effect must still be analyzed. Accumulation of reactive oxygen species damage is a known contributing factor to repeat expansion (233, 234). Age-dependent changes in oxidative metabolism must, however, have different effects in DM1 and DM2 muscles, as the affected fiber types differ in both forms of disease.
The shortening of telomeres is probably not a major contributor to muscle aging, although effects on premature senescence of DM2 satellite cells have been suggested (220). The situation in DM1 is less clear. Satellite cells in cDM patients did have a higher telomere shortening rate, but they entered senescence before reaching a critical length. This argues against a determining role of telomere shortening as an explanation for diminished differentiation capacity in cDM muscle (218, 235).
A more likely candidate mechanism for the premature growth arrest in DM1 muscle precursor cells is activation of the p16Ink4a-pathway that leads to CDK4 inhibition and cell cycle arrest. p16 accumulates in myoblasts from DM1 patients in response to (CTG)n-related stress (220, 235), resulting in impaired regeneration and atrophy. As mentioned, aging-like symptoms are not so apparent in DM2 patients and the p16 pathway appears not to be altered in DM2 satellite cells and fibers (220, 221). Finally, increased p38/MAPK signaling is a typical feature of aged satellite cells (236), but evidence for p38 signaling abnormalities in DM muscle is lacking. Also evidence for the involvement of apoptosis in DM muscle wasting is still limited (159, 163, 237).
An interesting test for the question how DM effects are superimposed on senescence of normal aging would be to study the effects of ablation of p16Ink4a-expressing cells in muscle of DM mouse models. This is possible with use of a genetic approach recently developed by Baker and co-workers in Van Deursen’s laboratory (238) and also with drug treatment (239). Any alteration in muscle health in the DM mice would provide us with novel insight in the causative effects of expanded repeats on the viability of progenitor cells in muscle.
Stress Signaling in Adaptation to Regenerative Failure, Effects of Disease, and Aging
Adaptation to cell-autonomous stress in muscle depends on a combination of intrinsic and extrinsic signaling mechanisms. Many intracellular pathways are known that protect cells against stress from for example DNA damage, proteotoxicity, and calcium-mediated excitotoxicity (240). Best-known are the P53, AKT, and NRF2 pathways, but these pathways have not yet been intensely studied in skeletal muscle of DM patients.
Changes in intercellular communication may also fulfill a central role. Many of the secreted hormones and factors that are exchanged between cells and orchestrate myogenesis and regeneration have been extensively discussed in some of the reviews mentioned above (208). Among these are the WNT proteins, HGF, FGFs, IGF-1 splice variants, myostatin, and TGF-β (241). Although the working mode of these secreted factors is reasonably well understood, it is not always clear what cell types in the muscle stem cell niche are in the secretory and/or the responding mode. Satellite cells from cDM patients secrete increased levels of prostaglandin E2 in vitro. This secretion is controlled via upregulation of cyclo-oxygenase 2, mPGES-1, and prostaglandin E2/EP4 receptors. A direct consequence of the prostaglandin E2 upregulation is a decrease in intracellular Ca2+ and impairment of fusogenic capacity of the satellite cells (242). It was also shown that cDM muscle and primary myoblast derived thereof produce a higher level of IL-6, indicative for increased activity of this myokine signaling pathway (52).
Another conspicuous observation was that variation in the level of CELF1, as seen in cDM muscles, causes imbalance in the production of subunits for the signal recognition particle in the ER-secretory pathway (243). CELF1 misregulation may thus be coupled to changes in the secretory route for extracellular matrix proteins. Others confirmed that production of ECM proteins is indeed altered in muscle of a mouse model for DM1 (244). Taken together, this is compelling evidence that the hormonal and ECM environment of progenitor cells in the DM muscle are changed. There is no doubt that this will compromise the “cry-for-help” communication in DM muscle and its adaptive regenerative capacity in response to accelerated fiber decay due to repeat stress.
MicroRNAs (miRNAs) and Other Non-Coding RNAs in Muscle Homeostasis
MicroRNAs have a critical role in cellular stress responses, differentiation, proliferation, and apoptosis in muscle (245, 246). MiRNAs are short, highly conserved non-coding RNAs that occur in all cell types, where they regulate the stability and the translational efficiency of target mRNAs (247). Multiple miRNAs that regulate differentiation and stress adaptation of skeletal muscle, referred to as myomiRs, exist (248). Among them are miR-1, -133a, -133b, -206 (the most abundant miRNA in skeletal muscle), and miR-208. Expression of these miRNAs is regulated by transcriptional networks involving MEF2, MYOD, SRF, and TWIST1 (249, 250). Non-muscle specific miRNAs that regulate differentiation and regeneration after muscle injury are miR-181, -221, and -222 (251).
Myoblasts and myofibers utilize exosome-clustered extracellular miRNAs as paracrine and endocrine communication signals to regulate homeostasis and regeneration. Extracellular myomiRs are elevated during perinatal muscle development and after exercise-induced muscle regeneration. Also in primary human myoblast and C2C12 cultures, these extracellular myomiRs were elevated and appeared to be released selectively as a consequence of the differentiation process (252).
Myotonic dystrophy type 1 and 2 profiling studies showed that deregulation of intracellular miRNA content in muscle, and extracellular extrusion via exosome secretion is a hallmark of disease. Eight miRNAs were found to be significantly deregulated in the serum of DM1 patients (i.e., miR-1, -27b, -133a, -133b, -140-3p, -206, -454, and -574) (253). Earlier work had shown upregulation of miR-1, -206, and -335 and downregulation of miR-29b, -29c, and -33 in DM1 biopsies compared with controls (254, 255). Moreover, cellular distribution of miR-1, -133b, and -206 was altered in DM1 skeletal muscles. Koutsoulidou et al. demonstrated that appearance of miR-1, -133a, -133b, and -206 in serum correlated with the progression of muscle wasting in DM1 patients. All four miRNAs were found encapsulated within exosomes in the circulation (256). Cell and animal model studies suggest that MBNL expression is controlled by miR-277 and -304 (257), and miR-30-5p (258), and that this regulatory network could be involved in inhibition of myogenic differentiation in DM1. In DM2 muscle biopsies, the levels of 11 miRNAs were found to be significantly modulated (259). Of these, three also showed modulation in DM1 patients (i.e., miR-193bp, -208a, and -381). Expression levels of the other eight (i.e., miR-34a-5p, -34b-3p, -34c-5p, -125b-5p, -146b-5p, -193a-3p, -221-3p, and -378a-3p) fitted in a unique DM2 profile. The differences in miRNA expression profiles might contribute to the differences in muscle pathobiology between DM1 and DM2 (259).
Long non-coding RNAs and circular RNAs (circRNAs) may also have a role as regulators of muscle homeostasis and gene expression (260, 261). LncRNAs are arbitrarily defined as RNAs >200 nts without overt protein-coding potential, of which at least 5,000 have been identified so far (262). CircRNAs are shaped as covalently closed molecules that lack 5′ and 3′ ends. They are expressed by a high number of genes and are highly conserved among species (263). Although little is known about the function of these RNA species, it has been shown that they can modulate gene expression by competing for miRNA or protein binding, or with regular mRNA production (264–267). Some lncRNAs are important players in muscle differentiation (268, 269) and involved in the pathomechanisms for Duchenne muscular dystrophy (270) and facioscapulohumeral muscular dystrophy (271). Malat1, one of the most abundant lncRNAs, was recently found to slow down myogenic differentiation in mice by interference with MyoD-binding loci and formation of a repressive histone-methylation complex. After the onset of differentiation, miR-181 targets Malat1 RNA for breakdown to release the repression (272). Our group has published evidence that DM1-AS transcripts belong to the class of lncRNAs. After alternative splicing and alternative polyadenylation, different (CAG)n repeat containing DM1-AS RNA isoforms are produced. Like many other lncRNAs, DM1-AS RNA is expressed at very low copy numbers per cell, in parallel with (CUG)n-containing DMPK mRNA (273). It remains to be seen whether expanded DM1-AS transcripts have an effect on DM1 myopathy, either in isolation or together with expanded DMPK transcripts. No circRNAs that are possibly linked to DM have so far been identified, but considering the fast developments, we might soon hear more from this field of research.
Regenerative Medicine for DM: Progenitor Cells as Source for Muscle Healing
Use of MuSCs
The satellite cells, the adult MuSCs located between the basal lamina and the sarcolemma of the multinucleated myofibers, form the main pool of progenitors for skeletal muscle regeneration in vivo (Table 2). A large body of research has been devoted to the isolation, propagation, and genome tailoring of these cells, as they are the most logical candidates for use in future cell-based therapies, capable of restoring tissue homeostasis, and enhancing muscle repair in patients with myopathies.
Identification and Isolation of MuSCs
For an ex vivo approach to gene therapy of DM in coupling with muscle cell transplantation the availability of sufficient quantities of MuSCs is a prerequisite. Use of these cells for regenerative medicine in DM, whereby different groups of skeletal muscles are differentially affected, will not be simple. Indeed, although all satellite cells should be considered remnants of embryonic development prepared to recapitulate muscle development in the event of muscle damage (197), it is only a fraction of this heterogeneous population that fully preserves the self-renewal potential and myogenic capacity, when brought in in vitro culture. This seemingly stochastic nature of fate adoption, which is associated with a high-degree of heterogeneity and plasticity of the satellite cell population in the natural environment of the muscle (203), is a complicating factor during the period that they regain proliferative activity as myoblasts.
Another complicating factor is that MuSCs have the same embryonic origin as the muscle in which they reside. Most skeletal muscles of the trunk and limb are derived from somites, but head muscles originate from cranial mesoderm. These distinct origins specify distinct genetic programs (274), which may be permanently associated with the intrinsic properties of MuSCs (275). More study is thus needed to verify whether the distinct origin is also a determining and retained factor for capacity to participate in regeneration of muscles in different locations of the body, or whether differences are smoothened out upon maintenance of cells in in vitro culture. Lastly, aging of the donor seems to render the MuSC pool increasingly dysfunctional, as MuSCs progressively lose their potency due to cell death and terminal differentiation. Hence, aging forms an extra problem in cases where the patient’s own progenitor cells must be used for cell therapy to circumvent immunological problems, and especially so in patients with late-onset genetic myopathies like in DM2 or certain cases of DM1.
Skuk and colleagues came up with three properties that cells used for repair of damaged and replacement of lost muscle fibers should have: (i) ability to fuse with pre-existing myofibers, (ii) ability to form new myofibers, and (iii) ability to produce myogenically committed stem cells (276). This means that the MuSC’s capacity to participate in all aspects of muscle homeostasis must be maintained during expansion ex vivo. Novel strategies for satellite cell culture and preservation of self-renewal capacity before transplantation into muscle have now become available. Cell culture on pliable soft hydrogel matrices, in combination with pharmacological inhibition of p38/MAPK signaling (277) or culture on natural biopolymeric films (278) simulate the conditions of the muscle stem cell niche and help to preserve MuSC quiescence and enhance their self-renewal capacity. Also modulation of PAX7 expression may thereby be of help (279).
Transplantation of MuSCs: Preclinical Studies Only
Currently, the use of MuSCs in cell-based therapies is almost impossible. As demonstrated in animal model studies, MuSCs cannot be delivered systematically to all muscles in the body (280). Upon intravenous delivery they accumulate in the lung, liver, spleen, and kidney but not in skeletal muscle. One of the largest technical hurdles that limit the feasibility of MuSC transplantation is, therefore, associated with the route of administration, i.e., intramuscular injection (Figure 4). Initial trials aiming to regenerate skeletal muscle by local injection of donor myoblasts failed due to their poor survival and limited ability to migrate more than a few millimeters away from the site of injection (281–283). Upon engraftment, these satellite-derived myoblasts could not efficiently repopulate the satellite cell niche, and therefore were not able to contribute significantly to muscle regeneration (284, 285). Future work is necessary to find out whether some of these issues might be overcome by increasing the numbers of engrafted cells, or by better preservation of their stemness during in vitro propagation as discussed above.
Figure 4.
