

Abstract
Early childhood infection with respiratory viruses, including human rhinovirus, respiratory syncytial virus (RSV) and influenza, is associated with an increased risk of allergic asthma and severe exacerbation of ongoing disease. Despite the long recognition of this relationship, the mechanism linking viral infection and later susceptibility to allergic lung inflammation is still poorly understood. We discuss the literature and provide new evidence demonstrating that these viruses induce the alternative activation of macrophages. Alternatively activated macrophages (AAM) induced by RSV or influenza infection persisted in the lungs of mice up to 90 days after initial viral infection. Several studies suggest that AAM contribute to allergic inflammatory responses, although their mechanism of action is unclear. In this commentary, we propose that virus-induced AAM provide a link between viral infection and enhanced responses to inhaled allergens.
Keywords: alternatively activated macrophages, Th2 cytokines, respiratory virus infection, allergic asthma
Commentary on the possibility that long-lived alternatively activated macrophages induced by respiratory virus infection contribute to the enhanced responses to allergens.
INTRODUCTION
Genetics and the environment contribute approximately equally to the development of asthma (Eder et al.2005; Martinez 2007; Yang et al.2007; Vercelli 2008). However, how sensing of the environment is linked to asthma inception and exacerbation is not well understood (Minnicozzi, Sawyer and Fenton 2011). Early childhood infection with respiratory viruses, including human rhinovirus (HRV) and respiratory syncytial virus (RSV) is associated with an increased risk of subsequent development of asthma (Jackson and Lemanske 2010) and HRV and influenza are associated with exacerbation of ongoing disease (Edwards et al.2012). Despite the long recognition of this relationship, the mechanism linking viral infection and later susceptibility to allergic lung inflammation remains to be elucidated.
Respiratory viruses first encounter lung epithelial cells and alveolar macrophages (AMs), initiating a cascade of innate immune responses (Schleimer et al.2007; Pribul et al.2008; Lambrecht and Hammad 2009; Gregory and Lloyd 2011). These viruses contain pathogen-associated molecular patterns (PAMPS) that directly stimulate pattern-recognition receptors (PRRs) and can result in release of host-derived damage-associated molecular patterns (DAMPS). Many groups have focused on the effects of viral infection on epithelial cells and the communication between activated epithelial cells, innate lymphoid cells, and dendritic cells (DCs) (Schleimer et al.2007; Pribul et al.2008; Lambrecht and Hammad 2009; Saenz, Noti and Artis 2010; Chang et al.2011; Holgate 2011). Virus-infected epithelial cells can produce cytokines including TSLP, IL-33 and IL-25 that establish a ‘Th2-promoting’ environment by programming lung DC (Lambrecht and Hammad 2009) and activating innate lymphoid cells (Lambrecht and Hammad 2009; Saenz, Noti and Artis 2010). Such DCs are thought to promote the differentiation of helper T cells to the Th2 type that produce high levels of IL-4, IL-5 and IL-13 (Lambrecht and Hammad 2009).
While these pathways clearly contribute to responses to respiratory insults, the role of the macrophage has been largely ignored. Macrophages are found in abundance in the lungs and airways, and are one of the first cells exposed to both viruses and aeroallergens. Macrophages exhibit great plasticity and can adopt different phenotypes that promote different immune environments (Anthony et al.2007; Mosser and Edwards 2008; Martinez, Helming and Gordon 2009; Murray et al.2014). Macrophages exposed to strong inflammatory stimuli, e.g. interferon-γ (IFN-γ) and LPS, differentiate into highly microbicidal, classically activated macrophages (CAMs, also termed M1). CAMs produce a strong pro-inflammatory cytokine profile that can result in tissue damage. Conversely, exposure of macrophages to IL-4 or IL-13 leads to development of alternatively activated macrophages (AAM, also called M2) that counter the strong pro-inflammatory CAMs and are associated with wound healing (Martinez, Helming and Gordon 2009). Several studies provide evidence suggesting that AAM actively participate in the severity of allergic lung inflammation (Kelly-Welch et al.2004; Dasgupta and Keegan 2012; Ford et al.2012); although, as we discuss below, their contribution remains controversial in the field. In addition, the mechanism by which increased numbers of AAM contribute to the severity of allergic asthma is unclear. AAM produce a number of enzymes, cytokines and chemokines that may play important roles in inflammation, but a single critical factor has not yet been identified in allergic lung inflammation. While enhanced allergic lung inflammation has been linked to AAM by others using mouse models and in studies of asthma patients (Kim et al.2008; Lee et al.2009; Subrata et al.2009; Melgert et al.2010, 2011; Nagarkar et al.2010), it is not yet known if AAM contribute to the initiation of allergic lung responses after viral infection or to exacerbation of allergic lung responses after viral infection.
BURDEN AND RISK FOR ASTHMA
Asthma is a chronic disease of the small airways resulting in wheezing, coughing and shortness of breath. Asthma ranks as the most common cause of non-communicable disease in children and poses a significant disease burden (Akinbami and Schoendorf 2002; Akinbami et al.2009). Asthma is the most common cause of pediatric hospitalizations and is the most common cause for missed school days in children aged 5–17 years (Akinbami and Schoendorf 2002). For children aged 0–17 years, 2% of all ambulatory visits and 2.3% of emergency room visits were attributed to asthma (Lasso-Pirot, Delgado-Villalta and Spanier 2015). Despite advances in drug delivery and standardization of management, ∼170 children die annually in the USA from asthma exacerbations/complications (Akinbami and Schoendorf 2002; Akinbami et al.2009). Weiss, Sullivan and Lyttle (2000) estimated that the annual healthcare cost of childhood asthma-related illnesses is 3.2 billion US dollars. Inner city children and low-income families are disproportionally affected and have higher morbidity and mortality due to asthma. In contrast, children living on farms have a reduced risk of asthma and allergies due to exposures to a greater diversity of microbes (Ege et al.2011). Similarly, children with more exposure to other children (siblings) at home or at a day care setting are less likely to develop asthma (Ball et al.2000).
Positive family history, prenatal exposure to cigarette smoke, prematurity and sensitization to allergens such as house dust mite (HDM) and molds have been associated with increased risk for childhood-onset asthma (Beasley, Semprini and Mitchell 2015). The role of interferons is considered vital in the development of wheezing and asthma in children, particularly after viral infections. Immature peripheral blood IFN-γ responses to viruses and mitogen have been linked with recurrent wheezing (Stein et al.1999; Gern et al.2006). Similarly, deficiency in the interferon response from airway epithelial cells has been implicated in asthma. Impairment of airway barrier, an important component of innate immunity, provides a potential mechanism by which environmental exposures can lead to asthma. Exposure to tobacco smoke or viruses in children with impaired airway barrier could lead to severe lower respiratory tract infection, enhanced viral replication and subsequent increased risk for asthma (Sigurs et al.2005; Fassl et al.2012).