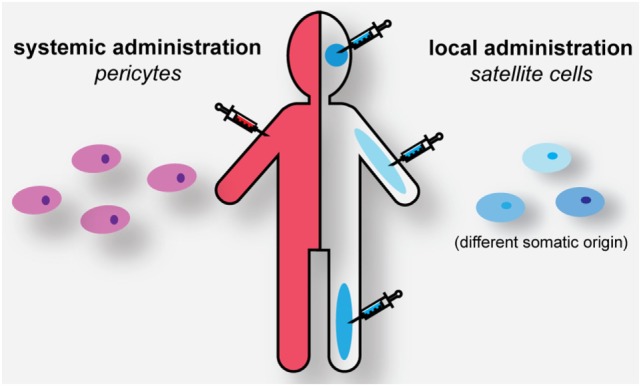
Strategies for cell-based muscle therapy in myotonic dystrophy. Genome-edited autologous or HLA-matched pericytes (PCs) can be administered systemically for muscle healing. Genome-edited or HLA-matched satellite cells need to be engrafted locally in the corresponding muscles to have a regenerative effect.
Further work on MuSCs in culture is, therefore, necessary. For application in basal and translational research in DM, immortalized myoblast cell lines are available. These lines have preserved the molecular hallmarks of disease, including splicing abnormalities and repeat RNA-MBNL foci (86) and were generated by lentiviral-mediated expression of the catalytic subunit of the human telomerase (TERT) and CDK4, the natural p16 ligand. Immortalized cells constitute an unlimited source of cells for evaluation of compounds with therapeutic potential. Immortalization per se may not be detrimental for the ability of muscle progenitor cells to serve in therapeutic engraftment experiments in mice, as already shown earlier for these type of cells and for SV40-TAgts immortalized cells (286, 287). However, for obvious reasons use of these transformed cells for human studies will probably remain restricted for in vitro work.
Use of Stem Cells From Non-Muscle Origin
The continuous search for stem cells with potency for transdifferentiation and adaptation from other sites than within the muscle basal lamina (288–295) has led to the identification of entirely unexpected cell types with muscle progenitor capacity. Among these are the vessel-associated MABs and PCs the best-known examples (Figure 3) (199).
Identification and Isolation of PCs and MABs
The participation of MABs and PCs in myogenic differentiation and regeneration in vivo is still a poorly understood phenomenon. There is, however, compelling evidence that these cell types have great potential for boosting muscle repair in regenerative medicine. One advantage, which MABs and PCs may have, is that they rapidly acquire unlimited lifespan and maintenance of multipotency, making them ideally suitable for the generation of replenishable pools of transplantable cells. Skeletal muscle tissue itself is the most effective source for PCs with this potential (200, 296). Their isolation can be accomplished by using explant culture methodology (297, 298), eventually in combination with enzymatic dissociation and FACS for surface markers (200, 299). PCs with skeletal myogenic potential can be distinguished by expression of ALP (200, 300) and new biomarkers for therapeutic potency, like PW1/Peg3, a regulator of myogenic ability and migration capacity in PCs, MABs and satellite cells, have recently been identified (301). Expression of PW1/Peg3 is high in both MABs and PCs and its level of expression correlates with their progenitor cell competence. Moreover, lack of PW1/Peg3 expression abrogates the cells ability to cross the vessel wall and to engraft into damaged myofibers through the modulation of the junctional adhesion molecule. PCs and MABs are expandable in vitro as a relatively homogeneous population and transducible with viral vectors for genomic editing.
Engraftment of PCs and MABs
Pericytes and MABs are able to systemically reach the target tissue, where they engraft and differentiate toward the myogenic lineage (Figure 4). One possible complication, however, is that adequate measures are necessary to ensure that myogenic commitment of these vessel-derived progenitor cells is appropriately stimulated, while adipose and fibrogenic commitment must be avoided. Several recent publications have implicated a role for a PC subtype in fibro-adipose infiltration of tissues (299, 302). Consistent with age-dependent changes in regeneration capacity seen before, this property seems to be more present in PCs isolated from aged individuals. PCs failed to differentiate or participate in myofiber repair following injury, but contributed to enhanced fibrous tissue deposition within the interstitial space in aged muscle (299, 303–305). Further work is thus necessary to see whether PCs and MABs are truly the ideal candidates for use in regenerative medicine in DM patients.
Translational studies in the GRMD dog model of myopathy demonstrated that ex vivo cultivated PCs can indeed adopt myogenic fate when exposed to injury factors in vivo and are able to directly differentiate into skeletal muscle or replenish the SC pool via activation of Pax7, Myf5, or MyoD at the onset of differentiation (200). For the GRMD model “a remarkable clinical amelioration and preservation of active motility” was seen (306). The first human clinical study with PCs was published in 2015, investigating primarily the safety of intra-arterial transplantation of HLA-matched donor cells. This exploratory clinical trial was performed in five Duchenne patients, in combination with immunosuppressive therapy. Clinical laboratory and MRI analysis revealed that the study was relatively safe. Unfortunately, the effects of the cellular therapy on muscle function were inconclusive.
Although the possibility for systemic administration is one of the strongest arguments for preference of vessel-associated progenitor cells over satellite cells, there is also concern, as blood flow in the artery of microvasculature downstream of the injection site might get disrupted (307). Moreover, a fraction of the injected cells might become trapped in filter organs decreasing the amount of cells available for engraftment into dystrophic muscle (200). Modification to improve homing to damaged muscles (308) or altering cell surface (309) needs to be studied in more detail to address these possible problems. For DM, research regarding the potential and use of MABs or PCs for therapy is entirely missing.
Induced Pluripotent Stem Cells (iPSCs)
Also whole new approaches toward deriving myogenic progenitor cells from pluripotent embryonic stem cells and iPSCs are now being developed (310–315). Generation of iPSCs from fibroblasts of DM1 and DM2 patients has been published (316–321). Recently, a revolutionary new method to direct human iPSCs to adopt muscle progenitor cell identity and create a renewable source of muscle progenitors for regenerative medicine was developed. Hicks et al. found that the use of FACS of cells for two cell surface markers, ERBB3 and NGFR, and treatment with a TGFβ inhibitor gave an enormous enrichment for progenitors with regenerative potential during engraftment (322). Further work is necessary to verify whether simple extrapolation of these animal model transplantation findings to the human situation is possible.
Cell-Based Therapy in Combination With Genome Editing
To prevent immunological problems linked to MuSC transplantation, the use of progenitor cells from HLA-matched donors or autologous cells from patients is strongly advisable. For DM1 and DM2 cells, this implicates that genome editing must be employed to normalize the length of the expanded repeats or the synthesis of the toxic RNAs must be otherwise permanently prevented. With the advent of gene editing tools such as ZFN, TALEN, and CRISPR/Cas9 this now has become a realistic goal. Specifically for DM1, a small number of gene editing studies have been published recently, all aiming at the prevention of the presence of toxic, expanded repeat-containing RNA.
Gao et al. inserted a poly(A) signal upstream of the expanded (CTG)n repeat in DMPK in iPSCs. This insertion led to premature termination of transcription and prevented production of (CUG)n repeat containing transcripts. As the DMPK mRNAs were now missing the repeat-containing 3′ end, a healthy stem-cell pool was created (320). Cardiomyocytes derived from these iPSCs reverted to normal splicing for a number of pre-mRNAs known to be misspliced in DM1.
Pinto et al. used a deactivated Cas9 variant to impede synthesis of expanded (CUG)n RNA during transcription (323), while Batra et al. (324) used an RNA-targeting Cas9 to eliminate toxic expanded RNA after production. Both studies showed efficient elimination of cellular hallmarks of disease, but the strategies used seem not well suited for permanent transformation of muscle progenitor cells and prevention of repeat RNA effects.
More permanent effects for use in cellular strategies may be expected from removal or trimming of the (CTG•CAG)n repeat expansion in the DM1 locus, creating permanently normalized DMPK/DM1-AS alleles. Our own group and others have published that excision of the repeat (and short flanking sequences) can be achieved by dual CRISPR/Cas9 cleavage at either side of the repeat (113, 325). Repeat removal had no adverse biological effects on DMPK isoform production and normalized splicing and myogenic capacity. Notably, CRISPR/Cas9 cleavage in the vicinity of the repeat was associated with a risk of uncontrollable DNA rearrangements across the area (113, 325). Also off-target alteration elsewhere in the genome is a known danger in the application of CRISPR/Cas9 technology. Hence, careful characterization and selection of cell clones with only the desired genome alterations should become routine steps in future cell-based therapeutic strategies.
Use of repeat-corrected cell therapy may serve to halt the degenerative process, or delay or prevent the onset of disease when applied upon first diagnosis with DM. In parallel, more work will be devoted to the development of modalities for direct in vivo treatment of DM, with vector-mediated gene-editing therapy. Finding ways for improvement of the quality of life of patients with DM will remain the goal of a large variety of future translational studies.
Author Contributions
LA and CA designed the figures. LA, CA, and BW drafted the contents of this review, and together with DW wrote the text. All authors contributed equally to critical reading of the final manuscript, including text and figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by ZonMw (TOP grant NL91212009) and by the Prinses Beatrix Spierfonds (grant numbers W.OR12-05 and W.OR16-09) with contribution from the Stichting Spieren voor Spieren. We apologize to all colleagues in the field whose findings could not be included due to size constraints.
References
- 1.Meola G, Cardani R. Myotonic dystrophy type 2 and modifier genes: an update on clinical and pathomolecular aspects. Neurol Sci (2017) 38:535–46. 10.1007/s10072-016-2805-5 [DOI] [PubMed] [Google Scholar]
- 2.Gourdon G, Meola G. Myotonic dystrophies: state of the art of new therapeutic developments for the CNS. Front Cell Neurosci (2017) 11:101. 10.3389/fncel.2017.00101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yum K, Wang ET, Kalsotra A. Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr Opin Genet Dev (2017) 44:30–7. 10.1016/j.gde.2017.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sicot G, Gourdon G, Gomes-Pereira M. Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: new findings and future challenges. Hum Mol Genet (2011) 20:116–23. 10.1093/hmg/ddr343 [DOI] [PubMed] [Google Scholar]
- 5.Mateos-Aierdi AJ, Goicoechea M, Aiastui A, Fernández-Torrón R, Garcia-Puga M, Matheu A, et al. Muscle wasting in myotonic dystrophies: a model of premature aging. Front Aging Neurosci (2015) 7:125. 10.3389/fnagi.2015.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wansink DG, Wieringa B. Transgenic mouse models for myotonic dystrophy type 1 (DM1). Cytogenet Genome Res (2003) 100:230–42. 10.1159/000072859 [DOI] [PubMed] [Google Scholar]
- 7.Gomes-Pereira M, Cooper TA, Gourdon G. Myotonic dystrophy mouse models: towards rational therapy development. Trends Mol Med (2011) 17:506–17. 10.1016/j.molmed.2011.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sicot G, Gomes-Pereira M. RNA toxicity in human disease and animal models: from the uncovering of a new mechanism to the development of promising therapies. Biochim Biophys Acta (2013) 1832:1390–409. 10.1016/j.bbadis.2013.03.002 [DOI] [PubMed] [Google Scholar]
- 9.Sabourin LA, Rudnicki MA. The molecular regulation of myogenesis. Clin Genet (2000) 57:16–25. 10.1034/j.1399-0004.2000.570103.x [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Conboy I. Embryonic vs. adult myogenesis: challenging the “regeneration recapitulates development” paradigm. J Mol Cell Biol (2010) 2:1–4. 10.1093/jmcb/mjp027 [DOI] [PubMed] [Google Scholar]
- 11.Ceafalan LC, Popescu BO, Hinescu ME. Cellular players in skeletal muscle regeneration. Biomed Res Int (2014) 2014:1–10. 10.1155/2014/957014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Theadom A, Rodrigues M, Roxburgh R, Balalla S, Higgins C, Bhattacharjee R, et al. Prevalence of muscular dystrophies: a systematic literature review. Neuroepidemiology (2014) 43:259–68. 10.1159/000369343 [DOI] [PubMed] [Google Scholar]
- 13.Mathieu J, Prévost C. Epidemiological surveillance of myotonic dystrophy type 1: a 25-year population-based study. Neuromuscul Disord (2012) 22:974–9. 10.1016/j.nmd.2012.05.017 [DOI] [PubMed] [Google Scholar]
- 14.López de Munain A, Blanco A, Emparanza JI, Poza JJ, Martí Massó JF, Cobo A, et al. Prevalence of myotonic dystrophy in Guipúzcoa (Basque country, Spain). Neurology (1993) 43:1573–6. 10.1212/WNL.43.8.1573 [DOI] [PubMed] [Google Scholar]
- 15.Gomes-Pereira M, Monckton DG. Chemical modifiers of unstable expanded simple sequence repeats: what goes up, could come down. Mutat Res (2006) 598:15–34. 10.1016/j.mrfmmm.2006.01.011 [DOI] [PubMed] [Google Scholar]
- 16.Llamusí B, Artero R. Molecular effects of the CTG repeats in mutant dystrophia myotonica protein kinase gene. Curr Genomics (2008) 9:509–16. 10.2174/138920208786847944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magaña J, Cisneros B. Myotonic Dystrophy Type 1 (DM1): From the Genetics to Molecular Mechanisms. (Vol. 1). (2007). p. 47–72. Available from: https://www.intechopen.com/books/muscular-dystrophy/myotonic-dystrophy-type-1-dm1-from-the-genetics-to-molecular-mechanisms (Accessed: February 15, 2018). [Google Scholar]
- 18.Lee J, Cooper T. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans (2009) 37:1281–6. 10.1042/BST0371281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Antonio M, Dogan C, Hamroun D, Mati M, Zerrouki S, Eymard B, et al. Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Rev Neurol (Paris) (2016) 172:572–80. 10.1016/j.neurol.2016.08.003 [DOI] [PubMed] [Google Scholar]
- 20.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science (1992) 255:1253–5. 10.1126/science.1546325 [DOI] [PubMed] [Google Scholar]
- 21.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell (1992) 68:799–808. 10.1016/0092-8674(92)90154-5 [DOI] [PubMed] [Google Scholar]
- 22.Ashizawa T, Dubel JR, Harati Y. Somatic instability of CTG repeat in myotonic dystrophy. Neurology (1993) 43:2674–8. 10.1212/WNL.43.12.2674 [DOI] [PubMed] [Google Scholar]
- 23.Morales F, Couto JM, Higham CF, Hogg G, Cuenca P, Braida C, et al. Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum Mol Genet (2012) 21:3558–67. 10.1093/hmg/dds185 [DOI] [PubMed] [Google Scholar]
- 24.Musova Z, Mazanec R, Krepelova A, Ehler E, Vales J, Jaklova R, et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am J Med Genet (2009) 149:1365–9. 10.1002/ajmg.a.32987 [DOI] [PubMed] [Google Scholar]
- 25.Braida C, Stefanatos RKA, Adam B, Mahajan N, Smeets HJM, Niel F, et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum Mol Genet (2010) 19:1399–412. 10.1093/hmg/ddq015 [DOI] [PubMed] [Google Scholar]
- 26.Axford MM, López-Castel A, Nakamori M, Thornton CA, Pearson CE. Replacement of the myotonic dystrophy type 1 CTG repeat with “non-CTG repeat” insertions in specific tissues. J Med Genet (2011) 48:438–43. 10.1136/jmg.2010.085944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groenen PJ, Wansink DG, Coerwinkel M, van den Broek W, Jansen G, Wieringa B. Constitutive and regulated modes of splicing produce six major myotonic dystrophy protein kinase (DMPK) isoforms with distinct properties. Hum Mol Genet (2000) 9:605–16. 10.1093/hmg/9.4.605 [DOI] [PubMed] [Google Scholar]
- 28.Gudde AEEG, van Heeringen SJ, de Oude AI, van Kessel IDG, Estabrook J, Wang ET, et al. Antisense transcription of the myotonic dystrophy locus yields low-abundant RNAs with and without (CAG)n repeat. RNA Biol (2017) 14:1374–88. 10.1080/15476286.2017.1279787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thornton CA, Griggs RC, Moxley RT, III. Myotonic dystrophy with no trinucleotide repeat expansion. Ann Neurol (1994) 35:269–72. 10.1002/ana.410350305 [DOI] [PubMed] [Google Scholar]
- 30.Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Otto M, Heine R, et al. Proximal myotonic myopathy: a new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology (1994) 44(8):1448–52. 10.1212/WNL.44.8.1448 [DOI] [PubMed] [Google Scholar]
- 31.Bachinski LL, Udd B, Meola G, Sansone V, Bassez G, Eymard B, et al. Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am J Hum Genet (2003) 73:835–48. 10.1086/378566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liquori CL, Ikeda Y, Weatherspoon M, Ricker K, Schoser BGH, Dalton JC, et al. Myotonic dystrophy type 2: human founder haplotype and evolutionary conservation of the repeat tract. Am J Hum Genet (2003) 73:849–62. 10.1086/378720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol (2012) 11:891–905. 10.1016/S1474-4422(12)70204-1 [DOI] [PubMed] [Google Scholar]
- 34.Bird TD. Myotonic Dystrophy Type 1. Seattle: University of Washington; (1993). [PubMed] [Google Scholar]
- 35.Turner C, Hilton-Jones D. Myotonic dystrophy: diagnosis, management and new therapies. Curr Opin Neurol (2014) 27:599–606. 10.1097/WCO.0000000000000128 [DOI] [PubMed] [Google Scholar]
- 36.Meola G, Cardani R. Myotonic dystrophy type 2: an update on clinical aspects, genetic and pathomolecular mechanism. J Neuromuscul Dis (2015) 2:S59–71. 10.3233/JND-150088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science (2001) 293:864–7. 10.1126/science.1062125 [DOI] [PubMed] [Google Scholar]
- 38.Schneider C, Ziegler A, Ricker K, Grimm T, Kress W, Reimers CD, et al. Proximal myotonic myopathy: evidence for anticipation in families with linkage to chromosome 3q. Neurology (2000) 55:383–8. 10.1212/WNL.55.3.383 [DOI] [PubMed] [Google Scholar]
- 39.Vihola A, Bassez G, Meola G, Zhang S, Haapasalo H, Paetau A, et al. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology (2003) 60:1854–7. 10.1212/01.WNL.0000065898.61358.09 [DOI] [PubMed] [Google Scholar]
- 40.Finsterer J. Myotonic dystrophy type 2. Eur J Neurol (2002) 9:441–7. 10.1046/j.1468-1331.2002.00453.x [DOI] [PubMed] [Google Scholar]
- 41.Day JW, Roelofs R, Leroy B, Pech I, Benzow K, Ranum LP. Clinical and genetic characteristics of a five-generation family with a novel form of myotonic dystrophy (DM2). Neuromuscul Disord (1999) 9:19–27. 10.1016/S0960-8966(98)00094-7 [DOI] [PubMed] [Google Scholar]
- 42.Eisenschenk S, Triggs WJ, Pearl GS, Rojiani AM. Proximal myotonic myopathy: clinical, neuropathologic, and molecular genetic features. Ann Clin Lab Sci (2001) 31:140–6. [PubMed] [Google Scholar]
- 43.Vihola A, Bachinski LL, Sirito M, Olufemi SE, Hajibashi S, Baggerly KA, et al. Differences in aberrant expression and splicing of sarcomeric proteins in the myotonic dystrophies DM1 and DM2. Acta Neuropathol (2010) 119:465–79. 10.1007/s00401-010-0637-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Day J, Ricker K, Jacobsen J, Rasmussen L, Dick K, Kress W, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology (2003) 60:657–64. 10.1212/01.WNL.0000054481.84978.F9 [DOI] [PubMed] [Google Scholar]
- 45.Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta (2015) 1852:594–606. 10.1016/j.bbadis.2014.05.019 [DOI] [PubMed] [Google Scholar]
- 46.Meola G, Biasini F, Valaperta R, Costa E, Cardani R. Biomolecular diagnosis of myotonic dystrophy type 2: a challenging approach. J Neurol (2017) 264:1705–14. 10.1007/s00415-017-8504-1 [DOI] [PubMed] [Google Scholar]
- 47.Westerlaken JHAM, Van Der Zee CEEM, Peters W, Wieringa B. The DMWD protein from the myotonic dystrophy (DM1) gene region is developmentally regulated and is present most prominently in synapse-dense brain areas. Brain Res (2003) 971:116–27. 10.1016/S0006-8993(03)02430-2 [DOI] [PubMed] [Google Scholar]
- 48.Jansen G, Mahadevan M, Amemiya C, Wormskamp N, Segers B, Hendriks W, et al. Characterization of the myotonic dystrophy region predicts multiple protein isoform-encoding mRNAs. Nat Genet (1992) 1:261–6. 10.1038/ng0792-261 [DOI] [PubMed] [Google Scholar]
- 49.Filippova GN, Thienes CP, Penn BH, Cho DH, Hu YJ, Moore JM, et al. CTCF-binding sites flank CTG/CAG repeats and form a methylation-sensitive insulator at the DM1 locus. Nat Genet (2001) 28:335–43. 10.1038/ng570 [DOI] [PubMed] [Google Scholar]
- 50.Otten AD, Tapscott SJ. Triplet repeat expansion in myotonic dystrophy alters the adjacent chromatin structure. Proc Natl Acad Sci U S A (1995) 92:5465–9. 10.1073/pnas.92.12.5465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barbé L, Lanni S, López-Castel A, Franck S, Spits C, Keymolen K, et al. CpG methylation, a parent-of-origin effect for maternal-biased transmission of congenital myotonic dystrophy. Am J Hum Genet (2017) 100:488–505. 10.1016/j.ajhg.2017.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakamori M, Hamanaka K, Thomas JD, Wang ET, Hayashi YK, Takahashi MP, et al. Aberrant myokine signaling in congenital myotonic dystrophy. Cell Rep (2017) 21:1240–52. 10.1016/j.celrep.2017.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yanovsky-Dagan S, Avitzour M, Altarescu G, Renbaum P, Eldar-Geva T, Schonberger O, et al. Uncovering the role of hypermethylation by CTG expansion in myotonic dystrophy type 1 using mutant human embryonic stem cells. Stem Cell Reports (2015) 5:221–31. 10.1016/j.stemcr.2015.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McMurray CT. Hijacking of the mismatch repair system to cause CAG expansion and cell death in neurodegenerative disease. DNA Repair (Amst) (2008) 7:1121–34. 10.1016/j.dnarep.2008.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmidt MHM, Pearson CE. Disease-associated repeat instability and mismatch repair. DNA Repair (Amst) (2016) 38:117–26. 10.1016/j.dnarep.2015.11.008 [DOI] [PubMed] [Google Scholar]