Viral infection and asthma
Infection with respiratory viruses has been associated with both the inception and exacerbation of asthma. It is well understood that wheezing in early life is a risk factor for asthma in later life, and the most common cause of wheezing in early life is viral infection. HRV and RSV infections in early childhood have been well-studied and their association with asthma has been widely reported (Garcia-Garcia et al.2007; Caliskan et al.2013). Recently, a link between human metapneumovirus infection and subsequent asthma has been demonstrated (Garcia-Garcia et al.2007). Furthermore, viral infections are the most common cause of asthma exacerbation in children. About 80% of exacerbations are attributed to viral infections and HRV accounts for nearly two-third of the cases. Children with asthma exacerbation due to influenza infection tend to have worse outcomes. The causative link between viral infection and subsequent development of asthma-like systems has been investigated in animal models.
A number of groups have established mouse models for virus-enhanced responses to later allergen exposure using adult or neonatal mice (You et al.2006; Kim et al.2008; Cormier, You and Honnegowda 2010; Kaiko et al.2010; Al-Garawi et al.2011; Schneider et al.2012). Infection of neonatal mice (7–8 days old) with RSV (You et al.2006), HRV (Schneider et al.2012) or influenza (Al-Garawi et al.2011) induced rapid production of IL-13, IL-4 and TSLP in the lungs that preceded enhanced responses to allergens administered 7–10 days after infection.
This enhancement can also occur in older mice. To demonstrate that RSV infection enhances the subsequent response to allergen, 4-week-old BALB/c mice were infected with RSV or PBS (as a control). Ten days later, mice were treated with HDM or PBS four times, and 48 h after the last exposure, bronchoalveolar lavage (BAL) cells and lungs were analyzed, representing 27 days between initial RSV infection and analysis (Fig. 1). RSV infection alone (RSV/PBS) induced a significant increase in total cell numbers found in the airways; these cells were predominantly macrophages (M) (Fig. 1A and B). As expected, HDM alone (PBS/HDM) induced an increase in the numbers of airway eosinophils (E). Strikingly, mice infected with RSV prior to HDM treatment (RSV/HDM) exhibited a 6-fold increase in the numbers of eosinophils in the airways compared to HDM exposure alone. We observed only few airway neutrophils (P) or lymphocytes (L) in the airways under these conditions (Fig. 1B). Prior exposure to RSV also significantly increased the percentage of eosinophils in the BAL (Fig. 1C)
Figure 1.
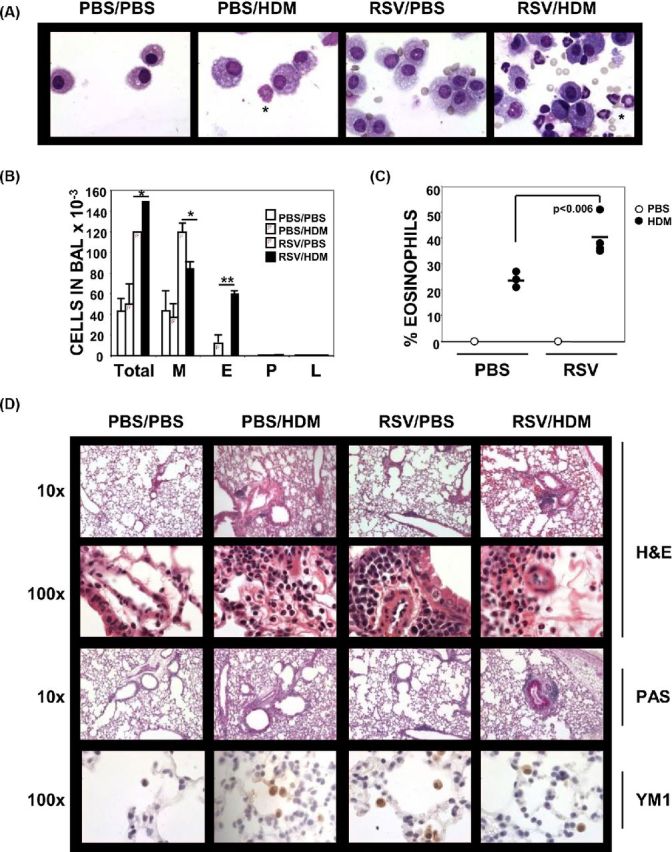
Prior infection with RSV enhances responses to allergen. Four-week old Balb/c mice were mock- or RSV-infected on day 0. Mice were treated 10 days later with PBS or HDM (50 μg) i.n. on days 10, 15, 20 and 25. Forty-eight hours later (day 27), BAL fluid and lungs were harvested. (A) Representative images of cytospins stained with Wright-Giemsa (100×). Examples of eosinophils are marked by asterisks. (B) Differential counts of total cells, macrophages (M), eosinophils (E), polymorphonuclear leukocytes (P) and lymphocytes (L) in the BAL (n = 3, mean ±SEM). (C) Percent eosinophils in the BAL of each mouse (n = 3, bar denotes the mean value). (D) Representative images of lung sections stained with H&E (10×, 100×), PAS (10×) and anti-YM1 (100×).
Enhanced responsiveness to allergen was also reflected in the analysis of lung sections (Fig. 1D). The HDM-induced inflammatory response was increased in mice previously infected with RSV. Prior infection with RSV increased the HDM-induced eosinophilia in the lung tissue and the development of PAS+, mucus-producing epithelial cells. RSV alone induced some inflammation predominantly surrounding blood vessels; however, this infiltrate was monocytic as observed under high power (100×). These results are consistent with those published by the Gelfand laboratory (Schwarze et al.1997). Importantly, we detected Ym1/2+ macrophages in the lungs of mice treated with either RSV or HDM indicating that RSV-induced AAM remain in the lungs for at least 27 days post-infection. Furthermore, our data show that a 10-day interval between infection and initial allergen exposure is sufficient to reveal enhanced responsiveness.
Viral infection and AAM
Respiratory syncytial virus: RSV infection of primary murine macrophages and the RAW 264.7 macrophage cell line in vitro leads to early (hours) induction of pro-inflammatory genes (e.g. IL-12, cyclooxygenase (COX)-2, etc.), followed by macrophage-derived IL-4 and IL-13 (measured by qRT-PCR, ELISA and intracellular IL-4 staining of F4/80+ BAL cells), and the anti-inflammatory cytokine, IL-10 (Shirey et al.2010). AAM marker expression (e.g. Arg1, Fizz1, Ym1, MR and others) peaked at 24–48 h in vitro and in vivo (Shirey et al.2010) as well as TSLP and IL-33 mRNA (unpublished observations), cytokines associated with allergic inflammation, a common sequella of RSV infection (Sigurs, Bjarnason and Sigurbergsson 1994; Sigurs et al.1995). The IL-4Rα chain mediates IL-4- and IL-13-induced, STAT6-dependent signaling (Heller et al.2008). RSV-infected IL-4Rα−/− and STAT6−/− macrophages or mice did not differentiate into AAM and exhibited enhanced and prolonged pro-inflammatory responses (e.g. IL-12, COX-2) (Shirey et al.2010). RSV-infected TLR4−/− macrophages failed to differentiate into AAM, and produced low levels of PPARγ (Shirey et al.2010) a transcription factor required for induction of AAM markers (Welch et al.2002; Odegaard et al.2007; Malur et al.2009; Xavier et al.2013). RSV-infected IFN-β−/− macrophages produced decreased levels of IL-4, IL-13 and IL-4Rα also failed to differentiate into AAM (Shirey et al.2010). Furthermore, the treatment of RSV-infected WT macrophages with anti-IL-4 and/or anti-IL-13 antibodies reduced Arg1 mRNA while enhancing COX-2 mRNA (Fig. 2). Thus, RSV-induced AAM differentiation requires macrophage-derived IL-4/IL-13 and IL-4Rα-, TLR4-, and IFN-β-mediated signaling. RSV-induced lung pathology (e.g. primarily alveolitis, peribronchiolitis) was significantly worse in IL-4Rα−/− than WT mice, due to the failure to produce AAM. Adoptive transfer of WT macrophages to IL-4Rα−/− mice reconstituted RSV-induced lung AAM, and mitigated IL-12 levels and pathology (Shirey et al.2010).