- 56.Mirkin SM. DNA structures, repeat expansions and human hereditary disorders. Curr Opin Struct Biol (2006) 16:351–8. 10.1016/j.sbi.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 57.Mazouzi A, Velimezi G, Loizou JI. DNA replication stress: causes, resolution and disease. Exp Cell Res (2014) 329:85–93. 10.1016/j.yexcr.2014.09.030 [DOI] [PubMed] [Google Scholar]
- 58.Recolin B, van der Laan S, Tsanov N, Maiorano D. Molecular mechanisms of DNA replication checkpoint activation. Genes (Basel) (2014) 5:147–75. 10.3390/genes5010147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Skourti-Stathaki K, Proudfoot NJ. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev (2014) 28:1384–96. 10.1101/gad.242990.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin Y, Wilson JH. Nucleotide excision repair, mismatch repair, and R-loops modulate convergent transcription-induced cell death and repeat instability. PLoS One (2012) 7:e46807. 10.1371/journal.pone.0046807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meola G. Clinical aspects, molecular pathomechanisms and management of myotonic dystrophies. Acta Myol (2013) 32:154–65. 10.1212/CON.0000000000000414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zu T, Cleary JD, Liu Y, Bañez-Coronel M, Bubenik JL, Ayhan F, et al. RAN translation regulated by muscleblind proteins in myotonic dystrophy type 2. Neuron (2017) 95:1292–305.e5. 10.1016/j.neuron.2017.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang G, Vasquez KM. Effects of replication and transcription on DNA structure-related genetic instability. Genes (Basel) (2017) 8:E17. 10.3390/genes8010017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rohilla KJ, Gagnon KT. RNA biology of disease-associated microsatellite repeat expansions. Acta Neuropathol Commun (2017) 5:63. 10.1186/s40478-017-0468-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet (2002) 11:805–14. 10.1093/hmg/11.7.805 [DOI] [PubMed] [Google Scholar]
- 66.Cardani R, Baldassa S, Botta A, Rinaldi F, Novelli G, Mancinelli E, et al. Ribonuclear inclusions and MBNL1 nuclear sequestration do not affect myoblast differentiation but alter gene splicing in myotonic dystrophy type 2. Neuromuscul Disord (2009) 19:335–43. 10.1016/j.nmd.2009.03.002 [DOI] [PubMed] [Google Scholar]
- 67.Wheeler TM, Krym MC, Thornton CA. Ribonuclear foci at the neuromuscular junction in myotonic dystrophy type 1. Neuromuscul Disord (2007) 17:242–7. 10.1016/j.nmd.2006.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem (2001) 276:7820–6. 10.1074/jbc.M005960200 [DOI] [PubMed] [Google Scholar]
- 69.Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J Biol Chem (2004) 279:13129–39. 10.1074/jbc.M312923200 [DOI] [PubMed] [Google Scholar]
- 70.Sallinen R, Vihola A, Bachinski LL, Huoponen K, Haapasalo H, Hackman P, et al. New methods for molecular diagnosis and demonstration of the (CCTG)n mutation in myotonic dystrophy type 2 (DM2). Neuromuscul Disord (2004) 14:274–83. 10.1016/j.nmd.2004.01.002 [DOI] [PubMed] [Google Scholar]
- 71.Pettersson OJ, Aagaard L, Jensen TG, Damgaard CK. Molecular mechanisms in DM1 – a focus on foci. Nucleic Acids Res (2015) 43:2433–41. 10.1093/nar/gkv029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Apponi LH, Corbett AH, Pavlath GK. RNA-binding proteins and gene regulation in myogenesis. Trends Pharmacol Sci (2011) 32:652–8. 10.1016/j.tips.2011.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ravel-Chapuis A, Belanger G, Yadava RS, Mahadevan MS, DesGroseillers L, Côté J, et al. The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J Cell Biol (2012) 196:699–712. 10.1083/jcb.201108113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bondy-Chorney E, Crawford Parks TE, Ravel-Chapuis A, Klinck R, Rocheleau L, Pelchat M, et al. Staufen1 regulates multiple alternative splicing events either positively or negatively in DM1 indicating its role as a disease modifier. PLoS Genet (2016) 12:e1005827. 10.1371/journal.pgen.1005827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bland CS, Wang ET, Vu A, David MP, Castle JC, Johnson JM, et al. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res (2010) 38:7651–64. 10.1093/nar/gkq614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rau F, Lainé J, Ramanoudjame L, Ferry A, Arandel L, Delalande O, et al. Abnormal splicing switch of DMD’s penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat Commun (2015) 6:7205. 10.1038/ncomms8205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hino S-I, Kondo S, Sekiya H, Saito A, Kanemoto S, Murakami T, et al. Molecular mechanisms responsible for aberrant splicing of SERCA1 in myotonic dystrophy type 1. Hum Mol Genet (2007) 16:2834–43. 10.1093/hmg/ddm239 [DOI] [PubMed] [Google Scholar]
- 78.Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol (2011) 18:840–5. 10.1038/nsmb.2067 [DOI] [PubMed] [Google Scholar]
- 79.Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell (2014) 56:311–22. 10.1016/j.molcel.2014.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Batra R, Manchanda M, Swanson MS. Global insights into alternative polyadenylation regulation. RNA Biol (2015) 12:597–602. 10.1080/15476286.2015.1040974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee JE, Lee JY, Wilusz J, Tian B, Wilusz CJ. Systematic analysis of cis-elements in unstable mRNAS demonstrates that CUGBP1 is a key regulator of mRNA decay in muscle cells. PLoS One (2010) 5:e11201. 10.1371/journal.pone.0011201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gonorazky H, Liang M, Cummings B, Lek M, Micallef J, Hawkins C, et al. RNAseq analysis for the diagnosis of muscular dystrophy. Ann Clin Transl Neurol (2016) 3:55–60. 10.1002/acn3.267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moshourab R, Palada V, Grunwald S, Grieben U, Lewin GR, Spuler S. A molecular signature of myalgia in myotonic dystrophy 2. EBioMedicine (2016) 7:205–11. 10.1016/j.ebiom.2016.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bachinski LL, Baggerly KA, Neubauer VL, Nixon TJ, Raheem O, Sirito M, et al. Most expression and splicing changes in myotonic dystrophy type 1 and type 2 skeletal muscle are shared with other muscular dystrophies. Neuromuscul Disord (2014) 24:227–40. 10.1016/j.nmd.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thomas JD, Sznajder ŁJ, Bardhi O, Aslam FN, Anastasiadis ZP, Scotti MM, et al. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev (2017) 31:1122–33. 10.1101/gad.300590.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Arandel L, Polay Espinoza M, Matloka M, Bazinet A, De Dea Diniz D, Naouar N, et al. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis Model Mech (2017) 10:487–97. 10.1242/dmm.027367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lueck JD, Lungu C, Mankodi A, Osborne RJ, Welle SL, Dirksen RT, et al. Chloride channelopathy in myotonic dystrophy resulting from loss of posttranscriptional regulation for CLCN1. Am J Physiol Cell Physiol (2007) 292:C1291–7. 10.1152/ajpcell.00336.2006 [DOI] [PubMed] [Google Scholar]
- 88.Bachinski LL, Sirito M, Böhme M, Baggerly KA, Udd B, Krahe R. Altered MEF2 isoforms in myotonic dystrophy and other neuromuscular disorders. Muscle Nerve (2010) 42:856–63. 10.1002/mus.21789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morrone A, Pegoraro E, Angelini C, Zammarchi E, Marconi G, Hoffman EP. RNA metabolism in myotonic dystrophy: patient muscle shows decreased insulin receptor RNA and protein consistent with abnormal insulin resistance. J Clin Invest (1997) 99:1691–8. 10.1172/JCI119332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet (2001) 29:40–7. 10.1038/ng704 [DOI] [PubMed] [Google Scholar]
- 91.Savkur RS, Philips AV, Cooper TA, Dalton JC, Moseley ML, Ranum LPW, et al. Insulin receptor splicing alteration in myotonic dystrophy type 2. Am J Hum Genet (2004) 74:1309–13. 10.1086/421528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tang ZZ, Yarotskyy V, Wei L, Sobczak K, Nakamori M, Eichinger K, et al. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum Mol Genet (2012) 21:1312–24. 10.1093/hmg/ddr568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Botta A, Malena A, Loro E, Del Moro G, Suman M, Pantic B, et al. Altered Ca2+ homeostasis and endoplasmic reticulum stress in myotonic dystrophy type 1 muscle cells. Genes (Basel) (2013) 4:275–92. 10.3390/genes4020275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol (2013) 74:862–72. 10.1002/ana.23992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Perfetti A, Greco S, Fasanaro P, Bugiardini E, Cardani R, Garcia Manteiga JM, et al. Genome wide identification of aberrant alternative splicing events in myotonic dystrophy type 2. PLoS One (2014) 9:e93983. 10.1371/journal.pone.0093983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Screen M, Jonson PH, Raheem O, Palmio J, Laaksonen R, Lehtimäki T, et al. Abnormal splicing of NEDD4 in myotonic dystrophy type 2: possible link to statin adverse reactions. Am J Pathol (2014) 184:2322–32. 10.1016/j.ajpath.2014.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thornton CA, Wang E, Carrell EM. Myotonic dystrophy: approach to therapy. Curr Opin Genet Dev (2017) 44:135–40. 10.1016/j.gde.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen W, Wang Y, Abe Y, Cheney L, Udd B, Li YP. Haploinsuffciency for Znf9 in Znf9±mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J Mol Biol (2007) 368:8–17. 10.1016/j.jmb.2007.01.088 [DOI] [PubMed] [Google Scholar]
- 99.Huichalaf C, Schoser B, Schneider-Gold C, Jin B, Sarkar P, Timchenko L. Reduction of the rate of protein translation in patients with myotonic dystrophy 2. J Neurosci (2009) 29:9042–9. 10.1523/JNEUROSCI.1983-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Benhalevy D, Gupta SK, Danan CH, Ghosal S, Sun H-W, Kazemier HG, et al. The human CCHC-type zinc finger nucleic acid binding protein binds G-rich elements in target mRNA coding sequences and promotes translation. Cell Rep (2017) 18:2979–90. 10.1016/j.celrep.2017.02.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Abdelmohsen K. Chapter 5: modulation of gene expression by RNA binding proteins: mRNA stability and translation. Binding Protein. London, UK: IntechOpen Ltd; (2012). [Google Scholar]
- 102.Timchenko L. Chapter 5: modulation of gene expression by RNA binding proteins: mRNA stability and translation. Int J Biochem Cell Biol (2013) 45:2280–7. 10.1016/j.biocel.2013.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol (2017) 18:285–98. 10.1038/nrm.2017.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Weber SCC, Brangwynne CPP. Getting RNA and protein in phase. Cell (2012) 149:1188–91. 10.1016/j.cell.2012.05.022 [DOI] [PubMed] [Google Scholar]
- 105.Brangwynne CP, Tompa P, Pappu RV. Polymer physics of intracellular phase transitions. Nat Phys (2015) 11:899–904. 10.1038/nphys3532 [DOI] [Google Scholar]
- 106.Jain A, Vale RD. RNA phase transitions in repeat expansion disorders. Nature (2017) 546:243–7. 10.1038/nature22386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cleary JD, Ranum LPW. Repeat-associated non-ATG (RAN) translation in neurological disease. Hum Mol Genet (2013) 22:45–51. 10.1093/hmg/ddt371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zu T, Gibbens B, Doty NS, Gomes-pereira M, Huguet A, Stone MD. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A (2010) 108:260–5. 10.1073/pnas.1013343108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pearson CE. Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities! PLoS Genet (2011) 7:e1002018. 10.1371/journal.pgen.1002018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Reid DW, Nicchitta CV. The enduring enigma of nuclear translation. J Cell Biol (2012) 197:7–9. 10.1083/jcb.201202140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.David A, Dolan BP, Hickman HD, Knowlton JJ, Clavarino G, Pierre P, et al. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J Cell Biol (2012) 197:45–57. 10.1083/jcb.201112145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell (2009) 136:731–45. 10.1016/j.cell.2009.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van Agtmaal EL, André LM, Willemse M, Cumming S, van Kessel IDG, van den Broek WJAA, et al. CRISPR/Cas9-induced (CTG•CAG)n repeat instability in the myotonic dystrophy type 1 locus: implications for therapeutic genome editing. Mol Ther (2017) 25:24–43. 10.1016/j.ymthe.2016.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schiaffino S, Rossi AC, Smerdu V, Leinwand LA, Reggiani C. Developmental myosins: expression patterns and functional significance. Skelet Muscle (2015) 5:1–14. 10.1186/s13395-015-0046-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scott W, Stevens J, Binder-Macleod SA. Human skeletal muscle fiber type classifications. Phys Ther (2001) 81:1810–6. 10.1093/ptj/81.11.1810 [DOI] [PubMed] [Google Scholar]
- 116.Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev (2011) 91:1447–531. 10.1152/physrev.00031.2010 [DOI] [PubMed] [Google Scholar]
- 117.Wang Y, Pessin JE. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care (2013) 16:243–50. 10.1097/MCO.0b013e328360272d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Goodman CA, Mayhew DL, Hornberger TA. Recent progress toward understanding the molecular mechanisms that regulate skeletal muscle mass. Cell Signal (2011) 23:1896–906. 10.1016/j.cellsig.2011.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wing SS, Lecker SH, Jagoe RT. Proteolysis in illness-associated skeletal muscle atrophy: from pathways to networks. Crit Rev Clin Lab Sci (2011) 48:49–70. 10.3109/10408363.2011.586171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Thornton CA. Myotonic dystrophy. Neurol Clin (2014) 32:705–19. 10.1016/j.ncl.2014.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bassez G, Chapoy E, Bastuji-Garin S, Radvanyi-Hoffman H, Authier F-JJ, Pellissier JF, et al. Type 2 myotonic dystrophy can be predicted by the combination of type 2 muscle fiber central nucleation and scattered atrophy. J Neuropathol Exp Neurol (2008) 67:319–25. 10.1097/NEN.0b013e31816b4acc [DOI] [PubMed] [Google Scholar]
- 122.Pisani V, Panico MB, Terracciano C, Bonifazi E, Meola G, Novelli G, et al. Preferential central nucleation of type 2 myofibers is an invariable feature of myotonic dystrophy type 2. Muscle Nerve (2008) 38:1405–11. 10.1002/mus.21122 [DOI] [PubMed] [Google Scholar]
- 123.Schoser BGH, Schneider-Gold C, Kress W, Goebel HH, Reilich P, Koch MC, et al. Muscle pathology in 57 patients with myotonic dystrophy type 2. Muscle Nerve (2004) 29:275–81. 10.1002/mus.10545 [DOI] [PubMed] [Google Scholar]
- 124.Argov Z, Gardner-Medwin D, Johnson MA, Mastaglia FL. Congenital myotonic dystrophy: fiber type abnormalities in two cases. Arch Neurol (1980) 37:693–6. 10.1001/archneur.1980.00500600041006 [DOI] [PubMed] [Google Scholar]
- 125.Tanabe Y, Nonaka I. Congenital myotonic dystrophy. Changes in muscle pathology with ageing. J Neurol Sci (1987) 77:59–68. 10.1016/0022-510X(87)90206-1 [DOI] [PubMed] [Google Scholar]
- 126.Farkas-Bargeton E, Barbet JP, Dancea S, Wehrle R, Checouri A, Dulac O. Immaturity of muscle fibers in the congenital form of myotonic dystrophy: its consequences and its origin. J Neurol Sci (1988) 83:145–59. 10.1016/0022-510X(88)90064-0 [DOI] [PubMed] [Google Scholar]
- 127.Furling D, Lemieux D, Taneja K, Puymirat J. Decreased levels of myotonic dystrophy protein kinase (DMPK) and delayed differentiation in human myotonic dystrophy myoblasts. Neuromuscul Disord (2001) 11:728–35. 10.1016/S0960-8966(01)00226-7 [DOI] [PubMed] [Google Scholar]
- 128.Iannaccone ST, Bove KE, Vogler C, Azzarelli B, Muller J. Muscle maturation delay in infantile myotonic dystrophy. Arch Pathol Lab Med (1986) 110:405–11. [PubMed] [Google Scholar]
- 129.Sarnat HB, Silbert SW. Maturational arrest of fetal muscle in neonatal myotonic dystrophy. A pathologic study of four cases. Arch Neurol (1976) 33:466–74. 10.1001/archneur.1976.00500070008002 [DOI] [PubMed] [Google Scholar]
- 130.Karpati G, Carpenter S, Watters GV, Eisen AA, Andermann F. Infantile myotonic dystrophy. Histochemical and electron microscopic features in skeletal muscle. Neurology (1973) 23:1066–77. 10.1212/WNL.23.10.1066 [DOI] [PubMed] [Google Scholar]
- 131.Ott MO, Bober E, Lyons G, Arnold H, Buckingham M. Early expression of the myogenic regulatory gene Myf-5 in precursor cells of skeletal muscle in the mouse embryo. Development (1991) 111:1097–107. [DOI] [PubMed] [Google Scholar]
- 132.Godinho RO. In vitro development of skeletal muscle fiber. Braz J Morphol Sci (2006) 23:173–86. [Google Scholar]
- 133.Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, et al. The formation of skeletal muscle: from somite to limb. J Anat (2003) 202:59–68. 10.1046/j.1469-7580.2003.00139.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Andrés V, Walsh K. Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J Cell Biol (1996) 132:657–66. 10.1083/jcb.132.4.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, et al. Correlation of terminal cell-cycle arrest of skeletal-muscle with induction of p21 by myoD. Science (1995) 267:1018–21. 10.1126/science.7863327 [DOI] [PubMed] [Google Scholar]
- 136.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell (1995) 81:323–30. 10.1016/0092-8674(95)90385-2 [DOI] [PubMed] [Google Scholar]
- 137.Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA, Gallie BL. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev (1996) 10:3051–64. 10.1101/gad.10.23.3051 [DOI] [PubMed] [Google Scholar]
- 138.Harrington EA, Bruce JL, Harlow E, Dyson N. pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc Natl Acad Sci U S A (1998) 95:11945–50. 10.1073/pnas.95.20.11945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fujio Y, Guo K, Mano T, Mitsuuchi Y, Testa JR, Walsh K. Cell cycle withdrawal promotes myogenic induction of Akt, a positive modulator of myocyte survival. Mol Cell Biol (1999) 19:5073–82. 10.1128/MCB.19.7.5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vlasova-St Louis I, Bohjanen PR. Feedback regulation of kinase signaling pathways by AREs and GREs. Cells (2016) 5:4. 10.3390/cells5010004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Salisbury E, Sakai K, Schoser B, Huichalaf C, Schneider-Gold C, Nguyen H, et al. Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1. Exp Cell Res (2008) 314:2266–78. 10.1016/j.yexcr.2008.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet (2005) 14:1539–47. 10.1093/hmg/ddi162 [DOI] [PubMed] [Google Scholar]
- 143.Schnorrer F, Dickson BJ. Muscle building mechanisms of myotube guidance and attachment site selection. Dev Cell (2004) 7:9–20. 10.1016/j.devcel.2004.06.010 [DOI] [PubMed] [Google Scholar]
- 144.Deng S, Azevedo M, Baylies M. Acting on identity: myoblast fusion and the formation of the syncytial muscle fiber. Semin Cell Dev Biol (2017) 72:45–55. 10.1016/j.semcdb.2017.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Harris AJ, Duxson MJ, Fttzsimons RB, Rieger F. Myonuclear birthdates distinguish the origins of primary and secondary myotubes in embryonic mammalian skeletal muscles. Development (1989) 107:771–84. [DOI] [PubMed] [Google Scholar]
- 146.Rochlin K, Yu S, Roy S, Baylies MK. Myoblast fusion: when it takes more to make one. Dev Biol (2010) 341:66–83. 10.1016/j.ydbio.2009.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hernández JM, Podbilewicz B. The hallmarks of cell-cell fusion. Development (2017) 144:4481–95. 10.1242/dev.155523 [DOI] [PubMed] [Google Scholar]
- 148.Quinn ME, Goh Q, Kurosaka M, Gamage DG, Petrany MJ, Prasad V, et al. Myomerger induces fusion of non-fusogenic cells and is required for skeletal muscle development. Nat Commun (2017) 8:1–9. 10.1038/ncomms15665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Millay DP, O’Rourke JR, Sutherland LB, Bezprozvannaya S, Shelton JM, Bassel-Duby R, et al. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature (2013) 499:301–5. 10.1038/nature12343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Millay DP, Sutherland LB, Bassel-duby R, Olson EN. Myomaker is essential for muscle regeneration. Genes Dev (2014) 28:1641–6. 10.1101/gad.247205.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gamage DG, Leikina E, Quinn ME, Ratinov A, Chernomordik LV, Millay DP. Insights into the localization and function of myomaker during myoblast fusion. J Biol Chem (2017) 292:17272–89. 10.1074/jbc.M117.811372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Di Gioia SA, Connors S, Matsunami N, Cannavino J, Rose MF, Gilette NM, et al. A defect in myoblast fusion underlies Carey-Fineman-Ziter syndrome. Nat Commun (2017) 8:16077. 10.1038/ncomms16077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bi P, Ramirez-Martinez A, Li H, Cannavino J, McAnally JR, Shelton JM, et al. Control of muscle formation by the fusogenic micropeptide myomixer. Science (2017) 356:323–7. 10.1126/science.aam9361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lee SH, Dominguez R. Regulation of actin cytoskeleton dynamics in cells. Mol Cells (2010) 29:311–25. 10.1007/s10059-010-0053-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sampath SC, Sampath SC, Millay DP. Myoblast fusion confusion: the resolution begins. Skelet Muscle (2018) 8:3. 10.1186/s13395-017-0149-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jansen G, Groenen PJTA, Bächner D, Jap PHK, Coerwinkel M, Oerlemans F, et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet (1996) 13:316–24. 10.1038/ng0796-316 [DOI] [PubMed] [Google Scholar]
- 157.Wansink DG, van Herpen RE, Coerwinkel-Driessen MM, Groenen PJ, Hemmings BA, Wieringa B. Alternative splicing controls myotonic dystrophy protein kinase structure, enzymatic activity, and subcellular localization. Mol Cell Biol (2003) 23:5489–501. 10.1128/MCB.23.16.5489-5501.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mulders SAM, van Horssen R, Gerrits L, Bennink MB, Pluk H, de Boer-van Huizen RT, et al. Abnormal actomyosin assembly in proliferating and differentiating myoblasts upon expression of a cytosolic DMPK isoform. Biochim Biophys Acta (2011) 1813:867–77. 10.1016/j.bbamcr.2011.01.024 [DOI] [PubMed] [Google Scholar]
- 159.Oude Ophuis RJ, Mulders SA, van Herpen RE, van de Vorstenbosch R, Wieringa B, Wansink DG. DMPK protein isoforms are differentially expressed in myogenic and neural cell lineages. Muscle Nerve (2009) 40:545–55. 10.1002/mus.21352 [DOI] [PubMed] [Google Scholar]
- 160.Pelletier R, Hamel F, Beaulieu D, Patry L, Haineault C, Tarnopolsky M, et al. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol Dis (2009) 36:181–90. 10.1016/j.nbd.2009.07.009 [DOI] [PubMed] [Google Scholar]
- 161.Timchenko NA, Iakova P, Cai Z, James R, Timchenko LT, Smith JR. Molecular basis for impaired muscle differentiation in myotonic dystrophy molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol Cell Biol (2001) 21:6927–38. 10.1128/MCB.21.20.6927-6938.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Buj-Bello A, Furling D, Tronchere H, Laporte J, Lerouge T, Butler-Browne GS, et al. Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum Mol Genet (2002) 11:2297–307. 10.1093/hmg/11.19.2297 [DOI] [PubMed] [Google Scholar]
- 163.Loro E, Rinaldi F, Malena A, Masiero E, Novelli G, Angelini C, et al. Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Differ (2010) 17:1315–24. 10.1038/cdd.2010.33 [DOI] [PubMed] [Google Scholar]
- 164.Kameda N, Ueda H, Ohno S, Shimokawa M, Usuki F, Ishiura S, et al. Developmental regulation of myotonic dystrophy protein kinase in human muscle cells in vitro. Neuroscience (1998) 85:311–22. 10.1016/S0306-4522(97)00602-7 [DOI] [PubMed] [Google Scholar]