Figure 2.

RSV infection of macrophages induces arginase-1 and suppresses COX-2 mRNA in an IL-4- and IL-13-dependent manner. Macrophages were prepared from highly purified thioglycollate-elicited C57BL/6J mice. Macrophages were infected with RSV (MOI = 2) for 48 h in the presence or absence of isotype control, anti-IL-4 and/or anti-IL-13 antibodies. Arginase-1 and COX-2 mRNA were measured by qRT-PCR 4 h post-infection.
We previously reported that treatment of RSV-infected cotton rats with indomethacin, a pan-COX inhibitor, reduced RSV-induced pathology (Richardson et al.2005) and that Parecoxib, a COX-2-specific inhibitor, increased levels of RSV-induced AAM in vivo (Shirey et al. 2010). Arachidonic acid is a substrate for both COX-2 and 5/15-lipoxygenase (5/15-LO). We observed a strong inverse relationship between COX-2 and 5/15-LO expression that is reflected in CAM or AAM responses, respectively (Shirey et al.2010, 2014b) (Fig. 3). RSV-infected 5-LO−/− mice and 5-LO−/− or 15-LO−/− macrophages, failed to develop AAM markers, but produced increased COX-2 mRNA (Shirey et al.2014b). 5/15-LO-specific inhibitors blocked RSV-induced AAM differentiation in vitro; conversely, 5/15-LO-derived lipoxins or resolvin partially restored to RSV-infected 5-LO−/− macrophages the ability to differentiate into AAM (Shirey et al.2014b) Indomethacin therapy significantly reduced lung pathology of RSV-infected 5-LO−/− mice (Shirey et al.2014b). Collectively, our data support the model proposed in Fig. 3: the extent of lung injury induced by RSV depends on the relative balance of COX-2 and 5/15-LO that controls CAM and AAM differentiation. More recently, we published that the administration of IL-4/anti-IL-4 complexes or azithromycin, agents shown to induce AAM in vivo (Finkelman et al.1993; Murphy et al.2008; Feola et al.2010), and rosiglitazone, a PPARγ agonist (Moller 2001), each increased RSV-induced AAM and mitigated pathology in mice (Shirey et al.2014a). This balance between viral-induced tissue damage and AAM differentiation may become dysregulated in some individuals leading to inflammatory diseases such as allergic inflammation and asthma.
Figure 3.

RSV infection induces a response balanced by pro-inflammatory and tissue repair pathways. RSV infects multiple cell types in the lung, including macrophages, which result in the initial production of pro-inflammatory cytokines and chemokines that also recruit more inflammatory cells such as monocytes. The production of inflammatory cytokines and COX-2 leads to the differentiation of CAM as well as the resulting observed pathology. Later, through a complex process that is IL-4Rα-, TLR4-, IFN-β- and LO-dependent, AAM differentiation occurs, counteracting the earlier inflammatory insults through (i) production of anti-inflammatory cytokines such as IL-10, (ii) induction and activation of PPARγ through TLR4, (iii) induction and activation of the 5- and 15-LO pathway preventing further production of COX-2 and the damaging prostaglandins and (iv) repair of damaged tissue through production of enzymes such as FIZZ1 and Ym1. Although the induction of AAM is beneficial to the host initially, if the AAM phenotype persists, it may lead to hypersensitivity to allergic triggers such as HDM.
Influenza: Imai et al. (2008) reported that chemical or microbial lung insults produce an oxidized phospholipid DAMP, OxPAPC, which elicits TLR4-, TRIF- and IL-6-dependent signaling in lung macrophages, leading to acute lung injury (ALI). This led to our finding that TLR4−/− mice are highly resistant to lethal infection by mouse-adapted influenza A/PR/8/34 (PR8) (Nhu et al.2010; Shirey et al.2013) and to our hypothesis that therapeutic blockade of TLR4 signaling should mitigate influenza-induced ALI and lethality. Eritoran (E5564; Eisai Inc.), the most potent, synthetic TLR4 antagonist known (Mullarkey et al. 2003; Kalil et al. 2011), strongly binds the TLR4 co-receptor, MD2 (Kim et al.2007), thereby blocking MD2-LPS interactions. Eritoran administered as late as 6 days p.i., protected mice significantly from PR8-induced ALI and lethality, and blocked cytokine production in lungs (Shirey et al.2013). Eritoran also protected cotton rats infected with non-adapted influenza from ALI. We recently confirmed the importance of TLR4 in PR8-induced lethality: mice treated on days 2 and 4 p.i. with anti-TLR4, but not control IgG, were significantly protected from lethality. In addition to blocking influenza-induced lethality when given therapeutically, Eritoran blocked not only the TLR4-mediated cytokine production induced by DAMPs, including OxPAPC and HMGB1, but also blunted accumulation of OxPAPC in the lungs and the release of HMGB1 into the serum (Shirey et al.2013, 2016).
Influenza infection may also predispose individuals to secondary bacterial pneumonia and is associated with increased mortality. Streptococcus pneumoniae and Staphylococcus aureus are frequently isolated (Klugman, Chien and Madhi 2009; Rynda-Apple, Robinson and Alcorn 2015). Influenza-induced ALI was much more severe compared to that induced by RSV infection, and we failed to detect AAM markers in mice that survived an LD90 of influenza (data not shown); however, a sublethal dose of influenza that resulted in reduced ALI led to an early wave of pro-inflammatory cytokine induction (e.g. TNF-α, IL-6) that overlapped with the induction of AAM markers (Chen et al.2012). Under these conditions, the mice exhibited increased susceptibility to an otherwise non-lethal dose of S. pneumonia (Chen et al.2012). This suggests that the hyper-susceptibility to S. pneumoniae after influenza infection may be mediated by long-lived AAM that have a reduced capacity to produce pro-inflammatory cytokines and chemokines that would normally contribute to clearance of the bacteria (Fig. 4). This hypothesis is consistent with the observations of Didierlaurent et al. (2008) who observed that AM exhibited a diminished sensitivity to TLR agonists as late as 60 days after infection with influenza or RSV. However, this study did not determine if the AM were, in fact, AAM. Thus, it is tempting to speculate that the development of AAM after influenza infection is intended to mitigate the host inflammatory response to virus infection, but in doing so, enhances susceptibility to secondary bacterial infection by diminishing cytokine production. Thus, we propose that such a population of long-lived AAM could also contribute to enhanced allergic responses to inhaled substances long after the initial respiratory viral infection is resolved.
Figure 4.

Model of influenza-induced AAM consequences. Infection with a sublethal dose of influenza virus induces an initial CAM response along with the production of various DAMPs. Over time, after viral clearance and during a recovery phase, the lung macrophages differentiate into AAM as shown by measurement of IL-4, IL-13 and markers associated with AAM (arginase-1, FIZZ1 and Ym1) that appear to be long-lived. Influenza-recovered mice that are challenged with a sublethal dose of S. pneumaniae serotype 3 within two weeks of the initial viral infection quickly develop a fatal pulmonary infection with rapid replication of S. pneumoniae observed along with a blunted inflammatory response.
To evaluate the longevity of AAM in the lung after RSV and influenza infection, BALB/ByJ mice were infected with RSV or a sublethal dose of influenza and cytokine and AAM marker mRNA measured over 120 days (Fig. 5). IL-1β, TNF-α and COX-2 mRNA were up-regulated at 6 days for both RSV and PR8; however, the PR8-infected mice exhibited sustained pro-inflammatory mRNA through 14 days post-infection (Fig. 5A). For AAM-associated genes, mannose receptor, Ym1 and Arginase-1 mRNA were up-regulated by RSV as early as 1 week post-infection and remained above background, even at 120 days. For influenza infection, induction of AAM gene expression was slightly delayed, with maximum expression at 30 days that was also sustained above background through 120 days. The abundance of AAM in the lungs of mice after viral infection was analyzed by immunohistochemistry for Ym1, a member of the chitinase family and marker for mouse AAM. We observed a significant increase in numbers of Ym1+ macrophages over PBS-treated mice within 6 days of RSV infection; this increase was maintained for 90 days post-infection. We also observed a significant increase in numbers of Ym1+ macrophages 14 days after infection with PR8 that lasted for 90 days post-infection.
Figure 5.
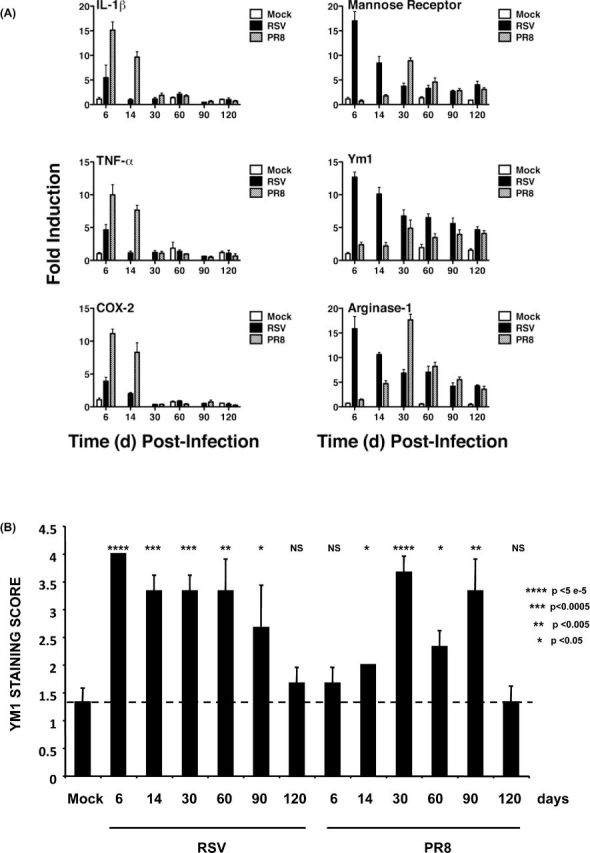
AAM remain in the lung long after virus infection. BALB/cJ mice were mock infected with PBS or infected with RSV (2 × 106 PFU/mouse i.n.) or influenza A/PR/8/34 (1000 TCID50/mouse i.n.). Mice were euthanized on various days post-infection up until day 120 (n = 3 mice/group). One lobe of each lung was harvested and processed for RNA. (A) Total RNA was isolated and the relative abundance of mRNA for specific genes was evaluated as relative gene expression normalized to mock-infected. (B) The second lobe was prepared for histology and scored for staining for Ym1+ macrophages in a blinded fashion on a scale of 0–4 as described in the Supplemental Materials, Supporting Information. Mean score ±SEM is shown.
Human rhinovirus: rhinovirus infections are associated with 60%–70% of the episodes of asthma exacerbations in school-age children (Johnston et al.1995; Rakes et al.1999; Holgate 2006), and in exacerbations of symptoms in chronic obstructive pulmonary disease patients (Seemungal et al.2000), and are a major cause of hospitalizations due to acute respiratory illness in young children in the USA (Iwane et al.2011). Until recently, animal models for studying HRV infection were very limited. Recent data suggested the BALB/c mouse as a model for rhinovirus-induced disease and exacerbation of allergic airway inflammation (Bartlett et al.2008). Using HRV1B, Bartlett et al. (2008) demonstrated that infection is localized in the lungs and induces airway and pulmonary inflammation and mucin production, albeit accompanied by low viral replication. In OVA-sensitized/challenged BALB/c mice, subsequent infection with RV1B increased lung infiltration with neutrophils, eosinophils and macrophages. This was associated with increased airway cholinergic hyperresponsiveness. Administration of anti-eotaxin-1, an eosinophil chemokine, attenuated the RV infection-induced eosinophilia. Eotaxin was present in lung macrophages and such cells stained for AAM markers including Arginase 1, Ym1, Mgl2 and IL-10 (Nagarkar et al.2010). The role of AAM in airway responsiveness was demonstrated by experiments in which macrophages were depleted from OVA-sensitized/challenged mice following RV infection (Nagarkar et al.2010). BALB/c mice were infected with RV. On day 35 post-infection, mice exhibited airway hyperresponsiveness and mucus metaplasia that was blocked by administration of anti-IL-13 antibody. In a similar model of allergic asthma, Hong et al. (2014) reported that OVA-treated IL-4Rα knockout mice had neutrophilic airway inflammation in contrast to wild-type mice that exhibited exacerbated airway hyperreactivity (AHR) associated with AAM. Blanco et al. (2014) recently reported that HRV-16 infection of cotton rats (Sigmodon hispidus) resulted in tracheal epithelial cell defoliation associated with peribronchiolar infiltration, bronchiolitis, perivascular infiltration alveolar septal infiltration and alveolar exudates, as well as mucus cell hypertrophy/hyperplasia.