- 165.Demonbreun AR, Biersmith BH, McNally EM. Membrane fusion in muscle development and repair. Semin Cell Dev Biol (2015) 45:48–56. 10.1016/j.semcdb.2015.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Berkes CA, Tapscott SJ. MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol (2005) 16:585–95. 10.1016/j.semcdb.2005.07.006 [DOI] [PubMed] [Google Scholar]
- 167.Shi X, Garry DJ. Muscle stem cells in development, regeneration, and disease. Genes Dev (2006) 20:1692–708. 10.1101/gad.1419406 [DOI] [PubMed] [Google Scholar]
- 168.Wang YX, Dumont NA, Rudnicki MA. Muscle stem cells at a glance. J Cell Sci (2014) 127:4543–8. 10.1242/jcs.151209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hernández-Hernández JM, García-González EG, Brun CE, Rudnicki MA. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin Cell Dev Biol (2017) 72:10–8. 10.1016/j.semcdb.2017.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Buckingham M, Rigby PWJ. Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev Cell (2014) 28:225–38. 10.1016/j.devcel.2013.12.020 [DOI] [PubMed] [Google Scholar]
- 171.Yajima H, Motohashi N, Ono Y, Sato S, Ikeda K, Masuda S, et al. Six family genes control the proliferation and differentiation of muscle satellite cells. Exp Cell Res (2010) 316:2932–44. 10.1016/j.yexcr.2010.08.001 [DOI] [PubMed] [Google Scholar]
- 172.Grifone R. Six1 and Six4 homeoproteins are required for Pax3 and Mrf expression during myogenesis in the mouse embryo. Development (2005) 132:2235–49. 10.1242/dev.01773 [DOI] [PubMed] [Google Scholar]
- 173.Inukai A, Doyu M, Kato T, Liang Y, Kuru S, Yamamoto M, et al. Reduced expression of DMAHP/SIX5 gene in myotonic dystrophy muscle. Muscle Nerve (2000) 23:1421–6. [DOI] [PubMed] [Google Scholar]
- 174.Klesert TR, Otten AD, Bird TD, Tapscott SJ. Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nat Genet (1997) 16:402–6. 10.1038/ng0897-402 [DOI] [PubMed] [Google Scholar]
- 175.Thornton CA, Wymer JP, Simmons Z, McClain C, Moxley RT. Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat Genet (1997) 16:407–9. 10.1038/ng0897-407 [DOI] [PubMed] [Google Scholar]
- 176.Alwazzan M, Newman E, Hamshere MG, Brook JD. Myotonic dystrophy is associated with a reduced level of RNA from the DMWD allele adjacent to the expanded repeat. Hum Mol Genet (1999) 8:1491–7. 10.1093/hmg/8.8.1491 [DOI] [PubMed] [Google Scholar]
- 177.Kirby RJ, Hamilton GM, Finnegan DJ, Johnson KJ, Jarman AP. Drosophila homolog of the myotonic dystrophy-associated gene, SIX5, is required for muscle and gonad development. Curr Biol (2001) 11:1044–9. 10.1016/S0960-9822(01)00319-0 [DOI] [PubMed] [Google Scholar]
- 178.Klesert TR, Cho DH, Clark JI, Maylie J, Adelman J, Snider L, et al. Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nat Genet (2000) 25:105–9. 10.1038/75490 [DOI] [PubMed] [Google Scholar]
- 179.Sarkar PS, Appukuttan B, Han J, Ito Y, Ai C, Tsai W, et al. Heterozygous loss of Six5 in mice is sufficient to cause ocular cataracts. Nat Genet (2000) 25:110–4. 10.1038/75500 [DOI] [PubMed] [Google Scholar]
- 180.Spletter ML, Schnorrer F. Transcriptional regulation and alternative splicing cooperate in muscle fiber-type specification in flies and mammals. Exp Cell Res (2014) 321:90–8. 10.1016/j.yexcr.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development (2007) 134:4131–40. 10.1242/dev.008367 [DOI] [PubMed] [Google Scholar]
- 182.Miller JW, Urbinati CR, Teng-Umnuay P, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J (2000) 19:4439–48. 10.1093/emboj/19.17.4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, et al. A muscleblind knockout model for myotonic dystrophy. Science (2003) 302:1978–80. 10.1126/science.1088583 [DOI] [PubMed] [Google Scholar]
- 184.Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet (2006) 15:2087–97. 10.1093/hmg/ddl132 [DOI] [PubMed] [Google Scholar]
- 185.Holt I, Jacquemin V, Fardaei M, Sewry CA, Butler-Browne GS, Furling D, et al. Muscleblind-like proteins: similarities and differences in normal and myotonic dystrophy muscle. Am J Pathol (2009) 174:216–27. 10.2353/ajpath.2009.080520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lee KS, Cao Y, Witwicka HE, Tom S, Tapscott SJ, Wang EH. RNA-binding protein muscleblind-like 3 (MBNL3) disrupts myocyte enhancer factor 2 (Mef2) β-exon splicing. J Biol Chem (2010) 285:33779–87. 10.1074/jbc.M110.124255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nikovits W, Stockdale F. Cellular and molecular bases of skeletal muscle fiber diversity. Basic Appl Myol (1996) 6:407–15. [Google Scholar]
- 188.Jackson HE, Ingham PW. Control of muscle fibre-type diversity during embryonic development: the zebrafish paradigm. Mech Dev (2013) 130:447–57. 10.1016/j.mod.2013.06.001 [DOI] [PubMed] [Google Scholar]
- 189.Braun T, Gautel M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat Rev Mol Cell Biol (2011) 12:349–61. 10.1038/nrm3118 [DOI] [PubMed] [Google Scholar]
- 190.Albert Y, Whitehead J, Eldredge L, Carter J, Gao X, Tourtellotte WG. Transcriptional regulation of myotube fate specification and intrafusal muscle fiber morphogenesis. J Cell Biol (2005) 169:257–68. 10.1083/jcb.200501156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tourtellotte WG, Keller-Peck C, Milbrandt J, Kucera J. The transcription factor Egr3 modulates sensory axon-myotube interactions during muscle spindle morphogenesis. Dev Biol (2001) 232:388–99. 10.1006/dbio.2001.0202 [DOI] [PubMed] [Google Scholar]
- 192.Oliveira Fernandes M, Tourtellotte WG. Egr3-dependent muscle spindle stretch receptor intrafusal muscle fiber differentiation and fusimotor innervation homeostasis. J Neurosci (2015) 35:5566–78. 10.1523/JNEUROSCI.0241-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Andrechek ER, Hardy WR, Girgis-Gabardo AA, Perry RLS, Butler R, Graham FL, et al. ErbB2 is required for muscle spindle and myoblast cell survival. Mol Cell Biol (2002) 22:4714–22. 10.1128/MCB.22.13.4714-4722.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Murgia M, Toniolo L, Nagaraj N, Ciciliot S, Vindigni V, Schiaffino S, et al. Single muscle fiber proteomics reveals fiber-type-specific features of human muscle aging. Cell Rep (2017) 19:2396–409. 10.1016/j.celrep.2017.05.054 [DOI] [PubMed] [Google Scholar]
- 195.Murgia M, Nagaraj N, Deshmukh AS, Zeiler M, Cancellara P, Moretti I, et al. Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep (2015) 16:387–95. 10.15252/embr.201439757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Eriksson M, Hedberg B, Carey N, Ansved T. Decreased DMPK transcript levels in myotonic dystrophy 1 type IIA muscle fibers. Biochem Biophys Res Commun (2001) 286:1177–82. 10.1006/bbrc.2001.5516 [DOI] [PubMed] [Google Scholar]
- 197.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol (1961) 9:493–5. 10.1083/jcb.9.2.493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ontell M, Feng KC, Klueber K, Dunn RF, Taylor F. Myosatellite cells, growth, and regeneration in murine dystrophic muscle: a quantitative study. Anat Rec (1984) 208:159–74. 10.1002/ar.1092080203 [DOI] [PubMed] [Google Scholar]
- 199.Pannérec A, Marazzi G, Sassoon D. Stem cells in the hood: the skeletal muscle niche. Trends Mol Med (2012) 18:599–606. 10.1016/j.molmed.2012.07.004 [DOI] [PubMed] [Google Scholar]
- 200.Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol (2007) 9:255–67. 10.1038/ncb1542 [DOI] [PubMed] [Google Scholar]
- 201.Messina G, Sirabella D, Monteverde S, Galvez BG, Tonlorenzi R, Schnapp E, et al. Skeletal muscle differentiation of embryonic mesoangioblasts requires pax3 activity. Stem Cells (2009) 27:157–64. 10.1634/stemcells.2008-0503 [DOI] [PubMed] [Google Scholar]
- 202.Esner M, Meilhac SM, Relaix F, Nicolas JF, Cossu G, Buckingham ME. Smooth muscle of the dorsal aorta shares a common clonal origin with skeletal muscle of the myotome. Development (2006) 133:737–49. 10.1242/dev.02226 [DOI] [PubMed] [Google Scholar]
- 203.Tierney MT, Sacco A. Satellite cell heterogeneity in skeletal muscle homeostasis. Trends Cell Biol (2016) 26:434–44. 10.1016/j.tcb.2016.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zammit PS. All muscle satellite cells are equal, but are some more equal than others? J Cell Sci (2008) 121:2975–82. 10.1242/jcs.019661 [DOI] [PubMed] [Google Scholar]
- 205.Chargé SBP, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev (2004) 84:209–38. 10.1152/physrev.00019.2003 [DOI] [PubMed] [Google Scholar]
- 206.Conboy IM, Rando TA. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell (2002) 3:397–409. 10.1016/S1534-5807(02)00254-X [DOI] [PubMed] [Google Scholar]
- 207.Troy A, Cadwallader AB, Fedorov Y, Tyner K, Tanaka KK, Olwin BB. Coordination of satellite cell activation and self-renewal by Par-complex-dependent asymmetric activation of p38α/β MAPK. Cell Stem Cell (2012) 11:541–53. 10.1016/j.stem.2012.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Thorley M, Malatras A, Duddy W, Le Gall L, Mouly V, Butler-Browne G, et al. Changes in communication between muscle stem cells and their environment with aging. J Neuromuscul Dis (2015) 2:205–17. 10.3233/JND-150097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zismanov V, Chichkov V, Colangelo V, Jamet S, Wang S, Syme A, et al. Phosphorylation of eIF2α is a translational control mechanism regulating muscle stem cell quiescence and self-renewal. Cell Stem Cell (2016) 18:79–90. 10.1016/j.stem.2015.09.020 [DOI] [PubMed] [Google Scholar]
- 210.Kassar-duchossoy L, Giacone E, Gayraud-morel B, Jory A, Gomès D, Tajbakhsh S. Pax3/Pax7 mark a novel population of primitive myogenic cells during development. Genes Dev (2005) 19:1426–31. 10.1101/gad.345505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell (2000) 102:777–86. 10.1016/S0092-8674(00)00066-0 [DOI] [PubMed] [Google Scholar]
- 212.Tajbakhsh S, Rocancourt D, Cossu G, Buckingham M. Redefining the genetic hierarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell (1997) 89:127–38. 10.1016/S0092-8674(00)80189-0 [DOI] [PubMed] [Google Scholar]
- 213.Beauchamp JR, Heslop L, Yu DS, Tajbakhsh S, Kelly RG, Wernig A, et al. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J Cell Biol (2000) 151:1221–34. 10.1083/jcb.151.6.1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Olguin HC, Olwin BB. Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self-renewal. Dev Biol (2004) 275:375–88. 10.1016/j.ydbio.2004.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Füchtbauer EM, Westphal H. MyoD and myogenin are coexpressed in regenerating skeletal muscle of the mouse. Dev Dyn (1992) 193:34–9. 10.1002/aja.1001930106 [DOI] [PubMed] [Google Scholar]
- 216.Hawke TJ, Meeson AP, Jiang N, Graham S, Hutcheson K, DiMaio JM, et al. P21 is essential for normal myogenic progenitor cell function in regenerating skeletal muscle. Am J Physiol Cell Physiol (2003) 285:C1019–27. 10.1152/ajpcell.00055.2003 [DOI] [PubMed] [Google Scholar]
- 217.Harper JW, Bennett EJ. Proteome complexity and the forces that drive proteome imbalance. Nature (2016) 537:328–38. 10.1038/nature19947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Thornell L-E, Lindstöm M, Renault V, Klein A, Mouly V, Ansved T, et al. Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1. Neuropathol Appl Neurobiol (2009) 35:603–13. 10.1111/j.1365-2990.2009.01014.x [DOI] [PubMed] [Google Scholar]
- 219.Vattemi G, Tomelleri G, Filosto M, Savio C, Rizzuto N, Tonin P. Expression of late myogenic differentiation markers in sarcoplasmic masses of patients with myotonic dystrophy. Neuropathol Appl Neurobiol (2005) 31:45–52. 10.1111/j.1365-2990.2004.00602.x [DOI] [PubMed] [Google Scholar]