Role of AAM in asthma inception and exacerbation
The potential for AAM to actively contribute to allergic lung inflammation has been long ignored, but recent reports have begun to shed light on their contribution to allergic disease. Asthmatics have greater numbers of macrophages in their lungs as compared to healthy controls (Viksman et al.2002; Melgert et al.2011). Asthmatics also demonstrate increased levels of chitinase family members in their blood and lung lavage fluid (Chupp et al.2007). Single-nucleotide polymorphisms found in CHI3L1 (YKL40), CHIA (AMCase) and MRC1 (mannose receptor), all AAM markers, have been associated with asthma (Bierbaum et al.2005; Ober et al.2008; Hattori et al.2009). Peripheral blood monocytes isolated from children undergoing viral-induced asthma exacerbations had elevated expression of AAM signature genes including MRC1 (Subrata et al. 2009). Kim et al. (2008) found increased numbers of IL-13+ macrophages in BAL samples from asthma patients as compared to healthy controls. Furthermore, immunohistochemistry of biopsy specimens taken from the lungs of asthma patients and healthy controls demonstrated an increase in AAM (defined by expression of mannose receptor, CD206, and stabilin-1) in the lungs of asthmatic patients (Melgert et al.2011). The percentage of AAM in the lung tissue correlated with peak expiratory flow variation, suggesting that AAM may regulate asthma severity.
Several reports using mouse models also support an important role for AAM in allergic lung inflammation. AAM were shown to contribute to the enhanced asthma response observed in female mice as compared to male mice (Melgert et al.2010). Female mice had greater numbers of AAM in lung tissue than male mice and showed enhanced allergic lung inflammation. Adoptive transfer of in vitro differentiated AAM by intranasal instillation enhanced allergic lung inflammation observed in male mice. Using a bone marrow chimeric model, the Keegan laboratory showed a strong correlation between the severity of allergic lung inflammation and the presence of IL-4Rα+ macrophages (Kelly-Welch et al.2004) that were also positive for Ym1/2, an AAM marker (Ford et al.2012). Furthermore, adoptive transfer of IL-4Rα+/+ but not IL-4Rα−/− bone marrow-derived macrophages (BMM) in the presence of Th2 effector cells was sufficient to enhance allergic lung inflammation in the OVA model (Ford et al.2012). Adoptive transfer of IL-4Rα+ BMM significantly enhanced the percentage of eosinophils, and levels of CCL11/eotaxin, CCL5/RANTES and CCL2/MCP-1 in the BAL fluid (Ford et al.2012). Ym1/2+ AAM were detected in the lungs and airways of mice receiving IL-4Rα+ BMM, but not IL-4Rα− BMM. Nieuwenhuizen et al. (2012) analyzed responses in a neutrophil- and macrophage-specific knock-out of the IL-4α and found that the ability to induce AAM during an allergic response was not absolutely necessary to support allergic lung inflammation. However, the LysM-cre-mediated deletion was later shown to be incomplete; IL-4Rα was not deleted on immature monocytes and macrophages (LysM low) during ongoing inflammation induced by Shistosoma mansoni infection while it was deleted on mature, resident lung macrophages (LysM-high) (Vannella et al.2014). Using an alternate macrophage deleting approach, diphtheria toxin (DT) treatment of Cd11b-DT receptor mice, Borthwick et al. (2016) recently showed that macrophage depletion with DT after priming of mice with the allergen HDM resulted in decreased AAM responses in the lung and decreased eosinophilic inflammation in response to HDM challenge.
These findings from the studies in human asthma patients and mouse models of allergic lung inflammation strongly suggest that AAM are important, active contributors to Th2-driven inflammation and are not just bystander cells responding to the Th2 cytokines. It is reasonable to propose that AAM contribute to asthma severity and, therefore, may play a role in asthma exacerbation by viral infection. The placement of AAM early in asthma inception is less established but should be carefully considered.
SUMMARY
As discussed above, viral infection induces AAM that are quite long-lived in the lung. We continued to detect AAM in the lungs of mice 90 days after infection (Fig. 5), long after virus infection is cleared. It is tempting to suggest that these long-lived macrophages may maintain ‘innate memory’ or training that leads them to respond vigorously to a subsequent stimulus (Netea et al.2015). If that stimulus happens to be an allergen, an overly robust Th2-type response could develop (Fig. 6). Thus, it is possible that the enhanced responsiveness to allergen exposure that occurs after viral infection is mediated by long-lived AAM.
Figure 6.

Role for AAM in virus-induced enhancement to allergen exposure. Initiation by virus infection. (1) PAMPS found in RSV, flu or RV stimulates PRRs on AMs and lung epithelial cells that stimulates rapid production of pro-inflammatory cytokines, followed by the production of Th2-promoting cytokines including IL-4, IL-13, IL-25, IL-33 and TSLP. These cytokines act on AMs as well as granulocytes (mast cells, basophils, eosinophils) and innate lymphoid cells such as iNKT or ILC2 to produce more IL-4 or IL-13. (2) IL-4 or IL-13, in combination with IL-33, drives AAM differentiation mediated by autocrine and paracrine mechanisms. (3) Long-lived AAM remain in the lung. Allergen exposure. (4) Molecular patterns found in allergens (AAMPS) stimulate PRR expressed on long-lived AAM leading to the rapid production of IL-4, IL-13, IL-25, IL-33, and TSLP and chemokines. Lung epithelial cells could also contribute to the production of a subset of these cytokines including IL-25, IL-33 and TSLP. These cytokines activate DCs, and other innate immune cells. (5) Activated DCs migrate to the draining lymph node, and with the help of IL-4-producing cells such as basophils, stimulate naive CD4+ T-cells to become allergen-specific Th2 effectors or memory cells. (6) Th2 effectors migrate to the site of inflammation, producing high levels of IL-4, IL-5 and IL-13 mediating the Type 2 inflammatory response.
In this scenario, viral infection would induce the formation of lung AAM. First, PAMPS found in RSV, influenza or RV would stimulate PRRs on AMs and lung epithelial cells that stimulate rapid production of pro-inflammatory cytokines, followed by production of Th2-promoting cytokines including IL-4, IL-13, IL-25, IL-33 and TSLP. These cytokines act on AMs as well as granulocytes (mast cells, basophils, eosinophils) and innate lymphoid cells such as invariant NK T-cells (iNKT) or the lineage negative innate helper cell (Ih2) to produce more IL-4 or IL-13. Second, IL-4 or IL-13, in combination with IL-33, would drive AAM differentiation mediated by autocrine and paracrine mechanisms. Third, these long-lived AAM remain in the lung with the potential to rapidly respond to innate activation.
A subsequent exposure to allergen could provide this innate activation. We propose that allergen-associated molecular patterns (AAMPS) (Dasgupta and Keegan 2012) stimulate PRR expressed on long-lived AAM leading to the rapid production of IL-4, IL-13, IL-25, IL-33 and TSLP and chemokines. Lung epithelial cells could also contribute to the production of a subset of these cytokines including IL-25, IL-33 and TSLP. These cytokines can activate DCs and other innate immune cells (step 4). Activated DCs migrate to the draining lymph node, and with the help of IL-4-producing cell such as basophils, stimulate naive CD4+ T-cells to become allergen-specific Th2 effectors or memory cells (step 5). Subsequently, Th2 effectors migrate to the site of inflammation producing high levels of IL-4, IL-5 and IL-13. These cytokines mediate mucus secretion, AHR, chemokine production, eosinophilic infiltration and further AAM differentiation (step 6). Lung epithelial cells and AAM produce chemokines and other factors that could recruit more inflammatory cells from the blood including eosinophils and monocytes. Further exposure to allergen or infection with virus would reactivate this cycle stimulating memory Th2 cells to produce high levels of IL-4, IL-5 and IL-13 (Guo et al.2015), thereby establishing a positive amplification loop. If this model turns out to be accurate, then new avenues for therapeutic intervention can be identified and explored.
Supplementary Material
SUPPLEMENTARY DATA
FUNDING
This work was supported by the University of Maryland School of Medicine Dean’s Challenge Award to Accelerate Innovation and Discovery in Medicine. The project was supported by Award Number I01BX0070080 (ADK) from the Biomedical Laboratory Research and Development Service of the Veteran’s Administration Office of Research and Development and the United States Public Health Services; and the National Institutes of Health [AI18797 to SNV, and AI104541 to SNV and JB].
Conflict of interest. None declared.
REFERENCES
- Akinbami LJ, Moorman JE, Garbe PL, et al. Status of childhood asthma in the United States, 1980-2007. Pediatrics. 2009;123:S131–45. doi: 10.1542/peds.2008-2233C. [DOI] [PubMed] [Google Scholar]
- Akinbami LJ, Schoendorf KC. Trends in childhood asthma: prevalence, health care utilization, and mortality. Pediatrics. 2002;110:315–22. doi: 10.1542/peds.110.2.315. [DOI] [PubMed] [Google Scholar]
- Al-Garawi A, Fattouh R, Botelho F, et al. Influenza A facilitates sensitization to house dust mite in infant mice leading to an asthma phenotype in adulthood. Mucosal Immunol. 2011;4:682–94. doi: 10.1038/mi.2011.35. [DOI] [PubMed] [Google Scholar]
- Anthony RM, Rutitzky LI, Urban JF, Jr, et al. Protective immune mechanisms in helminth infection. Nat Rev Immunol. 2007;7:975–87. doi: 10.1038/nri2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball TM, Castro-Rodriguez JA, Griffith KA, et al. Siblings, day-care attendance, and the risk of asthma and wheezing during childhood. New Engl J Med. 2000;343:538–43. doi: 10.1056/NEJM200008243430803. [DOI] [PubMed] [Google Scholar]
- Bartlett NW, Walton RP, Edwards MR, et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat Med. 2008;14:199–204. doi: 10.1038/nm1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley R, Semprini A, Mitchell EA. Risk factors for asthma: Is prevention possible? Lancet. 2015;386:1075–85. doi: 10.1016/S0140-6736(15)00156-7. [DOI] [PubMed] [Google Scholar]
- Bierbaum S, Nickel R, Koch A, et al. Polymorphisms and haplotypes of acid mammalian chitinase are associated with bronchial asthma. Am J Resp Crit Care. 2005;172:1505–9. doi: 10.1164/rccm.200506-890OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco JC, Core S, Pletneva LM, et al. Prophylactic antibody treatment and intramuscular immunization reduce infectious human rhinovirus 16 load in the lower respiratory tract of challenged cotton rats. Trials Vaccinol. 2014;3:52–60. doi: 10.1016/j.trivac.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthwick LA, Barron L, Hart KM, et al. Macrophages are critical to the maintenance of IL-13-dependent lung inflammation and fibrosis. Mucosal Immunol. 2016;9:38–55. doi: 10.1038/mi.2015.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliskan M, Bochkov YA, Kreiner-Moller E, et al. Rhinovirus wheezing illness and genetic risk of childhood-onset asthma. New Engl J Med. 2013;368:1398–407. doi: 10.1056/NEJMoa1211592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YJ, Kim HY, Albacker LA, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12:631–8. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WH, Toapanta FR, Shirey KA, et al. Potential role for alternatively activated macrophages in the secondary bacterial infection during recovery from influenza. Immunol Lett. 2012;141:227–34. doi: 10.1016/j.imlet.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chupp GL, Lee CG, Jarjour N, et al. A chitinase-like protein in the lung and circulation of patients with severe asthma. New Engl J Med. 2007;357:2016–27. doi: 10.1056/NEJMoa073600. [DOI] [PubMed] [Google Scholar]
- Cormier SA, You D, Honnegowda S. The use of a neonatal mouse model to study respiratory syncytial virus infections. Expert Rev Anti-Infe. 2010;8:1371–80. doi: 10.1586/eri.10.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta P, Keegan AD. Contribution of alternatively activated macrophages to allergic lung inflammation: a tale of mice and men. J Innate Immun. 2012;4:478–88. doi: 10.1159/000336025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didierlaurent A, Goulding J, Patel S, et al. Sustained desensitization to bacterial toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. 2008;205:323–9. doi: 10.1084/jem.20070891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder W, Klimecki W, Yu L, et al. Opposite effects of CD 14/-260 on serum IgE levels in children raised in different environments. J Allergy Clin Immun. 2005;116:601–7. doi: 10.1016/j.jaci.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Edwards MR, Bartlett NW, Hussell T, et al. The microbiology of asthma. Nat Rev Microbiol. 2012;10:459–71. doi: 10.1038/nrmicro2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ege MJ, Mayer M, Normand AC, et al. Exposure to environmental microorganisms and childhood asthma. New Engl J Med. 2011;364:701–9. doi: 10.1056/NEJMoa1007302. [DOI] [PubMed] [Google Scholar]
- Fassl BA, Nkoy FL, Stone BL, et al. The joint commission children's asthma care quality measures and asthma readmissions. Pediatrics. 2012;130:482–91. doi: 10.1542/peds.2011-3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feola DJ, Garvy BA, Cory TJ, et al. Azithromycin alters macrophage phenotype and pulmonary compartmentalization during lung infection with pseudomonas. Antimicrob Agents Ch. 2010;54:2437–47. doi: 10.1128/AAC.01424-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelman FD, Madden KB, Morris SC, et al. Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine-anti-cytokine antibody complexes. J Immunol. 1993;151:1235–44. [PubMed] [Google Scholar]