- 220.Renna LV, Cardani R, Botta A, Rossi G, Fossati B, Costa E, et al. Premature senescence in primary muscle cultures of myotonic dystrophy type 2 is not associated with p16 induction. Eur J Histochem (2014) 58:275–86. 10.4081/ejh.2014.2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Malatesta M. Skeletal muscle features in myotonic dystrophy and sarcopenia: do similar nuclear mechanisms lead to skeletal muscle wasting? Eur J Histochem (2012) 56:228–30. 10.4081/ejh.2012.e36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Malatesta M, Cardani R, Pellicciari C, Meola G. RNA transcription and maturation in skeletal muscle cells are similarly impaired in myotonic dystrophy and sarcopenia: the ultrastructural evidence. Front Aging Neurosci (2014) 6:196. 10.3389/fnagi.2014.00196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Wertz RL, Hartwig GB, Frost AP, Brophy JJ, Atwater SK, Roses AD. Patients with myotonic dystrophy, a possible segmental progeroid syndrome, and Duchenne muscular dystrophy have fibroblasts with normal limits for in vitro lifespan and growth characteristics. J Cell Physiol (1981) 107:255–60. 10.1002/jcp.1041070212 [DOI] [PubMed] [Google Scholar]
- 224.Edström E, Altun M, Bergman E, Johnson H, Kullberg S, Ramírez-León V, et al. Factors contributing to neuromuscular impairment and sarcopenia during aging. Physiol Behav (2007) 92:129–35. 10.1016/j.physbeh.2007.05.040 [DOI] [PubMed] [Google Scholar]
- 225.Malatesta M, Giagnacovo M, Costanzo M, Cisterna B, Cardani R, Meola G. Muscleblind-like1 undergoes ectopic relocation in the nuclei of skeletal muscles in myotonic dystrophy and sarcopenia. Eur J Histochem (2013) 57:e15. 10.4081/ejh.2013.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Thompson LV. Age-related muscle dysfunction. Exp Gerontol (2009) 44:106–11. 10.1016/j.exger.2008.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ohlendieck K. Proteomic profiling of fast-to-slow muscle transitions during aging. Front Physiol (2011) 2:105. 10.3389/fphys.2011.00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Welle S, Brooks AI, Delehanty JM, Needler N, Thornton CA. Gene expression profile of aging in human muscle. Physiol Genomics (2003) 14:149–59. 10.1152/physiolgenomics.00049.2003 [DOI] [PubMed] [Google Scholar]
- 229.García-Prat L, Sousa-Victor P, Muñoz-Cánoves P. Proteostatic and metabolic control of stemness. Cell Stem Cell (2017) 20:593–608. 10.1016/j.stem.2017.04.011 [DOI] [PubMed] [Google Scholar]
- 230.Blau HM, Cosgrove BD, Ho AT. The central role of muscle stem cells in regenerative failure with aging. Nat Med (2015) 21:854–62. 10.1038/nm.3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Sousa-Victor P, García-Prat L, Serrano AL, Perdiguero E, Muñoz-Cánoves P. Muscle stem cell aging: regulation and rejuvenation. Trends Endocrinol Metab (2015) 26:287–96. 10.1016/j.tem.2015.03.006 [DOI] [PubMed] [Google Scholar]
- 232.Vahidi Ferdousi L, Rocheteau P, Chayot R, Montagne B, Chaker Z, Flamant P, et al. More efficient repair of DNA double-strand breaks in skeletal muscle stem cells compared to their committed progeny. Stem Cell Res (2014) 13:492–507. 10.1016/j.scr.2014.08.005 [DOI] [PubMed] [Google Scholar]
- 233.Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature (2007) 447:447–52. 10.1038/nature05778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kumar A, Ratan RR. Oxidative stress and Huntington’s disease: the good, the bad, and the ugly. J Huntingtons Dis (2016) 5:217–37. 10.3233/JHD-160205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bigot A, Klein AF, Gasnier E, Jacquemin V, Ravassard P, Butler-Browne G, et al. Large CTG repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am J Pathol (2009) 174:1435–42. 10.2353/ajpath.2009.080560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Bentzinger CF, Rudnicki MA. Rejuvenating aged muscle stem cells. Nat Med (2014) 20:234–5. 10.1038/nm.3499 [DOI] [PubMed] [Google Scholar]
- 237.Bargiela A, Cerro-Herreros E, Fernandez-Costa JM, Vilchez JJ, Llamusi B, Artero R. Increased autophagy and apoptosis contribute to muscle atrophy in a myotonic dystrophy type 1 Drosophila model. Dis Model Mech (2015) 8:679–90. 10.1242/dmm.018127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature (2016) 530:184–9. 10.1038/nature16932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell (2017) 169:132–47.e16. 10.1016/j.cell.2017.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol (2010) 2010:214074. 10.1155/2010/214074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Bentzinger CF, von Maltzahn J, Rudnicki MA. Extrinsic regulation of satellite cell specification. Stem Cell Res Ther (2010) 1:27. 10.1186/scrt27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Beaulieu D, Thebault P, Pelletier R, Chapdelaine P, Tarnopolsky M, Furling D, et al. Abnormal prostaglandin E2 production blocks myogenic differentiation in myotonic dystrophy. Neurobiol Dis (2012) 45:122–9. 10.1016/j.nbd.2011.06.014 [DOI] [PubMed] [Google Scholar]
- 243.Russo J, Lee JE, López CM, Anderson J, Nguyen TMP, Heck AM, et al. The CELF1 RNA-binding protein regulates decay of signal recognition particle mRNAs and limits secretion in mouse myoblasts. PLoS One (2017) 12:e0170680. 10.1371/journal.pone.0170680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol (2010) 17:187–93. 10.1038/nsmb.1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Olejniczak M, Kotowska-Zimmer A, Krzyzosiak W. Stress-induced changes in miRNA biogenesis and functioning. Cell Mol Life Sci (2018) 75:177–91. 10.1007/s00018-017-2591-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Flynt AS, Li N, Thatcher EJ, Solnica-Krezel L, Patton JG. Zebrafish miR-214 modulates Hedgehog signaling to specify muscle cell fate. Nat Genet (2007) 39:259–63. 10.1038/ng1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, et al. A uniform system for microRNA annotation. RNA (2003) 9:277–9. 10.1261/rna.2183803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Wang H, Wang B. Extracellular vesicle microRNAs mediate skeletal muscle myogenesis and disease (Review). Biomed Rep (2016) 5(3):296–300. 10.3892/br.2016.725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.van Rooij E, Liu N, Olson EN. MicroRNAs flex their muscles. Trends Genet (2008) 24:159–66. 10.1016/j.tig.2008.01.007 [DOI] [PubMed] [Google Scholar]
- 250.Chen J-F, Callis TE, Wang D-Z. MicroRNAs and muscle disorders. J Cell Sci (2009) 122:13–20. 10.1242/jcs.041723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Ge Y, Chen J. MicroRNAs in skeletal myogenesis. Cell Cycle (2011) 10:441–8. 10.4161/cc.10.3.14710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Coenen-Stass AML, Betts CA, Lee YF, Mäger I, Turunen MP, El Andaloussi S, et al. Selective release of muscle-specific, extracellular microRNAs during myogenic differentiation. Hum Mol Genet (2016) 25:3960–74. 10.1093/hmg/ddw237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Perfetti A, Greco S, Cardani R, Fossati B, Cuomo G, Valaperta R, et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci Rep (2016) 6:38174. 10.1038/srep38174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Gambardella S, Rinaldi F, Lepore SM, Viola A, Loro E, Angelini C, et al. Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. J Transl Med (2010) 8:1–9. 10.1186/1479-5876-8-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Perbellini R, Greco S, Sarra-Ferraris G, Cardani R, Capogrossi MC, Meola G, et al. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul Disord (2011) 21:81–8. 10.1016/j.nmd.2010.11.012 [DOI] [PubMed] [Google Scholar]
- 256.Koutsoulidou A, Photiades M, Kyriakides TC, Georgiou K, Prokopi M, Kapnisis K, et al. Identification of exosomal muscle-specific miRNAs in serum of myotonic dystrophy patients relating to muscle disease progress. Hum Mol Genet (2017) 26:3285–302. 10.1093/hmg/ddx212 [DOI] [PubMed] [Google Scholar]
- 257.Cerro-Herreros E, Fernandez-Costa JM, Sabater-Arcis M, Llamusi B, Artero R. Derepressing muscleblind expression by miRNA sponges ameliorates myotonic dystrophy-like phenotypes in Drosophila. Sci Rep (2016) 6:1–13. 10.1038/srep36230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zhang BW, Cai HF, Wei XF, Sun JJ, Lan XY, Lei CZ, et al. miR-30-5p regulates muscle differentiation and alternative splicing of muscle-related genes by targeting MBNL. Int J Mol Sci (2016) 17:E182. 10.3390/ijms17020182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Greco S, Perfetti A, Fasanaro P, Cardani R, Capogrossi MC, Meola G, et al. Deregulated microRNAs in myotonic dystrophy type 2. PLoS One (2012) 7:e39732. 10.1371/journal.pone.0039732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Derrien T, Guigó R, Johnson R. The long non-coding RNAs: a new (p)layer in the “dark matter”. Front Genet (2012) 2:107. 10.3389/fgene.2011.00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell (2017) 66:22–37.e9. 10.1016/j.molcel.2017.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Palazzo AF, Lee ES. Non-coding RNA: what is functional and what is junk? Front Genet (2015) 6:2. 10.3389/fgene.2015.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Lasda E, Parker R. Circular RNAs: diversity of form and function. RNA (2014) 20:1829–42. 10.1261/rna.047126.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature (2013) 495:384–8. 10.1038/nature11993 [DOI] [PubMed] [Google Scholar]
- 265.Hentze MW, Preiss T. Circular RNAs: splicing’s enigma variations. EMBO J (2013) 32:923–5. 10.1038/emboj.2013.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell (2014) 56:55–66. 10.1016/j.molcel.2014.08.019 [DOI] [PubMed] [Google Scholar]
- 267.Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res (2016) 44:2846–58. 10.1093/nar/gkw027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Neguembor MV, Jothi M, Gabellini D. Long noncoding RNAs, emerging players in muscle differentiation and disease. Skelet Muscle (2014) 4:1–12. 10.1186/2044-5040-4-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zhu M, Liu J, Xiao J, Yang L, Cai M, Shen H, et al. Lnc-mg is a long non-coding RNA that promotes myogenesis. Nat Commun (2017) 8:1–11. 10.1038/ncomms14718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Bovolenta M, Erriquez D, Valli E, Brioschi S, Scotton C, Neri M, et al. The DMD locus harbours multiple long non-coding RNAs which orchestrate and control transcription of muscle dystrophin mRNA isoforms. PLoS One (2012) 7:e45328. 10.1371/journal.pone.0045328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Cabianca DS, Casa V, Bodega B, Xynos A, Ginelli E, Tanaka Y, et al. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in fshd muscular dystrophy. Cell (2012) 149:819–31. 10.1016/j.cell.2012.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Chen X, He L, Zhao Y, Li Y, Zhang S, Sun K, et al. Malat1 regulates myogenic differentiation and muscle regeneration through modulating MyoD transcriptional activity. Cell Discov (2017) 3:1–23. 10.1038/celldisc.2017.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Gudde AE, González-Barriga A, van den Broek WJ, Wieringa B, Wansink DG. A low absolute number of expanded transcripts is involved in myotonic dystrophy type 1 manifestation in muscle. Hum Mol Genet (2016) 25:1648–62. 10.1093/hmg/ddw042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Harel I, Nathan E, Tirosh-Finkel L, Zigdon H, Guimarães-Camboa N, Evans SM, et al. Distinct origins and genetic programs of head muscle satellite cells. Dev Cell (2009) 16:822–32. 10.1016/j.devcel.2009.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Ono Y, Boldrin L, Knopp P, Morgan JE, Zammit PS. Muscle satellite cells are a functionally heterogeneous population in both somite-derived and branchiomeric muscles. Dev Biol (2010) 337:29–41. 10.1016/j.ydbio.2009.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Skuk D, Paradis M, Goulet M, Chapdelaine P, Rothstein DM, Tremblay JP. Intramuscular transplantation of human postnatal myoblasts generates functional donor-derived satellite cells. Mol Ther (2010) 18:1689–97. 10.1038/mt.2010.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, et al. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med (2014) 20:255–64. 10.1038/nm.3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Monge C, DiStasio N, Rossi T, Sébastien M, Sakai H, Kalman B, et al. Quiescence of human muscle stem cells is favored by culture on natural biopolymeric films. Stem Cell Res Ther (2017) 8:104. 10.1186/s13287-017-0556-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Darabi R, Arpke RW, Irion S, Dimos JT, Grskovic M, Kyba M, et al. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell (2012) 10:610–9. 10.1016/j.stem.2012.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Crist C. Emerging new tools to study and treat muscle pathologies: genetics and molecular mechanisms underlying skeletal muscle development, regeneration, and disease. J Pathol (2017) 241:264–72. 10.1002/path.4830 [DOI] [PubMed] [Google Scholar]