- Ford AQ, Dasgupta P, Mikhailenko I, et al. Adoptive transfer of IL-4Ralpha+ macrophages is sufficient to enhance eosinophilic inflammation in a mouse model of allergic lung inflammation. BMC Immunol. 2012;13 doi: 10.1186/1471-2172-13-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Garcia ML, Calvo C, Casas I, et al. Human metapneumovirus bronchiolitis in infancy is an important risk factor for asthma at age 5. Pediatr Pulm. 2007;42:458–64. doi: 10.1002/ppul.20597. [DOI] [PubMed] [Google Scholar]
- Gern JE, Brooks GD, Meyer P, et al. Bidirectional interactions between viral respiratory illnesses and cytokine responses in the first year of life. J Allergy Clin Immun. 2006;117:72–8. doi: 10.1016/j.jaci.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Gregory LG, Lloyd CM. Orchestrating house dust mite-associated allergy in the lung. Trends Immunol. 2011;32:402–11. doi: 10.1016/j.it.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Huang Y, Chen X, et al. Innate immunological functions of TH2 cells in vivo. Nat Immunol. 2015;16:1051–9. doi: 10.1038/ni.3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori T, Konno S, Hizawa N, et al. Genetic variants in the mannose receptor gene (MRC1) are associated with asthma in two independent populations. Immunogenetics. 2009;61:731–8. doi: 10.1007/s00251-009-0403-x. [DOI] [PubMed] [Google Scholar]
- Heller NM, Qi X, Junttila IS, et al. Type I IL-4Rs selectively activate IRS-2 to induce target gene expression in macrophages. Sci Signal. 2008;1:ra17. doi: 10.1126/scisignal.1164795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holgate ST. Rhinoviruses in the pathogenesis of asthma: the bronchial epithelium as a major disease target. J Allergy Clin Immun. 2006;118:587–90. doi: 10.1016/j.jaci.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Holgate ST. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol Rev. 2011;242:205–19. doi: 10.1111/j.1600-065X.2011.01030.x. [DOI] [PubMed] [Google Scholar]
- Hong JY, Chung Y, Steenrod J, et al. Macrophage activation state determines the response to rhinovirus infection in a mouse model of allergic asthma. Respir Res. 2014;15:63. doi: 10.1186/1465-9921-15-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kuba K, Neely GG, et al. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–49. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwane MK, Prill MM, Lu X, et al. Human rhinovirus species associated with hospitalizations for acute respiratory illness in young US children. J Infect Dis. 2011;204:1702–10. doi: 10.1093/infdis/jir634. [DOI] [PubMed] [Google Scholar]
- Jackson DJ, Lemanske RF., Jr The role of respiratory virus infections in childhood asthma inception. Immunol Allergy Clin. 2010;30:513–22. doi: 10.1016/j.iac.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SL, Pattemore PK, Sanderson G, et al. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. Brit Med J. 1995;310:1225–9. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiko GE, Phipps S, Angkasekwinai P, et al. NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J Immunol. 2010;185:4681–90. doi: 10.4049/jimmunol.1001758. [DOI] [PubMed] [Google Scholar]
- Kalil AC, LaRosa SP, Gogate J, et al. Influence of severity of illness on the effects of eritoran tetrasodium (E5564) and on other therapies for severe sepsis. Shock. 2011;36:327–31. doi: 10.1097/SHK.0b013e318227980e. [DOI] [PubMed] [Google Scholar]
- Kelly-Welch AE, Melo ME, Smith E, et al. Complex role of the IL-4 receptor alpha in a murine model of airway inflammation: expression of the IL-4 receptor alpha on nonlymphoid cells of bone marrow origin contributes to severity of inflammation. J Immunol. 2004;172:4545–55. doi: 10.4049/jimmunol.172.7.4545. [DOI] [PubMed] [Google Scholar]
- Kim EY, Battaile JT, Patel AC, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. 2008;14:633–40. doi: 10.1038/nm1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, Park BS, Kim JI, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist eritoran. Cell. 2007;130:906–17. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Klugman KP, Chien YW, Madhi SA. Pneumococcal pneumonia and influenza: a deadly combination. Vaccine. 2009;27:C9–14. doi: 10.1016/j.vaccine.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31:412–24. doi: 10.1016/j.immuni.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Lasso-Pirot A, Delgado-Villalta S, Spanier AJ. Early childhood wheezers: identifying asthma in later life. J Asthma Allergy. 2015;8:63–73. doi: 10.2147/JAA.S70066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CG, Hartl D, Lee GR, et al. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J Exp Med. 2009;206:1149–66. doi: 10.1084/jem.20081271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malur A, Mccoy AJ, Arce S, et al. Deletion of PPAR gamma in alveolar macrophages is associated with a th-1 pulmonary inflammatory response. J Immunol. 2009;182:5816–22. doi: 10.4049/jimmunol.0803504. [DOI] [PubMed] [Google Scholar]
- Martinez FD. CD14, endotoxin, and asthma risk: actions and interactions. P Am Thorac Soc. 2007;4:221–5. doi: 10.1513/pats.200702-035AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- Melgert BN, Oriss TB, Qi Z, et al. Macrophages: regulators of sex differences in asthma? Am J Resp Cell Mol. 2010;42:595–603. doi: 10.1165/rcmb.2009-0016OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melgert BN, ten Hacken NH, Rutgers B, et al. More alternative activation of macrophages in lungs of asthmatic patients. J Allergy Clin Immun. 2011;127:831–3. doi: 10.1016/j.jaci.2010.10.045. [DOI] [PubMed] [Google Scholar]
- Minnicozzi M, Sawyer RT, Fenton MJ. Innate immunity in allergic disease. Immunol Rev. 2011;242:106–27. doi: 10.1111/j.1600-065X.2011.01025.x. [DOI] [PubMed] [Google Scholar]
- Moller DE. New drug targets for type 2 diabetes and the metabolic syndrome. Nature. 2001;414:821–7. doi: 10.1038/414821a. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullarkey M, Rose JR, Bristol J, et al. Inhibition of endotoxin response by e5564, a novel toll-like receptor 4-directed endotoxin antagonist. J Pharmacol Exp Ther. 2003;304:1093–102. doi: 10.1124/jpet.102.044487. [DOI] [PubMed] [Google Scholar]
- Murphy BS, Sundareshan V, Cory TJ, et al. Azithromycin alters macrophage phenotype. J Antimicrob Chemoth. 2008;61:554–60. doi: 10.1093/jac/dkn007. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarkar DR, Bowman ER, Schneider D, et al. Rhinovirus infection of allergen-sensitized and -challenged mice induces eotaxin release from functionally polarized macrophages. J Immunol. 2010;185:2525–35. doi: 10.4049/jimmunol.1000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Latz E, Mills KH, et al. Innate immune memory: a paradigm shift in understanding host defense. Nat Immunol. 2015;16:675–9. doi: 10.1038/ni.3178. [DOI] [PubMed] [Google Scholar]
- Nhu QM, Shirey K, Teijaro JR, et al. Novel signaling interactions between proteinase-activated receptor 2 and toll-like receptors in vitro and in vivo. Mucosal Immunol. 2010;3:29–39. doi: 10.1038/mi.2009.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuizen NE, Kirstein F, Jayakumar J, et al. Allergic airway disease is unaffected by the absence of IL-4Ralpha-dependent alternatively activated macrophages. J Allergy Clin Immun. 2012;130:743–50. doi: 10.1016/j.jaci.2012.03.011. [DOI] [PubMed] [Google Scholar]
- Ober C, Tan Z, Sun Y, et al. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. New Engl J Med. 2008;358:1682–91. doi: 10.1056/NEJMoa0708801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribul PK, Harker J, Wang B, et al. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J Virol. 2008;82:4441–8. doi: 10.1128/JVI.02541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakes GP, Arruda E, Ingram JM, et al. Rhinovirus and respiratory syncytial virus in wheezing children requiring emergency care. IgE and eosinophil analyses. Am J Resp Crit Care. 1999;159:785–90. doi: 10.1164/ajrccm.159.3.9801052. [DOI] [PubMed] [Google Scholar]
- Richardson JY, Ottolini MG, Pletneva L, et al. Respiratory syncytial virus (RSV) infection induces cyclooxygenase 2: a potential target for RSV therapy. J Immunol. 2005;174:4356–64. doi: 10.4049/jimmunol.174.7.4356. [DOI] [PubMed] [Google Scholar]
- Rynda-Apple A, Robinson KM, Alcorn JF. Influenza and bacterial superinfection: illuminating the immunologic mechanisms of disease. Infect Immun. 2015;83:3764–70. doi: 10.1128/IAI.00298-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz SA, Noti M, Artis D. Innate immune cell populations function as initiators and effectors in Th2 cytokine responses. Trends Immunol. 2010;31:407–13. doi: 10.1016/j.it.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Schleimer RP, Kato A, Kern R, et al. Epithelium: at the interface of innate and adaptive immune responses. J Allergy Clin Immun. 2007;120:1279–84. doi: 10.1016/j.jaci.2007.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider D, Hong JY, Popova AP, et al. Neonatal rhinovirus infection induces mucous metaplasia and airways hyperresponsiveness. J Immunol. 2012;188:2894–904. doi: 10.4049/jimmunol.1101391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze J, Hamelmann E, Bradley KL, et al. Respiratory syncytial virus infection results in airway hyperresponsiveness and enhanced airway sensitization to allergen. J Clin Invest. 1997;100:226–33. doi: 10.1172/JCI119516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemungal TA, Harper-Owen R, Bhowmik A, et al. Detection of rhinovirus in induced sputum at exacerbation of chronic obstructive pulmonary disease. Eur Respir J. 2000;16:677–83. doi: 10.1034/j.1399-3003.2000.16d19.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey KA, Lai W, Patel MC, et al. Novel strategies for targeting innate immune responses to influenza. Mucosal Immunol. 2016 doi: 10.1038/mi.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey KA, Lai W, Pletneva LM, et al. Agents that increase AAM differentiation blunt RSV-mediated lung pathology. J Leukocyte Biol. 2014a;96:951–5. doi: 10.1189/jlb.4HI0414-226R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey KA, Lai W, Pletneva LM, et al. Role of the lipoxygenase pathway in RSV-induced alternatively activated macrophages leading to resolution of lung pathology. Mucosal Immunol. 2014b;7:549–57. doi: 10.1038/mi.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey KA, Lai W, Scott AJ, et al. The TLR4 antagonist eritoran protects mice from lethal influenza infection. Nature. 2013;497:498–502. doi: 10.1038/nature12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey KA, Pletneva LM, Puche AC, et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol. 2010;3:291–300. doi: 10.1038/mi.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurs N, Bjarnason R, Sigurbergsson F. Eosinophil cationic protein in nasal secretion and in serum and myeloperoxidase in serum in respiratory syncytial virus bronchiolitis: relation to asthma and atopy. Acta Paediatr. 1994;83:1151–5. doi: 10.1111/j.1651-2227.1994.tb18269.x. [DOI] [PubMed] [Google Scholar]
- Sigurs N, Bjarnason R, Sigurbergsson F, et al. Asthma and immunoglobulin E antibodies after respiratory syncytial virus bronchiolitis: a prospective cohort study with matched controls. Pediatrics. 1995;95:500–5. [PubMed] [Google Scholar]
- Sigurs N, Gustafsson PM, Bjarnason R, et al. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Resp Crit Care. 2005;171:137–41. doi: 10.1164/rccm.200406-730OC. [DOI] [PubMed] [Google Scholar]
- Stein RT, Sherrill D, Morgan WJ, et al. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet. 1999;354:541–5. doi: 10.1016/S0140-6736(98)10321-5. [DOI] [PubMed] [Google Scholar]
- Subrata LS, Bizzintino J, Mamessier E, et al. Interactions between innate antiviral and atopic immunoinflammatory pathways precipitate and sustain asthma exacerbations in children. J Immunol. 2009;183:2793–800. doi: 10.4049/jimmunol.0900695. [DOI] [PubMed] [Google Scholar]
- Vannella KM, Barron L, Borthwick LA, et al. Incomplete deletion of IL-4Ralpha by LysM(cre) reveals distinct subsets of M2 macrophages controlling inflammation and fibrosis in chronic schistosomiasis. PLoS Pathog. 2014;10:e1004372. doi: 10.1371/journal.ppat.1004372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercelli D. Discovering susceptibility genes for asthma and allergy. Nat Rev Immunol. 2008;8:169–82. doi: 10.1038/nri2257. [DOI] [PubMed] [Google Scholar]
- Viksman MY, Bochner BS, Peebles RS, et al. Expression of activation markers on alveolar macrophages in allergic asthmatics after endobronchial or whole-lung allergen challenge. Clin Immunol. 2002;104:77–85. doi: 10.1006/clim.2002.5233. [DOI] [PubMed] [Google Scholar]
- Weiss KB, Sullivan SD, Lyttle CS. Trends in the cost of illness for asthma in the united states, 1985–1994. J Allergy Clin Immun. 2000;106:493–9. doi: 10.1067/mai.2000.109426. [DOI] [PubMed] [Google Scholar]
- Welch JS, Escoubet-Lozach L, Sykes DB, et al. TH2 cytokines and allergic challenge induce Ym1 expression in macrophages by a STAT6-dependent mechanism. J Biol Chem. 2002;277:42821–9. doi: 10.1074/jbc.M205873200. [DOI] [PubMed] [Google Scholar]
- Xavier MN, Winter MG, Spees AM, et al. PPARgamma-mediated increase in glucose availability sustains chronic brucella abortus infection in alternatively activated macrophages. Cell Host Microbe. 2013;14:159–70. doi: 10.1016/j.chom.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang IA, Savarimuthu S, Kim ST, et al. Gene-environmental interaction in asthma. Curr Opin Allergy Cl. 2007;7:75–82. doi: 10.1097/ACI.0b013e328012ce39. [DOI] [PubMed] [Google Scholar]
- You D, Becnel D, Wang K, et al. Exposure of neonates to respiratory syncytial virus is critical in determining subsequent airway response in adults. Respir Res. 2006;7:107. doi: 10.1186/1465-9921-7-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.