- 281.Fan Y, Maley M, Beilharz M, Grounds M. Rapid death of injected myoblasts in myoblast transfer therapy. Muscle Nerve (1996) 19:853–60. [DOI] [PubMed] [Google Scholar]
- 282.Qu Z, Balkir L, Van Deutekom JCT, Robbins PD, Pruchnic R, Huard J. Development of approaches to improve cell survival in myoblast transfer therapy. J Cell Biol (1998) 142:1257–67. 10.1083/jcb.142.5.1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Skuk D, Caron NJ, Goulet M, Roy B, Tremblay JP. Resetting the problem of cell death following muscle-derived cell transplantation: detection, dynamics and mechanisms. J Neuropathol Exp Neurol (2003) 62:951–67. 10.1093/jnen/62.9.951 [DOI] [PubMed] [Google Scholar]
- 284.Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, et al. Direct isolation of satellite cells for skeletal muscle regeneration. Science (2005) 309:2064–7. 10.1126/science.1114758 [DOI] [PubMed] [Google Scholar]
- 285.Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Self-renewal and expansion of single transplanted muscle stem cells. Nature (2008) 456:502–6. 10.1038/nature07384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Mamchaoui K, Trollet C, Bigot A, Negroni E, Chaouch S, Wolff A, et al. Immortalized pathological human myoblasts: towards a universal tool for the study of neuromuscular disorders. Skelet Muscle (2011) 1:34. 10.1186/2044-5040-1-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Morgan JE, Beauchamp JR, Pagel CN, Peckham M, Ataliotis P, Jat PS, et al. Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Dev Biol (1994) 162:486–98. 10.1006/dbio.1994.1103 [DOI] [PubMed] [Google Scholar]
- 288.LaBarge MA, Blau HM. Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell (2002) 111:589–601. 10.1016/S0092-8674(02)01078-4 [DOI] [PubMed] [Google Scholar]
- 289.Ferrari G, Cusella-De Angelis G, Coletta M, Paolucci E, Stornaiuolo A, Cossu G, et al. Muscle regeneration by bone marrow-derived myogenic progenitors. Science (1998) 279:1528–30. 10.1126/science.279.5356.1528 [DOI] [PubMed] [Google Scholar]
- 290.Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng (2001) 7:211–28. 10.1089/107632701300062859 [DOI] [PubMed] [Google Scholar]
- 291.Galli R, Borello U, Gritti A, Minasi MG, Bjornson C, Coletta M, et al. Skeletal myogenic potential of human and mouse neural stem cells. Nat Neurosci (2000) 3:986–91. 10.1038/79924 [DOI] [PubMed] [Google Scholar]
- 292.Tamaki T, Akatsuka A, Ando K, Nakamura Y, Matsuzawa H, Hotta T, et al. Identification of myogenic-endothelial progenitor cells in the interstitial spaces of skeletal muscle. J Cell Biol (2002) 157:571–7. 10.1083/jcb.200112106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.De Bari C, Dell’Accio F, Vandenabeele F, Vermeesch JR, Raymackers JM, Luyten FP. Skeletal muscle repair by adult human mesenchymal stem cells from synovial membrane. J Cell Biol (2003) 160:909–18. 10.1083/jcb.200212064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Camargo FD, Green R, Capetanaki Y, Jackson KA, Goodell MA. Single hematopoietic stem cells generate skeletal muscle through myeloid intermediates. Nat Med (2003) 9:1520–7. 10.1038/nm963 [DOI] [PubMed] [Google Scholar]
- 295.Corbel SY, Lee A, Yi L, Duenas J, Brazelton TR, Blau HM, et al. Contribution of hematopoietic stem cells to skeletal muscle. Nat Med (2003) 9:1528–32. 10.1038/nm959 [DOI] [PubMed] [Google Scholar]
- 296.Pierantozzi E, Vezzani B, Badin M, Curina C, Severi FM, Petraglia F, et al. Tissue-specific cultured human pericytes: perivascular cells from smooth muscle tissue have restricted mesodermal differentiation ability. Stem Cells Dev (2016) 25:674–86. 10.1089/scd.2015.0336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Quattrocelli M, Palazzolo G, Perini I, Crippa S, Cassano M, Sampaolesi M. Mouse and human mesoangioblasts: isolation and characterization from adult skeletal muscles. Methods Mol Biol (2012) 798:65–76. 10.1007/978-1-61779-343-1_4 [DOI] [PubMed] [Google Scholar]
- 298.Tonlorenzi R, Dellavalle A, Schnapp E, Cossu G, Sampaolesi M. Isolation and characterization of mesoangioblasts from mouse, dog, and human tissues. Curr Protoc Stem Cell Biol (2007) Chapter 2:Unit2B.1. 10.1002/9780470151808.sc02b01s3 [DOI] [PubMed] [Google Scholar]
- 299.Nirwane A, Gautam J, Yao Y. Isolation of type I and type II pericytes from mouse skeletal muscles. J Vis Exp (2017) (123). 10.3791/55904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Dellavalle A, Maroli G, Covarello D, Azzoni E, Innocenzi A, Perani L, et al. Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat Commun (2011) 2:499. 10.1038/ncomms1508 [DOI] [PubMed] [Google Scholar]
- 301.Bonfanti C, Rossi G, Tedesco FS, Giannotta M, Benedetti S, Tonlorenzi R, et al. PW1/Peg3 expression regulates key properties that determine mesoangioblast stem cell competence. Nat Commun (2015) 6:6364. 10.1038/ncomms7364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Greenhalgh SN, Iredale JP, Henderson NC. Origins of fibrosis: pericytes take centre stage. F1000Prime Rep (2013) 5:37. 10.12703/P5-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Birbrair A, Zhang T, Wang ZM, Messi ML, Enikolopov GN, Mintz A, et al. Role of pericytes in skeletal muscle regeneration and fat accumulation. Stem Cells Dev (2013) 22:2298–314. 10.1089/scd.2012.0647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O. Type-1 pericytes participate in fibrous tissue deposition in aged skeletal muscle. Am J Physiol Cell Physiol (2013) 305:C1098–113. 10.1152/ajpcell.00171.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Rotini A, Martínez-Sarrà E, Duelen R, Costamagna D, Di Filippo ES, Giacomazzi G, et al. Aging affects the in vivo regenerative potential of human mesoangioblasts. Aging Cell (2018) 17:e12714. 10.1111/acel.12714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Sampaolesi M, Blot S, D’Antona G, Granger N, Tonlorenzi R, Innocenzi A, et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature (2006) 444:574–9. 10.1038/nature05282 [DOI] [PubMed] [Google Scholar]
- 307.Vulliet PR, Greeley M, Halloran SM, MacDonald KA, Kittleson MD. Intra-coronary arterial injection of mesenchymal stromal cells and microinfarction in dogs. Lancet (2004) 363:783–4. 10.1016/S0140-6736(04)15695-X [DOI] [PubMed] [Google Scholar]
- 308.Giannotta M, Benedetti S, Tedesco FS, Corada M, Trani M, D’Antuono R, et al. Targeting endothelial junctional adhesion molecule-A/EPAC/Rap-1 axis as a novel strategy to increase stem cell engraftment in dystrophic muscles. EMBO Mol Med (2014) 6:239–58. 10.1002/emmm.201302520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Nystedt J, Anderson H, Tikkanen J, Pietila M, Hirvonen T, Takalo R, et al. Cell surface structures influence lung clearance rate of systemically infused mesenchymal stromal cells. Stem Cells (2013) 31:317–26. 10.1002/stem.1271 [DOI] [PubMed] [Google Scholar]
- 310.Chal J, Oginuma M, Al Tanoury Z, Gobert B, Sumara O, Hick A, et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat Biotechnol (2015) 33:962–9. 10.1038/nbt.3297 [DOI] [PubMed] [Google Scholar]
- 311.Abujarour R, Bennett M, Valamehr B, Lee TT, Robinson M, Robbins D, et al. Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl Med (2014) 3:149–60. 10.5966/sctm.2013-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Pawlowski M, Ortmann D, Bertero A, Tavares JM, Pedersen RA, Vallier L, et al. Inducible and deterministic forward programming of human pluripotent stem cells into neurons, skeletal myocytes, and oligodendrocytes. Stem Cell Reports (2017) 8:803–12. 10.1016/j.stemcr.2017.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Maffioletti SM, Gerli MFM, Ragazzi M, Dastidar S, Benedetti S, Loperfido M, et al. Efficient derivation and inducible differentiation of expandable skeletal myogenic cells from human ES and patient-specific iPS cells. Nat Protoc (2015) 10:941–58. 10.1038/nprot.2015.057 [DOI] [PubMed] [Google Scholar]
- 314.Roca I, Requena J, Edel MJ, Alvarez-Palomo AB. Myogenic precursors from iPS cells for skeletal muscle cell replacement therapy. J Clin Med (2015) 4:243–59. 10.3390/jcm4020243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Albini S, Coutinho P, Malecova B, Giordani L, Savchenko A, Forcales SV, et al. Epigenetic reprogramming of human embryonic stem cells into skeletal muscle cells and generation of contractile myospheres. Cell Rep (2013) 3:661–70. 10.1016/j.celrep.2013.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Xia G, Santostefano KE, Goodwin M, Liu J, Subramony SH, Swanson MS, et al. Generation of neural cells from DM1 induced pluripotent stem cells as cellular model for the study of central nervous system neuropathogenesis. Cell Reprogram (2013) 15:166–77. 10.1089/cell.2012.0086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Du J, Campau E, Soragni E, Jespersen C, Gottesfeld JM. Length-dependent CTG{middle dot}CAG triplet-repeat expansion in myotonic dystrophy patient-derived induced pluripotent stem cells. Hum Mol Genet (2013) 22:5276–87. 10.1093/hmg/ddt386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Martineau L, Racine V, Benichou SA, Puymirat J. Lymphoblastoids cell lines-derived iPSC line from a 26-year-old myotonic dystrophy type 1 patient carrying (CTG) 200 expansion in the DMPK gene: CHUQi001-A. Stem Cell Res (2018) 26:103–6. 10.1016/j.scr.2017.12.010 [DOI] [PubMed] [Google Scholar]
- 319.Ueki J, Nakamori M, Nakamura M, Nishikawa M, Yoshida Y, Tanaka A, et al. Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Sci Rep (2017) 7:42522. 10.1038/srep42522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Gao Y, Guo X, Santostefano K, Wang Y, Reid T, Zeng D, et al. Genome therapy of myotonic dystrophy type 1 iPS cells for development of autologous stem cell therapy. Mol Ther (2016) 24:1378–87. 10.1038/mt.2016.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Spitalieri P, Talarico RV, Botta A, Murdocca M, D’Apice MR, Orlandi A, et al. Generation of human induced pluripotent stem cells from extraembryonic tissues of fetuses affected by monogenic diseases. Cell Reprogram (2015) 17:275–87. 10.1089/cell.2015.0003 [DOI] [PubMed] [Google Scholar]
- 322.Hicks MR, Hiserodt J, Paras K, Fujiwara W, Eskin A, Jan M, et al. ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat Cell Biol (2018) 20:46–57. 10.1038/s41556-017-0010-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Pinto BS, Saxena T, Oliveira R, Méndez-Gómez HR, Cleary JD, Denes LT, et al. Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol Cell (2017) 68:479–90.e5. 10.1016/j.molcel.2017.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Batra R, Nelles DA, Pirie E, Blue SM, Marina RJ, Wang H, et al. Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell (2017) 170:899–912.e10. 10.1016/j.cell.2017.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Provenzano C, Cappella M, Valaperta R, Cardani R, Meola G, Martelli F, et al. CRISPR/Cas9-mediated deletion of CTG expansions recovers normal phenotype in myogenic cells derived from myotonic dystrophy 1 patients. Mol Ther Nucleic Acids (2017) 9:337–48. 10.1016/j.omtn.2017.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]